

Background: NAD biosynthesis was implicated as an antibacterial target pathway.
Results: Structure-functional analysis of Mycobacterium tuberculosis NadD revealed an over-closed conformation and a sequential kinetic mechanism.
Conclusion: M. tuberculosis NadD is a potentially druggable target suitable for the development of selective inhibitors.
Significance: This study describes a novel regulatory mechanism and points to a specific strategy for targeting mycobacterial NadD.
Keywords: Enzyme Inhibitor, Enzyme Structure, NAD Biosynthesis, Protein Engineering, Protein Targeting
Abstract
Nicotinate mononucleotide adenylyltransferase NadD is an essential enzyme in the biosynthesis of the NAD cofactor, which has been implicated as a target for developing new antimycobacterial therapies. Here we report the crystal structure of Mycobacterium tuberculosis NadD (MtNadD) at a resolution of 2.4 Å. A remarkable new feature of the MtNadD structure, compared with other members of this enzyme family, is a 310 helix that locks the active site in an over-closed conformation. As a result, MtNadD is rendered inactive as it is topologically incompatible with substrate binding and catalysis. Directed mutagenesis was also used to further dissect the structural elements that contribute to the interactions of the two MtNadD substrates, i.e. ATP and nicotinic acid mononucleotide (NaMN). For inhibitory profiling of partially active mutants and wild type MtNadD, we used a small molecule inhibitor of MtNadD with moderate affinity (Ki ∼ 25 μm) and antimycobacterial activity (MIC80) ∼ 40–80 μm). This analysis revealed interferences with some of the residues in the NaMN binding subsite consistent with the competitive inhibition observed for the NaMN substrate (but not ATP). A detailed steady-state kinetic analysis of MtNadD suggests that ATP must first bind to allow efficient NaMN binding and catalysis. This sequential mechanism is consistent with the requirement of transition to catalytically competent (open) conformation hypothesized from structural modeling. A possible physiological significance of this mechanism is to enable the down-regulation of NAD synthesis under ATP-limiting dormancy conditions. These findings point to a possible new strategy for designing inhibitors that lock the enzyme in the inactive over-closed conformation.
Introduction
Tuberculosis remains a major global health problem due to a prevalence of more than two billion latent infections, nine million active disease cases, and one and a half million deaths annually (1). Drug resistance, HIV co-infection, immunosuppression, ineffective health care delivery, and a protracted treatment regimen further complicate control measures (2). Tuberculosis is caused by Mycobacterium tuberculosis (Mtb),4 a bacterium that enters a dormant state in the human host to adapt to the pressures of the immune response and to tolerate drug treatment. This results in a long term chronic infection (3). Thus, there is a constant need to develop drugs that would efficiently kill drug-resistant strains and non-replicating mycobacteria as well as shorten the time of treatment (4, 5).
The recent approval of the ATP biogenesis inhibitor bedaquiline for the treatment of drug-resistant tuberculosis is an important milestone in this direction (6, 7). However, the dormancy triggered by decreasing ATP levels and the induction of alternative bioenergetic rescue pathways (8) emphasize the significance of targeting other aspects of respiratory energy metabolism that are essential for both actively replicating and dormant forms of mycobacteria. The biogenesis and homeostasis of NAD is strongly implicated as a target pathway in vitro and in vivo (9, 10). Nicotinic acid mononucleotide (NaMN) adenylyltransferase (NadD), and NAD synthase (NadE), indispensable and broadly conserved enzymes involved in the last two steps of NAD biogenesis (see Fig. 1), are actively explored as drug targets in a number of bacterial pathogens (11–16). Targeted degradation of mycobacterial NadD and NadE enzymes lead to a rapid depletion of the NAD cofactor pool followed by cell death, even under dormancy conditions (9, 17).
FIGURE 1.

NaMN adenylyltransferase NadD catalyzes the first indispensable step in the biogenesis and homeostasis of NAD(P) redox cofactors in mycobacteria. The NAD cofactor pool requires constant replenishing due to its degradative utilization by non-metabolic enzymes such as DNA ligase (LigA) and protein lysine deacetylase (CobB). Note that NadD substrate, NaMN, is a unique checkpoint intermediate shared by all, de novo, salvage, and recycling, pathways (shown by dotted arrows with enzyme names denoted as respective gene products).
NadE synthase enzymes were extensively characterized from many bacteria (18–20), including determination of the three-dimensional structure of the Mtb enzyme (10, 21). However, bacterial NadD enzymes have been less studied, and although the three-dimensional structures were reported for some bacterial pathogens (16, 22–24), it has not been reported for NadD from Mtb. Recently, we reported for the first time the successful expression, purification, and basic kinetic properties of MtNadD (17). As do all other members of the bacterial NadD family, the Mtb enzyme displays a strict preference for the NaMN substrate over its amidated analog (NMN) in the catalyzed transfer of the adenylyl moiety from ATP, releasing pyrophosphate (PPi) and forming nicotinic acid dinucleotide (NaAD), a last intermediate in the synthesis of NAD (Fig. 1). Notably, the analogous (but only distantly homologous) human enzymes (NMNAT1–3) have dual specificity with equal catalytic efficiency on both NMN and NaMN substrates (25, 26). These distinctive structure-functional features of the human NMNAT make MtNadD a potentially more selective target than MtNadE, which is far more similar to its human counterpart (NADSYN1).
Here we report for the first time the determination of MtNadD three-dimensional structure. The structure revealed both shared and unique features that provide the basis for the rational design of MtNadD inhibitors. Most notable is the observed “closed” conformation of the MtNadD active site imposed by a tightly packed α-helical segment in place of an extended coil that is commonly present in other NadD structures. This finding combined with site-directed mutagenesis, kinetic, and inhibitory analyses allowed us to refine the enzyme-substrate interactions and to propose a conformational regulatory mechanism of potential physiological significance. We hypothesize that the ATP-consuming NadD activity in Mtb cells is regulated by transition from the enzymatically incompetent closed conformation at low ATP (e.g. under conditions of metabolic dormancy) to fully active conformation, which could be triggered by the increase in ATP-producing metabolic activity. This hypothetical model points to a potential new strategy for rationally designing inhibitors that would specifically bind and lock MtNadD in the inactive closed conformation.
EXPERIMENTAL PROCEDURES
Materials
Enzymes for PCR and DNA manipulations were from New England Biolabs (Beverly, MA) and Agilent Technologies. Plasmid purification kits were from Promega (Madison, WI). PCR purification kits and nickel-nitrilotriacetic acid (Ni-NTA) resin were from Qiagen (Valencia, CA). Oligonucleotides for PCR and sequencing were synthesized by IDT (San Diego, CA).
Site-directed Mutagenesis
A full-size Mt nadD gene (17) cloned in pET-derived vector (27) was used as the template for site-directed mutagenesis to introduce the T12A, D14A, H17A, H20A, P44A, W45A, Q46A, K47A, T86A, W117A, W117F, L164A, and L164Q point mutations using standard procedures. The template DNA, dNTPs, and the appropriate mutagenic primers were incubated with Pfu DNA polymerase according to the instruction manual. The parental methylated and hemimethylated DNA were digested with Dpn1 for 10 min at 37 °C; then mutated molecules were transformed into DH5α competent cells (Stratagene) for nick repair. Individual plasmids with introduced mutations were verified by DNA sequencing and transformed into Escherichia coli BL21(DE3) for protein expression and purification.
Protein Expression and Purification
A wild type Mt nadD gene was expressed as a fusion protein with an N-terminal-cleavable His6-SUMO tag using pSMT3 expression vector in E. coli BL21(DE3) strain and purified as previously described (17). For the purpose of crystallization, the His6-SUMO tag was cleaved with Ulp1 protease at 4 °C overnight. The properly processed MtNadD was purified by capturing the protease, the cleaved tag, and the residual uncleaved protein from the reaction mixture on the Ni-NTA resin, and it was further polished on the Sepharose Q-HP column (FPLC, GE Healthcare) with a linear gradient of 0–500 mm NaCl in 20 mm Tris buffer, pH 7.8, 1 mm DTT, and 5% (v/v) glycerol. This procedure yielded up to 8 mg of homogenous MtNadD from 6 liters of culture. Mutant variants of MtNadD were expressed as fusions with His6-tobacco etch virus protease tag and purified from 50 ml of culture using our standard miniscale procedure (28) for the initial assessment of protein solubility and activity. Selected active mutant variants were expressed in larger scale (1 liter of culture) using a standard two-step procedure that included Ni-NTA chromatography and gel filtration (29), which also allowed us to assess their oligomeric state (typically dimeric). Protein concentration was determined using the Bradford protein assay kit (Bio-Rad), with a BSA standard, and the purity was assessed by SDS-PAGE. The presence of His6-tobacco etch virus tag had no effect on enzyme activity, and typically, it was not removed, except for the mutant W117A, which was selected for crystallization studies due to its substantially higher level of expression (∼10-fold) compared with wild type MtNadD. Upon incubation with tobacco etch virus-protease (1:100, overnight at 4 °C), the processed MtNadD(W117A) protein was purified by passing through Ni-NTA column followed by dialysis.
Crystallization
The initial crystallization conditions for the wild type and the W117A mutant variant of MtNadD were identified using commercial screens (Hampton Research) and robotic drop dispensers (Phoenix, Art Robbins). The proteins were crystallized by the vapor diffusion method in sitting drop plates at 20 °C. The obtained microcrystals of the WT protein were used as seeds in the following crystallization procedure. The hanging drops were mixtures of the protein at 10 mg/ml in 20 mm Tris, pH 7.8, 200 mm NaCl, and 5% v/v glycerol and a well solution containing 1.2–1.5 m MgSO4, 0.1 m MES buffer, pH 6–6.5. Micro-seeds were added to the drops 1 h later, and the diamond-shaped macrocrystals grew up within 3–5 days. The crystals were soaked with a well solution supplemented with 30% (w/v) glucose and flash-frozen in liquid nitrogen. The W117A mutant crystals from the robotic screen were big enough for the data collection. The investigated crystals grew up in a drop containing (at a 1:1 ratio) 7 mg/ml protein in 150 mm NaCl, 20 mm Tris, pH 8.0, 2.5% (v/v) DMSO, 0.7 mm Tris(2-carboxyethyl)phosphine, and a well solution consisting of 25% PEG 3350, 0.1 m HEPES, and 0.2 m MgCl2. The W117A crystals were flash-frozen in liquid nitrogen after a short soaking in a well solution containing 2.5% (v/v) DMSO and 20% (v/v) glycerol.
Diffraction Analysis and Structure Determination
Wild type crystals were twinned and belonged to space group P3121. Images were processed using autoPROC (30). The wild type structure was determined by molecular replacement using as an initial model the structure of MtNadD with a bound small molecule inhibitor solved by seleno-l-methionine phasing method (data not shown). Twinned refinement and model building were performed using REFMAC (31) as implemented in the CCP4 suite (32) and COOT (33), respectively. The W117A mutant structure was solved by molecular replacement using wild type MtNadD as the starting model. The W117A mutant structure was refined using Phenix-1.9 (34). Its space group (P212121) was different from that of the wild type structure, and no twinning was detected. Despite several attempts of co-crystallization with a substrate, product (up to 5 mm NaAD), or inhibitor, no ligands could be detected in the analyzed crystals. Relevant information and statistics are provided in Table 1.
TABLE 1.
Data collection and refinement statistics
| Wild type | W117A | |
|---|---|---|
| Data collection | ||
| Beam line | ALS 5.01 | SSRL BL11-1 |
| Wavelength (Å) | 0.97740 | 0.97945 |
| Space group | P3121 | P212121 |
| Cell dimensions; a, b, c (Å) | 67.54, 67.54, 187.37 | 49.0, 124.2, 140.4 |
| Resolution (Å) | 2.41 | 2.42a |
| Resolution highest shell (Å) | (2.48-2.41) | (2.51-2.42) |
| Rmerge | 0.066 (0.46) | 0.13 (0.78) |
| I/σI | 14.3 (2.8) | 5.6 (1.6) |
| Completeness (%) | 99.8 (100) | 97.5 (99.0) |
| Redundancy | 4.7 (4.9) | 3.8 (4.0) |
| Refinement | Refmac 5.8 | Phenix-1.9 |
| Reflections | 18,841 | 32,648 |
| Rwork/Rfree | 20.7/24.5 | 22.9/25.8 |
| No. of non-H atoms | ||
| Protein | 2877 | 6010 |
| Water | 8 | 168 |
| Non-water ligands | 12 | 11 |
| B-factors | ||
| Wilson | 52.8 | 37.4 |
| Protein | 66.1 | 48.6 |
| Waters | 48.9 | 39.5 |
| Non-water ligands | 47.0 | 47.5 |
| Root mean square deviation | ||
| Bond lengths (Å) | 0.007 | 0.004 |
| Bond angles (°) | 1.37 | 0.88 |
| Ramachandran favored/outliers (%) | 98.0/2.0 | 97.5/0.0 |
a The W117A data were slightly anisotropic. The estimates of resolution (from the I/σI > 2.0 analysis) in reciprocal lattice directions were 2.7, 2.4, and 2.4 Å.
Activity Assay and Steady-state Kinetic Analysis
NaMN adenylyltransferase activity of wild type and mutant variants of MtNadD was routinely monitored by coupling the NaAD product formation to the two-step generation of NADH by the action of ancillary NAD synthetase and alcohol dehydrogenase and was monitored at 340 nm as described (15). This assay was also used to assess the apparent Km for NaMN (which varied in the range of 0.1–3 mm) at saturating ATP (2 mm). The reaction mechanism of MtNadD was determined by varying one substrate concentration and fixing the concentration of the other substrate. The reaction rates were measured using an alternative end-point assay that coupled the formation of PPi product to colorimetric (malachite green) detection of free phosphate released upon action of added inorganic pyrophosphatase enzymatic hydrolysis as described (16). The activity of MtNadD was monitored at variable concentrations of ATP for a variety of fixed concentrations of NaMN ranging from 30 μm to 3 mm. The activity of MtNadD was also monitored at variable concentrations of NaMN for a variety of fixed concentrations of ATP ranging from 3 μm to 0.8 mm. The enzyme kinetic parameters were obtained by fitting the kinetic models to the experimental data using GraphPad Prism (GraphPad Software Inc., San Diego, CA). The best kinetic model for bisubstrate enzymatic reactions was selected based on visual inspection of the Lineweaver-Burk plots and the calculated r2 values. The initial velocities were fitted to the following equation for substrate inhibition in an ordered bi-reactant system (35),
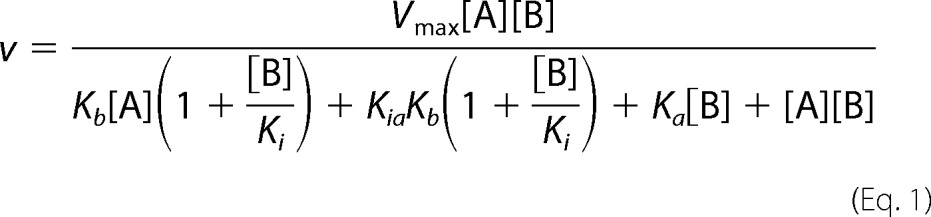 |
where Vmax is maximum velocity, Ka and Kb are Michaelis constants for corresponding substrates, Kia is the inhibition constant for A, and Ki is the dissociation constant of EB complex. In our model, A is the noninhibitory substrate ATP, whereas B is the inhibitory substrate NaMN. Initial velocity measurements for the bisubstrate enzyme kinetics and the inhibition studies were performed in duplicates and are presented as the average value (the variation between two parallel samples in these experiments was not >20%).
Protein Thermal Shift Assay
The assay was adopted from a previously published method (36). Briefly, the assay was carried out in a Framestar 384-well PCR plate with optical seals in the final assay volume of 10 μl. The assay mixture contained wild type MtNadD (2.5 μm) or W117A mutant (5 μm), 2 mm substrate, 10 mm MgCl2, and 5× Sypro Orange fluorescent dye in a 25 mm HEPES buffer, pH 7.3. The plate was heated and read on a Life Technologies ViiA7 RT-PCR instrument with a ramping speed of 0.05 °C/s. The melting temperature (Tm) was determined by taking the maximum point of the slope-derivative peak.
Inhibitory Analysis
A primary screening of potential MtNadD inhibitors was performed on the panel of 96 small molecule compounds originally identified as inhibitors of NadD from another bacterial pathogen, Staphylococcus aureus, with IC50 ≤ 10 μm, as reported in PubChem (Bioassay Identifier, AID: 624317) using the malachite green assay (at 80 μm NaMN, 300 μm ATP, and 50–100 μm concentration of compounds in the presence of 5% DMSO). Based on this analysis, 20 analogous compounds representing one identified inhibitory scaffold were selected for IC50 determination by the same assay. A kinetic inhibitory mechanism and Ki values were determined for selected compounds using the same assay at varying concentrations of substrates and inhibitors and fitting the data to the general inhibitory equation,
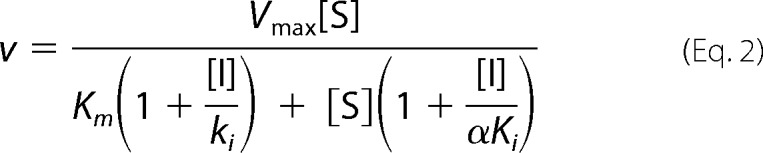 |
where the value of the empiric parameter α is indicative of the actual inhibitory mechanism: (i) truly competitive (α ≫ 1), (ii) non-competitive (α ≈ 1), or (iii) a mixed type (α > 1 or α < 1).
Mycobacterial Growth Suppression
Selected MtNadD inhibitors were tested in the model strain of Mycobacterium smegmatis in 7H9 medium at 37 °C using a 10–100 μm range of compound concentrations in 96-well plates and 2% DMSO, which did not interfere with the cell growth. The growth was monitored by optical density starting from A600 = 0.06. One of the few compounds (1594) displaying MIC values within the tested range was used to assess its effect on the NAD pool. Cells were grown for 22 h at a sublethal concentration of the compound, i.e. at 33 μm, harvested, washed by PBS, lysed, and analyzed using the Abcam NAD detection kit in comparison to a 2% DMSO-grown control (with normalization by total protein).
RESULTS
Quaternary Structure
The three-dimensional structure of the wild type enzyme and a mutant variant (W117A) of MtNadD was determined to a resolution of 2.4 Å (Table 1). This mutant was selected for structural analysis because of its significantly improved expression and stability that translated in a more complete structure. Thus, despite differences in the crystal packings (P3121 versus P212121), the two structures have identical quaternary structures (a tetramer with a point symmetry 222; Fig. 2A). In the wild type protein structure, ∼20% of residues are disordered, of which 4 residues are at the N terminus, 14–20 residues are internal, and 20 residues are at the C terminus. On the other hand, in the W117A mutant structure only 10% of residues are disordered, of which 3 residues are at the N terminus, 3–5 residues are internal, and 19 residues are at the C terminus. Two of four tetrameric interfaces (between subunits A and B or D and C) bury a surface area of 870 Å2 each and are likely to have biological significance, as predicted by the PISA server. The two other pairs of interfaces (between subunits A and D or B and C) bury a surface area of ∼580 Å2 each and are likely to form only under special conditions, such as an elevated protein concentration or the presence of interface-stabilizing ligands. Indeed, in both our structures, we observed non-water ligands bound at the tetramer interfaces (Fig. 2A). These observations suggest that the dimer is the active biological unit. Consistently, the analysis of apparent Mr of MtNadD by the size exclusion chromatography demonstrated that the protein elutes as a dimer. Superimposition of four monomers from the wild type and mutant MtNadD structures (Fig. 2B) illustrated that their conformations were almost identical (root mean square deviation for the ∼160 of 195 α-carbon atoms <0.25 Å) everywhere except for ∼20 residues around the helix α6. In the wild type protein, this helix is disordered, but in the mutant structure it has two alternative conformations (Fig. 2B). Thus, it is reasonable to conclude that the helix α6 is disordered in the apo form of MtNadD that appears to produce little effect on the conformation of the enzyme.
FIGURE 2.
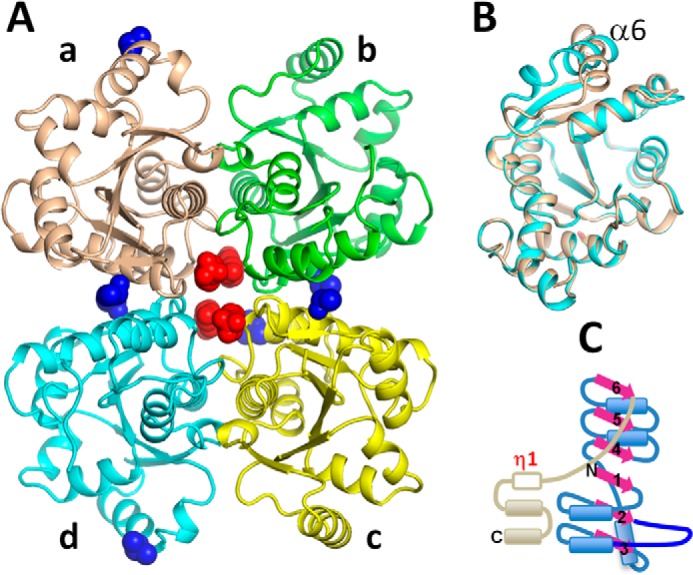
A, a tetramer that is observed in the NadD structure of both the wild type and the W117A mutant structures. The protein is shown as a schematic in which the monomeric subunits have different colors. Four HEPES molecules (red) and seven DMSO molecules (blue) acquired from the crystallization buffer are shown as van der Waals balls. B, superimposition of the tetramer subunits A and D. The helices α6 have different orientations. C, schematic representation of MtNadD secondary structure elements.
Overview of the Structure
The overall structure of MtNadD reveals a topological organization of the NAD(P) dinucleotide binding fold (Rossmann fold) with six-stranded parallel β sheets flanked by helices and connecting loops (Fig. 2C). This organization is characteristic of the nucleotidyltransferase superfamily of dinucleotide binding α/β-phosphodiesterases. A very similar structural organization was previously reported for several orthologous representatives of the bacterial NadD family (16, 22–24, 37, 38) as illustrated by two examples from Gram-positive and Gram-negative species (Fig. 3). A distantly homologous human NMNAT also shares a similar overall topology (Fig. 3), although there are substantial differences at the level of active site that translate into a radically different (dual for NMN and NaMN) substrate specificity. The key topologically distinctive feature of MtNadD is a short helix (η1), (residues 162–165) replacing a large portion of an extended coil (Fig. 4A and Fig. 3). In all other known NadD structures, this helix connects the C terminus of the β6 strand with another short and globally conserved helix (α7, residues 168–173 in MtNadD) that contains the C-terminal ATP binding motif (as highlighted in the multiple sequence alignment in Fig. 5). This 310 helix seems to play a critical role for the overall enzyme structure and function that distinguishes MtNadD from all other pyridine nucleotide adenylyltransferases reported to date. Indeed, as shown in Fig. 4A, the presence of this helix triggers an important structural re-organization, compared with other NadD structures, that ultimately affects substrate binding. First, the helix η1 pushes the adjacent β6 strand closer to the central cavity harboring the active site. In turn, the β6 strand brings the parallel strands β5 and β4 toward the same direction. Ultimately, the helix α5, which is contiguous to the β4 strand, is considerably shifted toward the active site cleft (∼6.4 Å). As a result, a tightly packed η1 helix enforces a compact conformation of the ligand binding sites of MtNadD that are significantly more compact than the corresponding sites in the product-bound forms of crystallographically investigated NadD members (Fig. 4A). Moreover, modeling substrates (ATP, NaMN) or a product (NaAD) in the MtNadD active site reveals steric hindrance that would not afford ligand binding without “opening” its apparently inactive conformation. In addition to mechanistic implications (discussed below), this unusual closed conformation provides a plausible explanation of the many failed attempts to co-crystallize MtNadD with substrates or a product. This is in contrast with our previous experience on co-crystallization of other members of the NaMN/NMN adenylyltransferase family, which have consistently yielded better results compared with crystallization of their respective apoenzymes.
FIGURE 3.
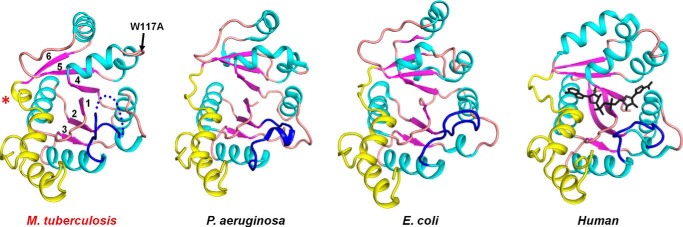
Structural topology of MtNadD, E. coli NadD (PDB code 1K4K), P. aeruginosa NadD (PDB code 1YUL), and human NMNAT (PDB code 1KKU). The overall topology of MtNadD is highly similar to other representatives of the bacterial NadD family illustrated by examples from Gram-negative and Gram-positive species and to a distantly homologous human NMNAT. For comparison, all apo forms of the protein family were used. The position of the active site is indicated by the NaAD product (black) from PDB code 1KQO superimposed on the human enzyme. Secondary elements of the core Rossman fold are depicted in cyan helices, purple strands, and an orange coil. The variable C-terminal domain is in yellow. The flexible loop that has been implicated in NaMN binding is in blue. Disordered segments are depicted with dotted traces. The asterisk indicates the 310 helix η1, a unique feature of MtNadD, replacing an extended coil in all other characterized NaMN/NMN adenylyltransferases. When present, the product molecule is shown in a stick representation.
FIGURE 4.

A, closed conformation of apoNadD from Mtb (yellow) as revealed by superposition with a canonical open conformation of apoNadD from B. anthracis (magenta, PDB code 2QTM) and its NaAD-bound form (cyan, PDB code 2QTR, with a bound NaAD product shown in orange sticks). Major conformational changes (movement of flexible loops and α-helix shifts) are highlighted by dotted arrows. The tighter packing observed in the active site of the Mtb apoenzyme is mostly due to the α5 helix shift. The circle shows a zoomed-in surface area of MtNadD around the adenosine portion of modeled NaAD from a superimposed BaNadD-NaAD complex form. This portion contains the η1 structural element. It can be seen that the adenosine portion of NaAD cannot fit into mtNadD active site pocket (see panel B). B, close-up view of ATP and NaMN active subsites. Key residues of the MtNadD active site are rendered as cyan sticks. Their potential H-bonds with the modeled ATP substrate (pink), based on superimposition with the PaNadD-ATP complex form (PDB code 1YUN), and with the NaMN portion of NaAD (gold), based on superimposition with the BaNadD-NaAD complex form (PDB code 2QTR), are represented by dashed lines. The steric clash of Leu-164 with the adenine ring is marked by a dotted circle.
FIGURE 5.

Structure-based sequence alignment of MtNadD with representative members of the bacterial NadD family and with human NMNAT isozymes. Secondary structure elements of the MtNadD are represented by spirals (α and 310 helices) and arrows (β strands). The 310 helix is labeled η. Partially conserved residues (numbered by MtNadD sequence) are shown on a gray background, and universally conserved residues are highlighted by a solid background color. Mutated residues are marked by a red triangle. The N-terminal and C-terminal ATP binding motifs are indicated under the sequences.
Active Site
A close inspection of the active site area, aided by a structure-based sequence alignment, highlighted both conserved and divergent structural features from other characterized members of NadD that warranted further investigation. As shown in Fig. 4A, the MtNadD adenosine binding pocket exhibits a significant deviation from the same site in BaNadD due to a higher rigidity and tightness around the AMP moiety of the NaAD product. Most notably, the side chain of Leu-164, the central amino acid of helix η1, would clash with the adenine base (Fig. 4). Another interesting feature of MtNadD resides in the characteristic flexible loop connecting the β2 and α2 helices (residues 44–53) that in other NadDs is supposed to bend over NaMN in the process of substrate recognition. This loop is mainly disordered in our structures and in several other apo forms of the enzyme (Figs. 3 and 4). Furthermore, it displays a rather divergent sequence in MtNadD that may explain the low affinity of the MtNadD versus the pyridine mononucleotide substrate (see below). Besides these apparently characteristic features of MtNadD structure, a visual inspection and structure-based sequence alignment (Fig. 5) points to a general conservation of the catalytic core architecture (see conservation of signature motifs GXFXPX(H/T)XXH and SXTXXR, mainly implicated in ATP binding, and highly conserved residues Thr-85 and Trp-117, responsible for pyridine ring stabilization). Importantly, the position of Trp-177 in our structures appears to be far off from a modeled NaMN and is thus in a nonproductive position (see Fig. 4A). To elucidate the role of these key residues, we carried out an extensive site-directed mutagenesis that included several highly conserved residues characteristic of the NadD family. The latter effort seemed to be justified also because they have not been systematically tested in the NadD family.
Site-directed Mutagenesis
Mt nadD-pODC was used as a template to generate the following 13 mutants involving 12 protein residues: T12A, D14A, H17A, H20A, P44A, W45A, Q46A, K47A, T86A, W117A, W117F, L164A, and L164Q. Mutants were overexpressed in E. coli cells and purified to homogeneity with a two-step purification procedure as described under “Experimental Procedures.” SDS-PAGE of purified enzymes showed that all of the mutants were expressed as soluble proteins and migrated at the same position as the wild type MtNadD (about 27 kDa). Furthermore, the wild type MtNadD and all mutants except L164A, L164Q, and T12A showed the same elution profile at gel filtration corresponding to an ∼48-kDa protein dimer (not shown). The L164A and L164Q mutants eluted as both a monomer and a dimer, whereas the T12A mutant eluted as a dimer and as an aggregated protein. Table 2 lists the apparent kinetic parameters obtained for the 13 mutant enzymes. It can be observed that replacement with alanine of invariably or highly conserved signature residues Thr-12, Asp-14, His-17, His-20, Thr-86, and Trp-117, directly interacting with the substrate/product, provoke dramatic effects ranging from complete enzyme inactivation to a nearly 30-fold drop in kcat. Conversely, mutation of partially conserved amino acids yielded a more complex and diverse perturbation of enzyme activity, including opposite effects toward the substrate ATP and NaMN. For instance, alanine scanning of the β2-α2 connecting loop confirmed a key role of this structural element in NaMN substrate recognition with a significant depression of kcat and affinity Km for ATP. Similarly, replacement of Leu-164 with an alanine or glutamine was largely detrimental for enzymatic efficiency but had an inverse effect on kcat and Km. Taken together, these data combined with structural modeling (in comparison to other liganded structures) allowed us to dissect the two ATP and NaMN binding subsites of the enzyme.
TABLE 2.
Kinetic parameters of wild-type and mutant variants of MtNadD
Apparent kinetic parameters kcat and Km NaMN were determined in the presence of saturation ATP (1.5 mm) using the discontinuous coupled assay; the apparent Km, ATP was determined at saturating NaMN (2–3 mm) using the end-point malachite green assay.
| Mutations in MtNadD | kcat | Km NaMN | Km ATP | kcat/Km, NaMN | kcat/Km, ATP | IC50, 1594 |
|---|---|---|---|---|---|---|
| s−1 | mm | mm | s−1mm−1 | s−1mm−1 | μm | |
| None | 5.5 ± 0.5 | 0.48 ± 0.06 | 0.041 ± 0.007 | 12 | 134 | 35 ± 11 |
| T12A | 0.22 ± 0.02 | 0.53 ± 0.13 | 0.042 ± 0.006 | 0.4 | 5 | 24 ± 2 |
| D14A | <LODa | |||||
| H17A | <LOD | |||||
| H20A | <LOD | |||||
| P44A | 0.36 ± 0.02 | 0.84 ± 0.13 | 0.017 ± 0.003 | 0.4 | 21 | 37 ± 12 |
| W45A | 0.37 ± 0.03 | 0.98 ± 0.20 | 0.011 ± 0.003 | 0.4 | 34 | 56 ± 15 |
| Q46Ab | 4.7 ± 0.4 | 0.33 ± 0.09 | 0.033 ± 0.007 | 14 | 142 | NDc |
| K47A | 0.02 ± 0.002 | 2.70 ± 0.38 | 0.006 ± 0.001 | 0.007 | 3 | 94 ± 17 |
| T86A | 0.2 ± 0.02 | 2.80 ± 0.72 | 0.011 ± 0.2 | 0.07 | 18 | 114 ± 12 |
| W117A | 0.002 ± 0.0002 | 2.67 ± 0.50 | -d | 0.0008 | 30 ± 13 | |
| W117F | 0.03 ± 0.008 | 1.13 ± 0.70 | 0.023 ± 0.006 | 0.03 | 1.30 | ND |
| L164A | 0.13 ± 0.01 | 0.052 ± 0.027 | 0.17 ± 0.03 | 2.5 | 0.8 | ND |
| L164Q | 0.015 ± 0.002 | 0.43 ± 0.13 | 0.19 ± 0.07 | 0.04 | 0.08 | ND |
a LOD, limit of detection, is calculated as ∼0.0002 s−1 under assay conditions.
b Mutant enzyme preparation was only partially purified.
c ND, not determined.
d Value could not be reliably measured.
ATP Subsite
Inspection of residues surrounding the ATP binding site revealed an overall preservation of this active site portion with other members of NMN/NaMN adenylyltransferases. A modeled ATP based on superimposition of MtNadD with a PaNadD-ATP complex (Fig. 4B) reveals that the five highly conserved residues comprising the two ATP binding motifs HXXH and SXTXXR are in a productive position to make H-bond contacts with the ATP triphosphate group. The essential role of the two signature histidines was confirmed by mutagenesis (Table 2). The ATP adenosine binding portion of the active site is quite divergent due to the presence of a unique η1 helix that appears to prevent the access of the ATP substrate. In particular, as also shown for the NaAD product modeled into the active site (Fig. 4A), Leu-164 would clash with the adenine base (Fig. 4B). To investigate the role of Leu-164, we generated two mutants: L164Q, where leucine is converted into a polar amino acid, and L164A, which replaces the bulky leucine with small hydrophobic alanine. Kinetic analysis showed that both L164Q and L164A mutants displayed a 4-fold increase in Km for ATP compared with wild type MtNadD, but although the L164Q mutant is a barely active enzyme (∼700-fold drop of kcat), the fall of L164A activity is 40-fold. Collectively, these results point to a key role of Leu-164 in ATP substrate recognition and stabilization.
NaMN Subsite
To explore the key contacts of MtNadD to NaMN substrate, we mutated the two key residues that have been proposed to facilitate the binding of nicotinate mononucleotide. These residues, corresponding to MtNadD Trp-117 and Thr-86, are conserved across bacteria and human (Fig. 5). Trp-117, responsible for a stacking interaction against the pyridine ring of nicotinic acid, is crucial for mammalian NMNAT activity. When we mutated Trp-117 to alanine, a nearly 3000-fold drop in kcat was observed, and Km for the NaMN substrate decreased over 4-fold. Moreover, the transition of Trp-117 to Phe, an aromatic amino acid that can guarantee a π-stacking interaction to some extent, is less dramatic, yielding a 10-fold larger turnover number than W117A and just a 2-fold increase in Km for NaMN. Interestingly, Trp-117 resides at the end of a tight α5 helix, which, in other apoNadD structures, e.g. from Pseudomonas aeruginosa and Bacillus anthracis, but also human, is largely bent outward (not shown). Moreover, although this key tryptophan is already in a productive conformation in the apo form of other NadDs, in MtNadD it is in a divergent position (Fig. 4A). Taken together, the mutagenesis and structural analysis lead to two important conclusions: (i) they support a key role for Trp-117 in nicotinate stabilization, although its function is probably carried out upon a large conformational change implying an opening of the NaMN binding site, and (ii) they reveal important differences between MtNadD and other NadD structures, including human, thus providing a unique opportunity for selective drug design. Two conserved threonines, Thr-86 and Thr-12, are additional key residues stabilizing the NaMN substrate. A modeled NaAD based on superimposition of MtNadD with a BaNadD-NaAD complex shows that Thr-86 and Thr-12 could stabilize the pyridine carboxylate and the phosphate group through H-bonds involving both the main and side chains, respectively (Fig. 4B). Such roles are confirmed by a 30-fold drop of kcat and increased Km for NaMN of corresponding mutants (Table 2). Last, Fig. 4B indicates the position and H-bond network of Asp-14, a partially conserved residue of the GXFXPX(H/T)XXH motif shown by mutagenesis to have a critical role for NadD activity (Table 2). We speculate that Asp-14 might play an important structural rather than catalytic role given its outward orientation and the absence of direct interactions with substrate/product.
The Role of β2-α2 Connecting Loop
Next, we analyzed the role of a flexible loop connecting the β2 strand with the α2 helix (residues 44–53). This region contains a partially conserved motif PPHK(R) (Fig. 5) that has been reported to act as an arm to recognize the NaMN substrate or, more importantly, to bind the NaAD product. This loop, usually disordered in the apo form of bacterial NadDs, becomes ordered upon contact with the substrate or product (22, 23, 37, 38). Because this loop was not fully resolved in our structure, which is not uncommon in NadD structure apo forms, and to better elucidate its mechanistic role, we carried out a full alanine-scanning mutagenesis of the partially conserved motif Pro-44–Lys-47. The results clearly indicate a key role of this structural element in catalysis and in NaMN binding as reflected by increased values of Km for NaMN (Table 2). In particular, although Pro-44 contribution is likely to be only structural, Trp-45 might form a favorable stacking interaction with the nicotinate ring that would become sandwiched between two tryptophan aromatic systems. Last, in contrast to a fairly neutral effect of the Q46A mutant, K47A manifested an ∼6-fold increase in Km for NaMN and nearly abolished the enzyme activity (Table 2). Such a remarkable effect is attainable by disrupting a strong electrostatic interaction such as that between the negatively charged phosphate of NaMN moiety and the positively charged lysine. We, therefore, confirmed the role that was proposed for Lys/Arg of the PH(K/R) loop motif in other NadDs. The P44A and K47A mutants seem to have opposite effects on recognition of the two substrates, as they show an appreciable decrease in Km for ATP (at times below the detection limit of assay conditions). Strikingly, this seems to be a common trend for all the mutations that compromise NaMN substrate binding (see Table 2). A detailed steady-state kinetic analysis allowed us to reconcile these results with the model of competitive substrate inhibition exerted by NaMN (see the next paragraph). Indeed, if one assumes that NaMN might compete with the ATP binding site, all detrimental mutations for NaMN binding should at the same time generate an improved affinity toward the ATP site.
Kinetic Mechanism and Substrate Inhibition
A detailed bi-reactant initial velocity study of MtNadD kinetics was performed to gain insight into the MtNadD enzyme kinetic mechanism. Such an analysis was warranted by the unique structural properties displayed by this representative of the bacterial NadD family. Initial rate plots were best fitted by the substrate inhibition model in an ordered bi-reactant system (see Fig. 6). The downward curvature of hyperbolic plots (Fig. 6A) observed at high NaMN concentrations (>0.1 mm) and low ATP concentrations (<30 μm) is indicative of the enzyme inhibition exerted by the NaMN substrate. The observed NaMN substrate inhibition at a low ATP concentration range also suggests a preferred sequential mechanism where ATP binds first (see Fig. 6C for a proposed reaction course diagram). The values of Km ATP and Km NaMN are 5 ± 2 μm and 0.48 ± 0.03 mm, respectively. Although the unusually high Km for NaMN is consistent with its apparent Km value previously obtained for MtNadD (17) and further confirmed in this study (Table 2), the Km for ATP resulted in a 10-fold decrease as compared with its corresponding apparent value (∼40 μm). This discrepancy is reconciled by our two-step kinetic model that takes into account the NaMN substrate inhibition. Furthermore, MtNadD mutants with impaired NaMN binding exhibit a higher affinity for ATP than the wild type enzyme. This is also consistent with our model of competitive substrate inhibition of the NaMN substrate. The inhibitory constant (Ki) of the NaMN substrate was assessed to be 0.51 mm.
FIGURE 6.

Steady-state kinetic analysis of MtNadD. Initial rate plots as a function of NaMN substrate concentration (mm) measured at varied fixed concentration of ATP substrate (A) and vice versa (B). Details of the assay setup are provided “Experimental Procedures.” Of note, due to the dynamic range of the enzymatic assay, it was not possible to provide initial rate values at an ATP concentration below its calculated Km. Thus, Km for ATP is an approximate value. C, simplified reaction course diagram. The binding of NaMN in the absence of ATP-induced enzyme activation generates a nonproductive E·NaMN complex.
Substrate and Product Binding by Protein Thermal Shift Assay
Protein thermal shift provides an assessment of the stability of protein and protein-ligand complexes based on melting temperature (36). We used this approach to assess interactions of MtNadD with substrates and products. The protein thermal shift analysis revealed a substantial difference between MtNadD binding with the two substrates ATP (ΔTm = 10.7 °C) and NaMN (ΔTm = 0.5 °C) (at saturating concentration, 2 mm). This is consistent with the proposed mechanistic model assuming high affinity ATP binding and stabilization of the enzyme in its active conformation. The Tm shift of 10.1 °C for PPi is comparable with ATP, pointing to the importance of the interaction with the pyrophosphoryl moiety of product/substrate. The product NaAD had modest but statistically significant stabilization with ΔTm = 2.7 °C (Fig. 7A). The relatively lower binding energy of the main product (NaAD) would be beneficial for product release and the reaction proceeding in the forward direction. Finally, we used this assay to compare wild type MtNadD with the W117A mutant, which was used for additional structural analysis. The mutant enzyme exhibited a somewhat higher melting temperature with respect to the wild type MtNadD (Fig. 7B), which is consistent with its improved stability (and ease of crystallization). However, the major effects of ATP (and PPi) binding to W117A mutant were essentially the same as that observed for the wild type MtNadD, substantiating the relevance of structural data obtained for the mutant enzyme.
FIGURE 7.
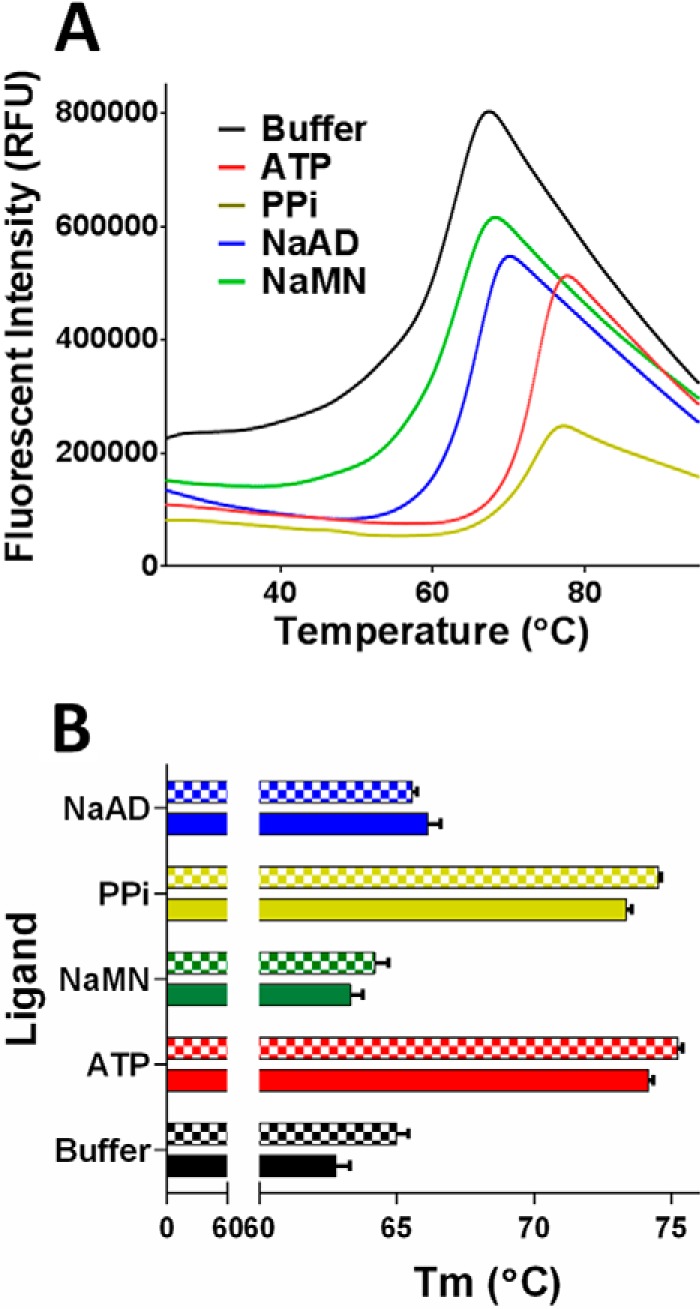
Protein thermal shift analysis with ligands ATP, PPi, NaAD, and NaMN. A, denaturation curves of the wild type MtNadD in the absence (buffer sample) and in the presence of ligands. B, comparison of Tm values for wild type MtNadD and W117A mutant (checked bars). RFU, relative fluorescence units.
Assessment of MtNadD Druggability
Inhibitory profiling of MtNadD was assessed by using a subset of small molecule compounds that were originally selected by a high throughput screening campaign targeting another member of this family, NadD from S. aureus (PubChem AID: 624317). Of the 96 compounds tested (with IC50 for S. aureus NadD < 10 μm), representing 10 distinct scaffolds, only one scaffold displayed appreciable (albeit far less efficient) inhibition of MtNadD. This was not surprising given the structural differences of these enzymes. A comparison of ∼20 analogs representing this scaffold revealed a broad range of IC50 values from 17 to >180 μm on MtNadD (Table 3) and no detectable inhibition (IC50 > 250 μm) of human NMNAT1. Several compounds showed moderate growth inhibition of M. smegmatis with MIC values in the range of 20–80 μm (see Fig. 8D and Table 3).
TABLE 3.
Inhibitory profiling of MtNadD
IC50 and MIC80 for MtNadD were obtained using a series of analogous compounds previously identified as inhibitors of NadD from S. aureus (Sa NadD) PubChem AID: 624317. IC50 values for SaNadD, originally obtained for the same compounds, are shown in parallel.
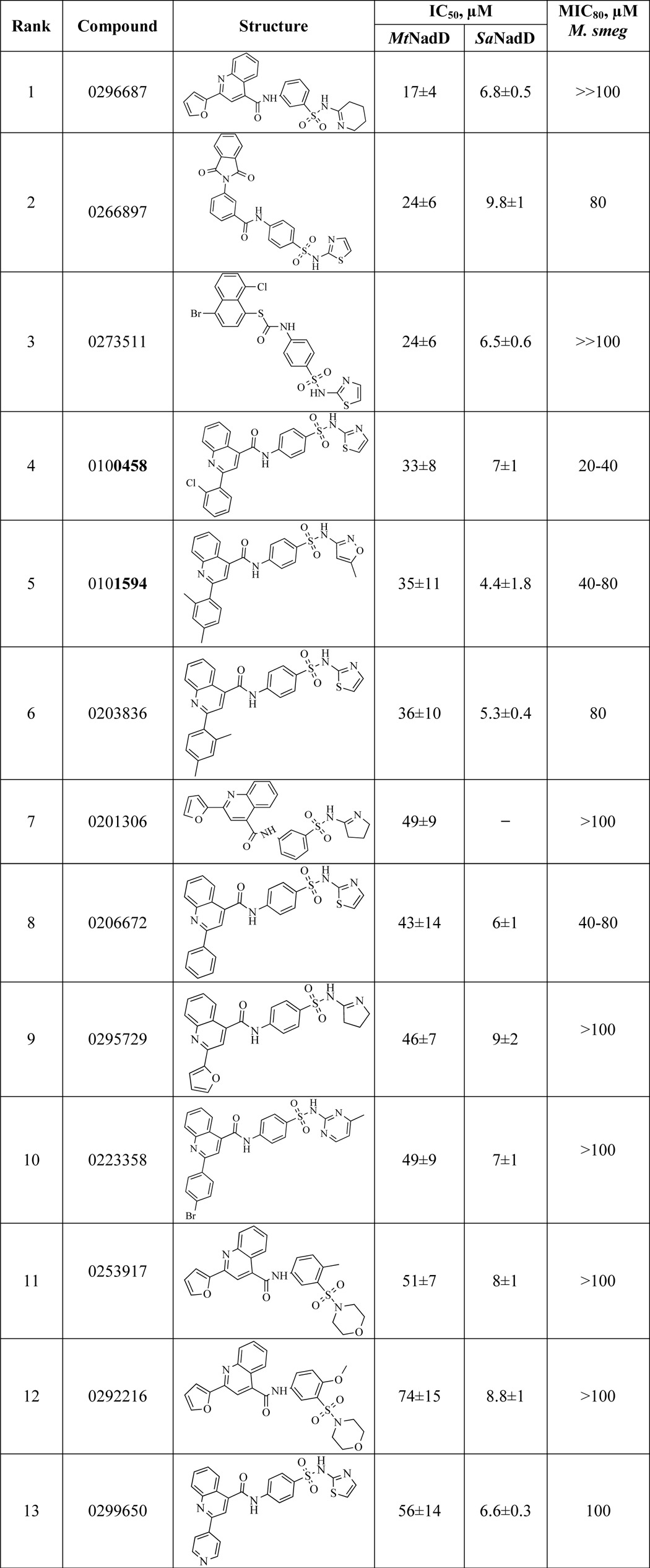
FIGURE 8.

Inhibitory profiling of compound 1549 (shown in the inset). Competitive inhibitory mechanism with respect to the NaMN substrate (apparent Ki = 25 μm) at a fixed concentration of ATP 300 μm. Inhibitor concentration ranged from 0 to 62 μm (A). Inhibition of MtNadD by compound 1594 at varying ATP concentrations is shown. Appreciable inhibition could be detected only above 100 μm inhibitor concentration (B). Effect of 1594 inhibitor on selected MtNadD mutants (C) is shown. The apparent values of Km for the NaMN substrate (blue bars) and the IC50 (at half-saturating concentration of NaMN for each mutant) for compound 1594 are presented as -fold change compared with the respective wild type values (absolute values are provided in Table 2). Two of the three mutants with the largest increase in Km for NaMN (∼5.5-fold) also show the largest increase in the observed IC50 for compound 1594 (∼3-fold). D, M. smegmatis growth inhibition at 20 μm by compounds 1594 and 0458.
Under conditions of partial growth suppression elicited by compound 1594 (PubChem SID 17409160, see “Experimental Procedures” for details), the estimated NAD intracellular concentration dropped from 7.4 to 4 mm. This ∼2-fold decrease of the NAD level provides supporting evidence that the compound 1594 had specific on-target activity.
A steady-state inhibitory kinetic analysis (Fig. 8, A and B) revealed a competitive mechanism of this inhibitor with respect to NaMN (apparent Ki = 25 μm) and a mixed-type inhibition for ATP (apparent Ki ≫ 150 μm). Notably, two of the three mutants (K47A and T86A but not W117A) with the highest apparent Km for NaMN (2.7–2.8 mm) also showed the largest increase (∼3-fold) of the observed IC50 for compound 1594 (see Fig. 8C and Table 2). These results provide additional evidence that there is a specific interaction of this inhibitor with the NaMN binding subsite.
DISCUSSION
The MtNadD enzyme has been validated as an attractive antimycobacterial drug target by genetic methods in vitro and in vivo (9, 10, 17). In this study we used a combination of structural and steady-state kinetic analyses with directed mutagenesis and inhibitory profiling to provide guidelines for the rational design of inhibitors targeting this enzyme.
One of the most distinctive features of the MtNadD structure, which was solved in apo form at 2.4 Å resolution, is the closed conformation of its active site. This conformation is implemented by the tight packing of an unusual 310 helix that hinders substrate access to the active site and, thus, renders this conformation catalytically incompetent. This is in contrast to an open apoenzyme conformation characteristic of all other known structures within the bacterial NadD family as well as distantly related human NMNAT and archaeal NadM families. In all these enzymes, the respective structural segment exists as an extended coil, providing unconstrained access to the active site.
A unique, closed conformation of the Mtb apoenzyme likely dictates its rather unusual kinetic properties: (i) relatively high Km for NaMN (480 μm as compared with 50 μm Km for ATP) and (ii) pronounced substrate (NaMN) inhibition at a low ATP concentration (Fig. 6). A possible interpretation is that when ATP first binds to the enzyme, there is a transient opening of the ATP subsite that is obscured by the 310 helix (Fig. 4). This shifts the active site toward a conformation that is functionally competent for productive binding of the second substrate, NaMN, and thus for catalysis. However, a less favorable binding of NaMN first apparently yields a nonproductive complex (as observed at high NaMN/low ATP concentrations), which is not conducive for the interaction with the second substrate (ATP). This hypothetical mechanism (Fig. 6C) is corroborated by some of the results of site-directed mutagenesis. For example, the replacement of a bulky side chain of Leu-164 in the 310 helix with the short-chained Ala has opposite effects on Km values of the two substrates. A 4-fold increase in Km for ATP may reflect a net loss of favorable interactions with ATP, which also impairs the catalytic rate (40-fold drop in kcat). However, an observed ∼10-fold improvement of the Km for NaMN suggests that a relaxation of the original closed conformation in the L164A mutant may allow NaMN binding in either order. Notably, a replacement of Leu-164 with a hydrophilic, but also bulky Gln has a similar negative effect on Km for ATP (with an even more profound >350-fold drop in kcat), whereas it does not improve the Km for NaMN.
A multiple sequence alignment of the nearly 70 mycobacterial NadDs currently available from genomic databases indicated >98% sequence identity for the vast majority of the orthologs. In particular, a strict conservation of all distinct features analyzed for M. tuberculosis NadD, i.e. the moveable arm (Pro-44–Lys-47) and the η1 helix (Pro-161–Ala-164), are conserved throughout, enabling us to project the proposed reaction mechanism to other mycobacterial NadDs.
The established distinctive features of MtNadD may have important physiological implications. It is tempting to speculate that the closed, catalytically inactive conformation of the enzyme is functionally adequate (or at least consistent) with a metabolic dormancy state of Mtb cells. Indeed, under low ATP production reflecting low respiratory activity of dormant cells, MtNadD would have a low enzymatic activity even at relatively high levels of NaMN. This would prevent depletion of the limiting ATP pool for producing NAD beyond the minimal requirements of nonreplicating cells. Moreover, even under enhanced ATP supply, NAD synthesis would remain limited (due to intrinsically elevated Km for NaMN) before sufficient accumulation of the NaMN precursor from the increased de novo synthesis and/or NAD degradation and recycling. Notably, previous studies have established several functional routes yielding NaMN in Mtb cells (Fig. 1). Beyond de novo synthesis from aspartate, the salvage of vitamin B3 (nicotinamide or nicotinate) from the medium, inferred from the presence of respective genes (pncA and pncB) in all mycobacterial genomes, has been documented for M. tuberculosis (10). These enzymes also drive recycling of intracellular nicotinamide resulting from NAD utilization by CobB (and possibly other, yet unknown, N-glycosyl hydrolases). Recycling of NMN, another NAD degradation product generated by the NAD-dependent DNA ligase, is likely mediated by a recently characterized enzyme NMN deamidase, directly converting it to NaMN (39, 40).
A hypothesized substrate-level regulation of NAD biogenesis and homeostasis may play an important role in the well known ability of Mtb to transition between dormant and proliferating states during the course of an infection. Interestingly, the inferred accumulation of NaMN in non-replicating Mtb may provide a rationale for the well known presence of nicotinic acid in the breath of tuberculosis patients (41). Indeed, in our recent study we observed an appreciable excretion of nicotinic acid after accumulation and degradation of NaMN (e.g. by reverse reaction of PncB and/or N-glycosyl hydrolases) as a result of NadD knockdown in the model system of M. smegmatis (17).
Finally, the elucidated structural basis of MtNadD “enzymatic dormancy” hints at a previously unexplored strategy for the development of small molecule inhibitors that would target and further stabilize the catalytically inactive closed form of the enzyme. A rational implementation of this strategy would include in silico screening for small molecule compounds selectively binding and locking the apoenzyme. Identification of such bioactive inhibitors would provide the first step toward developing new drugs against actively replicating and dormant forms of Mtb (9, 10, 17).
Acknowledgments
Identification of S. aureus NadD inhibitors from National Institutes of Health (NIH) Molecular Libraries Screening Centers Network (MLSCN) collection used in this study was supported by NIH Grant R03 MH095597-02 (to A. O.). We are grateful to Dr. A. Pinkerton and other colleagues at Conrad Prebys Center for Chemical Genomics (Sanford-Burnham Medical Research Institute (SBMRI)) for invaluable contributions to this effort. We thank Dr. Dongyang Li, Yuzhou Xu, and Yannan Wang of Shanghai ChemPartner for protein expression, protein purification, and crystallography support and Dr. John A. Thomson of Vertex Pharmaceuticals for support of the project. We thank Brian Schuster for help on mycobacterial genetics and physiology during his graduate work at Harvard Medical School. The Berkeley Center for Structural Biology is supported in part by the NIGMS, National Institutes of Health and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract DE-AC02-05CH11231.
This study was funded for the most part by Vertex Pharmaceuticals.
The atomic coordinates and structure factors (codes 4X0E and 4RPI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Mtb
- M. tuberculosis
- NaMN
- nicotinic acid mononucleotide
- NMN
- nicotinamide mononucleotide
- NaAD
- nicotinic acid adenine dinucleotide
- NMNAT
- nicotinamide mononucleotide adenylyltransferase
- Ni-NTA
- nickel-nitrilotriacetic acid
- MIC
- minimal inhibitory concentration
- MtNadD
- M. tuberculosis NadD
- NadD
- nicotinate mononucleotide adenylyltransferase
- NadE
- NAD synthase
- BaNadD
- B. anthracis NadD.
REFERENCES
- 1. Organization W. H. (2014) Global Tuberculosis Report 2014, p. 1, World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Zumla A., George A., Sharma V., Herbert N., and Baroness Masham of, I. (2013) WHO's 2013 global report on tuberculosis: successes, threats, and opportunities. Lancet 382, 1765–1767 [DOI] [PubMed] [Google Scholar]
- 3. Gengenbacher M., Kaufmann S. H. (2012) Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 36, 514–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fattorini L., Piccaro G., Mustazzolu A., Giannoni F. (2013) Targeting dormant bacilli to fight tuberculosis. Mediterr. J. Hematol. Infect. Dis. 5, e2013072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zumla A. I., Gillespie S. H., Hoelscher M., Philips P. P., Cole S. T., Abubakar I., McHugh T. D., Schito M., Maeurer M., Nunn A. J. (2014) New antituberculosis drugs, regimens, and adjunct therapies: needs, advances, and future prospects. Lancet Infect. Dis. 14, 327–340 [DOI] [PubMed] [Google Scholar]
- 6. Andries K., Verhasselt P., Guillemont J., Göhlmann H. W., Neefs J. M., Winkler H., Van Gestel J., Timmerman P., Zhu M., Lee E., Williams P., de Chaffoy D., Huitric E., Hoffner S., Cambau E., Truffot-Pernot C., Lounis N., Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227 [DOI] [PubMed] [Google Scholar]
- 7. Haagsma A. C., Podasca I., Koul A., Andries K., Guillemont J., Lill H., Bald D. (2011) Probing the interaction of the diarylquinoline TMC207 with its target mycobacterial ATP synthase. PLoS ONE 6, e23575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koul A., Vranckx L., Dhar N., Göhlmann H. W., Özdemir E., Neefs J. M., Schulz M., Lu P., Mørtz E., McKinney J. D., Andries K., Bald D. (2014) Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat. Commun. 5, 3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim J. H., O'Brien K. M., Sharma R., Boshoff H. I., Rehren G., Chakraborty S., Wallach J. B., Monteleone M., Wilson D. J., Aldrich C. C., Barry C. E., 3rd, Rhee K. Y., Ehrt S., Schnappinger D. (2013) A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad. Sci. U.S.A. 110, 19095–19100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boshoff H. I., Xu X., Tahlan K., Dowd C. S., Pethe K., Camacho L. R., Park T. H., Yun C. S., Schnappinger D., Ehrt S., Williams K. J., Barry C. E., 3rd. (2008) Biosynthesis and recycling of nicotinamide cofactors in Mycobacterium tuberculosis. An essential role for NAD in nonreplicating bacilli. J. Biol. Chem. 283, 19329–19341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerdes S. Y., Scholle M. D., D'Souza M., Bernal A., Baev M. V., Farrell M., Kurnasov O. V., Daugherty M. D., Mseeh F., Polanuyer B. M., Campbell J. W., Anantha S., Shatalin K. Y., Chowdhury S. A., Fonstein M. Y., Osterman A. L. (2002) From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J. Bacteriol. 184, 4555–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang N., Kolhatkar R., Eyobo Y., Sorci L., Rodionova I., Osterman A. L., Mackerell A. D., Zhang H. (2010) Complexes of bacterial nicotinate mononucleotide adenylyltransferase with inhibitors: implication for structure-based drug design and improvement. J. Med. Chem. 53, 5229–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sorci L., Blaby I., De Ingeniis J., Gerdes S., Raffaelli N., de Crécy Lagard V., Osterman A. (2010) Genomics-driven reconstruction of acinetobacter NAD metabolism: insights for antibacterial target selection. J. Biol. Chem. 285, 39490–39499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sorci L., Blaby I. K., Rodionova I. A., De Ingeniis J., Tkachenko S., de Crécy-Lagard V., Osterman A. L. (2013) Quinolinate salvage and insights for targeting NAD biosynthesis in group A streptococci. J. Bacteriol. 195, 726–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sorci L., Martynowski D., Rodionov D. A., Eyobo Y., Zogaj X., Klose K. E., Nikolaev E. V., Magni G., Zhang H., Osterman A. L. (2009) Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 106, 3083–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sorci L., Pan Y., Eyobo Y., Rodionova I., Huang N., Kurnasov O., Zhong S., MacKerell A. D., Jr., Zhang H., Osterman A. L. (2009) Targeting NAD biosynthesis in bacterial pathogens: structure-based development of inhibitors of nicotinate mononucleotide adenylyltransferase NadD. Chem. Biol. 16, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodionova I. A., Schuster B. M., Guinn K. M., Sorci L., Scott D. A., Li X., Kheterpal I., Shoen C., Cynamon M., Locher C., Rubin E. J., Osterman A. L. (2014) Metabolic and bactericidal effects of targeted suppression of NadD and NadE Enzymes in mycobacteria. MBio 5, e00747–00713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Ingeniis J., Kazanov M. D., Shatalin K., Gelfand M. S., Osterman A. L., Sorci L. (2012) Glutamine versus ammonia utilization in the NAD synthetase family. PLoS ONE 7, e39115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sorci L., Kurnasov O., Rodionov D., Osterman A. (2010) Genomics and enzymology of NAD biosynthesis. Comprehensive Natural Products, II Chemistry and Biology, pp. 213–257, Elsevier, Oxford [Google Scholar]
- 20. LaRonde-LeBlanc N., Resto M., Gerratana B. (2009) Regulation of active site coupling in glutamine-dependent NAD(+) synthetase. Nat. Struct. Mol. Biol. 16, 421–429 [DOI] [PubMed] [Google Scholar]
- 21. Hegymegi-Barakonyi B., Székely R., Varga Z., Kiss R., Borbély G., Németh G., Bánhegyi P., Pató J., Greff Z., Horváth Z., Mészáros G., Marosfalvi J., Erôs D., Szántai-Kis C., Breza N., Garavaglia S., Perozzi S., Rizzi M., Hafenbradl D., Ko M., Av-Gay Y., Klebl B. M., Orfi L., Kéri G. (2008) Signalling inhibitors against Mycobacterium tuberculosis: early days of a new therapeutic concept in tuberculosis. Curr. Med. Chem. 15, 2760–2770 [DOI] [PubMed] [Google Scholar]
- 22. Sershon V. C., Santarsiero B. D., Mesecar A. D. (2009) Kinetic and x-ray structural evidence for negative cooperativity in substrate binding to nicotinate mononucleotide adenylyltransferase (NMAT) from Bacillus anthracis. J. Mol. Biol. 385, 867–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han S., Forman M. D., Loulakis P., Rosner M. H., Xie Z., Wang H., Danley D. E., Yuan W., Schafer J., Xu Z. (2006) Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Staphylococcus aureus: structural basis for NaAD interaction in functional dimer. J. Mol. Biol. 360, 814–825 [DOI] [PubMed] [Google Scholar]
- 24. Yoon H. J., Kim H. L., Mikami B., Suh S. W. (2005) Crystal structure of nicotinic acid mononucleotide adenylyltransferase from Pseudomonas aeruginosa in its apo and substrate-complexed forms reveals a fully open conformation. J. Mol. Biol. 351, 258–265 [DOI] [PubMed] [Google Scholar]
- 25. Sorci L., Cimadamore F., Scotti S., Petrelli R., Cappellacci L., Franchetti P., Orsomando G., Magni G. (2007) Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry 46, 4912–4922 [DOI] [PubMed] [Google Scholar]
- 26. Berger F., Lau C., Dahlmann M., Ziegler M. (2005) Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 280, 36334–36341 [DOI] [PubMed] [Google Scholar]
- 27. Osterman A. L., Lueder D. V., Quick M., Myers D., Canagarajah B. J., Phillips M. A. (1995) Domain organization and a protease-sensitive loop in eukaryotic ornithine decarboxylase. Biochemistry 34, 13431–13436 [DOI] [PubMed] [Google Scholar]
- 28. Yang C., Rodionov D. A., Rodionova I. A., Li X., Osterman A. L. (2008) Glycerate 2-kinase of Thermotoga maritima and genomic reconstruction of related metabolic pathways. J. Bacteriol. 190, 1773–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daugherty M., Vonstein V., Overbeek R., Osterman A. (2001) Archaeal shikimate kinase, a new member of the GHMP-kinase family. J. Bacteriol. 183, 292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 34. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Segel I. H. (1975) Enzyme Kinetics, pp. 819–826, John Wiley & Sons, Inc., New York [Google Scholar]
- 36. Lo M. C., Aulabaugh A., Jin G., Cowling R., Bard J., Malamas M., Ellestad G. (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 332, 153–159 [DOI] [PubMed] [Google Scholar]
- 37. Olland A. M., Underwood K. W., Czerwinski R. M., Lo M. C., Aulabaugh A., Bard J., Stahl M. L., Somers W. S., Sullivan F. X., Chopra R. (2002) Identification, characterization, and crystal structure of Bacillus subtilis nicotinic acid mononucleotide adenylyltransferase. J. Biol. Chem. 277, 3698–3707 [DOI] [PubMed] [Google Scholar]
- 38. Zhang H., Zhou T., Kurnasov O., Cheek S., Grishin N. V., Osterman A. (2002) Crystal structures of E. coli nicotinate mononucleotide adenylyltransferase and its complex with deamido-NAD. Structure 10, 69–79 [DOI] [PubMed] [Google Scholar]
- 39. Galeazzi L., Bocci P., Amici A., Brunetti L., Ruggieri S., Romine M., Reed S., Osterman A. L., Rodionov D. A., Sorci L., Raffaelli N. (2011) Identification of nicotinamide mononucleotide deamidase of the bacterial pyridine nucleotide cycle reveals a novel broadly conserved amidohydrolase family. J. Biol. Chem. 286, 40365–40375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sorci L., Brunetti L., Cialabrini L., Mazzola F., Kazanov M. D., D'Auria S., Ruggieri S., Raffaelli N. (2014) Characterization of bacterial NMN deamidase as a Ser/Lys hydrolase expands diversity of serine amidohydrolases. FEBS Lett. 588, 1016–1023 [DOI] [PubMed] [Google Scholar]
- 41. Syhre M., Manning L., Phuanukoonnon S., Harino P., Chambers S. T. (2009) The scent of Mycobacterium tuberculosis: part II breath. Tuberculosis 89, 263–266 [DOI] [PubMed] [Google Scholar]