

Background: GASP-1 and GASP-2 are highly specific antagonists for the TGF-β ligand myostatin, a negative regulator of muscle growth.
Results: GASP-1 and GASP-2 form asymmetric and symmetric complexes with myostatin, respectively.
Conclusion: Despite the different binding modes, the GASP proteins retain a high specificity for myostatin.
Significance: Inhibition of myostatin can be achieved using different binding modes and may facilitate future development of novel anti-myostatin therapeutics.
Keywords: Analytical Ultracentrifugation, Myostatin, Small Angle X-ray Scattering (SAXS), Structural Biology, Transforming Growth Factor Beta (TGF-B), Growth and Differentiation-associated Serum Protein, Growth and Differentiation factor-8 (GDF-8), WFIKKN
Abstract
Myostatin, a member of the TGF-β family of ligands, is a strong negative regulator of muscle growth. As such, it is a prime therapeutic target for muscle wasting disorders. Similar to other TGF-β family ligands, myostatin is neutralized by binding one of a number of structurally diverse antagonists. Included are the antagonists GASP-1 and GASP-2, which are unique in that they specifically antagonize myostatin. However, little is known from a structural standpoint describing the interactions of GASP antagonists with myostatin. Here, we present the First low resolution solution structure of myostatin-free and myostatin-bound states of GASP-1 and GASP-2. Our studies have revealed GASP-1, which is 100 times more potent than GASP-2, preferentially binds myostatin in an asymmetrical 1:1 complex, whereas GASP-2 binds in a symmetrical 2:1 complex. Additionally, C-terminal truncations of GASP-1 result in less potent myostatin inhibitors that form a 2:1 complex, suggesting that the C-terminal domains of GASP-1 are the primary mediators for asymmetric complex formation. Overall, this study provides a new perspective on TGF-β antagonism, where closely related antagonists can utilize different ligand-binding strategies.
Introduction
Myostatin, also known as growth and differentiation factor-8 (GDF-8),2 is a member of the TGF-β family. It is well established that myostatin is a strong negative regulator of muscle. Myostatin-null mice have three times more functional muscle mass than WT mice (1, 2). Further, the same phenotype can be recapitulated using naturally occurring myostatin antagonists and myostatin dominant negatives (3). These findings have prompted the development of anti-myostatin therapeutics to treat disorders in which loss of muscle mass is either a complication or a comorbidity.
Most TGF-β ligands, including myostatin, are covalently linked dimers with a propeller-like morphology. Their signaling activity is regulated by a number of extracellular binding proteins or antagonists. Antagonists can range from small single domain proteins to large multidomain proteins but appear to utilize similar paradigms for ligand inhibition where they block ligand-receptor interactions (4–9). However, significant differences occur in both the structures of the antagonist and the specific mechanisms or strategies used to block ligand-receptor interactions. For example, the antagonist noggin is a covalent dimer that binds ligands through a single disulfide-rich domain and an extended N terminus (8). In contrast, the antagonist follistatin (FS) utilizes multiple domains and two FS molecules to encircle both myostatin and activin ligands (4, 6). Despite these structural differences, both noggin and FS bind dimeric ligands in a symmetrical fashion. This ensures full inhibition of the TGF-β ligand, because all receptor-binding sites are occupied.
Of the known ligand antagonists, only certain ones antagonize myostatin including its pro-domain, FS, FSTL3 (follistatin-like-3), decorin, and GASP-1 and GASP-2 (growth and differentiation factor-associated serum proteins 1 and 2, respectively) (10–15). GASP-1 and GASP-2 are multidomain proteins consisting of an N-terminal whey acidic protein domain (W), follistatin domain, Ig-like domain, two tandem kunitz domains (K1 and K2), and a C-terminal netrin-like domain (C) (see Fig. 1A) (16). Despite their similar domain architecture, they only exhibit ∼56% identity. Binding experiments have shown that both GASP-1 and GASP-2 have high affinity for myostatin (pm for GASP-1 and nm for GASP-2) and the closely related TGF-β ligand GDF-11 (90% identical to myostatin) (17). However, differences arise in their temporal and spatial expression during development and into adulthood (12, 16, 18–20).
FIGURE 1.
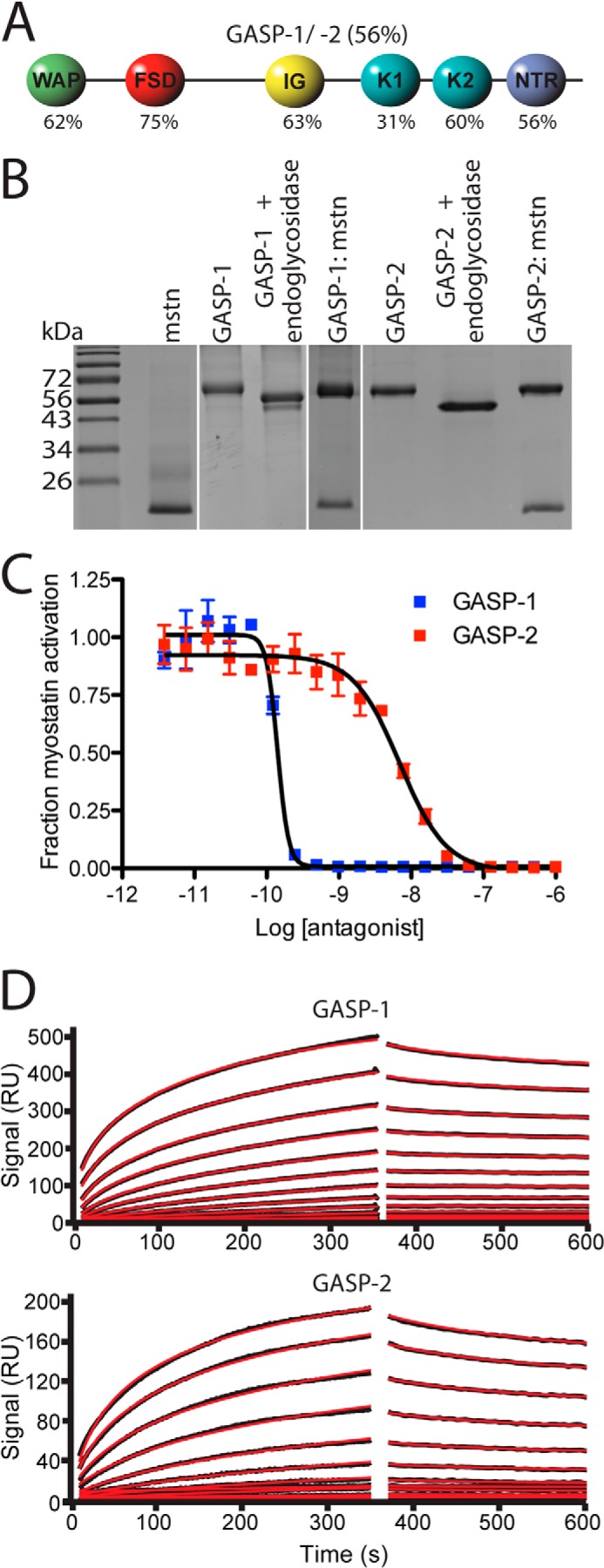
Domain structure, purification, and biological activity of GASP-1 and GASP-2. A, schematic diagram of GASP-1 and GASP-2. The percentages listed are representative of sequence identity between GASP-1 and GASP-2. B, nonreduced SDS-PAGE gel of purified myostatin (mstn), GASP-1, GASP-2, and respective complexes between myostatin and GASP proteins. Note the shift in molecular weight of both GASP-1 and GASP-2 following treatment with endoglcyosidases. C, luciferase reporter assay showing the inhibitory activity of GASP-1 and GASP-2 tested against a constant concentration of myostatin using HEK293 (CAGA)12 cells (error bars represent ± standard deviation). The curves shown are representative of three independent assays. D, surface plasmon resonance sensorgrams for GASP-1 and GASP-2 binding to immobilized myostatin. GASP-1 binds to myostatin with an apparent Kd value of 28.5 pm, and GASP-2 binds to myostatin with an apparent Kd of 19 nm. The fitted line is shown in red.
GASP-1 is primarily expressed in many adult tissues, including skeletal muscle, whereas GASP-2 only has limited expression in adult tissues (12, 16, 18–20). Despite this, GASP-1−/− and GASP-2−/− mice showed a small but significant increase in muscle mass, but much less significant than FS−/− mice. Moreover, following cardiotoxin-induced injury, mice lacking GASP-1 and/or GASP-2 displayed significant muscle regenerative impairments compared with WT mice. The reciprocal effect is observed in mice lacking myostatin (21, 22). On the other hand, during embryological development, GASP-2−/− mice had significant impairments in skeletal axial patterning, suggesting that GASP-2 may modulate GDF-11 activity because these defects are seen in GDF-11−/− mice (19, 23).
Analysis of GASP-2 identified the FS domain as the predominant domain responsible for myostatin binding, whereas the C-terminal domains alone (K2 and netrin-like domain) showed limited binding (17). However, the full-length protein was necessary for the nm affinity, suggesting that multiple domains contribute to ligand binding (17). It should be noted that unlike FS, which inhibits multiple ligands (e.g. activin A, activin B, BMP-7, and myostatin), both GASP-1 and GASP-2 selectively inhibit myostatin and GDF-11 (17, 24–27). Therefore, apart from the myostatin pro-domain, GASP-1 and GASP-2 are the only known molecules to be highly specific for myostatin. Despite this, little is known about how GASP molecules interact with myostatin at the molecular level and, further, how they compare with the other ligand antagonists such as FS and noggin.
To address this, we present the first low resolution solution structure of GASP-1 and GASP-2 in complex with myostatin. Our evidence shows that although GASP-1 and GASP-2 are structurally similar, they use two different binding modes to antagonize myostatin. Through biophysical characterization, we show that GASP-1 preferentially binds myostatin with a 1:1 stoichiometry, whereas GASP-2 preferentially binds myostatin with a 2:1 stoichiometry. Finally, we show that the progressive truncation of domains from the C terminus of GASP-1 result in less potent molecules with a shift to a 2:1 stoichiometric complex with myostatin, similar to GASP-2.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
CHO cells stably overexpressing myostatin, GASP-1 and GASP-2 were used as previously published (4, 19). Myostatin conditioned medium (CM) was concentrated ∼10-fold using tangential flow and concomitantly buffer exchanged into 50 mm Tris pH 7.4, 500 mm NaCl and applied to a Lentil Lectin-Sepharose 4B (Amersham Biosciences) column. Myostatin was eluted with the same buffer with the addition of 500 mm methyl mannose. Eluted protein was then dialyzed against 20 mm trisodium citrate pH 5.0, 20 mm NaCl. Myostatin was then applied to a HiPrep SP FF 16/10 column (GE Life Sciences) and eluted using the same buffer with the addition of 1 m NaCl. The eluted protein was then dialyzed against 20 mm trisodium citrate pH 5.0, 20 mm NaCl. Next, the protein was adjusted to 5% acetonitrile, 0.1% trifluoroacetic acid, 4 m guanidinium HCl and applied to a Sepax C4 reverse phase HPLC column. Myostatin was eluted using an acetonitrile gradient.
GASP-1 and GASP-2
GASP-1 and GASP-2 was expressed and purified as published earlier with some minor modifications (19). Following application to butyl-Sepharose and heparin columns, the heparin eluent containing either GASP-1 or GASP-2 was dialyzed extensively into 50 mm Tris pH 7.4, 20 mm NaCl, 1 mm EDTA, applied to a MonoQ 10/100 GL column and eluted with a linear NaCl gradient.
GASP-1 C-terminal Truncation Mutants
The full-length mouse GASP-1 cDNA fragment was inserted into pFastBac1 followed by subsequent insertion of a stop codon at the desired location for C-terminal truncation. The truncations consisted of the following amino acids (a.a.): WF (30–198), WFI (30–314), WFIK (30–375). The maltose binding protein (MBP)-WFIK fusion construct consisted of MBP positioned on the N terminus linked to WFIK (30–375) by three alanines. Baculovirus production and protein expression was performed according to the manufacture's protocol (Invitrogen). Following expression in SF9 insect cells, conditioned medium was adjusted to 750 mm ammonium sulfate and applied to a butyl-Sepharose column. The eluent was subsequently applied to a Nickel-Sepharose HiTrap column (GE) followed by extensive dialysis into 50 mm Tris pH 7.4, 20 mm NaCl, 1 mm EDTA, applied to a MonoQ 10/100 GL column and eluted with a linear NaCl gradient. MBP-WFIK was purified using the same strategy except that the buffers utilized contained 5 mm maltose as an additive.
Luciferase Reporter Assays
The luciferase reporter assays were performed as previously described (28, 29). Briefly, HEK293 CAGA12 cells were plated in a 96-well plate and grown for ∼24 h. Subsequently, the growth medium was removed and myostatin at a concentration of 0.62 nm was mixed with the antagonist (GASP-1, GASP-2, or GASP-1 truncations) in serum free medium and applied to the cells for ∼18 h. The cells were lysed and luminescence was recorded immediately using a Synergy H1 Hybrid plate reader (BioTek). The inhibition data were imported into GraphPad Prism and fit using nonlinear regression with a variable slope. Inhibition curves shown are one representative experiment of three independent conducted assays.
Surface Plasmon Resonance (SPR)
SPR analysis was performed similar to previous studies (5). Briefly, Surface plasmon resonance (SPR) measurements were carried out in HBS-EP buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.005% P-20 surfactant (BIAcore AB)) on a BIAcore 3000 optical biosensor system operated with BIAevaluation 4.1 software. Myostatin was immobilized on a CM4 research grade sensor chip (BIAcore AB) by amine coupling chemistry using the manufacturer's protocol at 25 °C (2785 RUs). For kinetic measurements, GASP-1 and GASP-2 were diluted in HBS-EP buffer to a concentration of 2 μm. The proteins were then diluted in a 2-fold dilution series and applied to the chip at a flow rate of 20 μl/min. Protein was injected for an association time of 6 min, and then dissociation was monitored for 9 min. After each measurement, the chip surface was regenerated with four 15 μl pulses of 2 m guanidine HCl at a flow rate of 100 μl/min. SPR sensorgrams were globally analyzed using a distribution model for continuous affinity and rate constant analysis using the program EVILFIT (30).
Size Exclusion Analysis
GASP-1, GASP-2, GASP-1·myostatin, and GASP-2·myostatin were applied independently to a Phenomenex HPLC S2000 size exclusion column equilibrated with 20 mm HEPES pH 7.4, 500 mm NaCl, 1 mm EDTA to obtain their retention volume. Sizing standards were run under identical conditions for determination of apparent molecular weight. Fractions from each peak were analyzed using SDS-PAGE followed by Western analysis to ensure both proteins were present.
Analytical Ultracentrifugation (AUC) Sedimentation Velocity
Experiments were performed with a Beckman ProteomeLab XL-1 fitted with absorbance optics and a four-hole rotor. Samples and matched buffer (20 mm HEPES pH 7.4, 500 mm NaCl, 1 mm EDTA; 5 mm maltose was added to the buffer for the GASP-1 MBP-WFIK:myostatin experiment) for buffer subtraction were loaded in a two-channel, carbon-filled, epon centerpieces at 48,000 rpm at 20 °C. Absorbance was monitored at 230 nm. Absorbance data were processed using the program Sedfit (31) to determine the sedimentation coefficient (c(s)) and sedimentation coefficient-frictional ratio c(s,f/fo) distributions (32). Values for buffer density and viscosity and protein partial specific volume were calculated using Sednterp. Because of the presence of both N-linked and O-linked glycosylation of GASP-1 and GASP-2, the partial specific volume was calculated as a weight-averaged value.
Small Angle X-ray Scattering (SAXS)
SAXS data were collected at beam line 12-ID-B at the Advanced Photon Source at Argonne National Laboratory and using the SIBYLS beam line mail-in program (Berkeley, CA) (33). Purified GASP-1, GASP-2, GASP-1·myostatin, GASP-2·myostatin and GASP-1 MBP-WFIK:myostatin were purified as described above. Three different concentrations of each sample were analyzed to determine whether concentration dependent effects exist. Data were collected in 20 mm HEPES pH 7.4, 500 mm NaCl, 1 mm EDTA at 10 °C. Additionally, for the GASP-1 MBP-WFIK construct, the buffer contained 1 mm maltose. Four exposure times of 0.5 s, 1 s, 2 s, and 5 s were collected. Exposures exhibiting radiation damage were discarded. Buffer matched controls were used for buffer subtraction. ScÅtter (SIBYLS) and the ATSAS program suite (EMBL) were used for data analysis. The online DAMMIF server (EMBL-Hamburg) was used for generation of 20 independent ab initio molecular envelope reconstructions (34). P1 symmetry operators were employed to improve the quality of the fit for GASP-1, GASP-2, GASP-1·myostatin whereas a P2 symmetry operator was used for GASP-2·myostatin. Final models were then averaged using DAMAVER pipeline (35). Graphics were generated using UCSF Chimera and Pymol.
RESULTS
Characterization of Purified GASP-1 and GASP-2
For structural analysis, we produced and purified murine GASP-1 and GASP-2 as previously described (Fig. 1B) (19). Analysis by SDS-PAGE shows that GASP proteins migrate at higher than expected molecular weight. Subsequent analysis by mass spectrometry reveals that GASP-1 and GASP-2 exhibit mass increases of ∼8.7 and 5.3 kDa, respectively, over their predicted molecular mass (Table 1). The majority of this mass is attributed to both O- and N-linked glycosylation and is consistent with previous studies describing that GASP-1 is both O-linked and N-linked glycosylated (Fig. 1B and Table 1) (36). Because of a significant decrease in solubility upon deglycosylation, all structural analysis was performed on the fully glycosylated form of the GASP proteins.
TABLE 1.
Theoretical and experimentally determined molecular weights for ligand-free and ligand-bound complexes
| Protein | Theoretical molecular mass | Mass spectrometry | Predicted molecular massa | SEC | Sedimentation velocity | SAXS |
|---|---|---|---|---|---|---|
| kDa | kDa | kDa | kDa | kDa | kDa | |
| Myostatin | 24.8 | 24.8 | NDb | NDb | NDb | |
| GASP-1 | 60 | 68.7 (62.2)c | 41 | 61.3 | 76.4 | |
| GASP-1·myostatin (1:1) | 84.8 | 93.5 | 114 | 97.1 | 103.6 | |
| GASP-1·myostatin (2:1) | 144.8 | 162.2 | ||||
| GASP-2 | 57 | 62.3 (58.1+) | 28 | 60.4 | 74.5 | |
| GASP-2·myostatin (1:1) | 81.8 | 87.1 | ||||
| GASP-2·myostatin (2:1) | 138.8 | 149.4 | 160 | 137 | 187.3 |
a Values indicate the predicted molecular mass based on the molecular mass determined by mass spectrometry.
b ND, not determined.
c Mass following treatment with endoglycosidases.
Purified GASP proteins were biologically active and maintained specificity for myostatin. Using a HEK293-CAGA12 luciferase reporter assay, we determined the myostatin IC50 values for GASP-1 and GASP-2 (Fig. 1C). Similar to previous studies, both GASP-1 and GASP-2 potently blocked myostatin signaling with IC50 values of 4.4 ± 2.6 × 10−10 and 1.2 ± 0.5 × 10−8 m, respectively (25). The nearly 100-fold difference in inhibitory activity is consistent with previous studies (17, 25). SPR analysis confirms that our purified GASP-1 and GASP-2 bind myostatin with an apparent low pm and nm Kd values, respectively, and both display a nearly irreversible off rate (Fig. 1D). GASP-1 and GASP-2 did not inhibit activin A or activin B signaling (data not shown), consistent with previous reports that GASP proteins are highly selective for myostatin (17, 24, 25).
Size Exclusion Chromatography (SEC) Reveals Differences in GASP-1·Myostatin and GASP-2·Myostatin Complexes
We next analyzed the GASP proteins alone and in the presence of myostatin by SEC. GASP-1 and GASP-2 had similar retention times with symmetrical peaks but eluted at smaller than expected molecular masses, suggesting that there may be nonspecific interactions with the SEC resin (Fig. 2A).
FIGURE 2.
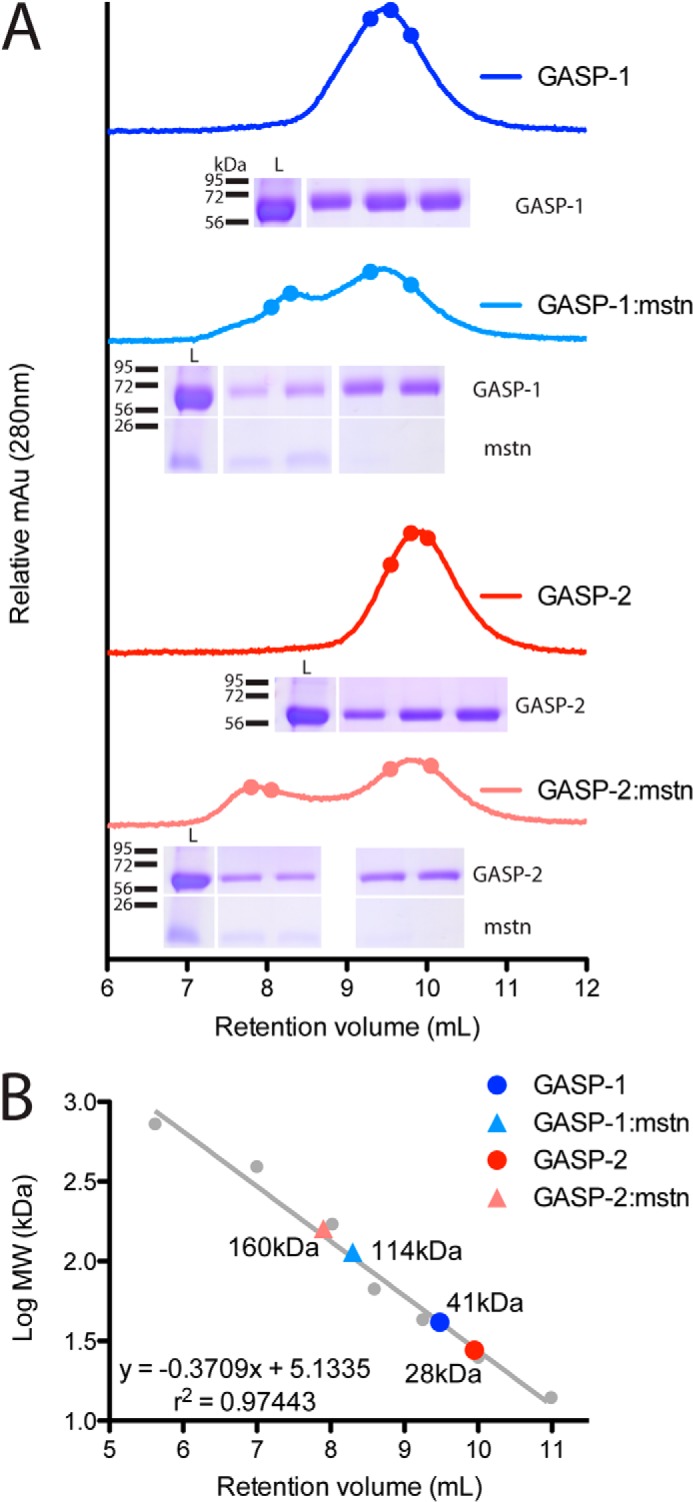
Size exclusion analysis on GASP proteins and GASP·myostatin complexes. A, representative chromatograms for GASP-1 (dark blue), GASP-1·myostatin (light blue), GASP-2 (red), and GASP-2·myostatin (pink). The closed circles represent the fractions loaded on the nonreduced SDS-PAGE gel shown below each chromatogram. The SEC load is shown in the leftmost lane. The fraction loading order is from left to right. mstn, myostatin. B, regression analysis for determination of the apparent molecular weight for each protein. Proteins of known molecular weight (closed gray circles) were used for the regression analysis. The proteins used as standards are as follows: thyroglobulin, 725 kDa; ferritin, 391 kDa; aldolase, 171 kDa; albumin, 67 kDa; ovalbumin, 43 kDa; chymotrypsin, 25 kDa; and ribonuclease, 14 kDa.
To form the GASP·myostatin complex, GASP proteins were mixed at a 2.25:1 (GASP·myostatin) molar ratio and analyzed by SEC. Excess GASP was used to ensure full saturation of the myostatin dimer. In both cases, a new peak formed that contained both myostatin and GASP, indicative of complex. In addition, the traces also contained a peak from unbound GASP, indicating that myostatin in the complex peak is saturated with GASP. Interestingly, the peak from the GASP-1·myostatin complex shifted to the left by 1.1 ml and overlapped with the free GASP-1 peak, whereas there was a distinct separation of the GASP-2·myostatin peak from the unbound GASP-2. As such, regression analysis revealed significant differences in the apparent molecular mass of the complexes between GASP-1 and GASP-2 (Fig. 2B). However, because of the inherent limitations of SEC, analysis of these differences required further, more rigorous investigation. Nonetheless, the qualitative difference in retention volume between GASP-1·myostatin and GASP-2·myostatin suggested differences in the two complexes.
Analytical Ultracentrifugation (AUC) of Purified Components
To more accurately define the molecular mass of the complexes, we turned to analytical ultracentrifugation (Fig. 3 and Table 2). Sedimentation velocity showed that GASP-1 and GASP-2 contained single species with sedimentation coefficients of 3.6 and 3.36 S, respectively. Each exhibited a similar frictional ratio of ∼1.47, suggesting that they are more elongated than globular in shape (Fig. 3A and Table 2). Importantly, the apparent molecular mass for GASP-1 and GASP-2 were more consistent with the mass spectrometry results than the SEC-derived molecular masses (Tables 1 and 2).
FIGURE 3.
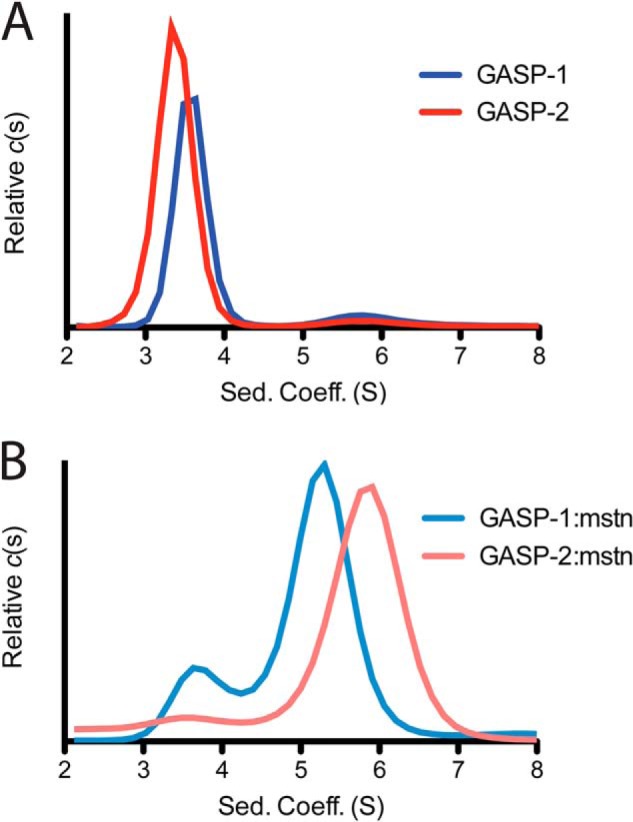
Sedimentation velocity analytical ultracentrifugation on myostatin-free and myostatin-bound GASP proteins. A, sedimentation velocity results for the GASP-1 (dark blue) and GASP-2 (red). B, sedimentation velocity results for the GASP-1·myostatin (light blue) and GASP-2·myostatin (pink) complexes. See also Table 2.
TABLE 2.
Sedimentation velocity values and statistics for ligand-free and ligand-bound GASP proteins
| Protein |
c(s) |
RMSD |
c(s,*) |
f/f0 |
Molecular mass |
||||
|---|---|---|---|---|---|---|---|---|---|
| Antagonist | Complex | Antagonist | Complex | Antagonist | Complex | Antagonist | Complex | ||
| kDa | |||||||||
| GASP-1 | 3.58 (84%) | NA | 0.0048 | 3.58 | NA | 1.48 | NA | 61.3 | NA |
| GASP-1·myostatin | 3.73 (17%) | 5.24 (68.8%) | 0.0093 | 3.48 | 5.21 | 1.37 | 1.39 | 51.4 | 97.1 |
| GASP-2 | 3.36 (92.4%) | NA | 0.0076 | 3.37 | NA | 1.46 | NA | 60.4 | NA |
| GASP-2·myostatin | 3.57 (8%) | 5.81 (82%) | 0.0089 | 3.97 | 5.8 | 1.47 | 1.46 | 78 | 137 |
Samples containing the GASP·myostatin complexes were pooled from SEC (high molecular weight peak) and analyzed by sedimentation velocity. For GASP-1·myostatin the c(s) profile exhibited two peaks with sedimentation coefficients of 3.73 and 5.24 S, indicative of excess GASP-1 and the GASP-1·myostatin complex, respectively (Fig. 3B and Table 2). Derivation of molecular mass from sedimentation velocity data can be less accurate when multiple species are present. Therefore, we utilized a recently developed method known as c(s, f/f0) analysis to more accurately determine the molecular mass for the GASP-1·myostatin complex (32). The c(s, f/f0) analysis separately fits both the frictional ratio and sedimentation coefficient for each species to provide a more accurate estimation of molecular mass (32). Applying this analysis, the apparent molecular mass for the GASP-1·myostatin complex is 97.1 kDa (Table 2). This indicates that GASP-1 preferentially binds myostatin in a 1:1 complex. We suspect the excess GASP-1 detected in our AUC experiment is linked to our inability to fully resolve the GASP-1·myostatin complex from free GASP-1 using SEC.
In contrast, analysis of the GASP-2·myostatin complex by sedimentation velocity resulted in a more homogenous sample with a major species at 5.81 S (Fig. 3B and Table 2). A minor peak at 3.97 S was also detected, suggestive of unbound GASP-2; however, this only accounted for ∼8% of the total signal (Fig. 3B). Again, using c(s, f/f0) analysis, we determined the apparent molecular mass for the GASP-2·myostatin complex to be 137 kDa (Table 2), consistent with a 2:1 complex stoichiometry. These data suggest that GASP-1 and GASP-2 preferentially form different stochiometric complexes with myostatin. In fact, closer inspection of the SDS-PAGE gels by densitometry analysis also suggests a difference in stoichiometry where the intensity of GASP-2 (3.1) is almost twice that of GASP-1 (1.8) when normalized to myostatin (Fig. 2A).
SAXS Analysis of GASP-1 and GASP-2
The SAXS scattering profiles for GASP-1 and GASP-2 are shown in Fig. 4A, along with the corresponding analysis in Table 3. Samples were well behaved in solution and did not show evidence of interparticle repulsion or aggregation over multiple protein concentrations (Fig. 4, A and B). The data are of high quality because (a) the I(0) increases proportionally to the concentrations tested, (b) the Gunier range has a linear appearance at low scattering angles, and (c) the radius of gyration (Rg) does not significantly change in response to changes in protein concentration (Fig. 4 and Table 3).
FIGURE 4.

SAXS analysis on GASP and GASP·myostatin complexes. A and F, intensity distribution of the SAXS scattering function for myostatin-free and myostatin-bound GASP proteins. B, Gunier plot for GASP-1 (dark blue) and GASP-2 (red) showing a linear, unbiased distribution. G, Gunier plot for GASP-1·myostatin (light blue) and GASP-2·myostatin (pink) showing a linear, unbiased distribution. Residuals are shown below each plot. C and H, pairwise distribution function for myostatin-free and myostatin-bound GASP proteins. D, q4·I(q) versus q4(Å−1)4 plot on GASP-1 (dark blue) and GASP-2 (red) showing a distinct increase in slope as q increases. D and I, q4·I(q) versus q4(Å−1)4 plot on GASP-1·myostatin (light blue) and GASP-2·myostatin (pink) showing a gradual increase in slope as q increases. E, q3·I(q) versus q3(Å−1)3 plot on GASP-1 (dark blue) and GASP-2 (red) showing a distinct plateau as q increases. J, q3·I(q) versus q3(Å−1)3 plot on GASP-1·myostatin (light blue) and GASP-2·myostatin (pink) showing a distinct plateau as q increases.
TABLE 3.
Experimentally determined parameters from SAXS analysis of ligand-free and ligand-bound GASP proteins
| Sample | Concentration | I(0) |
Rg |
Dmax | Volume | Molecular mass | DAMMIF NSDa | |
|---|---|---|---|---|---|---|---|---|
| Gunier | Real space | |||||||
| mg/ml | cm−1 | Å | Å | Å3 | Da | Å | ||
| GASP-1 | 2.6 | 682 | 53.1 | 53.85 | 177 | 347,796 | 86,000 | |
| 1.6 | 393 | 51.9 | 52.06 | 172 | 341,665 | 75,600 | 0.945 ± 0.06 | |
| 1 | 240 | 51 | 50.97 | 168 | 341,668 | 67,500 | ||
| GASP-1·myostatin | 1.4 | 396 | 49.1 | 48.12 | 163 | 432,793 | 112,000 | |
| 1.2 | 303 | 47.5 | 46.52 | 160 | 433,075 | 96,700 | 1.034 ± 0.057 | |
| 1 | 252 | 48.8 | 46.33 | 161 | 422,324 | 102,000 | ||
| GASP-2 | 3 | 556 | 46.3 | 46.19 | 151 | 321,500 | 77,600 | |
| 2 | 369 | 45.6 | 43.51 | 147 | 326,803 | 71,300 | 0.828 ± 0.047 | |
| 1 | 191 | 44.8 | 41.19 | 141 | 316,598 | 61,000 | ||
| GASP-2·myostatin | 3 | 1130 | 62.6 | 64.83 | 205 | 595,832 | 211,000 | |
| 2 | 661 | 60.5 | 63.32 | 210 | 589,810 | 189,000 | 1.488 ± 0.478 | |
| 1 | 336 | 58.1 | 59.97 | 200 | 528,526 | 162,000 | ||
| MBP GASP-1 WFIK·myostatin | 3 | 357 | 56.7 | 56.5 | 173 | 376,242 | 179,000 | |
| 2 | 249 | 55.6 | 55.1 | 167 | 337,781 | 167,000 | ||
| 1 | 127 | 52.7 | 54.5 | 168 | 349,291 | 174,000 | ||
a NSD, normalized spatial discrepancy.
From the Gunier analysis, we determined that GASP-1 has a slightly higher Rg than GASP-2, 52 ± 1.1 versus 45.6 ± 0.75 Å, respectively (Fig. 4B and Table 3). This suggests that GASP-1 is more elongated in solution than GASP-2. In support, this observation is readily apparent in the pairwise distribution plot (P(r); Fig. 4C). Further transformation of the SAXS data indicates that both GASP-1 and GASP-2 are inherently flexible as shown by a plateau in the q3·I(q) versus q3(Å−1)3 plot compared with a plateau observed in the q4·I(q) versus q4(Å−1)4, which would indicated a globular, rigid structure (Fig. 4, D and E). The apparent molecular masses for GASP-1 and GASP-2 derived from SAXS are 76.4 ± 9.2 and 70 ± 8.4 kDa and are in agreement with the expected molecular mass (Tables 1 and 3).
The SAXS data were then used to perform ab initio modeling to generate molecular envelopes as shown in Fig. 5. Both GASP-1 and GASP-2 have acceptable normalized spatial discrepancy values (0.94 ± 0.06 and 0.83 ± 0.047, respectively; Table 3). In agreement with the P(r) analysis, the generated GASP-1 molecular envelope has an elongated and rather featureless shape (Fig. 5A). Along these same lines, the ab initio results for the GASP-2 molecular envelope revealed a slightly less elongated shape that adopted a more compact structure (Fig. 5B), suggesting that there may be interdomain contacts within GASP-2, resulting in a more globular-like appearance.
FIGURE 5.
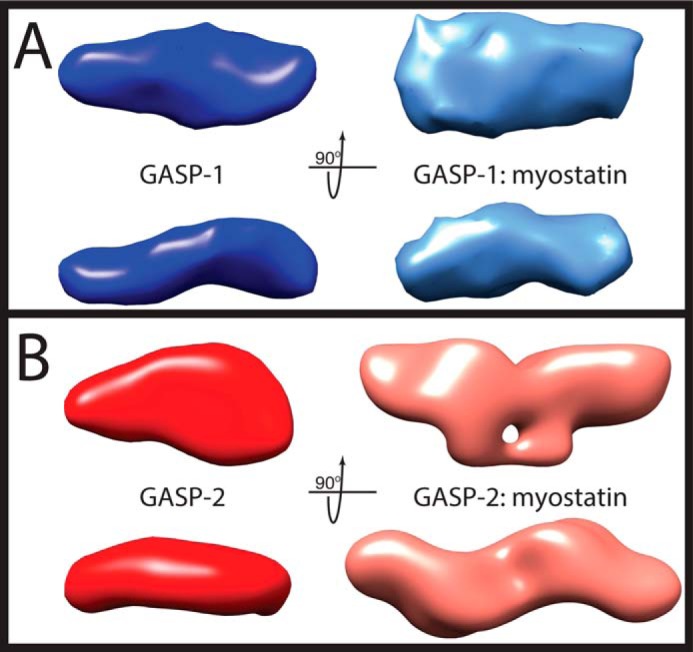
SAXS ab initio reconstructions of myostatin-free and myostatin-bound GASP proteins. A, ab intio model generated from SAXS data of GASP-1 (dark blue, left) and GASP-1·myostatin (light blue, right). A P1 symmetry operator was used for both GASP-1 and GASP-1·myostatin. B, ab intio model generated from SAXS data of GASP-2 (red, left) and GASP-2·myostatin (pink, right). A P1 symmetry operator was used for GASP-2 and P2 symmetry operator for GASP-2·myostatin. All envelopes were generated using DAMMIF and averaged using DAMAVER. The averaged envelope is shown. See also Table 3.
SAXS Analysis of the GASP-1·Myostatin and GASP-2·Myostatin Complexes Supports Differences in Binding Stoichiometry
Because previous methods to purify the GASP-1·myostatin complex resulted in contamination of unbound antagonist, we attenuated the ratio of GASP-1 to reflect the 1:1 stoichiometry identified by sedimentation velocity. Therefore, GASP-1 was mixed with myostatin in a 1.1:1 molar ratio, which resulted in the loss of the excess GASP-1 AUC peak (data not shown). No change to the GASP-2·myostatin ratio was required, and the complex was purified using a mixing ratio of 2.25:1.
Scattering profiles for GASP-1·myostatin and GASP-2·myostatin are shown in Fig. 4F. The data are of high quality and free of aggregation (Fig. 4, F and G, and Table 3). Gunier analysis and inspection of the P(r) plot reveals that the GASP-1·myostatin complex is more compact than GASP-1 alone as indicated by a significant decrease in both the Rg and the maximum particle distance (Dmax; Fig. 4, F and G, and Table 3). This is in stark contrast to the GASP-2 complex, which has a significantly larger Rg and Dmax than GASP-2 alone (Table 3).
Similar to the flexibility analysis applied to the myostatin-free GASP proteins, both complexes display a plateau in the q3·I(q) versus q3(Å−1)3 plot compared with the q4·I(q) versus q4(Å−1)4 plot, indicating that both complexes are flexible in solution (Fig. 4, I and J). However, the plateau appeared more prominent for the complexes than the GASP proteins alone, suggesting that some rigidity is gained when complex is formed (37).
Analysis of the scattering profiles provides several lines of evidence to support that GASP-1 and GASP-2 form different stoichiometric complexes with myostatin (Table 3). For example, the GASP-2·myostatin complex had ∼25% more overall scattering intensity compared with GASP-1·myostatin, denoting a significant difference in overall particle mass. In addition, the Rg of the GASP-1·myostatin complex is significantly smaller (48.5 ± 0.85 Å) as compared with the GASP-2·myostatin (60.4 ± 2.25 Å) complex (Table 3). Furthermore, the P(r) plot shows that the GASP-1·myostatin complex is globular with a smaller Dmax, whereas the GASP-2·myostatin complex is much more extended (Fig. 4H and Table 3). Finally, the SAXS derived apparent molecular mass for the GASP-1·myostatin complex was determined to be 103 ± 8.0 kDa compared with the much larger GASP-2·myostatin complex (187 ± 25 kDa; Table 3).
A summary of the theoretical molecular masses and experimentally determined molecular masses for each technique is displayed in Table 1. Taken together, it is evident that GASP-1 preferentially binds myostatin in a 1:1 configuration, whereas GASP-2 preferentially binds myostatin in a 2:1 configuration.
The SAXS data were subsequently used to perform ab initio modeling to derive low resolution molecular envelopes for both the GASP-1·myostatin and GASP-2·myostatin complexes (Fig. 5). The envelopes for each complex had acceptable normalized spatial discrepancy values of 1.034 ± 0.057 and 1.488 ± 0.478, respectively (Table 3). The GASP-1·myostatin molecular envelope resembles the featureless characteristics of myostatin-free GASP-1 but is more compact with a slightly more “full” shape (Fig. 5A). The additional volume contribution is most likely from the bound myostatin. In striking contrast, the GASP-2·myostatin complex adopts a symmetrical conformation and has a more featured appearance (Fig. 5B), whereas the molecular envelope has a symmetrical resemblance to previously determined antagonist·ligand structures (e.g. two molecules of FS wrapping around the ligand myostatin). However, it appears that the GASP-2 domains are splayed away from a centrally positioned ligand as opposed to the compact FS·ligand structures (4–7).
Truncation of GASP-1 C-terminal Domains Changes Binding Stoichiometry
Previous truncation analysis of GASP-2 revealed that the whey acidic protein and FS domains of GASP-2 are the primary mediators for myostatin binding, suggesting that the C-terminal domains are expendable (17). Given the differences in stoichiometry, we next wanted to determine whether GASP-1 exhibits a similar behavior. Therefore, we generated a number of C-terminal truncations of GASP-1 and tested their ability to inhibit myostatin in the luciferase reporter assay (Fig. 6A). For simplicity, we will use the following abbreviations to describe the domain composition of the GASP-1 C-terminal truncation constructs: W indicates whey acidic protein domain, F indicates follistatin domain, I indicates immunoglobulin-like domain, and K indicates kunitz domain 1, unless otherwise stated.
FIGURE 6.
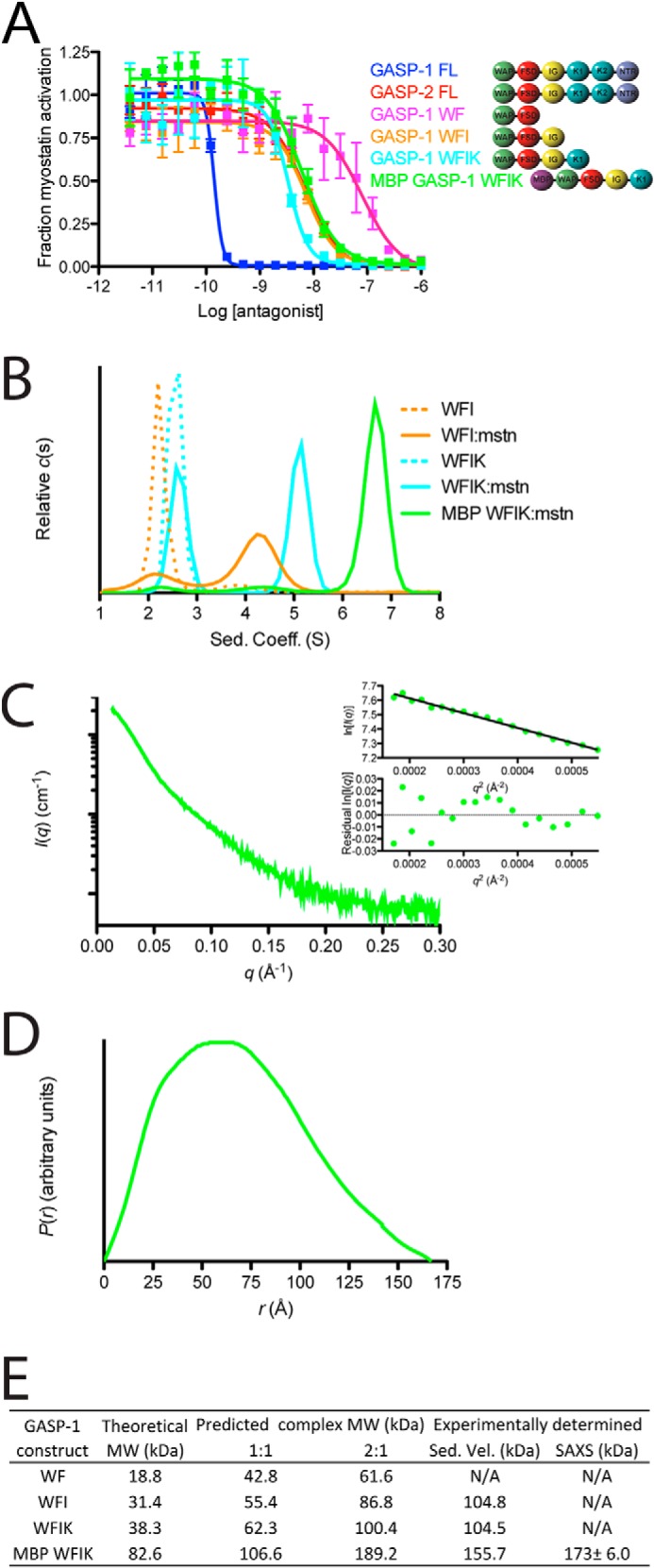
Biological activity, sedimentation velocity analysis, and SAXS analysis of GASP-1 C-terminal truncations. A, luciferase reporter inhibition assay for GASP-1 C-terminal truncations tested against a constant concentration of myostatin using HEK293 (CAGA)12 cells (error bars represent ± standard deviation). A schematic representation depicting the domain architecture of each construct shown next to the curve labels. B, sedimentation velocity for the GASP-1 constructs WFI (dotted orange) and WFIK (dotted cyan), and complexes WFI·myostatin (solid orange), WFIK·myostatin (solid cyan), and MBP-WFIK·myostatin (solid green). C, SAXS scattering profile of the MBP-WFIK·myostatin complex. The Gunier plot and residuals shown in the inset demonstrate that the sample is free of aggregation. D, pairwise distribution function of the MBP-WFIK·myostatin complex. E, summary of the theoretical, expected, and experimentally determined molecular weights for GASP-1 C-terminal constructs in complex with myostatin.
Following progressive removal of the C-terminal domains, we observed a corresponding decrease in the IC50 of GASP-1 for myostatin (Fig. 6A). Interestingly, the C-terminal truncation constructs WFIK (IC50 = 2.6 ± 1.0 × 10−9 m) and WFI (IC50 = 5.1 ± 1.7 × 10−9 m) displayed an inhibitory curve strikingly similar to full-length GASP-2, whereas the WF construct showed a significantly weaker IC50 (6.5 ± 2.3 × 10−8 m; Fig. 6A). Furthermore, the GASP-1 truncations did not inhibit activin A or activin B (data not shown).
We next wanted to determine whether the truncated versions of GASP-1 maintained the 1:1 stoichiometry. WFI or WFIK was mixed with myostatin at 2.5:1 (antagonist·ligand) ratio to ensure full saturation of myostatin and analyzed by sedimentation velocity. As expected, we observed two peaks in the c(s) profile. The larger peak is consistent with complex formation, whereas the smaller peak is consistent with sedimentation analysis of WFI or WFIK alone (Fig. 6B). The sedimentation coefficients for the WFI·myostatin and WFIK·myostatin complexes were determined to be 4.2 and 5.1 S, respectively (Fig. 6B). These results are similar to the large shift in the sedimentation coefficient of the GASP-2·myostatin complex versus unbound GASP-2. Analysis of the c(s,f/f0) distribution revealed that both complexes share an apparent molecular mass of 104 kDa, consistent with a 2:1 binding stoichiometry (Fig. 6, B and E). We would expect that the WFI·myostatin complex to be of lower molecular mass than the WFIK·myostatin complex; however, the exact molecular mass for the WFI·myostatin is difficult to accurately determine because of the breadth of the peak in the c(s) profile (Fig. 6B).
We further analyzed a fusion protein of MBP-WFIK in complex with myostatin by AUC and SAXS (Fig. 6 and Table 3). The fusion partner was used to enhance production and solution behavior necessary for SAXS. Importantly, the MBP-WFIK fusion construct maintained a similar myostatin inhibitory activity as WFIK (IC50 = 7.2 ± 2.8 × 10−9 m; Fig. 6A). Similar to GASP-2·myostatin complex preparation, we mixed MBP-WFIK at a 2.5:1 (antagonist·ligand) ratio with myostatin and purified the complex using SEC. Fractions from the peak were pooled, concentrated, and subjected to AUC and SAXS. Consistent with the other GASP-1 C-terminal truncations, both AUC (Fig. 6B) and SAXS (Fig. 6, C and D, and Table 3) analysis revealed a 2:1 MBP-WFIK·myostatin complex (Fig. 6E and Table 3).
Taken together, we have shown that progressive removal of the C-terminal domains of GASP-1 results in decreased inhibitory potential, similar to that of GASP-2, a significant increase in the sedimentation coefficient upon complex formation with myostatin, and an apparent molecular mass for each complex that is consistent with formation of a 2:1 antagonist·ligand complex.
DISCUSSION
At the onset of our study, we operated under the assumption that GASP would bind myostatin in a similar fashion to previous known antagonists (FS, FSTL3, noggin, etc.), where two antagonist molecules would interact with one ligand dimer (4–8). However, upon complex isolation using SEC and AUC analysis, we were surprised by the dramatic difference in apparent molecular mass of the two GASP·myostatin complexes. This prompted us to structurally characterize these differences using SAXS and provided the first low resolution solution structures of GASP-1 and GASP-2 in their myostatin-free and myostatin-bound states. To extend our findings, we progressively truncated the C-terminal domains from GASP-1, which resulted in less potent GASP-1 molecules that preferentially bind myostatin in a 2:1 fashion similar to full-length GASP-2. Taken together, these data support that, although GASP-2 binds in a similar manner to other known antagonist by using 2 molecules to sequester the ligand, GASP-1 preferentially binds using only one molecule.
Over the years, efforts have started to unravel the different mechanisms of TGF-β antagonism. Interestingly, extracellular antagonists have evolved different approaches to regulate ligand signaling. Not surprisingly, because of the dimeric architecture of the ligand, antagonists prefer a 2:1 binding mode (e.g. FS-type·ligand, noggin·BMP-7, CV2·BMP-2) (4–9). Although this paradigm is consistent with GASP-2, we have uncovered that GASP-1 diverges from the more standard 2:1 complex and forms an asymmetric complex with the ligand.
GASP-1 is clearly a more potent myostatin inhibitor than GASP-2. However, when the C-terminal domains of GASP-1 were removed, myostatin inhibition was reduced. Accordingly, the truncated versions of GASP-1 exhibited a 2:1 binding stoichiometry. This suggests that the very potent anti-myostatin activity of GASP-1 could be coupled with its ability to form a 1:1 complex (e.g. through a cooperative binding mechanism or interaction using multiple binding surfaces). Certainly future experiments will be needed to determine the mechanistic differences in GASP-1 versus GASP-2 myostatin complex formation.
It should be noted that similar to GASP-1, the BMP antagonist chordin also binds to its ligand in a asymmetrical 1:1 fashion (38, 39). However, unlike GASP, Chordin is composed of four highly conserved von Willebrand C (vWC) domains that are shown to occupy receptor sites in a symmetrical fashion (i.e. vWC1 binds to one side, whereas vWC3 binds to the other side of the ligand). Although we have shown that GASP-1 interacts with myostatin in a 1:1 fashion, the lack of repetitive binding domains within GASP-1 emphasizes a completely asymmetric mechanism where unrelated domains within GASP-1 are likely to bind each side of the symmetrical dimer. Overall, the results of this study extend our understanding of the different mechanisms utilized for TGF-β inhibition. Furthermore, to our knowledge, this is the first example of where closely related extracellular TGF-β antagonists interact with ligands utilizing different binding modes. With myostatin inhibitors in high demand, knowledge that alternative binding strategies exist yet are still able to achieve high ligand specificity may offer additional therapeutic development options.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AR060636 (to S.-J. L.) and R01GM105404 (to Project MINOS). This work was also supported by the Integrated Diffraction Analysis Technologies Program of the Department of Energy Office of Basic Energy Sciences (to Advanced Light Source at Lawrence Berkeley National Laboratory), American Heart Association Grant 12PRE11790027 (to R. G. W.), and Muscular Dystrophy Association Grant 240087 (to T. B. T.).
- GDF
- growth and differentiation factor
- SEC
- size exclusion chromatography
- SAXS
- small angle x-ray scattering
- FS
- follistatin
- AUC
- analytical ultracentrifugation.
REFERENCES
- 1. McPherron A. C., Lee S. J. (1997) Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. U.S.A. 94, 12457–12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McPherron A. C., Lawler A. M., Lee S.-J. (1997) Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387, 83–90 [DOI] [PubMed] [Google Scholar]
- 3. Lee S. J. (2010) Extracellular regulation of myostatin: a molecular rheostat for muscle mass. Immunol. Endocr. Metab. Agents Med. Chem. 10, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cash J. N., Rejon C. A., McPherron A. C., Bernard D. J., Thompson T. B. (2009) The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J. 28, 2662–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cash J. N., Angerman E. B., Kattamuri C., Nolan K., Zhao H., Sidis Y., Keutmann H. T., Thompson T. B. (2012) Structure of myostatin:follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J. Biol. Chem. 287, 1043–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thompson T. B., Lerch T. F., Cook R. W., Woodruff T. K., Jardetzky T. S. (2005) The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev. Cell 9, 535–543 [DOI] [PubMed] [Google Scholar]
- 7. Stamler R., Keutmann H. T., Sidis Y., Kattamuri C., Schneyer A., Thompson T. B. (2008) The structure of FSTL3.activin A complex: differential binding of N-terminal domains influences follistatin-type antagonist specificity. J. Biol. Chem. 283, 32831–32838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Groppe J., Greenwald J., Wiater E., Rodriguez-Leon J., Economides A. N., Kwiatkowski W., Affolter M., Vale W. W., Izpisua Belmonte J. C., Choe S. (2002) Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature 420, 636–642 [DOI] [PubMed] [Google Scholar]
- 9. Zhang J.-L., Qiu L.-Y., Kotzsch A., Weidauer S., Patterson L., Hammerschmidt M., Sebald W., Mueller T. D. (2008) Crystal structure analysis reveals how the chordin family member crossveinless 2 blocks BMP-2 receptor binding. Dev. Cell 14, 739–750 [DOI] [PubMed] [Google Scholar]
- 10. Thies R. S., Chen T., Davies M. V., Tomkinson K. N., Pearson A. A., Shakey Q. A., Wolfman N. M. (2001) GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors 18, 251–259 [DOI] [PubMed] [Google Scholar]
- 11. Hill J. J., Davies M. V., Pearson A. A., Wang J. H., Hewick R. M., Wolfman N. M., Qiu Y. (2002) The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J. Biol. Chem. 277, 40735–40741 [DOI] [PubMed] [Google Scholar]
- 12. Hill J. J., Qiu Y., Hewick R. M., Wolfman N. M. (2003) Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: a novel protein with protease inhibitor and follistatin domains. Mol. Endocrinol. 17, 1144–1154 [DOI] [PubMed] [Google Scholar]
- 13. Amthor H., Nicholas G., McKinnell I., Kemp C. F., Sharma M., Kambadur R., Patel K. (2004) Follistatin complexes myostatin and antagonises myostatin-mediated inhibition of myogenesis. Dev. Biol. 270, 19–30 [DOI] [PubMed] [Google Scholar]
- 14. Sidis Y., Mukherjee A., Keutmann H., Delbaere A., Sadatsuki M., Schneyer A. (2006) Biological activity of follistatin isoforms and follistatin-Like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 147, 3586–3597 [DOI] [PubMed] [Google Scholar]
- 15. Lee S.-J., McPherron A. C. (2001) Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. U.S.A. 98, 9306–9311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trexler M., Bányai L., Patthy L. (2001) A human protein containing multiple types of protease-inhibitory modules. Proc. Natl. Acad. Sci. U.S.A. 98, 3705–3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kondás K., Szláma G., Trexler M., Patthy L. (2008) Both WFIKKN1 and WFIKKN2 have high affinity for growth and differentiation factors 8 and 11. J. Biol. Chem. 283, 23677–23684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monestier O., Brun C., Cocquempot O., Petit D., Blanquet V. (2012) GASP/WFIKKN proteins: evolutionary aspects of their functions. PLoS One 7, e43710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee Y. S., Lee S. J. (2013) Regulation of GDF-11 and myostatin activity by GASP-1 and GASP-2. Proc. Natl. Acad. Sci. U.S.A. 110, E3713–E3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trexler M., Bányai L., Patthy L. (2002) Distinct expression pattern of two related human proteins containing multiple types of protease-inhibitory modules. Biol. Chem. 383, 223–228 [DOI] [PubMed] [Google Scholar]
- 21. McCroskery S. (2005) Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J. Cell Sci. 118, 3531–3541 [DOI] [PubMed] [Google Scholar]
- 22. Wagner K. R., Liu X., Chang X., Allen R. E. (2005) Muscle regeneration in the prolonged absence of myostatin. Proc. Natl. Acad. Sci. U.S.A. 102, 2519–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McPherron A. C., Lawler A. M., Lee S.-J. (1999) Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat. Genet. 22, 260–264 [DOI] [PubMed] [Google Scholar]
- 24. Szláma G., Trexler M., Patthy L. (2013) Latent myostatin has significant activity and this activity is controlled more efficiently by WFIKKN1 than by WFIKKN2. FEBS J. 280, 3822–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szláma G., Kondás K., Trexler M., Patthy L. (2010) WFIKKN1 and WFIKKN2 bind growth factors TGFβ1, BMP2 and BMP4 but do not inhibit their signalling activity. FEBS J. 277, 5040–5050 [DOI] [PubMed] [Google Scholar]
- 26. Iemura S., Yamamoto T. S., Takagi C., Uchiyama H., Natsume T., Shimasaki S., Sugino H., Ueno N. (1998) Direct binding of follistatin to a complex of bone-morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proc. Natl. Acad. Sci. U.S.A. 95, 9337–9342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakatani M., Takehara Y., Sugino H., Matsumoto M., Hashimoto O., Hasegawa Y., Murakami T., Uezumi A., Takeda S., Noji S., Sunada Y., Tsuchida K. (2008) Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 22, 477–487 [DOI] [PubMed] [Google Scholar]
- 28. Cash J. N., Angerman E. B., Keutmann H. T., Thompson T. B. (2012) Characterization of follistatin-type domains and their contribution to myostatin and activin A antagonism. Mol. Endocrinol. 26, 1167–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cash J. N., Angerman E. B., Kirby R. J., Merck L., Seibel W. L., Wortman M. D., Papoian R., Nelson S., Thompson T. B. (2013) Development of a small-molecule screening method for inhibitors of cellular response to myostatin and activin A. J. Biomol. Screen. 18, 837–844 [DOI] [PubMed] [Google Scholar]
- 30. Svitel J., Balbo A., Mariuzza R. A., Gonzales N. R., Schuck P. (2003) Combined affinity and rate constant distributions of ligand populations from experimental surface binding kinetics and equilibria. Biophys. J. 84, 4062–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brown P. H., Schuck P. (2006) Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 90, 4651–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dyer K. N., Hammel M., Rambo R. P., Tsutakawa S. E., Rodic I., Classen S., Tainer J. A., Hura G. L. (2014) High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol. Biol. 1091, 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brun C., Monestier O., Legardinier S., Maftah A., Blanquet V. (2012) Murine GASP-1 N-glycosylation is not essential for its activity on C2C12 myogenic cells but alters its secretion. Cell Physiol. Biochem. 30, 791–804 [DOI] [PubMed] [Google Scholar]
- 37. Rambo R. P., Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang J. L., Huang Y., Qiu L. Y., Nickel J., Sebald W. (2007) von Willebrand factor type C domain-containing proteins regulate bone morphogenetic protein signaling through different recognition mechanisms. J. Biol. Chem. 282, 20002–20014 [DOI] [PubMed] [Google Scholar]
- 39. Troilo H., Zuk A. V., Tunnicliffe R. B., Wohl A. P., Berry R., Collins R. F., Jowitt T. A., Sengle G., Baldock C. (2014) Nanoscale structure of the BMP antagonist chordin supports cooperative BMP binding. Proc. Natl. Acad. Sci. U.S.A. 111, 13063–13068 [DOI] [PMC free article] [PubMed] [Google Scholar]