

Background: MAFA expression is markedly decreased in islet β-cells of type 2 diabetes mellitus.
Results: Mis-expression of Mafa in mouse diabetic db/db β-cells ameliorated glucose-stimulated insulin secretion and β-cell mass.
Conclusion: Mafa alone is sufficient to improve β-cell function and mass under diabetic conditions.
Significance: These results establish how consequential this transcription factor is to islet β-cells under pathological conditions.
Keywords: Diabetes, Glucose Metabolism, Insulin Synthesis, Islet, Oxidative Stress, Mafa, Insulin Transcription Factor
Abstract
The murine Mafa transcription factor is a key regulator of postnatal islet β-cell activity, affecting insulin transcription, insulin secretion, and β-cell mass. Human MAFA expression is also markedly decreased in islet β-cells of type 2 diabetes mellitus (T2DM) patients. Moreover, levels are profoundly reduced in db/db islet β-cells, a mouse model of T2DM. To examine the significance of this key islet β-cell-enriched protein to glycemic control under diabetic conditions, we generated transgenic mice that conditionally and specifically produced Mafa in db/db islet β-cells. Sustained expression of Mafa resulted in significantly lower plasma glucose levels, higher plasma insulin, and augmented islet β-cell mass. In addition, there was increased expression of insulin, Slc2a2, and newly identified Mafa-regulated genes involved in reducing β-cell stress, like Gsta1 and Gckr. Importantly, the levels of human GSTA1 were also compromised in T2DM islets. Collectively, these results illustrate how consequential the reduction in Mafa activity is to islet β-cell function under pathophysiological conditions.
Introduction
Type 2 diabetes mellitus (T2DM)2 is caused by insufficient insulin production from pancreatic islet β-cells in the setting of insulin resistance, with the latter principally reflecting the inability of cells in muscle, liver, and fat to respond adequately to normal insulin levels. Precisely why β-cells fail to produce sufficient quantities of insulin under these conditions is unclear. Notably, a subset of β-cell-enriched transcription factors essential to β-cell development and/or function were recently shown to be inactivated under T2DM stress conditions in rodent models and human islet β-cells (1–3), specifically MAFA, PDX1, and NKX6.1. Compelling evidence indicates that reactive oxygen species generated by increased glucose metabolism causes β-cell inactivation and even death in T2DM. Significantly, islet β-cells have unusually low antioxidant enzyme levels (e.g. glutathione peroxidase-1 and catalase) (4–6), with antioxidant treatment improving β-cell function in human T2DM islets (7–9) and T2DM animal models (10–12). For example, transgenic β-cell-specific expression of glutathione peroxidase-1 improved Mafa, Nkx6.1, and blood glucose levels in db/db mice, a model of T2DM (2, 12).
The change in Mafa was found to occur earlier than Nkx6.1 in mouse db/db β-cells, correlating closely with decreased expression of essential regulators involved in cell proliferation, glucose sensing, and insulin secretion (2, 13). Reduced levels of such effectors were also found in pancreas-specific MafaΔpanc deletion mutant mice (e.g. Insulin, CyclinD2, and Munc18-1 (14)). In addition, Mafa is only produced in embryonic insulin+ cells destined to populate the adult, which represents an unusually late and highly specific expression pattern in relationship to other islet-enriched transcription factors (15). Islet β-cell dysfunction under T2DM stress conditions likely results from the gradual loss of MAFA followed by either PDX1 or NKX6.1, because MafaΔpanc mice are only glucose intolerant (14), whereas islet β-cell-specific loss of Pdx1 or Nkx6.1 almost immediately causes overt hyperglycemia (16–19).
In the present study, we directly evaluated the impact of Mafa insufficiency in T2DM by generating transgenic db/db mice that conditionally expressed this transcription factor in only islet β-cells. The Mafa producing db/db mice demonstrated improved glycemic control and β-cell function, with restoration coinciding with expression of proteins that reduce oxidative stress. These studies not only provide keen insight into the prominence of Mafa activity in vivo, but also shed light on the significance of developing T2DM therapeutics to ameliorate β-cell function by preventing transcription factor inactivation.
EXPERIMENTAL PROCEDURES
Human Pancreas Samples
Pancreatic tissue was obtained from patients at Osaka University Hospital who were undergoing a partial pancreatectomy to remove pancreatic or distal bile duct tumor cells. Glucagon tolerance tests were performed before the surgery. The pancreas sample was fixed in 4% paraformaldehyde and 4-μm sections were prepared using routine procedures. The study protocol was approved by the Osaka University Hospital Ethics Committee, and informed consent was obtained from each patient. The clinical donor data are provided in supplemental Table 1.
Generation of Pdx1PB-CreERTM;CAG-CAT-Mafamyc;db/db Mice (termed βMafamyc:db/db)
pCAG-CAT-Mafamyc was constructed from pCAG-CAT-lacZ (20) by replacing the lacZ sequences with a fragment containing mouse Mafa coding sequences linked to a myc tag and the bovine growth hormone polyadenylation signal. A 5.0-kb SalI-SacI CAG-CAT-Mafamyc spanning fragment of this plasmid was purified and microinjected into fertilized eggs of BDF1 mice. A total of 13 lines of CAG-CAT-Mafamyc mice were generated, and the high TM-inducible signal to sham-treated Mafamyc expression properties of the b, d, and f lines were selected for further analysis. Pdx1PB-CreERTM transgenic mice (21), which express TM-activated Cre recombinase under the control of the islet β-cell specific Pdx1Area I/II enhancer, were crossed with the b, d, and f CAG-CAT-Mafamyc lines to generate Pdx1PB-CreERTM;CAG-CAT-Mafamyc (i.e. βMafamyc) mice. These mice were then back-crossed with C57BL/KsJ-db/m (db/m) mice for more than 10 generations to eventually obtain βMafamyc;db/db mice. Subcutaneous injections of 0.1 mg/1.0 g of BW TM were performed three times within 5 days for the induction of Mafamyc expression. The efficacy of islet β-cell expression was determined by anti-myc epitope staining. Because all of three lines demonstrated a similar improvement of plasma glucose levels after crossing with db/db mice, we mainly used the b line of CAG-CAT-Mafamyc. All animal procedures were approved by the Ethics Review Committee for Animal Experimentation of Osaka University Graduate School of Medicine.
Glucose Tolerance Tests
Glucose tolerance tests (0.5 g/kg of BW) were performed 4 and 8 weeks after TM injection on mice fasted overnight. Glucose and insulin levels were measured from tail vein sampled blood with a portable glucose meter and the insulin ELISA Kit (Morinaga Biochemicals, Yokohama, Japan).
Immunohistochemical and Islet β-Cell Mass Analysis
Mouse pancreata were dissected and fixed overnight in 4% paraformaldehyde and 4-μm thick sections were prepared under standard procedures. After blocking with 3% donkey serum, immunolabeling was performed with the following antibodies: rabbit Mafa, 1:500 (Bethyl Laboratories, Inc., Montgomery, TX); rabbit myc tag, 1:200 (Cell Signaling Technology, Inc., Danvers, MA); guinea pig insulin, 1:1000 (DAKO, Glostrup, Denmark); mouse Ki-67, 1:500 (BD Biosciences); rabbit cleaved caspase-3, 1:500 (Cell Signaling Technology, Inc., Danvers, MA); mouse 4–4-hydroxy-2-nonenal, 1:200 (Japan Institute for the Control of Aging, Shizuoka, Japan); and rabbit GSTA1 (Thermo Fisher Scientific, Rockford, IL). The secondary antibodies used for fluorescent imaging on an Olympus FV1000-D confocal microscope were 488- or 555-conjugated donkey anti-guinea pig and anti-rabbit (Jackson ImmunoResearch Laboratories, 1:200). Biotinylated goat anti-guinea pig, donkey anti-rabbit, or anti-mouse antibodies were diluted 1:200 for 3,3′-diaminobenzidine staining. Sections were counterstained with Mayer's hemalum solution. Insulin+ islet β-cell mass was determined by insulin and 3,3′-diaminobenzidine staining as reported previously (22) using WinRoof® (Mitani Corporation, Japan) and ImageJ software.
Islet Perifusion Analysis
Isolated islets were first cultured overnight in 10% FCS RPMI medium containing 5 mm glucose, and then 20 islets were placed in a chamber and perifused for 1 h with 40 mg/dl of glucose, followed by 30 min with 400 mg/dl of glucose. The effluent was collected every 30 s for 5 min, followed by 1 min for 5 min, and 2 min for 8 min. The sample insulin concentration was normalized to that of the whole cell protein.
Real-time PCR Analysis
Real-time RT-PCR analysis was performed as described previously (23) with the following primer sets: mouse insulin 1 (mRNA numbering relative to ATG, forward, −47 GACCAGCTATAATCAGAGACC; reverse +331 AGTTGCAGTAGTTCTCCAGCTG, 378 bp product), mouse insulin 2 (forward, −57 AGCCCTAAGTGATCCGCTACAA; reverse, +331 AGTTGCAGTAGTTCTCCAGCTG, 388 bp), mouse total Mafa (forward, +757 TTCAGCAAGGAGGAGGTCAT; reverse, +973 CCGCCAACTTCTCGTATTTC; 217 bp), mouse Pdx1 (forward, +192 CATCTCCCCATACGAAGTGC; reverse, +526 GGGGCCGGGAGATGTATTTG; 335 bp), mouse Gck (forward, +1893 CTTTCCAGGCCACAAAACATT; reverse, +2079 TGAGTGTTGAAGCTGCCATC; 187 bp), mouse Gckr (forward, +1429 CAGCGTGAGTTAAGCACCAA; reverse, +1649 TCAGTGATGGAGCACCTGAG; 221 bp), mouse Gsta1 (forward, +225 CGCCACCAAATATGACCTCT; reverse, +456 CCTGTTGCCCACAAGGTAGT; 232 bp), and mouse β-actin (forward, +778 GCTCTTTTCCAGCCTTCCTT; reverse, +945 CTTCTGCATCCTGTCAGCAA; 168 bp). To quantify only endogenous Mafa mRNA levels, the TaqMan MGB Gene Expression Kit (Applied Biosystems, Foster City, CA) was used with primers spanning unique 3′-flanking region sequences (forward, +1360 TCCGAGCCAGGTCTGACTTC; reverse, +1414 TGCGCTCCACGTCTGTACA; 55 bp, probe +1381 TCGGCAGCGTCCAC).
Preparation of shMafa-expressing Adenoviruses
Recombinant adenoviruses expressing short hairpin RNA against Mafa (Ad-shMafa) was constructed using the pAdEasy system and the following oligonucleotides: 5′-GTTTAGTGGGACTTGTACAGGGAACGTGTGCTGTCCGTTCCTTGTACAGGTCCCGCTTTTTT-3′ and 5′-ATGCAAAAAAGCGGGACCTGTACAAGGAACGGACAGCACACGTTCCCTGTACAAGTCCCACT-3′ (wild type Mafa sequences are underlined and mutant in italics). These oligonucleotides or control oligonucleotides (5′-GTTTTTTTTTT-3′ and 5′-ATGCAAAAAAA-3′; T7stop) were inserted downstream of the mouse U6 promoter of piGENETMmU6 (iGENE Therapeutics, Inc., Tokyo, Japan).
Microarray Analysis
The quality of islet RNAs was determined using an Agilent Bioanalyzer (Agilent Technologies, Palo Alto, CA), and samples with RNA integrity number more than 7.0 were used for microarray analysis. Total RNA was amplified with the WT-OvationTM RNA Amplification system (NuGEN Technologies, San Carlos, CA) and labeled with cyanine 3. Each hybridization contained 1.65 μg of fragmented cyanine 3-labeled cDNA, and was hybridized at 65 °C for 17 h to the Agilent Mouse GE 4 × 44K v2 microarray (Design ID 026655). Signal intensity was determined with an Agilent DNA microarray scanner. Normalization was performed using Agilent GeneSpring GX version 11.0.2 (per chip, normalization to 75 percentile shift; per gene, normalization to median of all samples). Data filtration was performed, resulting in a total of 30,161 probes as a valid probe set where at least one of the four total samples had a present flag. A 2-fold or greater change in signal intensity was considered a significant difference.
Gene Ontology (GO) Analysis
Gene ontogeny analyses on microarray data were performed with GeneSpring GX software using the annotations provided in the database of the Gene Ontology Consortium. The data were processed with Fisher's exact test and multiple test correction to identify significant over-representation of Gene Ontology annotations belonging to Molecular Functions, and the top 20 of significantly up-regulated (≥ 2.0-fold) GO terms were listed in Table 1.
TABLE 1.
GO terms up-regulated in double-TG db/db islets
| GO Term | Counts in selectiona | Counts in totalb | Corrected p value |
|---|---|---|---|
| % | |||
| Endopeptidase inhibitor activity | 7.94 | 0.53 | 3.67.E-16 |
| Peptidase inhibitor activity | 7.94 | 0.57 | 1.45.E-15 |
| Serine-type endopeptidase inhibitor activity | 6.50 | 0.39 | 4.06.E-14 |
| Enzyme inhibitor activity | 8.66 | 0.86 | 4.36.E-14 |
| Oxidoreductase activity | 13.72 | 2.76 | 4.90.E-13 |
| Monooxygenase activity | 6.50 | 0.48 | 1.64.E-12 |
| Catalytic activity | 40.07 | 20.03 | 9.26.E-12 |
| Iron ion binding | 6.50 | 0.70 | 7.06.E-10 |
| Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen | 3.25 | 0.11 | 7.18.E-09 |
| Enzyme regulator activity | 11.19 | 2.72 | 1.46.E-08 |
| Heme binding | 4.69 | 0.43 | 8.54.E-08 |
| Aromatase activity | 2.89 | 0.11 | 1.66.E-07 |
| Tetrapyrrole binding | 4.69 | 0.46 | 1.90.E-07 |
| Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen | 5.05 | 0.55 | 1.92.E-07 |
| Lipid binding | 6.86 | 1.34 | 2.80.E-06 |
| Lipid transporter activity | 2.89 | 0.16 | 4.31.E-06 |
| Electron carrier activity | 3.61 | 0.42 | 6.53.E-05 |
| Glutathione transferase activity | 2.17 | 0.11 | 9.97.E-05 |
| Cofactor binding | 5.05 | 0.95 | 1.11.E-04 |
| Fatty acid transporter activity | 1.44 | 0.03 | 1.31.E-04 |
a Refers to the percentage of genes in the input entity list that have that GO term.
b Refers to the percentage of genes in the all entities list that have that GO term.
Statistical Analysis
Data are expressed as mean ± S.D. Statistical analysis was performed using one-way analysis of variance followed by the Scheffe's test. A value of p < 0.05 was considered to be statistically significant.
RESULTS
Transgenic Islet β-cell Mafamyc Improved Glycemic Control in Insulin-resistant and Diabetic βMafamyc:db/db Mice
To obtain insight into significance of the reduction of human MAFA levels in T2DM islets (supplemental Fig. S1) (2, 24), we generated CAG-CAT-Mafa-myc epitope-tagged (Mafamyc) expressing mice to conditionally and specifically produce sustained factor activity in db/db β-cells (Fig. 1A). Significantly, the gel-shift binding and insulin-reporter driven properties of Mafamyc was indistinguishable from the wild type protein (supplemental Fig. 2). CAG-CAT-Mafamyc mice were crossed with Pdx1PB-CreERTM mice (21) to produce Mafamyc in pancreatic β-cells upon tamoxifen (TM) treatment (βMafamyc in Fig. 1A). Mafamyc was detected in 59% of islet β-cells in TM-induced βMafamyc mice (supplemental Fig. S3, A and B). Moreover, immunoblot analysis of nuclear extract from double transgenic islets demonstrated that Mafamyc was produced at roughly non-diabetic, control levels (52%, supplemental Fig. S3C).
FIGURE 1.

Generating islet β-cell-specific Mafamyc expressing db/db mice. A, schematic representation of transgenic strategy used to specifically express Mafamyc in islet db/db β-cells. Exogenous Mafamyc is expressed via Cre-mediated excision of the loxP flanked stuffer CAT cassette of CAG-CAT-Mafamyc, with TM injection at 9 weeks causing activation of the ER-fused Cre recombinase produced from the islet β-cell-specific Pdx1Pst-Bst promoter of Pdx1PB-CreERTM. B, pancreatic db/db islets from 18-week-old βMafamyc, CAG-CAT-Mafamyc, Pdx1PB-CreERTM mice were immunostained with myc (red), insulin (green), and/or Mafa (red) antibodies. The percent of insulin+ cells synthesizing Mafamyc or Mafa is shown as average ± S.D. Scale bars: 50 μm.
βMafamyc mice were crossed more than 10 generations into the C57BL/6KsJ db/db background, a T2DM model characterized by insulin resistance and β-cell failure due to severe obesity (25). TM injections were performed in 9-week-old Pdx1PB-CreERTM;db/db, CAG-CAT-Mafamyc;db/db, and βMafamyc;db/db mice, roughly when Mafa levels decline in db/db β-cells (1, 2). In contrast, Pdx1 expression was unaffected under these circumstances, another key islet-enriched transcription factor sensitive to glucotoxic conditions associated with T2DM (supplemental Fig. S4). Transgenic Mafamyc in βMafamyc;db/db mice was clearly detectable in insulin+ cells within 1 week of TM induction (Fig. 1B), and expression was sustained during the remaining 8 weeks of analysis. In contrast, Mafamyc was not produced in non-diabetic (db/m) and diabetic (Pdx1PB-CreERTM;db/db, CAG-CAT-Mafamyc;db/db) control islets. The total number of Mafa+ cells was not only much greater than in diabetic control db/db β-cells, but exceeded the number of Mafamyc producing cells (Fig. 1B). These results imply that Mafamyc restored expression of endogenous Mafa.
Blood glucose levels in βMafamyc;db/db mice were significantly lower than either control diabetic Pdx1PB-CreERTM;db/db or CAG-CAT-Mafamyc;db/db mice (Fig. 2A). Notably, this improvement was observed after only 1 week of TM treatment, which was associated with Mafamyc production in β-cells and the reduction in fed blood glucose levels in βMafamyc;db/db mice to 327.0 ± 59.4 mg/dl, whereas control diabetic and non-diabetic (db/m) were 471.3 ± 89.1 and 130.2 ± 26.8 mg/dl, respectively. This resulted from higher peripheral and portal plasma insulin levels in the double transgenic db/db mice (Fig. 2, C and D), which also manifested reduced HbA1c values (Fig. 2B).
FIGURE 2.

Islet β-cell Mafamyc production improves glycemic control in βMafamyc;db/db mice. A, fed blood glucose levels were measured every week at 4 p.m. before and after TM injections. Control, db/m (○); Pdx1PB-CreERTM;db/db (■); CAG-CAT-Mafamyc;db/db (▴); βMafamyc;db/db (●). *, p < 0.05; **, p < 0.01 versus control db/db mice. †, p < 0.01 versus db/m mice (n = 6–8). B, HbA1c levels were measured at 9 and 17 weeks. *, p < 0.01 versus control db/db mice. †, p < 0.01 versus db/m mice (n = 6–8). C, fed plasma insulin levels. *, p < 0.01 versus control db/db mice. †, p < 0.05; ††, p < 0.01 versus db/m mice (n = 6–8). D, plasma blood insulin levels sampled from the hepatic portal vein of 13 week-old mice. *, p < 0.001 versus control db/db mice (n = 4). All of error bars represent S.D.
Improved plasma glucose clearance and higher insulin secretion levels were observed in βMafamyc;db/db mice in intra-peritoneal glucose tolerance tests (Fig. 3, A and B), whereas insulin sensitivity (Fig. 3C) and body weight (supplemental Fig. 5) did not differ between the transgenic Mafamyc expressing and non-expressing db/db mice. In addition, both 1st and 2nd phase insulin secretion increased in isolated βMafamyc;db/db islets (Fig. 3D). Collectively, these results show that Mafamyc synthesis in 9-week-old db/db mice enabled quick recovery of β-cell function even in the context of persistent insulin insensitivity and obesity, resulting in better glucose utilization in peripheral tissues.
FIGURE 3.

Improved glucose-stimulated insulin secretion in βMafamyc;db/db mice. Intraperitoneal glucose tolerance tests were performed on 13- and 17-week-old mice after a 14-h fast. Blood glucose (A) and plasma insulin (B) levels in βMafamyc;db/db (●), Pdx1PB-CreERTM;db/db (■), CAG-CAT-Mafamyc;db/db (▴), and db/m (○) mice. *, p < 0.05; **, p < 0.01 versus control db/db mice. †, p < 0.05; ††, p < 0.01 versus db/m mice (n = 4–5). C, blood glucose levels in 17-week-old mice injected intraperitoneally with insulin (2.0 units/kg) after a 2-h fast. †, p < 0.05; ††, p < 0.01 versus db/m mice. D, islets from 13-week-old βMafamyc;db/db (●), Pdx1PB-CreERTM;db/db (■), and db/m mice (○) perifused with normal and high glucose. *, p < 0.05; **, p < 0.01 versus Pdx1PB-CreERTM;db/db, †, p < 0.05 versus βMafamyc;db/db (n = 4). All of error bars represent S.D.
Islet β-Cell Mass Increases in Mafamyc Producing db/db Mice
Insulin resistance and elevated blood glucose levels are first observed at around 4 weeks in db/db mice, whereas hyperglycemia plateaus at around 12 weeks. High-level β-cell proliferation occurs at 4 weeks in response (2), with sustained hyperglycemia decreasing islet insulin+ cell mass (25). Significantly, β-cell mass improved in βMafamyc;db/db islets relative to diabetic controls (Fig. 4A and supplemental Fig. S6). This resulted from reduced cellular caspase-3-mediated apoptosis, and not a change in proliferation rate of islet β-cells (Fig. 4, B and C). These findings illustrated the importance of Mafa in preserving β-cell levels in T2DM islets.
FIGURE 4.

Mafamyc preserves islet β-cell mass in db/db mice. A, diaminobenzidine peroxidase immunodetection was used to measure the islet insulin+ cell area in the 18-week-old βMafamyc;db/db, Pdx1PB-CreERTM;db/db, CAG-CAT-Mafamyc;db/db, and db/m pancreas. *, p < 0.01 versus db/db or db/m (n = 3–4). Pancreatic sections from 18-week-old mice immunostained for (B) insulin and Ki-67 or (C) cleaved caspase-3. The percent of Ki-67+ or caspase-3+ cells per total insulin+ β-cells. Arrows mark Ki-67+ or caspase-3+ cells. Scale bars: 50 μm. *, p < 0.05; **, p < 0.01 versus db/m mice. †, p < 0.01 versus control db/db mice (n = 3–4). All of error bars represent S.D.
Identification of Gene Products Impacted by Maintaining Mafa Expression in βMafamyc;db/db Islet β-Cells
We first investigated how Mafamyc influenced the expression of genes known to be regulated by this factor in βMafamyc;db/db islets, specifically insulin I, insulin II, Pdx1, and glucokinase (Gck), and Mafa itself (13). As expected from immunostaining (Fig. 1B), total Mafa mRNA was greatly induced over the non-transgenic diabetic controls, essentially now equivalent to euglycemic db/m islets (Fig. 5). Moreover, insulin 1, insulin 2, and Slc2a2 were also increased in Mafamyc expressing db/db samples, with Pdx1 trending toward recovery. In contrast, Gck expression was unaffected by diabetic conditions. These results demonstrated that functionally important Mafa-regulated gene products were limiting in diabetic db/db islet β-cells.
FIGURE 5.

Sustained expression of bona fide Mafa target genes in βMafamyc;db/db islet β-cells. Real-time RT-PCR was used to measure Gckr, GSTa1, total Mafa (Mafamyc + endogenous Mafa), Slc2a2, insulin 1, insulin 2, Pdx1, Gck, and endogenous Mafa expression with islets at (A) 14 weeks, and (B) 9 weeks or 3 days after TM injections. *, p < 0.05; **, p < 0.01 (n = 3–4). All of error bars represent S.D.
To identify other significant targets of Mafamyc control, Agilent Mouse GE 4 × 44K v2 microarray analysis was performed with RNA extracted from 14-week-old βMafamyc;db/db and diabetic control Pdx1PB-CreERTM;db/db islets. Approximately 340 genes had a more than 2-fold level increase in Mafamyc producing islets (supplemental Appendix S1), including, as expected, Mafa and Slc2a2. Notably, gene ontology analysis indicated that proteins involved in reducing oxidative stress in β-cells were up-regulated, like the glucokinase regulatory protein (Gckr) that inactivates Gck by translocating the enzyme into the nucleus (26, 27), and glutathione S-transferase α1 (Gsta1), which catalyzes the conjugation of glutathione to various molecules for the purpose of detoxification (28, 29).
The selective increase in Gckr and Gsta1 expression levels was confirmed upon comparing RNA expression in 14-week-old Mafamyc db/db islets to control db/db and db/m islets (Fig. 5A). This is likely caused by Mafa directly activating Gckr and Gsta1 transcription, because expression of both was significantly reduced upon shRNA-mediated knock-down of Mafa in mouse MIN6 β-cells (supplemental Fig. S7). These results imply that factors reducing db/db β-cell stress, such as Gckr and Gsta1, were important effectors of βMafamyc;db/db recovery.
Induction of Gckr and Gsta1 Levels Coincides with Mafamyc Production and Reduced Oxidative Stress in βMafamyc;db/db β-Cells
Further support of a direct role for Mafamyc in recovery of db/db β-cells, Gckr, and Gsta1 expression increased within 3 days of TM treatment in βMafamyc;db/db islets (Fig. 5B). Insulin 1, insulin 2, and Slc2a2 levels were also elevated within this early period, although there was not yet a change in blood glucose levels (supplemental Fig. S8). Because elevated Gckr and Gsta1 could reduce oxidative stress in β-cells, for example, by the Gckr protein sequestering Gck in the nucleus and reducing glucose flux (27), production of the oxidative stress marker 4-hydroxy-2-nonenal was compared in non-diabetic, diabetic, and Mafamyc containing db/db islets (supplemental Fig. S9). As predicted from elevated Gckr and Gsta1, 4-hydroxy-2-nonenal staining levels were decreased in Mafamyc islets in comparison to diabetic CAG-CAT-Mafamyc;db/db and Pdx1PB-CreERTM islets. However, Mafamyc islet staining was still elevated in relationship to euglycemic db/m islets, implying that restoration of Mafa alone is unable to completely prevent glucotoxicity in db/db β-cells. Such was expected from the difference in blood glucose and HbA1c levels between βMafamyc;db/db and db/m mice (Fig. 2B). Collectively, these results strongly suggest that Mafamyc improves db/db β-cell by reducing toxic stress conditions, presumably by elevating expression of factors like Gckr and Gsta1.
There Is a Marked Decrease of GSTA1 in Human T2DM Islets
The presence of MAFA in insulin+ cells is markedly decreased in T2DM islets (see supplemental Fig. S1 and Refs. 2 and 24). Immunostaining for GSTA1 was performed in normal and T2DM pancreatic samples to determine whether compromised expression was also associated with the disease state in humans. Gsta1 was clearly detected in normal human β-cells, whereas almost undetectable in T2DM islet (Fig. 6). Collectively, our analysis in db/db mice and human T2DM islets illustrates that reduction of MAFA in T2DM not only influences normal mediators of β-cell function and mass, but also stress effector levels in islet cells.
FIGURE 6.
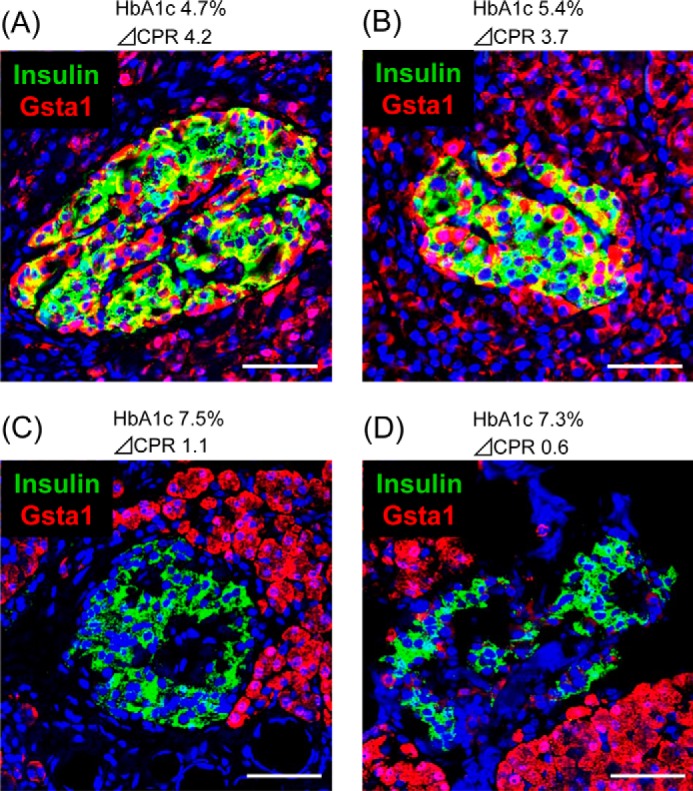
GSTA1 levels are compromised in human T2DM islets. Pancreatic section from (A and B) normal non-diabetic and (C and D) T2DM donors were costained with GSTA1 (red) and insulin (green). Nuclei (blue) were stained with DAPI. Scale bars: 50 μm.
DISCUSSION
Transcription factors have been demonstrated to play essential roles in both the embryonic formation and adult function of islet cells in mice. The association of many of these islet-enriched proteins to MODY implies that their function is also conserved within humans (30). Notably, Mafa is distinct among this group of transcription factors in being uniquely produced at the onset of islet β-cell formation during development and exclusively within this embryonic and adult islet population. Moreover, gene knock-out studies have demonstrated that Mafa only affects islet β-cell maturation, but not euglycemia. Human MAFA has also been found to be markedly decreased in T2DM β-cells, with overt cell dysfunction likely caused by decreased NKX6.1 and/or PDX1, whose loss from mouse islets quickly results in hyperglycemia (2, 16, 19). However, because the expression and/or activity of a variety of factors were affected in islet β-cells under these glucotoxic conditions, it is difficult to elucidate what factor(s) is pathophysiological. In this report, we directly examined the significance of Mafa activity in insulin-resistant, diabetic db/db mice by conditionally and specifically producing Mafamyc in islet β-cells. Expression levels comparable with endogenous normoglycemic Mafa improved β-cell function, β-cell mass, and blood glucose levels. These results support MAFA in the molecular underpinnings of islet β-cell inactivation during T2DM.
The induction of Mafamyc in βMafamyc;db/db mice was initiated at 9 weeks of age to maintain production during the period when endogenous stressors would normally cause Mafa inactivation. The expression of many target genes of Mafa increased in Mafamyc β-cells relative to diabetic β-cells. Improvement of glycemic control by Mafamyc β-cells was produced in the context of continued production of obesity- and dyslipidemia-induced stress signals instigated by the leptin receptor mutation in db/db mice. Microarray studies were performed to identify the proteins enhancing β-cell activity. As expected, a variety of gene products normally required in glucose sensing and insulin secretion were elevated in Mafamyc β-cells, like the Slc2a1 and Slc2a2 (supplemental Fig. S10), glucose transporters essential to rodent and human glucose-stimulated insulin secretion (31). Interestingly, genes encoding proteins protective to the β-cell were also found, with Gsta1 and Gckr representative examples (supplemental Appendix S1). Notably, Gsta1 and Gckr expression increased immediately after Mafamyc production, prior to the reduction in blood glucose levels. Moreover, their expression was sustained throughout the time course of experimentation, as predicted of important neutralizing effectors of β-cell stress.
Islet β-cells have very low hydrogen peroxide scavenging enzyme levels relative to other cell types, like glutathione peroxidase-1 and catalase (4–6). Significantly, transgenic β-cell-specific glutathione peroxidase-1 expression profoundly increased β-cell function in db/db mice, coinciding with recovery of nuclear Mafa and Nkx6.1 (2). The insulin secretion defects in human T2DM islets are also improved upon in vitro treatment with reactive oxygen species scavengers (7). Induction of Gsta1 and Gckr by Mafamyc would reduce oxidative stress in β-cells, for example, by preventing oxidation of cysteine 277 and 293 in Mafa that cause loss in cis-acting DNA binding activity (2). This could explain why endogenous Mafa levels were also increased in βMafamyc;db/db islets (Fig. 1B).
Interestingly, the mechanisms of action of these antioxidants are distinct. Thus, the Gsta1 enzyme detoxifies reactive oxygen species by direct conjugation with glutathione, whereas Gckr inhibits glucokinase by binding non-covalently to form an inactive complex (26). As glucokinase is the principal regulator of glucose flux and insulin secretion in β-cells (32), Gckr binding would reduce cellular metabolism, and consequentially the generation of active oxygen species. Gckr was identified as a T2DM susceptible gene in genome-wide association studies (33). The increased oxoreductase activity in βMafamyc;db/db islet β-cells would also reduce thioredoxin-interacting protein levels, a cellular redox regulator that both induces cell death and inhibits Mafa mRNA expression (34).
GSTA1 levels were decreased in T2DM islets (Fig. 6). Consequently, it will be important to investigate precisely how expression of antioxidant factors like Gckr and Gsta1 influence β-cell activity. These findings suggest that screening of pharmacological agents to either enhance antioxoreductase levels is warranted for treatment of pre- and established T2DM patients. For example, could induction of antioxidant factors be protective in islet β-cells of obese individuals, as the majority will not become T2DM (35)? Perhaps expression is also limiting within the insulin+ cells produced during in vitro differentiation of human embryonic stem cells, which express many transcriptional regulators associated with mature islet cells, yet remain dysfunctional with regards to glucose responsiveness and high insulin production until able to express MAFA (36). Notably, oxidative destruction is still evident in βMafamyc;db/db islets (supplemental Fig. S9), presumably limiting β-cell activity and the complete restoration of blood glucose levels. Presumably, the failure to produce normal Nkx6.1 levels in βMafamyc;db/db β-cells results in relative inactivity, and prevents the return to euglycemia.
Interestingly, improved blood glucose levels and islet β-cell function are observed upon treatment of db/db mice with either phloridzin (37) or insulin (38), which act by effecting glucose utilization (i.e. insulin) or clearance (phloridzin) in peripheral tissues. The consequential reduction in blood glucose levels likely enhances β-cell mass and activity by decreasing cellular glucose flux and stress effector levels. The present work strongly suggests that Mafa is a direct mediator of improved β-cell activity under these conditions. Thus, sustaining Mafa expression in diabetic βMafamyc;db/db β-cells led to lowered blood glucose levels, increased β-cell mass, and improved β-cell function. Future efforts will be directed at determining how to prevent MAFA inactivation in T2DM β-cells.
Supplementary Material
Acknowledgments
We thank Satomi Takebe and Yuko Sasaki for excellent technical assistance, and Chikayo Yokogawa for secretarial assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant DK090570 (to R. S.), Juvenile Diabetes Research Foundation Career Development Award 2-2005-946 (to T. Matsuoka), and JSPS KAKENHI Grant 10379258 (to T. Matsuoka).

This article contains supplemental Figs. S1–S10, Table S1, and Appendix S1.
- T2DM
- type 2 diabetes mellitus
- GO
- gene ontology
- TM
- tamoxifen
- Gckr
- glucokinase regulatory protein
- GSTA1
- glutathione S-transferase α1.
REFERENCES
- 1. Matsuoka T. A., Kaneto H., Miyatsuka T., Yamamoto T., Yamamoto K., Kato K., Shimomura I., Stein R., Matsuhisa M. (2010) Regulation of MafA expression in pancreatic beta-cells in db/db mice with diabetes. Diabetes 59, 1709–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guo S., Dai C., Guo M., Taylor B., Harmon J. S., Sander M., Robertson R. P., Powers A. C., Stein R. (2013) Inactivation of specific beta cell transcription factors in type 2 diabetes. J. Clin. Investig. 123, 3305–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mahadevan J., Parazzoli S., Oseid E., Hertzel A. V., Bernlohr D. A., Vallerie S. N., Liu C. Q., Lopez M., Harmon J. S., Robertson R. P. (2013) Ebselen treatment prevents islet apoptosis, maintains intranuclear Pdx-1 and MafA levels, and preserves beta-cell mass and function in ZDF rats. Diabetes 62, 3582–3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lenzen S., Drinkgern J., Tiedge M. (1996) Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 20, 463–466 [DOI] [PubMed] [Google Scholar]
- 5. Tiedge M., Lortz S., Drinkgern J., Lenzen S. (1997) Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 46, 1733–1742 [DOI] [PubMed] [Google Scholar]
- 6. Grankvist K., Marklund S. L., Täljedal I. B. (1981) CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 199, 393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Del Guerra S., Lupi R., Marselli L., Masini M., Bugliani M., Sbrana S., Torri S., Pollera M., Boggi U., Mosca F., Del Prato S., Marchetti P. (2005) Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 54, 727–735 [DOI] [PubMed] [Google Scholar]
- 8. Lupi R., Del Guerra S., Mancarella R., Novelli M., Valgimigli L., Pedulli G. F., Paolini M., Soleti A., Filipponi F., Mosca F., Boggi U., Del Prato S., Masiello P., Marchetti P. (2007) Insulin secretion defects of human type 2 diabetic islets are corrected in vitro by a new reactive oxygen species scavenger. Diabetes Metab. 33, 340–345 [DOI] [PubMed] [Google Scholar]
- 9. D'Aleo V., Del Guerra S., Martano M., Bonamassa B., Canistro D., Soleti A., Valgimigli L., Paolini M., Filipponi F., Boggi U., Del Prato S., Lupi R. (2009) The non-peptidyl low molecular weight radical scavenger IAC protects human pancreatic islets from lipotoxicity. Mol. Cell. Endocrinol. 309, 63–66 [DOI] [PubMed] [Google Scholar]
- 10. Kaneto H., Kajimoto Y., Miyagawa J., Matsuoka T., Fujitani Y., Umayahara Y., Hanafusa T., Matsuzawa Y., Yamasaki Y., Hori M. (1999) Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 48, 2398–2406 [DOI] [PubMed] [Google Scholar]
- 11. Tanaka Y., Gleason C. E., Tran P. O., Harmon J. S., Robertson R. P. (1999) Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. U.S.A. 96, 10857–10862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harmon J. S., Bogdani M., Parazzoli S. D., Mak S. S., Oseid E. A., Berghmans M., Leboeuf R. C., Robertson R. P. (2009) β-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 150, 4855–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang H., Brun T., Kataoka K., Sharma A. J., Wollheim C. B. (2007) MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 50, 348–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hang Y., Yamamoto T., Benninger R. K., Brissova M., Guo M., Bush W., Piston D. W., Powers A. C., Magnuson M., Thurmond D. C., Stein R. (2014) The MafA transcription factor becomes essential to islet beta-cells soon after birth. Diabetes 63, 1994–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsuoka T. A., Zhao L., Artner I., Jarrett H. W., Friedman D., Means A., Stein R. (2003) Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol. Cell. Biol. 23, 6049–6062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao T., McKenna B., Li C., Reichert M., Nguyen J., Singh T., Yang C., Pannikar A., Doliba N., Zhang T., Stoffers D. A., Edlund H., Matschinsky F., Stein R., Stanger B. Z. (2014) Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 19, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang C., Moriguchi T., Kajihara M., Esaki R., Harada A., Shimohata H., Oishi H., Hamada M., Morito N., Hasegawa K., Kudo T., Engel J. D., Yamamoto M., Takahashi S. (2005) MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 25, 4969–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahlgren U., Jonsson J., Jonsson L., Simu K., Edlund H. (1998) Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 12, 1763–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor B. L., Liu F. F., Sander M. (2013) Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 4, 1262–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Araki K., Araki M., Miyazaki J., Vassalli P. (1995) Site-specific recombination of a transgene in fertilized eggs by transient expression of Cre recombinase. Proc. Natl. Acad. Sci. U.S.A. 92, 160–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang H., Fujitani Y., Wright C. V., Gannon M. (2005) Efficient recombination in pancreatic islets by a tamoxifen-inducible Cre-recombinase. Genesis 42, 210–217 [DOI] [PubMed] [Google Scholar]
- 22. Waguri M., Yamamoto K., Miyagawa J. I., Tochino Y., Yamamori K., Kajimoto Y., Nakajima H., Watada H., Yoshiuchi I., Itoh N., Imagawa A., Namba M., Kuwajima M., Yamasaki Y., Hanafusa T., Matsuzawa Y. (1997) Demonstration of two different processes of beta-cell regeneration in a new diabetic mouse model induced by selective perfusion of alloxan. Diabetes 46, 1281–1290 [DOI] [PubMed] [Google Scholar]
- 23. Matsuoka T. A., Kaneto H., Stein R., Miyatsuka T., Kawamori D., Henderson E., Kojima I., Matsuhisa M., Hori M., Yamasaki Y. (2007) MafA regulates expression of genes important to islet beta-cell function. Mol. Endocrinol. 21, 2764–2774 [DOI] [PubMed] [Google Scholar]
- 24. Butler A. E., Robertson R. P., Hernandez R., Matveyenko A. V., Gurlo T., Butler P. C. (2012) Beta cell nuclear musculoaponeurotic fibrosarcoma oncogene family A (MafA) is deficient in type 2 diabetes. Diabetologia 55, 2985–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hummel K. P., Coleman D. L., Lane P. W. (1972) The influence of genetic background on expression of mutations at the diabetes locus in the mouse: I. C57BL-KsJ and C57BL-6J strains. Biochem. Genet. 7, 1–13 [DOI] [PubMed] [Google Scholar]
- 26. Shiota C., Coffey J., Grimsby J., Grippo J. F., Magnuson M. A. (1999) Nuclear import of hepatic glucokinase depends upon glucokinase regulatory protein, whereas export is due to a nuclear export signal sequence in glucokinase. J. Biol. Chem. 274, 37125–37130 [DOI] [PubMed] [Google Scholar]
- 27. de la Iglesia N., Mukhtar M., Seoane J., Guinovart J. J., Agius L. (2000) The role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyte. J. Biol. Chem. 275, 10597–10603 [DOI] [PubMed] [Google Scholar]
- 28. Nebert D. W., Vasiliou V. (2004) Analysis of the glutathione S-transferase (GST) gene family. Hum. Genomics 1, 460–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayes J. D., Pulford D. J. (1995) The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 30, 445–600 [DOI] [PubMed] [Google Scholar]
- 30. Steck A. K., Winter W. E. (2011) Review on monogenic diabetes. Curr. Opin. Endocrinol. Diabetes Obesity 18, 252–258 [DOI] [PubMed] [Google Scholar]
- 31. Ohtsubo K., Chen M. Z., Olefsky J. M., Marth J. D. (2011) Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat. Med. 17, 1067–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matschinsky F. M. (2002) Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes 51, S394–S404 [DOI] [PubMed] [Google Scholar]
- 33. Dupuis J., Langenberg C., Prokopenko I., Saxena R., Soranzo N., Jackson A. U., Wheeler E., Glazer N. L., Bouatia-Naji N., Gloyn A. L., et al. (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 42, 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu G., Chen J., Jing G., Shalev A. (2013) Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 19, 1141–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nolan C. J., Damm P., Prentki M. (2011) Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet 378, 169–181 [DOI] [PubMed] [Google Scholar]
- 36. Kroon E., Martinson L. A., Kadoya K., Bang A. G., Kelly O. G., Eliazer S., Young H., Richardson M., Smart N. G., Cunningham J., Agulnick A. D., D'Amour K. A., Carpenter M. K., Baetge E. E. (2008) Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 26, 443–452 [DOI] [PubMed] [Google Scholar]
- 37. Kjørholt C., Akerfeldt M. C., Biden T. J., Laybutt D. R. (2005) Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of beta-cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes 54, 2755–2763 [DOI] [PubMed] [Google Scholar]
- 38. Kawashima S., Matsuoka T. A., Kaneto H., Tochino Y., Kato K., Yamamoto K., Yamamoto T., Matsuhisa M., Shimomura I. (2011) Effect of alogliptin, pioglitazone and glargine on pancreatic beta-cells in diabetic db/db mice. Biochem. Biophys. Res. Commun. 404, 534–540 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.