

Background: There are seven histone H1 variants in somatic mammalian cells, two of which are replication-independent, H1.0 and H1X.
Results: In breast cancer cells, H1.0 is enriched at nucleolus-associated domains, whereas H1X is associated with RNA polymerase II-enriched regions.
Conclusion: Most H1 variants show great redundancy across the genome, but there is also some specificity.
Significance: Some H1 variants may have specific functions.
Keywords: Chromatin, Epigenetics, Histone, Nucleolus, RNA Polymerase II, H1.0, H1X, Histone H1, Variants
Abstract
Unlike core histones, the linker histone H1 family is more evolutionarily diverse, and many organisms have multiple H1 variants or subtypes. In mammals, the H1 family includes seven somatic H1 variants; H1.1 to H1.5 are expressed in a replication-dependent manner, whereas H1.0 and H1X are replication-independent. Using ChIP-sequencing data and cell fractionation, we have compared the genomic distribution of H1.0 and H1X in human breast cancer cells, in which we previously observed differential distribution of H1.2 compared with the other subtypes. We have found H1.0 to be enriched at nucleolus-associated DNA repeats and chromatin domains, whereas H1X is associated with coding regions, RNA polymerase II-enriched regions, and hypomethylated CpG islands. Further, H1X accumulates within constitutive or included exons and retained introns and toward the 3′ end of expressed genes. Inducible H1X knockdown does not affect cell proliferation but dysregulates a subset of genes related to cell movement and transport. In H1X-depleted cells, the promoters of up-regulated genes are not occupied specifically by this variant, have a lower than average H1 content, and, unexpectedly, do not form an H1 valley upon induction. We conclude that H1 variants are not distributed evenly across the genome and may participate with some specificity in chromatin domain organization or gene regulation.
Introduction
There are five major classes of histones that participate in the correct folding of eukaryotic DNA into chromatin: the core histones H2A, H2B, H3, and H4, which form an octamer and constitute the nucleosome core particle, and the linker histone H1, which binds to the nucleosomes near the entry/exit sites of linker DNA. Stabilization of the condensed states of chromatin is the function most commonly attributed to the linker histone (1, 2), in addition to its inhibitory effect in vitro on nucleosome mobility (3) and transcription (4).
Histone H1 in humans is a family of closely related, single gene-encoded proteins, including seven somatic subtypes (H1.1 to H1.5, H1.0, and H1X), three testis-specific variants (H1t, H1T2, and HILS1), and one restricted to oocytes (H1oo) (5, 6). Among the somatic histone H1 variants, H1.1 to H1.5 are expressed in a replication-dependent manner, whereas H1.0 and H1X are replication-independent. The H1.1 to H1.5-encoding genes are clustered in a region of chromosome 6 together with the core histone genes, whereas the H1X and H1.0 genes are on chromosomes 3 and 22, respectively. H1.2 to H1.5 and H1X are ubiquitously expressed, H1.1 is restricted to certain tissues, and H1.0 accumulates in terminally differentiated cells. There are few studies characterizing the most recently identified and distantly related human variant, H1X, and its specific function in the cell remains unknown. Like H1.0, it has been suggested that H1X is enriched in a less accessible region of chromatin, but expression of the two variants is regulated differently (7). It has been shown previously that H1X accumulates in nucleoli in G1 and is distributed across the entire nucleus in the S phase (8). The same year, Takata et al. (9) found that H1X was preferentially located at the chromosome periphery in mitosis, and they observed defects in chromosome alignment and segregation after H1X knockdown (KD).5 Taken together, these findings indicate that H1X may have functions that differ from those of the other variants.
Because it participates in the formation of higher order chromatin structures, H1 is seen as a structural component related to chromatin compaction and inaccessibility to transcription factors and to RNA polymerase. Nonetheless, it has also been suggested that histone H1 plays a more dynamic and gene-specific role, participating in the regulation of gene expression. Previous studies on the effect of H1 depletion on global gene expression have found no effect on the vast majority of genes but rather have detected up- or down-regulation of small groups of genes (10–13). It is not clear whether the different variants have specific roles or regulate specific promoters. In mice, single or double H1 variant knockouts have no apparent phenotype due to compensatory up-regulation of other subtypes (14). These reports have favored the view that H1 variants are redundant.
On the other hand, we reported that depletion of single H1 subtypes by inducible RNA interference in breast cancer cells produced a range of phenotypic effects (10), suggesting different functions for the various H1 variants in somatic cells. Furthermore, H1 subtypes can be post-translationally modified, and these modifications modulate their interaction with various other proteins. This could explain some reported specific functions for certain H1 variants (15–24). Moreover, H1 subtypes have cell type- and tissue-specific expression patterns, and their expression is regulated over the course of differentiation and development (25–30). Different H1 subtypes have also been differentially related to cancer processes (31–34).
To fully understand the function of histone H1 and its variants, several studies have explored the genomic distribution of H1 in vivo. Initial biochemical and microscopy-based approaches suggested a non-uniform distribution of H1 in the cell nucleus and found differences between variants (35–37). However, due to the lack of specific ChIP-grade antibodies for most H1 variants, it has been challenging to identify the precise mapping of H1 variants in the genome until recently. Two reports, using ChIP of tagged H1 variants in mouse embryonic stem cells and DamID technology in human IMR90 cells, respectively, showed depletion of H1c and H1d from guanine-cytosine (GC)- and gene-rich regions as well as an overrepresentation in major satellites (38) and depletion of H1.2 to H1.5 from CpG-dense and regulatory regions, only H1.1 having a distinct profile (39). Moreover, it has previously been shown that when a gene is transcriptionally active, there is depletion of H1 (an H1 valley) at the TSS of its promoter (40).
Using variant-specific antibodies against H1 and hemagglutinin (HA)-tagged recombinant H1 variants expressed in breast cancer cells, we investigated the distribution of six H1 variants in promoters (ChIP-chip) and genome-wide (ChIP-seq), including H1.0 and H1X, for the first time (41). In short, we reported that histone H1 is not uniformly distributed across the genome, and there are differences between variants, H1.2 showing the most specific pattern and strongest correlation with low gene expression. H1.2 is enriched at chromosomal domains with low GC content and is associated with gene-poor chromosomes, intergenic DNA, and lamina-associated domains (LADs). Meanwhile, other variants are associated with higher GC content, CpG islands, and gene-rich domains. Overall, the distribution of H1.2 along chromosomes differed from that of other variants, including H1.0 and H1X, the two variants most structurally distant within the somatic H1 family.
In this new work, we have further analyzed the distribution of H1 variants in other genomic regions, including repetitive DNA, nucleolus-associated chromatin domains (NADs), and ribosomal DNA (rDNA), and their association with methylated CpG sites and RNA polymerase II-enriched regions. This analysis has revealed that H1.0 and H1X are enriched at particular regions compared with the other variants. H1.0 is the variant that is most abundant at NADs, rDNA, and certain satellite repeats related to nucleolus organizer regions. The association of H1.0 with nucleolar chromatin has been confirmed by immunoblotting on fractionated cellular extracts. In contrast, H1X is associated with RNA polymerase II-enriched sites, coding regions, and hypomethylated CpG islands. Notably, the H1X content at coding regions is higher at active genes, especially toward the 3′ end of genes, and more abundant at exons and intron-exon junctions than within introns themselves. We have also further investigated the functionality of H1X by testing the effect of an inducible KD of this H1 variant on cell proliferation and global gene expression.
EXPERIMENTAL PROCEDURES
Cell Lines and Culture Conditions
T47D-MTVL (carrying one stably integrated copy of luciferase reporter gene driven by the murine mammary tumor virus promoter) (42) and MCF7 breast cancer cells were separately grown at 37 °C with 5% CO2. T47D-derivative cells were grown in RPMI 1640 medium, supplemented with 10% FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. MCF7 cells were grown in minimum Eagle's medium containing 10% FBS, 1% penicillin/streptomycin, 1% glutamine, and 1% sodium pyruvate. Doxycycline (Sigma) was added at 2.5 μg/ml when required.
Drug-inducible RNA Interference
H1X KD cell lines were established from T47D-MTVL and MCF7 breast cancer cells. Plasmids for the lentivirus vector-mediated drug-inducible RNA interference system (pLVTHM, ptTR-KRAB-Red, pCMC-R8.91, and pVSVG) were provided by Dr. D. Trono (University of Geneva) (58). After testing five shRNAs against H1X from the MISSION library (Sigma-Aldrich), the 21-mer H1X-specific target sequence 5′-CAACGGTTCCTTCAAGCTCAA-3′ was chosen to generate the inducible system. The 71-mer oligonucleotides for shRNA cloning into Mlu/ClaI-digested pLVTHM were designed, annealed, and phosphorylated as recommended by Dr. Trono (see the Tronolab Web site). For the production of viral particles containing the lentiviral vector and infections, see Sancho et al. (10). The inducible knocked down cell lines were sorted in a FACSCalibur machine (BD Biosciences) for RedFP-positive and GFP-positive fluorescence after 3 days of doxycycline treatment. Then, cells were amplified in the absence of doxycycline until an experiment was performed. Over a 6-day treatment with doxycycline, cells were passaged on day 3. When required, serum-containing medium was replaced with serum-free medium on day 4 to arrest growth.
Histone H1 Extraction, Gel Electrophoresis, and Immunoblotting
Histone H1 was purified by lysis with 5% perchloric acid for 1 h at 4 °C. Soluble acid proteins were precipitated with 30% trichloroacetic acid overnight at 4 °C, washed twice with 0.5 ml of acetone, and reconstituted in water. Protein concentration was determined with the Micro BCA protein assay (Pierce). Purified histones were exposed to SDS-PAGE (10%), transferred to a PVDF membrane, blocked with Odyssey blocking buffer (LI-COR Biosciences) for 1 h, and incubated with primary antibodies overnight at 4 °C and with secondary antibodies conjugated to fluorescence (IRDye 680 goat anti-rabbit IgG, LI-COR) for 1 h at room temperature. Bands were visualized in an Odyssey infrared imaging system (LI-COR). Polyclonal antibodies specifically recognizing human H1 variants, including those generated in our laboratory (10), are available from Abcam: H1.0 (ab11079), H1.2 (ab17677), H1.3 (ab24174), H1.4-T146p (ab3596), H1.5 (ab24175), and rabbit antiH1X (ab31972). Mouse anti-H1X was obtained from Sigma (SAB1400328). Other antibodies used were β-tubulin (Sigma, nrT4026), nucleophosmin (Abcam, ab15440), nucleolin (Abcam, ab22758), H3K4me3 (Millipore, 07-473), and H3K9me3 (Abcam, ab8898).
Cell Fractionation for Purification of Nucleoli
Cell fractionation was performed as described by Andersen et al. (43). Briefly, 30 million cells were resuspended in 1 ml of Buffer A (10 mm HEPES-KOH, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT, and protease inhibitors: 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 0.1 units/ml aprotinin, 1 mm orthovanadate, and 50 mm NaF) and incubated for 10 min on ice. Then the cell pellet was homogenized, by passing the cell suspension through a 23-gauge needle 15 times and through a 25-gauge needle 10 times. From this, we collected the total protein fraction. The homogenized suspension was pelleted at 228 × g for 5 min at 4 °C, and the supernatant was taken as the cytoplasmic fraction. The remaining pellet was resuspended in Buffer B (0.25 m sucrose, 10 mm MgCl2, and protease inhibitors) and was homogenized again by passing the suspension through a 23-gauge needle 10 times. Then it was centrifuged at 1,430 × g for 5 min at 4 °C on a sucrose cushion (Buffer C: 0.35 m sucrose, 0.5 mm MgCl2, and protease inhibitors). The remaining pellet was resuspended with Buffer C and sonicated for six cycles of 10 s on ice. The sonicated sample was centrifuged at 2,800 × g for 10 min at 4 °C on a sucrose pillow (Buffer D: 0.88 m sucrose, 0.5 mm MgCl2, and protease inhibitors). The supernatant was collected as the nucleoplasm fraction. The nucleoli pellet was washed with Buffer C and centrifuged at 200 × g for 2 min at 4 °C. Then it was resuspended with lysis buffer (SDS (2%), 67 mm Tris-HCl, pH 6.8). Protein concentration in all fractions was determined with the Micro BCA protein assay kit (Pierce). Fractionated extracts were exposed to SDS-PAGE (10%), transferred to a PVDF membrane, and immunoblotted as described above. Immunoblot band intensities were measured using ImageJ (version 1.48) software and normalized by Coomassie staining.
Immunostaining
Cells were grown over coverslips, washed twice with PBS, and fixed with 4% formaldehyde for 15 min at room temperature. After three washes, they were permeabilized with Triton X-100 for 15 min at room temperature and blocked with bovine serum albumin for 1 h. Then the cells were incubated with primary antibodies diluted with bovine serum albumin for 1 h at room temperature in darkness. After the pertinent washes, the secondary antibodies Alexa-555 and Alexa-647 were added for 1 h at room temperature in darkness. The nucleus was stained with DAPI. The coverslips were mounted on the glass slides using Mowiol mounting medium. The samples were visualized by confocal laser scanning microscopy using a Leica TCS SPE system.
Cell Cycle Analysis
Cells were washed with cold 1× PBS, fixed in 70% ethanol, and stained with analysis solution: 3% ribonuclease A (Sigma) (10 mg/ml) and 3% solution A (38 mm sodium citrate, 500 μg/ml propidium iodide) in 1× PBS. Samples were analyzed with a FACSCalibur machine, using CellQuest Pro Analysis software (both from BD Biosciences) and ModFit LT software (Verity Software House).
Chromatin Immunoprecipitation
Immunoprecipitation of chromatin was performed according to the Upstate (Millipore) standard protocol. Briefly, cells were fixed using 1% formaldehyde for 10 min at 37 °C, harvested, and sonicated to generate chromatin fragments of 200–500 bp. Then 20 μg of sheared chromatin was immunoprecipitated overnight with 2 μg of antibody. Immunocomplexes were recovered using 20 μl of protein A magnetic beads, washed, and eluted. Cross-linking was reversed at 65 °C overnight, and immunoprecipitated DNA was recovered using the PCR purification kit from Qiagen. Genomic regions of interest were identified by real-time quantitative PCR (qPCR) using SYBR Green Master Mix (Invitrogen) and specific oligonucleotides in a Roche Applied Science 480 light cycler machine. Each value was corrected by the corresponding input chromatin sample. Oligonucleotide sequences used for the amplifications are shown in Table 1.
TABLE 1.
Primer sets used for RT-PCR and ChIP-qPCR
| Name of gene | Forward primer (5′–3′) | Backward primer (5′–3′) |
|---|---|---|
| ChIP-qPCR | ||
| KNG1 TSS | TCCCAGTTGGCTCTTGATTC | TTTCCTCGGACTGTGATTCC |
| KNG1 −3 kb | GTGCAGGATGGGTGATTTTT | CCTGTGCTTCAACACCATTC |
| KRT37 TSS | AATCAAGGCAGGAGGTCAAA | CTTCAGATCAGCTGGGAAGG |
| KRT37 −3 kb | GGCACTTGTAGTGACCTGGAT | CCGAAGTCCTCAAAGTCCAT |
| UGT2B10 TSS | AAGGATGGCTCTGAAATGGA | CTGTATTCTGCGGCCCATAC |
| UGT2B10 −3 kb | GGCATTGGATATTGGCTGTC | TCACCCAGATTTCCCTTTTG |
| AMTN TSS | CGTGGACCCAAAGGTAACAT | TGTTGAAACTGGCTGGCATA |
| AMTN −3 kb | TGACATGTGCATTCAATCAGC | GCCCTTTAGTTCCAGGCATT |
| SPINK9 TSS | CGGACACCAGGTCACTTCTT | TTGCAAGTGTCAGAGCCAAG |
| SPINK9 −3 kb | TCAAGTTCACCAGGCTTTTTG | CCTCTATGATGAGTCCAGCTC |
| ALOX15B TSS | TAACCAGGGGCAATAACCAG | CCACGATGCTGACAGACACT |
| ALOX15B −3 kb | TTGAAAACGTGTGGGTCTTG | CACCTTTGGAGCAATGTCTG |
| CDK2 TSS | GCGGCAACATTGTTTCAAGT | GTCGGGATGGAACGCAGTAT |
| CDK2 −3 kb | CAGCGAGGAAAGTCACATCA | TGGGGTGAGGGTAGTTTCTG |
| FOXB2 TSS | GTGCGGAGAGATTCTGTGGT | AGAGTAGGGCGGTTTTTGGT |
| FOXB2 −3 kb | CTGTAGCGAGCTCACCCAGT | ACAAATCTTGGGCGCATAAC |
| TBKBP1 TSS | AGGCCCGAGAGAAGTACACA | CGAAAGCAGGAGTAGGCAGT |
| TBKBP1 −3 kb | TGCAATGAGATCAGGTCCAG | GTTGGTGGCAAAAGTCCATT |
| ACTL7B TSS | AGGTGGGGGATCTCATTTCT | CTTGCTCCCCTTCTCACATC |
| ACTL7B −3 kb | GGTCCCAAGACTGTGTCCAT | AGACAGCTCCTCTCCCTTCC |
| JUN TSS | GGGTGACATCATGGGCTATT | GCCCGAGCTCAACACTTATC |
| JUN −10 kb | CCTTTTTGTCCCTCCAAACA | TCTAGGAACTGAGCCCTCCA |
| RT-PCR | ||
| KNG1 | GTGGTGGCTGGATTGAACTT | CGCAAATCTTGGTSGGTGGT |
| KRT37 | TGGGGAGATGATTCTGAAGG | TGCTACCGGTTGATTTAGGG |
| UGT2B10 | GACCTGCTGAATGCACTGAA | ACTGGAACCAGGTGAGGTTG |
| AMTN | AGCAGGAGGAGCAGGTGTAA | CCAAATTCGAGGCAGCTTAG |
| ALOX15B | GAAGTGGCTGCCAAAGAGAC | GCTGGCCTTGAACTTCTGAC |
| SPINK9 | GAATGTGCCAAACAGACGAA | GTTTTGCCATCAGATCCACA |
| H1X | TTCCTTCAAGCTCAACCG | TGCCTTCTTCGCTTTGTG |
| H1.0 | CCTGCGGCCAAGCCCAAGCG | AACTTGATCTGCGAGTCAGC |
| H1.1 | CTCCTCTAAGGAGCGTGGTG | GAGGACGCCTTCTTGTTGAG |
| H1.2 | GGCTGGGGGTACGCCT | TTAGGTTTGGTTCCGCCC |
| H1.3 | CTGCTCCACTTGCTCCTACC | GCAAGCGCTTTCTTAAGC |
| H1.4 | GTCGGGTTCCTTCAAACTCA | CTTCTTCGCCTTCTTTGGG |
| H1.5 | CATTAAGCTGGGCCTCAAGA | TCACTGCCTTTTTCGCCCC |
RNA Extraction, Reverse Transcriptase qPCR, and Expression Microarrays
Total RNA was extracted using the High Pure RNA isolation kit (Roche Applied Science). Then cDNA was generated from 100 ng of RNA using the Superscript first strand synthesis system (Invitrogen). Gene products were analyzed by qPCR, again using SYBR Green master mix (Invitrogen) and specific oligonucleotides in a Roche Applied Science 480 light cycler machine. Each value was corrected by human GAPDH and represented as relative units. Each experiment was performed in duplicate. Gene-specific oligonucleotide sequences are shown in Table 1. The procedures for microarray hybridization using an Agilent platform (SurePrint G3 Human Gene Expression 8x60K version 2) and data analysis are described elsewhere (41). Gene ontology analysis was performed using the DAVID software (Database for Annotation, Visualization, and Integrated Discovery).
Analysis of ChIP-seq Data
Because there are a limited number of H1 variant-specific ChIP-grade antibodies (only H1.2 and H1X being available to us), we developed T47D-derived cell lines stably expressing hemagglutinin (HA)-tagged versions of each of the five somatic H1 variants expressed in most cell types (H1.0, H1.2, H1.3, H1.4, and H1.5) (10). Therefore, in addition to using H1.2 and H1X antibodies to pull down these variants in parental T47D cells, an anti-HA antibody was used to specifically pull-down H1-associated chromatin fragments in cells expressing H1-HAs. ChIP-chip and ChIP-seq data on the occurrence of H1 variants at promoters and genome-wide in T47D-derivative cells, respectively, were generated in previous research, and the analysis was reported elsewhere (41). Briefly, ChIP-seq libraries were prepared with the ChIP-seq Sample Preparation Kit (Illumina), and sequencing was performed with the Illumina HiSeq 2000 system. Read mapping and peak detection methods have been described before (41). Other types of analysis used were as follows.
Publicly Available Genome-wide Location Data Analysis
Genomic locations of CpG islands and LADs (in hg18) were taken from the University of California Santa Cruz (UCSC) database (44), genomic locations of NADs from Nemeth et al. (45), and RNA polymerase II (RNAPII) binding sites from Ballaré et al. (46). Further, acromeric satellite 1 (ACRO1) genomic locations (in hg 18) were taken from the UCSC database. Repetitive sequences were taken from RepBase database (47). The mean methylation levels at individual CpG islands were calculated by assessing the overlap between the methylation levels from Vanderkraats et al. (48) and the CpG islands using BedTools (49). The genomic locations of hyper- and hypomethylated regions from the T47D cell line were recalculated as in Ref. 50, the source of the raw data.
RNA-sequencing data from the T47D cell line was taken from Vanderkraats et al. (48). Reads were mapped to the hg18 genome using the TopHat algorithm (version 2.0.12) (51). Next, we extracted the database of “cassette” exons and retained introns included in the MISO software package (52). The inclusion levels (Ψ) of alternatively spliced exons (ASEs) and retained introns were estimated using the MISO algorithm (52) with default parameters. Exons with inclusion level Ψ ≥ 0.9 were considered to be included ASEs, and those with Ψ ≤ 0.1 were considered excluded ASEs. Retained introns with Ψ ≥ 0.9 were considered to be retained in the T47D cell line.
H1 Occupancy at Genomic Features
Input-subtracted normalized average H1 variant read density was calculated at each location enriched in CpG islands, NADs, hyper- and hypomethylated regions, ACRO1, repetitive elements, exons, introns, ASEs, retained introns, and RNAPII and represented in box plots using in-house R scripts. As a control, a random sample of bulk genomic windows with equal width was used to perform the significance test (Kolmogorov-Smirnov test). In addition, for H1 abundance at ACRO1, a second method was used, namely mapping to sequences in RepBase (47), with the bowtie aligner (53) allowing for multiple positions.
ChIP signals around the center of RNAPII binding sites were calculated using normalized input-subtracted average tag numbers in each 50-bp bin in a set window. Relative distances of each tag from the aforementioned positions and average signals were determined using the Sitepro script from the CEAS package (54) and plotting using R. Continuous ChIP signal profile distribution of reads along the metagene, exons, and introns were performed using CEAS (54). Correlation analysis between NAD content and H1.0 abundance on individual chromosomes was performed using in-house R scripts.
H1 Occupancy at Individual Chromosomes
Occupancy of H1 variants at all human chromosomes is measured in terms of the mean of the input-subtracted ChIP-seq signal in 50-bp windows. LAD and NAD occupancy at all chromosomes was calculated as the number of bases coinciding with LADs or NADs divided by the length of the chromosome. Expression on each chromosome is the mean of the expression of all genes in that particular chromosome. Heat maps and dendrograms were created with in-house R scripts.
H1 Occupancy at rDNA
The abundance of H1 variants on rDNA was assessed as described previously (55). In short, because the rDNA sequence is not included in the reference genome, a custom hg18 assembly was constructed with the bowtie-build tool (53), adding a human rDNA repeat (GenBankTM accession number U13369). Alignment was carried out with the bowtie aligner (53), allowing up to two mismatches, and only unique hits were kept. The input-subtracted ChIP-seq signal in the rDNA sequence was calculated in reads/kilobase/million mapped and plotted using in-house R scripts.
Overlap Analysis of H1 Islands
The number of enriched and depleted H1 islands that overlapped NADs and RNAPII binding sites was calculated using BedTools (49) and plotted using in-house R scripts. Box plots showing the methylation levels of CpG islands overlapping enriched and depleted islands were calculated in the same way. Features were considered to overlap if the genomic intervals shared at least one base. Chromatin states based on the combined presence of H1 variant-enriched regions were calculated with a multivariate hidden Markov model using the chromHMM software (56).
Human H1 Variant Nomenclature
The correspondence of the nomenclature of the human H1 variants with their gene names is as follows: H1.0, HIF0; H1.1, HIST1H1A; H1.2, HIST1H1C; H1.3, HIST1H1D; H1.4, HIST1H1E; H1.5, HIST1H1B; and H1X, HIFX.
RESULTS
Human H1 Variants Are Differentially Associated with NADs and Repetitive DNA
To further explore whether the distribution of H1 variants is heterogeneous along several genome features or chromatin domains, we used our previously reported ChIP-seq data on endogenous H1.2, H1X, and H3 and HA-tagged H1.0, H1.2, and H1.4 (41). DNA sequences associated with the human nucleolus have recently been identified and used to define NADs by Németh et al. (45). Different gene families and certain satellite repeats are the major building blocks of NADs, which constitute about 4% of the genome. Using the input-subtracted ChIP-seq signal, we investigated the occupancy of H1 variants within NADs. H1.0 was significantly enriched at NADs (Fig. 1A). Furthermore, H1.0 was the variant that had the largest number of H1-enriched regions overlapping NADs (Fig. 1B).
FIGURE 1.

H1 variant abundance at NADs and across human chromosomes. A, box plots showing the occupancy of H1 variants (input-subtracted ChIP-seq signal) within NADs. Significance was assessed using the Kolmogorov-Smirnov test. Significant enrichment of H1.0 compared with other variants is marked with asterisks (p < 0.001). NAD data determined in HeLa cells were obtained from Németh et al. (45). The labels for the different ChIP-seq data sets are consistent with those used elsewhere (41), H1.2e and H1Xe referring to endogenous H1 variants immunoprecipitated with variant-specific antibodies and H1.2_r2 and H1.2_r3 being two independent ChIP-seq replicates. B, number of H1 variant-enriched regions overlapping with NADs. Areas were considered to show enrichment of H1 variants if there was a -fold change greater than or equal to 2 compared with inputs derived from ChIP-seq data. C, correlation scatter plot between the occupancy of H1.0 at all chromosomes and the NAD content. The table below shows Pearson's correlation coefficient (R) between the occupancy of H1 variants at all chromosomes and the NAD or LAD content or mean gene expression. Correlation between NAD content, LAD content, and gene expression of all chromosomes is also shown. D, heat map and dendrogram of the occupancy of H1 variants (mean input-subtracted ChIP-seq signal over 50-bp genomic windows) at individual chromosomes. Mean gene expression as well as NAD and LAD contents of all chromosomes are shown as heat maps.
A large part of chromosome 19 is associated with the nucleolus and is reported to be located in central regions of the interphase nucleus, being close to the nucleoli (45). We have previously reported that H1.0 is highly enriched at this chromosome (41). Correlation analysis between H1 variant ChIP-seq signals and NAD content at each chromosome confirmed that H1.0 is the most abundant variant at chromosomes with a higher NAD content, whereas H1.2 signals were negatively correlated with NAD content (Fig. 1, C and D). As predicted, there was a negative association between the content of NADs and LADs at chromosomes, the former being located within the inner part of the nucleus and the latter at the periphery. We previously reported that H1.2 overlaps with LADs (41).
Ribosomal DNA encoding the 45 S single transcription unit giving rise to the 18 S, 5.8 S, and 28 S rRNA and flanked by non-transcribed spacers is present as repetitive DNA at the short arms of acrocentric chromosomes, called nucleolus organizer regions, within NADs. We aligned the input-subtracted H1 variant ChIP-seq signal to the rDNA complete repeating unit and found that H1.0 was enriched in the rDNA, mostly in the non-transcribed spacers (Fig. 2A). Instead, at the single transcription unit, H1X was locally enriched. H1.0 was also enriched at the 5 S ribosomal RNA subunit (encoded in tandem arrays, the largest one on chromosome 1), whereas H1X was slightly enriched at microRNAs and small nucleolar RNAs, compared with other variants (Fig. 2B).
FIGURE 2.

H1 variant abundance at rDNA, non-coding RNA genes, and repetitive DNA. A, alignment of the input-subtracted H1 variant ChIP-seq signal to the human ribosomal DNA complete repeating unit obtained from GenBankTM (U13369.1). To avoid bias in the alignment, the rDNA sequence was added to the human reference genome (hg18), and alignment was only allowed to a single position. A schematic representation of the rDNA repeating unit is shown above, with the rRNA transcription unit on the left and the non-transcribed spacer (NTS) on the right. ITS/ETS, internal/external transcribed spacer. B, box plots showing the occupancy of H1 variants (ChIP-seq signal) within some human non-coding RNA genes: 5 S ribosomal RNA subunit (rRNA), microRNA (miRNA), and small nucleolar RNA (snoRNA). C, box plots showing the occupancy of H1 variants (ChIP-seq signal) within human acromeric satellite 1, performing single mapping to the reference genome (top) or multiple mapping to a repeat database (RepBase) (bottom). D, box plots showing the occupancy of H1 variants (ChIP-seq signal) within some human DNA repeats: SINE-VNTR-Alu (SVA; non-autonomous, hominid-specific non-LTR retrotransposons) and telomeric satellites. Significance was assessed using the Kolmogorov-Smirnov test. Significant enrichment of H1.0 or H1X (marked with an arrow) compared with other variants is marked with asterisks (*, p < 0.05; **, p < 0.001).
Next, we aligned the input-subtracted H1 variant ChIP-seq signal to many repetitive DNA categories found in RepBase. One of the few categories that showed differential occupancy was ACRO1 (a 147-bp satellite found in the short arm of acrocentric chromosomes, where nucleolus organizer regions are located), which presented H1.0 enrichment (Fig. 2C). H1.0 was also enriched at SINE-VNTR-Alus (SVAs, non-autonomous, hominid-specific non-LTR retrotransposons) and telomeric satellites (Fig. 2D).
In summary, H1.0 is found to be enriched at DNA associated with nucleoli, including NADs, rDNAs, and acrocentric and telomeric satellites, suggesting that it could be involved in the stabilization of perinucleolar late-replicating heterochromatin. In contrast, H1X is overrepresented in the coding region of non-coding RNAs, such as 45 S rRNA, miRNA, and small nucleolar RNA, possibly related to the association of this variant with transcribed genes (see below).
H1.0 Is the H1 Variant Most Closely Associated with the Nucleolus
Because previous studies have shown enrichment of H1X and H1.0 in the nucleolus of cells by immunostaining (8, 9, 54), we further explored the localization of H1 variants at nucleoli by cellular fractionation and immunoblotting. Total, cytoplasmic, nucleoplasmic, and nucleolar extracts were prepared from T47D cells. Immunoblots were performed with specific antibodies for the six somatic H1 variants as well as tubulin as the cytoplasmic control and nucleophosmin and nucleolin as nucleolar controls, and the band intensity was quantified (Fig. 3). The cytoplasm was devoid of histone H1, whereas all variants were abundant in the nucleoplasm. At nucleoli, H1.0 was the most enriched variant, compared with total or nucleoplasm extracts. H1X and H1.5 were also enriched to some extent compared with the other variants. Overall, cellular fractionation confirmed our ChIP-seq results, pointing toward a specific association of H1.0 with nucleolar chromatin, although other variants, such H1X and possibly H1.5, are also present. In agreement with this, it has recently been reported that H1.0 interacts with an extensive network of proteins, many of them functioning in RNA metabolism in the nucleolus (55). Additionally, H1.5 was pulled down with H1.0 in one of the cell types used.
FIGURE 3.
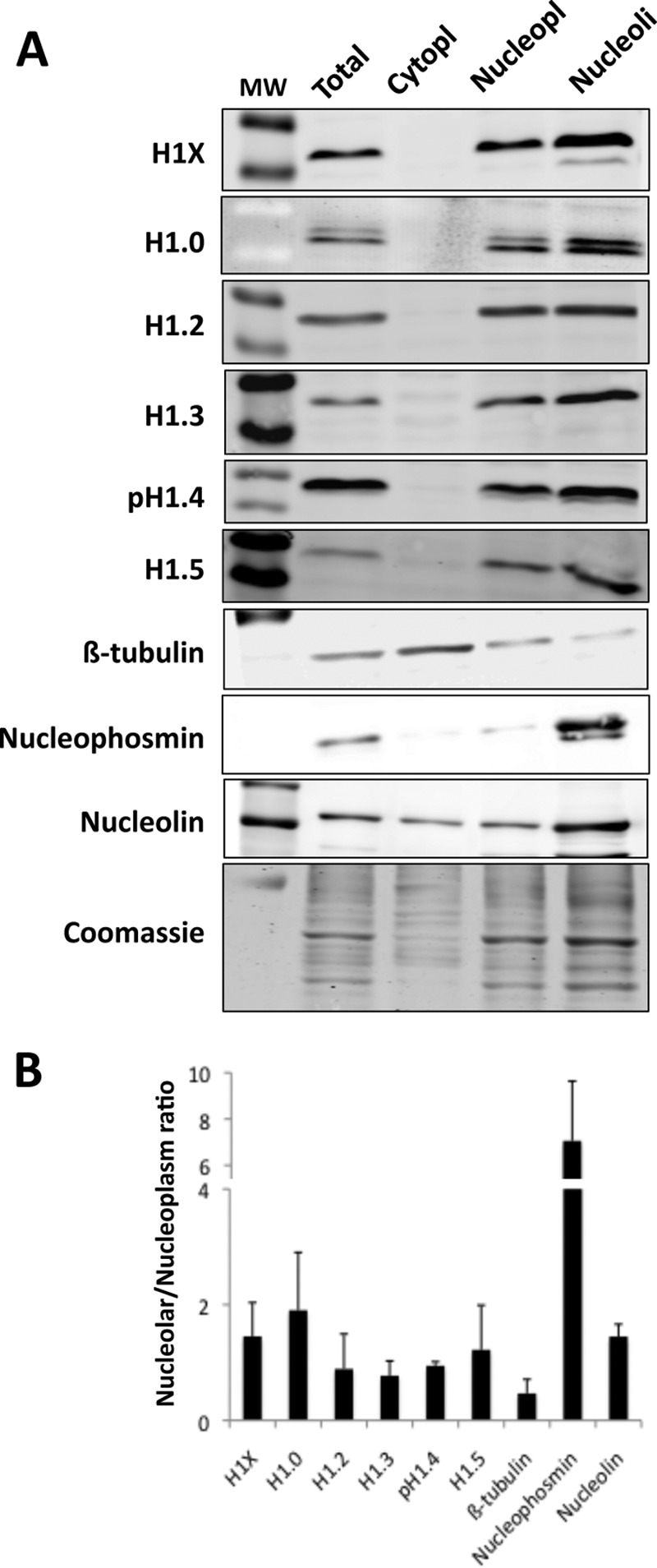
H1 variant distribution within different cell compartments. A, immunoblot of H1 variant abundance in total cell lysate, cytoplasm, nucleoplasm, and nucleolar fractions of T47D cells. 30 μg of protein extract was resolved in SDS-PAGE (10%) and immunoblotted with antibodies specific for H1X, H1.0, H1.2, H1.3, H1.4-T146p, H1.5, β-tubulin, nucleophosmin, and nucleolin. An acrylamide gel was stained with Coomassie solution and used as loading control and for normalization upon quantification of bands. Molecular weight marker (MW) bands shown in the H1 blots correspond to 34,000 and 26,000. B, representation of the normalized relative units of variants H1X, H1.0, H1.2, H1.3, H1.4, and H1.5, β-tubulin, nucleophosmin, and nucleolin present in the nucleolar fraction divided by the relative units in the nucleoplasm fraction. Immunoblot band intensities were measured using ImageJ version 1.48 software and normalized by Coomassie staining. The means and S.D. values (error bars) are shown for three independent fractionation experiments.
H1X Is Highly Associated with RNA Polymerase II-enriched Regions, Exons, Hypomethylated CpG Islands, and Active Transcription
We have described here and elsewhere (41) that different chromosomes have different abundances of H1 variants and that this is related to their gene content, mean gene expression, NAD and LAD content, and ultimately their position within the nucleus. Nonetheless, chromosomes are not uniform, and there may be territories or domains with different forms of chromatin organization. Using our ChIP-seq-derived data on genomic regions enriched in each individual H1 variant (41), we identified chromatin states based on the combined presence of H1-enriched regions, and we analyzed the presence of specific associated features (Fig. 4A). Because the number of islands of H1 enrichment identified was limited (ranging between 7,000 for H1.2 and 49,000 for H1.0 (41)), most of the genome was in a chromatin state without H1 islands. The next most abundant states contained islands enriched in H1.0, H1X, or H1.0 and H1X simultaneously. As reported previously, chromatin states containing H1.2 were associated with lamina, as were states containing H1.4. States containing other variants or combinations of variants were associated with genes and CpG islands, especially those containing both H1.0 and H1X.
FIGURE 4.

H1X is enriched at RNA polymerase II-enriched regions. A, identification of chromatin states based on the combined presence of H1 variant-enriched regions. The 10 most likely states obtained from a multivariate hidden Markov model are presented in the table, listing the frequency of each state. The abundance of different genomic features within each state is shown as a heat map. Darker blue corresponds to greater enrichment. B, normalized number of H1 variant-enriched and -depleted regions overlapping with RNA polymerase II binding sites (data from T47D cells obtained from Ballaré et al. (46)). C, box plots showing the occupancy of H1 variants (input-subtracted ChIP-seq signal) within RNA polymerase II binding sites. Significance was assessed using the Kolmogorov-Smirnov test, taking as a control a random sample of bulk genome windows with equal width to the RNA polymerase II sites. Enrichment and depletion are marked with red and blue asterisks, respectively. *, p value<0.001. D, mean input-subtracted ChIP-seq signal of H1 variants around the center of RNA polymerase II binding sites.
Because, in this work and elsewhere, we have found that the association with gene promoters and coding regions differs between H1 variants, we explored the overlap of H1 variant-enriched or -depleted regions with RNAPII binding sites, using data from T47D cells obtained by Ballaré et al. (46). H1X was the variant showing the greatest overlap of enriched regions and least overlap of depleted regions with RNAPII peaks (Fig. 4B). Genes with an RNAPII peak had a higher mean expression level than other genes (data not shown), in agreement with our previous observation that H1X-enriched target genes are highly expressed and H1X-depleted target genes are repressed (see Fig. 7 in Ref. 41). Next, we analyzed the strength of input-subtracted H1 variant ChIP-seq signals within RNAPII peaks, and we again found that H1X was enriched, unlike other variants, which were depleted, compared with random control samples representing regions of the bulk genome (Fig. 4C). This was also observed when H1 occupancy was explored around the center of RNAPII binding sites (Fig. 4D). Previously, we reported that H1X is the variant most enriched at DNase-hypersensitive sites and FAIRE regions as well as being associated with various histone H3 post-translational modifications and the one least depleted at p300 and CTCF sites (41). Additionally, H1X shows the highest correlation with GC content. Taken together, these findings suggest that H1X may have a role in transcriptional regulation of gene expression.
FIGURE 7.

Depletion of histone H1X in breast cancer cells does not alter cell proliferation. A, inducible depletion of H1X in T47D and MCF7 breast cancer cells. T47D- and MCF7-derived cells stably infected with a lentiviral inducible system for the expression of an shRNA against H1X were treated for 6 days with doxycycline or left untreated. Total chromatin or H1 extracts, respectively, were analyzed by immunoblot against H1 variant-specific antibodies or Coomassie-stained as a loading control. Phospho-Thr-146 antibody was used to detect H1.4. B, expression of H1 variants after induced depletion of H1X in T47D cells. Reverse transcription real-time PCR of shH1X T47D cells treated or not with doxycycline for 6 days was performed with specific oligonucleotides for H1 variants. Each value was corrected by GAPDH and the value of real-time PCR amplification with the same primer set of genomic DNA extracted from the same cell line. C, indirect immunofluorescence detection of H1X in T47D cells. T47D cells expressing the shH1X or a random shRNA (shRd) as a control, grown in the absence or presence of doxycycline, were fixed and stained with an H1X antibody. DNA was labeled with DAPI. D, cell cycle profile after propidium iodide staining of H1X knockdown T47D and MCF7 cells grown for 6 days in the absence or presence of doxycycline. Data are expressed as the percentage of cells in G1, S, and G2/M cell cycle phases. The means and S.D. values (error bars) for two independent experiments are shown. T47D cells harboring a random shRNA (shRd) were used as a control.
Notably, whereas the H1X content at distal promoters and TSS is lower at active than at inactive genes (an H1 valley), it is higher at coding regions of active genes, especially toward the 3′ end (Fig. 5A). This is not true for the other somatic H1 variants (41), the opposite trend being observed for H1.2, and this may be related to the association of H1X with RNAPII described herein. Moreover, H1X is the most abundant variant within exons (Fig. 5B). Furthermore, this variant is far more abundant at retained introns than the mean across all introns and is more abundant at included ASEs than excluded exons. Core histone H3 was also found to be more abundant at exons than introns, reflecting the reported higher nucleosome occupancy of exons (56), but this was not seen for all H1 variants (Fig. 5B). Overall, H1X is more abundant at exons and intron-exon junctions than within introns themselves and always higher at active genes (Fig. 5C). H1X accumulation toward the 3′ end of coding regions and at transcribed exons resembles H3K36me3 distribution (57).
FIGURE 5.

H1X is enriched in constitutive and included alternatively spliced exons within active coding regions. A, H1 abundance (mean input-subtracted ChIP-seq signal) around the body of the top and bottom 10% expressed genes, compared with the mean H1 abundance for all genes (shown in black). Gene regions are represented as a 3-kb-long metagene surrounded by a 1-kb region upstream TSS and 1-kb downstream transcription termination site (TTS). B, box plots showing the occupancy of H1 variants (mean input-subtracted ChIP-seq signal) at exons, retained introns (RI), and total introns. In the right panel, H1X occupancy is also shown at included and excluded ASEs. Significance was assessed using the Kolmogorov-Smirnov test to compare exons or retained introns with total introns or ASEs with total exons in the right panel. Enrichment and depletion is marked with red and blue asterisks, respectively. *, p < 0.001. C, H1 abundance as in A around exon (left) or intron (right) profiles.
Furthermore, we have reported that H1 variant abundance at CpG islands is heterogeneous across the genome, H1.0 and H1X being clearly overrepresented and H1.2 underrepresented (Fig. 6A). Approximately 12% of H1X-enriched regions overlap with CpG sites (41). Next, we investigated the methylation state of CpG islands overlapping H1 variant-enriched or -depleted regions. Although CpG islands overlapping H1-enriched islands are more methylated than those in H1-depleted regions, in agreement with the general consideration that H1-containing chromatin is repressive, H1X-enriched CpGs are less methylated than those overlapping other H1 variants (Fig. 6B). In summary, H1X is abundant at CpG islands, but the methylation level of these islands is lower than average, further confirming the relationship of H1X with active transcription. In contrast, H1.2 is disfavored at CpG sites, but the sites at which it is found are highly methylated, related to repressed chromatin.
FIGURE 6.

H1 abundance at methylated CpG islands. A, box plots showing the occupancy of H1 variants (mean input-subtracted ChIP-seq signal) at CpG islands. Significance was assessed using the Kolmogorov-Smirnov test taking as a control a random sample of windows with equal width to the CpGs. Enrichment and depletion are marked with red and blue asterisks, respectively. *, p < 0.001. B, box plots showing the methylation level of CpG islands overlapping H1 variant-enriched and -depleted regions. Data on the methylation levels at individual CpG islands were calculated by assessing the overlap between the genome methylation levels from Vanderkraats et al. (48) and the CpG islands from the UCSC database. Significance was assessed using the Kolmogorov-Smirnov test, taking the methylation level of all CpGs in the genome as a control. Significantly increased or decreased methylation at H1-enriched or -depleted regions is marked with red and blue asterisks, respectively. Differential methylation between H1X- and H1.2-enriched regions was also tested. *, p < 0.01; **, p < 0.001. C, box plots showing the occupancy of H1 variants (mean input-subtracted ChIP-seq signal) at hypermethylated (n = 500) and hypomethylated (n = 5,000) regions in breast cancer cells (T47D) compared with normal human mammary epithelial cells (data obtained from Ruike et al. (50)). Significance was assessed using the Kolmogorov-Smirnov test, taking as a control a random sample of windows with equal width to the hyper- and hypomethylated regions. Enrichment and depletion is marked with red and blue asterisks, respectively. **, p < 0.001.
The pattern of DNA methylation is known to be altered in cancer cells. In general, there is genome-wide intergenic hypomethylation and localized hypermethylation at particular promoters (including tumor-suppressor genes) and CpG-rich and gene-related regions. We have analyzed the occupancy of H1 variants at two subsets of the genome, defined as hyper- or hypomethylated regions in breast cancer cells (T47D) compared with normal human mammary epithelial cells (data obtained from Ruike et al. (50)). Hypermethylated regions in cancer cells were enriched in H1.0 and H1X, whereas hypomethylated regions were enriched in H1.2 (Fig. 6C). In other words, both sets of analysis confirm the preferential association of H1X with coding regions (hypermethylated in cancer) and active promoters containing hypomethylated CpG islands and the preferential association of H1.2 with intergenic regions (hypomethylated in cancer) and inactive promoters (containing methylated CpGs).
H1X Depletion in Breast Cancer Cells Does Not Alter Proliferation but Does Alter the Expression of Certain Genes
Given the specific association of H1X with RNAPII and active transcription in general, we decided to knock down this variant to explore the effect on cell proliferation and gene expression, in a manner comparable with the inducible depletion of the other five somatic H1 variants that we reported elsewhere (10). We used an inducible shRNA lentiviral expression system based on a Tet-ON strategy (58) to infect T47D and MCF7 breast cancer cells, and stable cell lines were established as described previously (10). Specific H1X depletion upon doxycycline treatment (6 days) was confirmed by immunoblotting, reverse transcription coupled with real-time PCR (RT-qPCR), and immunofluorescence (Fig. 7, A–C). No changes in the expression of other H1 variants were detected upon H1X depletion. Moreover, expression of a so-called H1X antisense gene (H1X-AS1) located upstream of the H1X gene in the human genome was not affected either by H1X KD or treatment with trichostatin A, a histone deacetylase inhibitor that induced H1X expression (data not shown), ruling out any functional association between H1X-AS and H1X expression.
Depletion of H1.2 or H1.4 in T47D cells affected cell proliferation and promoted arrest in the G1 phase of the cell cycle but not the depletion of H1.0, H1.3, or H1.5 (10). H1X-induced depletion slightly slowed down cell proliferation, although the change was not significant (data not shown), and no effect was detected on the cell cycle profile (Fig. 7D) in T47D or MCF7 cells compared with untreated cells or control cells expressing random shRNA. Overall, these results suggest that H1X does not play a role in cell proliferation and that this variant is dispensable for the cell lines analyzed under normal growth conditions.
Next, we studied the consequences of H1X depletion on global gene expression using a genome-wide Agilent microarray platform containing ∼28,000 mRNAs and 7,400 long non-coding RNAs (see “Experimental Procedures”). T47D cells harboring H1X shRNA were treated or not with doxycycline for 6 days and serum-starved for 48 h for synchronization in G1 prior to RNA extraction to avoid differences between H1 KDs that produce G1 arrest, as reported elsewhere (10). Applying a -fold change threshold of 1.4 and false discovery rate of q ≤ 0.05, we found 149 genes to be up-regulated and 45 down-regulated upon H1X KD. The basal expression level, without doxycycline, of up-regulated genes (mean ± S.D. = 8.011 ± 1.99) was lower than the mean expression level across the entire transcriptome (8.723 ± 2.57), whereas that of down-regulated genes was higher than the mean (8.796 ± 1.97) (p < 0.005). Some genes, such as KNG1 and KRT37 were up-regulated more than 50-fold in H1X KDs (Fig. 8A). Gene deregulation by H1X KD was confirmed by RT-qPCR of selected genes in independent samples (Fig. 8B). Gene ontology analysis of both up- and down-regulated genes was performed, and interestingly, the most significant functions identified were related to cell movement and transport, and common functions were found between up- and down-regulated genes (data not shown).
FIGURE 8.

Genes deregulated by H1X KD are not enriched in specific H1 variants. A, expression levels of the top six up-regulated genes in H1X knockdown T47D cells treated or not with doxycycline for 6 days. Expression data were obtained by hybridization with an Agilent microarray in duplicate, and log2 values are represented. B, expression of genes up-regulated upon H1X KD measured by real-time PCR in H1X and random shRNA-expressing cells treated or not with doxycycline for 6 days. Expression of H1X was measured to test its inhibition by the inducible shRNA. GAPDH was measured for normalization. Expression data are presented relative to the maximal value for each gene. The means and S.D. values (error bars) are shown from a representative experiment measured in duplicate. C, box plots of H1.0, H1.2, H1.4, and H1X abundance (input-subtracted ChIP-seq signal) at distal promoter regions (−3,200 to −2,000 bp relative to TSS) for the genes up- or down-regulated upon H1X knockdown, compared with the H1 abundance of total genes. D, H1X abundance (mean input-subtracted ChIP-seq signal) around exon (left) or intron (right) profiles of genes up- or down-regulated upon H1X knockdown, compared with the mean H1 abundance for all genes (shown in black). E, H1X and H1.2 abundance at the up-regulated gene promoters in H1X KD cells. ChIP with specific antibodies for H1X and H1.2 was performed in H1X KD T47D cells treated (+D) or not (−D) with doxycycline, and the abundance of immunoprecipitated material was quantified by real-time PCR with oligonucleotides for the indicated promoters (−3 kb distal promoter or TSS) and corrected by input DNA amplification with the same primer pair. Genes that did not change their expression in the H1X KD microarray were also analyzed for comparison. The means and S.D. values are shown from a representative experiment measured in duplicate.
Genes Specifically Deregulated by Knockdown of a Particular H1 Variant Are Not Enriched in That Variant at the Promoter
We have previously shown that KD of individual H1 variants deregulates a small subset of genes (≤2%) specific for each variant, including up- or down-regulated genes in similar proportions (10). Here, we have confirmed this for H1X KD. One hypothesis would be that these subsets contain genes specifically targeted by or with prevalence of some of the H1 variants. We explored the occupancy of H1 variant at promoters specifically deregulated by particular variant KDs, and we found no differences in their abundance compared with the mean abundance across all genes (data not shown). For example, the H1X content at distal promoters of genes up- or down-regulated by inducible H1X KD was similar to or at most only slightly lower than the H1X content distribution across all genes (Fig. 8C). H1X content was lower at down-regulated genes (high basal expression) than at up-regulated genes (low basal expression), in agreement with the lower H1X content at the distal promoter of genes expressed above average (Fig. 5A) (41). Up-regulated genes, despite having below average basal expression, do not have above average H1X content (i.e. they present less H1X than expected). Moreover, genes dysregulated at H1X KD cells also were observed to have below average H1 content at distal promoters for the other H1 variants (i.e. H1.0, H1.2, and H1.4) (Fig. 8C).
As mentioned above, whereas the H1X content at the promoter of active genes is lower than at inactive genes, the H1X content at coding regions is higher at active genes. Accordingly, genes down-regulated in H1X KD cells, which present higher basal levels than average or up-regulated genes, showed higher H1X content along their coding region and at collapsed exons and introns (Fig. 8D).
Promoters of Up-regulated Genes Become Deprived of H1X upon H1X Knockdown but Do Not Show an H1 Valley or Active Histone Marks
Next, we explored changes in H1 and histone marks at the promoters of six genes up-regulated upon H1X KD by ChIP-qPCR. In all promoters, H1X was removed upon doxycycline treatment of inducible H1X KD cells. In parallel, no significant changes in the promoter occupancy by H1.2 (Fig. 8E) or total H1 or H3 (data not shown) were observed. Moreover, these ChIP results confirmed that H1X was not the only H1 variant occupying these H1X-responsive promoters.
We and others have reported elsewhere that there is a valley in H1 occupancy at active promoters compared with that in surrounding regions (40, 41). We compared H1 occupancy at TSS and −3 kb upstream (the distal promoter). Only UGT2B10 showed a small decrease in H1 at TSS in the absence of doxycycline. This is in agreement with the limited basal expression of these genes, as described above. Nonetheless, these genes were strongly up-regulated upon H1X KD. Hence, an H1 valley was expected under doxycycline treatment, but this was not observed, whereas some of the active genes tested as controls (CDK2, TBKBP1, and JUN) did show an H1 valley at TSS (Fig. 8E). Furthermore, enrichment of H3K4me3, a mark of active transcription, was not stronger at TSS of the up-regulated genes under stimulatory conditions, whereas it was present at constitutively active promoters tested as a control (data not shown). In summary, H1X KD up-regulated a limited number of genes in a manner that does not seem to involve regular mechanisms leading to transcription initiation, such as H1 removal and histone H3 Lys-4 methylation at promoters; nor was it linked to removal of a specific H1 variant targeting a promoter for repression.
DISCUSSION
H1.0 Is Enriched at Nucleolus-associated Chromatin
We previously reported that the distribution of H1 variants in the breast cancer cell line T47D is not uniform and that H1.2 is the variant that shows the most distinctive pattern. Specifically, H1.2 was found to be non-abundant at genes but enriched at chromosomal domains with low GC content and associated with gene-poor chromosomes, intergenic DNA, and LADs. In contrast, other variants are associated with a relatively high GC content, CpG islands, and gene-rich domains (41). Because not all repetitive DNA is included in the conventional alignment of ChIP-seq data with the human genome, we further investigated the abundance of H1 variants at repetitive features. Strikingly, we found that H1.0 is associated with several repetitive DNA elements related to nucleoli, including ribosomal DNA and acrocentric satellites, as well as NADs. Furthermore, cell fractionation followed by immunoblotting with variant-specific antibodies provided further data suggesting that H1.0 is the variant most enriched at nucleoli.
The nucleolus is the site of ribosome biogenesis and is surrounded by a shell of late replicating condensed heterochromatic DNA (59, 60). DNA associated with this nucleolar chromatin has recently been identified (45, 61) and, in addition to the rDNA repeat units located at human acrocentric chromosomes, includes specific sequences from most chromosomes in a reproducible and heritable manner. NADs have in common a low amount of AT-rich sequence elements, low gene density, and enrichment in silent genes. Some of these sequences may also be associated with the nuclear envelope forming the LADs (61). Both the nucleolus-associated and nuclear periphery-associated chromatin domains exhibit common features, being highly condensed and known to replicate preferentially at late stages of the S phase. We have found different H1 variants, H1.0 and H1.2, to be associated with NADs and LADs, respectively, suggesting that these two compartments may have different components, and specific H1 variants may contribute to their organization or stabilization.
H1.0 has been previously reported to be mainly located in chromatin regions that are not affected by micrococcal nuclease digestion, in condensed chromatin, and in perinucleolar regions in certain types of tissue, although it is not fully excluded from active chromatin (54). Recently, a network of proteins interacting with H1.0 in four different cell lines was identified, including splicing factors and proteins involved in rRNA biogenesis, ribosome function/translation, and cellular transport, most of them identified as components of the nucleolus (55). As a consequence, it has been proposed that H1.0 could be a key regulator of nucleolar function and that nucleoli may be the source of the slower exchanging fraction of H1 in the cell. Nonetheless, other H1 variants have also been identified in proteomic profiling of the human nucleolus (62), and phosphorylated H1.2 and H1.4 are associated with RNA polymerase I activity and rRNA biogenesis and have been localized to the nucleolus (63).
It was previously reported that H1X associates with nucleoli in the G1 phase of the cell cycle (8, 9), although it was suggested that H1X is not located directly at the sites of rDNA transcription but rather at inactive ribosomal genes. Here we also show H1X to be located at nucleoli by immunostaining, but our data indicated that H1.0 is the main nucleolar H1 in the breast cancer cells analyzed. Nonetheless, neither H1.0 nor H1X are restricted to nucleoli, both being found overlapping with somatic H1 variants other than H1.2 throughout the genome of T47D cells (41). H1.0 and, probably, H1X are synthesized independently of DNA replication, and hence, they accumulate when cells stop proliferating and start to differentiate and, consequently, may replace replication-dependent variants. High-throughput mapping of H1.0 and H1X in the genome of differentiated cells has not yet been performed. Additionally, because, for instance, H1.0 content is highly reduced in HeLa cells (data not shown), it would be interesting to analyze whether the association of H1.0 with nucleoli is conserved through different cell types or H1X becomes prominent in other cells as well as whether oncogenic transformation influences the specificity of H1 variant localization.
H1X Associates with RNAPII-enriched Regions, Included Exons, Hypomethylated CpG Islands, and Actively Transcribed Coding Regions
Although histone H1 is often regarded as a basic component of chromatin, growing evidence suggests that particular H1 variants are involved in regulating gene expression at a more specific level. We have found that H1X is the variant that best colocalizes with RNAPII in T47D cells and accumulates at coding regions, mainly exons, of expressed genes. Moreover, we previously reported that expressed genes are devoid of H1, including H1X, at promoters (an H1 valley). Accordingly, the association of H1X with RNAPII might be related to the elongation process, as suggested by the accumulation of H1X toward the 3′ end of coding regions. Besides distinct patterns of expression and localization in different types of tissues and cells, it has also been postulated that H1 associates with regulatory proteins or chromatin components to control their activity. The question of whether H1X is occupying the expected position at nucleosomes at these gene regions or interacts with members of the elongating complex is intriguing. Interestingly, H1X was first found in a two-hybrid screen with the WD40 repeat region of the transcription factor TFIID as the bait (64), although this association was not further explored functionally. In relation to this, it has recently been described that H1.2 functionally interacts with Cul4A E3 ubiquitin ligase, PAF1 elongation complexes, and the serine 2-phosphorylated form of RNAPII that potentiates core histone modifications and targets gene transcriptional elongation in HeLa cells (65). Moreover, in the same study, WDR5, a substrate adaptor for Cul4A E3 ligase, was found to co-purify with six of the somatic H1 variants (H1.0 to H1.5); H1X was not explored. H1X playing a role in T47D cells similar to that of H1.2 in HeLa, while H1.2 is excluded from actively transcribed regions in T47D cells, is an intriguing possibility, compatible with the view that the distinct patterns of expression and localization of H1 variants in different types of tissues and cell types may provide an important regulatory mechanism of gene expression.
H1X accumulation toward the 3′ end of coding regions and at transcribed exons resembles H3K36me3 distribution. H3K36me3 is co-transcriptionally deposited in a splicing-dependent manner and represses internal initiation. Moreover, this histone mark seems to associate with exons included by the splicing machinery more than with ASEs (56, 57, 66, 67). Included exons show higher nucleosome occupancy, either to protect fidelity or to slow down transcription to ensure inclusion. It would be interesting to investigate a hypothetical association of H1X with factors that set or recognize H3K36me3 and to investigate whether H1X plays a specific role in marking or protecting exons to be expressed. Alternatively, H1X-mediated compaction could slow RNAPII elongation, favoring alternative exon inclusion. In relation to this, we have found that H1X is more abundant at included ASEs than at excluded exons. Interestingly, H1X is also enriched in retained introns. It has also been reported that DNA methylation is enriched in included exons and that inhibition of DNA methylation, as well as inhibition of histone deacetylase activity, results in aberrant alternative splicing (68). Regarding our observations, H1X could be an additional player in the functional interconnections between chromatin structure, transcriptional elongation, and splicing, raising the intriguing possibility of the existence of an epigenetic memory for splicing patterns that could be inherited.
We have shown that CpG islands overlapping H1-enriched regions are hypermethylated compared with the mean for CpG islands across the genome, whereas those overlapping H1-depleted regions are hypomethylated, further supporting the general notion that H1-containing chromatin is repressive, at least at gene promoters where most of the CpG islands are located. Similar results were reported by Izzo et al. (39). Interestingly, there are differences between variants, in agreement with further observations relating H1X to active transcription and H1.2 to repressive chromatin. In other words, H1X is more abundant than H1.2 at CpG islands, and those islands coinciding with H1X enrichment are significantly less methylated than H1.2-occupied islands. It remains to be explored in human cells whether there is a direct interplay between H1 variants and DNA methylation at CpG islands or just co-localization of features related to active or repressed chromatin. In mouse embryonic stem cells, it has been reported that there is interaction of DNA methyltransferases with H1 variants and recruitment to two imprinting control loci for their repression, except for H1c (H1.2) that did not interact (69). Because H1.2 is disfavored at coding regions, according to our data, it is plausible that it is not involved in the regulation of gene expression by associating to DNA methyltransferases, but further investigation is required to clarify this hypothetical interplay between histone H1 and DNA methylation.
Cancer cells have an altered methylation pattern compared with that in healthy cells, namely general hypomethylation and localized hypermethylation at certain promoters containing CpG islands. By analyzing the H1 variant content at regions described to be significantly hypo- or hypermethylated in T47D cells compared with normal mammary epithelial cells, we found hypomethylated DNA to be enriched in H1.2 and found enrichment of H1.0 and H1X at hypermethylated DNA. In turn, regions hypomethylated in cancer have been related to repressive chromatin, transcriptional inactivation, and large genomic structures, such as LADs (70–72), features associated with H1.2 according to our results. Thus, we hypothesize that different H1 variants may be involved in establishing or maintaining altered DNA methylation patterns in the course of cancer. At the same time, we have shown that different H1 variants are associated with different nuclear chromatin structures, such as LADs or NADs, which are developmentally regulated and altered in cancer (73, 74). The view that the origin of cancer may lay in an epigenetic dysregulation that would increase transcriptional noise, variability, and gene expression plasticity is gaining strength (75). Altered expression of H1 variants during the onset of cancer (31, 32) could participate in global chromatin rearrangements by altering the formation of such chromatin structures or chromatin spatial interactions.
H1X Is Dispensable in Breast Cancer Cells but Alters Gene Expression through an Unknown Mechanism
We reported elsewhere that H1.2 and H1.4 KD in T47D cells slowed cell proliferation and, in the H1.2 KD at least, caused the arrest of the cell cycle in G1 (10). Using the same methodology, no growth phenotype was observed for H1.0, H1.3, or H1.5 KD, as observed here for H1X KD in T47D and MCF7 cells. It is noteworthy that H1X expression was detected not only in T47D and MCF7, but also in HeLa, 293T, and Jurkat cells, all of the cell lines tested to date (data not shown) (7). Elsewhere, H1X was found to be expressed in all tissues examined (64). This is also true for H1.2 and H1.4 but not for the other variants (H1.1 being undetected in most cell types, whereas there is a low level of expression of H1.0 and H1.3 in HeLa and of H1.5 in 293T) and can be considered an indication of the prominent role of H1.2 and H1.4 in human cells. Moreover, regulation of H1X expression differs not only from that of the replication-dependent variants but also from that of replication-independent H1.0 (7). Taken together, these observations suggest that H1X may have a specific and prominent role among H1 functions in human cells, although this has not become apparent upon H1X KD. One possibility could be that shRNA-mediated H1X depletion was not complete or that other H1 variants undertake some of its functions or localization. For this, H1.0 seems to be the stronger candidate, because the two variants show considerable overlap throughout our analysis despite specific enrichment at RNAPII binding regions and nucleolar chromatin, respectively. As yet, no attempt has been made to develop a double H1.0/H1X KD, but such an approach would clarify this possibility.
H1X KD affected expression of a small number of genes, either up- or down-regulated, as observed when some of the other H1 variants were knocked down with the doxycycline-inducible system in the same cells (10). Moreover, small changes in gene expression upon H1 depletion have also been reported in many other systems, including knock-out mice (11–13). Because H1X localization is enriched at RNAPII sites and at gene exons and correlates with high expression within the body of genes, we could expect that H1X KD would have a larger effect on global gene expression than what we observed. Alternatively, H1X could play a role in the coupling of elongation and splicing. Again, insufficient depletion or redundancy between variants could explain the lack of major effects.
Our hypothesis was that genes dysregulated upon H1 KD might have a higher content of that particular H1 variant at the promoter and, consequently, be more exposed to its depletion if H1 plays a regulatory role, but this was not confirmed. Instead, the H1X (and general H1) content under uninduced conditions of genes up-regulated upon H1X KD was lower than expected according to its low basal expression level. Moreover, the content of H1X along the coding region of these genes was normal, and the abundance of H1X-enriched or -depleted islands within the coding region of genes down-regulated in H1X KD was not significantly different from the abundance of other H1 variants (data not shown). ChIP showed that variants other than H1X were present at the promoter of up-regulated genes, and their abundance did not increase upon H1X depletion.
Surprisingly, genes up-regulated upon H1X KD did not present features of promoter clearance and transcription initiation, such as an H1 valley or increased methylation of histone H3 Lys-4 at TSS, and this cannot be attributed to a lack of nucleosomes because H1 and H3 were clearly observed. Expression of genes in the absence of histone marking has been reported elsewhere (76–78). Maybe H1X clearance is sufficient for these particular promoters to allow recruitment of transcription factors and machinery. Alternatively, gene induction is not caused by increased initiation but rather by a later step that was blocked by H1X, perhaps related to its hypothetical role in elongation or splicing. There is no doubt that further studies are required to understand how particular H1 variants regulate a limited subset of genes and whether this relates to the specific localization of variants across genomic features, chromatin domains, and nuclear territories that we are starting to envisage.
This work was supported by funding from the Spanish Ministry of Science and Innovation (MICINN), European Regional Development Fund Grant BFU2011-23057, and Generalitat de Catalunya Grant 2009-SGR-1222.
The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession nos. GSE49345 and GSE62766).
- KD
- knockdown
- TSS
- transcription start site
- H3K4me3
- H3K9me3, and H3K36me3, histone H3 Lys-4, -9, and -36 trimethylation, respectively
- ChIP-seq
- ChIP-sequencing
- LAD
- lamina-associated domain
- NAD
- nucleolus-associated chromatin domain
- qPCR
- quantitative PCR
- RNAPII
- RNA polymerase II
- ASE
- alternatively spliced exon.
REFERENCES
- 1. Thoma F., Koller T., Klug A. (1979) Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol. 83, 403–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bednar J., Horowitz R. A., Grigoryev S. A., Carruthers L. M., Hansen J. C., Koster A. J., Woodcock C. L. (1998) Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. U.S.A. 95, 14173–14178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pennings S., Meersseman G., Bradbury E. M. (1994) Linker histones H1 and H5 prevent the mobility of positioned nucleosomes. Proc. Natl. Acad. Sci. U.S.A. 91, 10275–10279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laybourn P. J., Kadonaga J. T. (1991) Role of nucleosomal cores and histone H1 in regulation of transcription by RNA polymerase II. Science 254, 238–245 [DOI] [PubMed] [Google Scholar]
- 5. Happel N., Doenecke D. (2009) Histone H1 and its isoforms: contribution to chromatin structure and function. Gene 431, 1–12 [DOI] [PubMed] [Google Scholar]
- 6. Izzo A., Kamieniarz K., Schneider R. (2008) The histone H1 family: specific members, specific functions? Biol. Chem. 389, 333–343 [DOI] [PubMed] [Google Scholar]
- 7. Happel N., Schulze E., Doenecke D. (2005) Characterisation of human histone H1x. Biol. Chem. 386, 541–551 [DOI] [PubMed] [Google Scholar]
- 8. Stoldt S., Wenzel D., Schulze E., Doenecke D., Happel N. (2007) G1 phase-dependent nucleolar accumulation of human histone H1x. Biol. Cell. 99, 541–552 [DOI] [PubMed] [Google Scholar]
- 9. Takata H., Matsunaga S., Morimoto A., Ono-Maniwa R., Uchiyama S., Fukui K. (2007) H1.X with different properties from other linker histones is required for mitotic progression. FEBS Lett. 581, 3783–3788 [DOI] [PubMed] [Google Scholar]
- 10. Sancho M., Diani E., Beato M., Jordan A. (2008) Depletion of human histone H1 variants uncovers specific roles in gene expression and cell growth. PLoS Genet. 4, e1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fan Y., Nikitina T., Zhao J., Fleury T. J., Bhattacharyya R., Bouhassira E. E., Stein A., Woodcock C. L., Skoultchi A. I. (2005) Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212 [DOI] [PubMed] [Google Scholar]
- 12. Shen X., Gorovsky M. A. (1996) Linker histone H1 regulates specific gene expression but not global transcription in vivo. Cell 86, 475–483 [DOI] [PubMed] [Google Scholar]
- 13. Lin Q., Inselman A., Han X., Xu H., Zhang W., Handel M. A., Skoultchi A. I. (2004) Reductions in linker histone levels are tolerated in developing spermatocytes but cause changes in specific gene expression. J. Biol. Chem. 279, 23525–23535 [DOI] [PubMed] [Google Scholar]
- 14. Fan Y., Sirotkin A., Russell R. G., Ayala J., Skoultchi A. I. (2001) Individual somatic H1 subtypes are dispensable for mouse development even in mice lacking the H1(0) replacement subtype. Mol. Cell Biol. 21, 7933–7943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Daujat S., Zeissler U., Waldmann T., Happel N., Schneider R. (2005) HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J. Biol. Chem. 280, 38090–38095 [DOI] [PubMed] [Google Scholar]
- 16. Hale T. K., Contreras A., Morrison A. J., Herrera R. E. (2006) Phosphorylation of the linker histone H1 by CDK regulates its binding to HP1α. Mol. Cell 22, 693–699 [DOI] [PubMed] [Google Scholar]
- 17. Hergeth S. P., Dundr M., Tropberger P., Zee B. M., Garcia B. A., Daujat S., Schneider R. (2011) Isoform-specific phosphorylation of human linker histone H1.4 in mitosis by the kinase Aurora B. J. Cell Sci. 124, 1623–1628 [DOI] [PubMed] [Google Scholar]
- 18. Kamieniarz K., Izzo A., Dundr M., Tropberger P., Ozretic L., Kirfel J., Scheer E., Tropel P., Wisniewski J. R., Tora L., Viville S., Buettner R., Schneider R. (2012) A dual role of linker histone H1.4 Lys 34 acetylation in transcriptional activation. Genes Dev. 26, 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim K., Jeong K. W., Kim H., Choi J., Lu W., Stallcup M. R., An W. (2012) Functional interplay between p53 acetylation and H1.2 phosphorylation in p53-regulated transcription. Oncogene 31, 4290–4301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuzmichev A., Jenuwein T., Tempst P., Reinberg D. (2004) Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol. Cell 14, 183–193 [DOI] [PubMed] [Google Scholar]
- 21. Lee H., Habas R., Abate-Shen C. (2004) MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science 304, 1675–1678 [DOI] [PubMed] [Google Scholar]
- 22. Vaquero A., Scher M., Lee D., Erdjument-Bromage H., Tempst P., Reinberg D. (2004) Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 16, 93–105 [DOI] [PubMed] [Google Scholar]
- 23. Weiss T., Hergeth S., Zeissler U., Izzo A., Tropberger P., Zee B. M., Dundr M., Garcia B. A., Daujat S., Schneider R. (2010) Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics Chromatin 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Terme J. M., Millán-Ariño L., Mayor R., Luque N., Izquierdo-Bouldstridge A., Bustillos A., Sampaio C., Canes J., Font I., Sima N., Sancho M., Torrente L., Forcales S., Roque A., Suau P., Jordan A. (2014) Dynamics and dispensability of variant-specific histone H1 Lys-26/Ser-27 and Thr-165 post-translational modifications. FEBS Lett. 588, 2353–2362 [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y., Cooke M., Panjwani S., Cao K., Krauth B., Ho P. Y., Medrzycki M., Berhe D. T., Pan C., McDevitt T. C., Fan Y. (2012) Histone h1 depletion impairs embryonic stem cell differentiation. PLoS Genet. 8, e1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lennox R. W., Cohen L. H. (1983) The histone H1 complements of dividing and nondividing cells of the mouse. J. Biol. Chem. 258, 262–268 [PubMed] [Google Scholar]
- 27. Piña B., Martínez P., Suau P. (1987) Changes in H1 complement in differentiating rat-brain cortical neurons. Eur. J. Biochem. 164, 71–76 [DOI] [PubMed] [Google Scholar]
- 28. Meergans T., Albig W., Doenecke D. (1997) Varied expression patterns of human H1 histone genes in different cell lines. DNA Cell Biol. 16, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 29. Parseghian M. H., Hamkalo B. A. (2001) A compendium of the histone H1 family of somatic subtypes: an elusive cast of characters and their characteristics. Biochem. Cell Biol. 79, 289–304 [PubMed] [Google Scholar]
- 30. Terme J. M., Sesé B., Millán-Ariño L., Mayor R., Izpisúa Belmonte J. C., Barrero M. J., Jordan A. (2011) Histone H1 variants are differentially expressed and incorporated into chromatin during differentiation and reprogramming to pluripotency. J. Biol. Chem. 286, 35347–35357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sato S., Takahashi S., Asamoto M., Nakanishi M., Wakita T., Ogura Y., Yatabe Y., Shirai T. (2012) Histone H1 expression in human prostate cancer tissues and cell lines. Pathol. Int. 62, 84–92 [DOI] [PubMed] [Google Scholar]
- 32. Medrzycki M., Zhang Y., McDonald J. F., Fan Y. (2012) Profiling of linker histone variants in ovarian cancer. Front. Biosci. 17, 396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sjöblom T., Jones S., Wood L. D., Parsons D. W., Lin J., Barber T. D., Mandelker D., Leary R. J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S. D., Willis J., Dawson D., Willson J. K., Gazdar A. F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B. H., Bachman K. E., Papadopoulos N., Vogelstein B., Kinzler K. W., Velculescu V. E. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 [DOI] [PubMed] [Google Scholar]
- 34. Telu K. H., Abbaoui B., Thomas-Ahner J. M., Zynger D. L., Clinton S. K., Freitas M. A., Mortazavi A. (2013) Alterations of histone H1 phosphorylation during bladder carcinogenesis. J. Proteome Res. 12, 3317–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Orthaus S., Klement K., Happel N., Hoischen C., Diekmann S. (2009) Linker histone H1 is present in centromeric chromatin of living human cells next to inner kinetochore proteins. Nucleic Acids Res. 37, 3391–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Th'ng J. P., Sung R., Ye M., Hendzel M. J. (2005) H1 family histones in the nucleus: control of binding and localization by the C-terminal domain. J. Biol. Chem. 280, 27809–27814 [DOI] [PubMed] [Google Scholar]
- 37. Parseghian M. H., Newcomb R. L., Winokur S. T., Hamkalo B. A. (2000) The distribution of somatic H1 subtypes is non-random on active versus inactive chromatin: distribution in human fetal fibroblasts. Chromosome Res. 8, 405–424 [DOI] [PubMed] [Google Scholar]
- 38. Cao K., Lailler N., Zhang Y., Kumar A., Uppal K., Liu Z., Lee E. K., Wu H., Medrzycki M., Pan C., Ho P. Y., Cooper G. P., Jr., Dong X., Bock C., Bouhassira E. E., Fan Y. (2013) High-resolution mapping of h1 linker histone variants in embryonic stem cells. PLoS Genet. 9, e1003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Izzo A., Kamieniarz-Gdula K., Ramírez F., Noureen N., Kind J., Manke T., van Steensel B., Schneider R. (2013) The genomic landscape of the somatic linker histone subtypes H1.1 to H1.5 in human cells. Cell Rep 3, 2142–2154 [DOI] [PubMed] [Google Scholar]
- 40. Krishnakumar R., Gamble M. J., Frizzell K. M., Berrocal J. G., Kininis M., Kraus W. L. (2008) Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 319, 819–821 [DOI] [PubMed] [Google Scholar]
- 41. Millán-Ariño L., Islam A. B., Izquierdo-Bouldstridge A., Mayor R., Terme J. M., Luque N., Sancho M., López-Bigas N., Jordan A. (2014) Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2. Nucleic Acids Res. 42, 4474–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Truss M., Bartsch J., Schelbert A., Haché R. J., Beato M. (1995) Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J. 14, 1737–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andersen J. S., Lyon C. E., Fox A. H., Leung A. K., Lam Y. W., Steen H., Mann M., Lamond A. I. (2002) Directed proteomic analysis of the human nucleolus. Curr. Biol. 12, 1–11 [DOI] [PubMed] [Google Scholar]
- 44. Guelen L., Pagie L., Brasset E., Meuleman W., Faza M. B., Talhout W., Eussen B. H., de Klein A., Wessels L., de Laat W., van Steensel B. (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951 [DOI] [PubMed] [Google Scholar]
- 45. Németh A., Conesa A., Santoyo-Lopez J., Medina I., Montaner D., Péterfia B., Solovei I., Cremer T., Dopazo J., Längst G. (2010) Initial genomics of the human nucleolus. PLoS Genet. 6, e1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ballaré C., Castellano G., Gaveglia L., Althammer S., González-Vallinas J., Eyras E., Le Dily F., Zaurin R., Soronellas D., Vicent G. P., Beato M. (2013) Nucleosome-driven transcription factor binding and gene regulation. Mol. Cell 49, 67–79 [DOI] [PubMed] [Google Scholar]
- 47. Jurka J., Kapitonov V. V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 [DOI] [PubMed] [Google Scholar]
- 48. Vanderkraats N. D., Hiken J. F., Decker K. F., Edwards J. R. (2013) Discovering high-resolution patterns of differential DNA methylation that correlate with gene expression changes. Nucleic Acids Res. 41, 6816–6827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinlan A. R., Hall I. M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruike Y., Imanaka Y., Sato F., Shimizu K., Tsujimoto G. (2010) Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics 11, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zentner G. E., Saiakhova A., Manaenkov P., Adams M. D., Scacheri P. C. (2011) Integrative genomic analysis of human ribosomal DNA. Nucleic Acids Res. 39, 4949–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Langmead B., Trapnell C., Pop M., Salzberg S. L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ernst J., Kellis M. (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gorka C., Fakan S., Lawrence J. J. (1993) Light and electron microscope immunocytochemical analyses of histone H1(0) distribution in the nucleus of Friend erythroleukemia cells. Exp. Cell Res. 205, 152–158 [DOI] [PubMed] [Google Scholar]
- 55. Kalashnikova A. A., Winkler D. D., McBryant S. J., Henderson R. K., Herman J. A., DeLuca J. G., Luger K., Prenni J. E., Hansen J. C. (2013) Linker histone H1.0 interacts with an extensive network of proteins found in the nucleolus. Nucleic Acids Res. 41, 4026–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hnilicová J., Staněk D. (2011) Where splicing joins chromatin. Nucleus 2, 182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kolasinska-Zwierz P., Down T., Latorre I., Liu T., Liu X. S., Ahringer J. (2009) Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 41, 376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wiznerowicz M., Trono D. (2003) Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 77, 8957–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sadoni N., Langer S., Fauth C., Bernardi G., Cremer T., Turner B. M., Zink D. (1999) Nuclear organization of mammalian genomes: polar chromosome territories build up functionally distinct higher order compartments. J. Cell Biol. 146, 1211–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ferreira J., Paolella G., Ramos C., Lamond A. I. (1997) Spatial organization of large-scale chromatin domains in the nucleus: a magnified view of single chromosome territories. J. Cell Biol. 139, 1597–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van Koningsbruggen S., Gierlinski M., Schofield P., Martin D., Barton G. J., Ariyurek Y., den Dunnen J. T., Lamond A. I. (2010) High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol. Biol. Cell 21, 3735–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jarboui M. A., Wynne K., Elia G., Hall W. W., Gautier V. W. (2011) Proteomic profiling of the human T-cell nucleolus. Mol. Immunol. 49, 441–452 [DOI] [PubMed] [Google Scholar]
- 63. Zheng Y., John S., Pesavento J. J., Schultz-Norton J. R., Schiltz R. L., Baek S., Nardulli A. M., Hager G. L., Kelleher N. L., Mizzen C. A. (2010) Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. J. Cell Biol. 189, 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamamoto T., Horikoshi M. (1996) Cloning of the cDNA encoding a novel subtype of histone H1. Gene 173, 281–285 [DOI] [PubMed] [Google Scholar]
- 65. Kim K., Lee B., Kim J., Choi J., Kim J. M., Xiong Y., Roeder R. G., An W. (2013) Linker Histone H1.2 cooperates with Cul4A and PAF1 to drive H4K31 ubiquitylation-mediated transactivation. Cell Rep. 5, 1690–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Braunschweig U., Gueroussov S., Plocik A. M., Graveley B. R., Blencowe B. J. (2013) Dynamic integration of splicing within gene regulatory pathways. Cell 152, 1252–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gómez Acuña L. I., Fiszbein A., Alló M., Schor I. E., Kornblihtt A. R. (2013) Connections between chromatin signatures and splicing. Wiley Interdiscip Rev. RNA 4, 77–91 [DOI] [PubMed] [Google Scholar]
- 68. Maunakea A. K., Chepelev I., Cui K., Zhao K. (2013) Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang S. M., Kim B. J., Norwood Toro L., Skoultchi A. I. (2013) H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone H3 methylation. Proc. Natl. Acad. Sci. U.S.A. 110, 1708–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hon G. C., Hawkins R. D., Caballero O. L., Lo C., Lister R., Pelizzola M., Valsesia A., Ye Z., Kuan S., Edsall L. E., Camargo A. A., Stevenson B. J., Ecker J. R., Bafna V., Strausberg R. L., Simpson A. J., Ren B. (2012) Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 22, 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Berman B. P., Weisenberger D. J., Aman J. F., Hinoue T., Ramjan Z., Liu Y., Noushmehr H., Lange C. P., van Dijk C. M., Tollenaar R. A., Van Den Berg D., Laird P. W. (2012) Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 44, 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hansen K. D., Timp W., Bravo H. C., Sabunciyan S., Langmead B., McDonald O. G., Wen B., Wu H., Liu Y., Diep D., Briem E., Zhang K., Irizarry R. A., Feinberg A. P. (2011) Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 43, 768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Peric-Hupkes D., Meuleman W., Pagie L., Bruggeman S. W., Solovei I., Brugman W., Gräf S., Flicek P., Kerkhoven R. M., van Lohuizen M., Reinders M., Wessels L., van Steensel B. (2010) Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 38, 603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McDonald O. G., Wu H., Timp W., Doi A., Feinberg A. P. (2011) Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 18, 867–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Timp W., Feinberg A. P. (2013) Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 13, 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hodl M., Basler K. (2012) Transcription in the absence of histone H3.2 and H3K4 methylation. Curr. Biol. 22, 2253–2257 [DOI] [PubMed] [Google Scholar]
- 77. Nègre N., Brown C. D., Ma L., Bristow C. A., Miller S. W., Wagner U., Kheradpour P., Eaton M. L., Loriaux P., Sealfon R., Li Z., Ishii H., Spokony R. F., Chen J., Hwang L., Cheng C., Auburn R. P., Davis M. B., Domanus M., Shah P. K., Morrison C. A., Zieba J., Suchy S., Senderowicz L., Victorsen A., Bild N. A., Grundstad A. J., Hanley D., MacAlpine D. M., Mannervik M., Venken K., Bellen H., White R., Gerstein M., Russell S., Grossman R. L., Ren B., Posakony J. W., Kellis M., White K. P. (2011) A cis-regulatory map of the Drosophila genome. Nature 471, 527–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang H., Gao L., Anandhakumar J., Gross D. S. (2014) Uncoupling transcription from covalent histone modification. PLoS Genet. 10, e1004202. [DOI] [PMC free article] [PubMed] [Google Scholar]