

Background: The enzyme, Δ1-pyrroline-5-carboxylate dehydrogenase (P5CDH), is a key enzyme involved in proline catabolism.
Results: The parasite Trypanosoma cruzi up-regulates P5CDH during host infection.
Conclusion: T. cruzi uses P5C to produce energy, resist metabolic stress, and invade host cells through this mitochondrion-bound enzyme.
Significance: The oxidation of P5C is sufficient to supply energy and enhance the pathogenicity of T. cruzi.
Keywords: Bioenergetics, Enzyme Mechanism, Host-Pathogen Interaction, Mitochondrial Metabolism, Parasite Metabolism, Trypanosoma cruzi
Abstract
Proline is crucial for energizing critical events throughout the life cycle of Trypanosoma cruzi, the etiological agent of Chagas disease. The proline breakdown pathway consists of two oxidation steps, both of which produce reducing equivalents as follows: the conversion of proline to Δ1-pyrroline-5-carboxylate (P5C), and the subsequent conversion of P5C to glutamate. We have identified and characterized the Δ1-pyrroline-5-carboxylate dehydrogenase from T. cruzi (TcP5CDH) and report here on how this enzyme contributes to a central metabolic pathway in this parasite. Size-exclusion chromatography, two-dimensional gel electrophoresis, and small angle x-ray scattering analysis of TcP5CDH revealed an oligomeric state composed of two subunits of six protomers. TcP5CDH was found to complement a yeast strain deficient in PUT2 activity, confirming the enzyme's functional role; and the biochemical parameters (Km, kcat, and kcat/Km) of the recombinant TcP5CDH were determined, exhibiting values comparable with those from T. cruzi lysates. In addition, TcP5CDH exhibited mitochondrial staining during the main stages of the T. cruzi life cycle. mRNA and enzymatic activity levels indicated the up-regulation (6-fold change) of TcP5CDH during the infective stages of the parasite. The participation of P5C as an energy source was also demonstrated. Overall, we propose that this enzymatic step is crucial for the viability of both replicative and infective forms of T. cruzi.
Introduction
Protozoan parasites include several species responsible for a number of major health problems affecting humans and livestock as well as wild and domesticated animals. Organisms belonging to the genera Trypanosoma, Leishmania, and Plasmodium are clinically relevant and serve as models for studying the adaptation strategies of unicellular organisms to different environments. Changes in organelle composition, the regulation of gene expression and “metabolic architecture,” among other adaptations, can greatly affect the success of the survival strategies of these organisms. Trypanosoma cruzi is a vector-borne hemoflagellate that can infect humans and cause Chagas disease, one of the 13 diseases classified by the World Health Organization as “neglected diseases” that affect 8–10 million people and cause 10 thousand deaths per year worldwide (1–3). Natural transmission of T. cruzi occurs via its parasitic colonization of a blood-sucking triatomine insect (popularly known as the kissing bug), followed by its passage into the insect excreta, which subsequently passes the infective form of the parasite to a mammalian host (e.g. humans) (2, 4). Additionally, evidence of outbreaks attributed to vector-excreta-contaminated food indicates the oral transmission of T. cruzi (5, 6). Recently, the World Health Organization warned that Chagas disease has become a more serious health problem due to its spread from endemic areas (south of the United States to Argentina and Chile) to nonendemic countries (3).
Because T. cruzi lives in different environments throughout its life cycle (i.e. the different regions of the insect host digestive tube and the intra- and extracellular media in different mammalian tissues), it is exposed to various temperatures, ion compositions, redox states, pH, and metabolic conditions (7). Therefore, the parasite must adapt to these changing habitats for survival, proliferation, and eventually differentiation. For example, the ability to switch from a carbohydrate-based to an amino acid-based metabolism requires cellular versatility to enable the organism to cope with the challenges associated with the composition of available food sources (8). In T. cruzi, as well as in other organisms, proline has been shown to sufficiently provide cellular energy requirements. Specifically, the proline metabolism of T. cruzi has been found to be involved in oxygen consumption, resistance to osmotic and metabolic stresses, and oxidative imbalance (9–11). In addition, in this parasite, proline has been shown to be involved in several energy-consuming processes, such as cellular differentiation and host cell invasion (12–15).
Proline breakdown occurs via two enzymatic redox steps and one nonenzymatic reaction, producing glutamate and reduced equivalents (16, 17). First, proline is converted to Δ1-pyrroline-5-carboxylate (P5C)2 through a FAD (cofactor)-dependent reaction that is catalyzed by a proline dehydrogenase (TcProDH) (EC 1.5.99.8) (10). The subsequent step involves a nonenzymatic reaction in which the cyclic P5C ring is spontaneously opened, producing glutamate-γ-semialdehyde (γGS) (18). The carbonyl moiety of γGS is further oxidized to glutamic acid by Δ1-pyrroline-5-carboxylate dehydrogenase (TcP5CDH) (EC 1.5.1.12) with a concomitant reduction of NAD(P)+ to NAD(P)H (19). P5CDH has been characterized in distinct prokaryote and eukaryote species. However, no direct evidence supports the existence of this key enzyme in protozoan organisms. In other organisms, both enzymes (ProDH and P5CDH) have been reported to be associated with the mitochondria (20, 21). In yeast, rat, and human cells, these enzymes are localized within the mitochondrial matrix (22–24). In the case of Zea mays, these enzymes appear to be bound to the mitochondrial inner membrane (21). In addition, structural studies have revealed that these types of enzymes undergo oligomerization that can vary from two to six protomers (25–27).
The hemoflagellate cells of the Kinetoplastid order harbor a single well developed mitochondrion that spans the entire cell body and is distributed in branches under the subpellicular microtubules (28). This particular organelle is composed of classical mitochondrial compartments (the outer and inner membranes, the intermembrane space, and the matrix) that exhibit a particular structure known as the kinetoplast, which includes an intercatenated DNA network that includes the mitochondrial genome (29, 30). Depending on the environmental conditions, the mitochondrion can occupy a variable amount of the total cellular volume (29). With regard to the electron transport machinery, the presence of functional complexes II to IV, as well as the F0/F1-ATPase, has been consistently demonstrated (7).
The connection between proline metabolism and mitochondrial activity is well established (20, 21, 24). This relationship has also been confirmed in several pathogenic trypanosomatids (10, 31). However, there is no evidence to indicate a role for P5CDH in mitochondrial functions. In plant and yeast cells, altered P5C metabolism triggers a redox imbalance that results in cell death (20, 32). In particular, Saccharomyces cerevisiae acetylates P5C via an MPR1-mediated pathway as a detoxifying mechanism against oxidative injury (32). In CHO-K1 cells, P5C was shown to be involved in redox metabolism by up-regulating the hexose monophosphate shunt pathway (33). Farrant et al. (35) assessed the response of patients with P5CDH deficiency, which was previously reported as an inborn error of metabolism (hyperprolinemia type II), and showed that an increased intracellular P5C pool chemically inhibits vitamin B6 (pyridoxal 5′-phosphate) (34), which is an essential prosthetic group of transaminases. Taken together, these data suggest that the proline-glutamate interconversion pathway participates in relevant physiological processes and that the altered equilibrium of their components might elicit detrimental effects beginning at the mitochondrial level and leading to cell death.
Previous biochemical data suggested that glutamate is biosynthesized via a proline oxidation pathway (9). The first enzymatic step of this pathway consists of the conversion of proline into P5C, as has been recently elucidated (10). In this study, we provide conclusive biochemical, mechanistic, and molecular evidence of the functionality of TcP5CDH in several aspects of the T. cruzi life cycle. We show the involvement of TcP5CDH in mitochondrial processes and its developmental expression in distinct parasite stages. These data provide new insights into the proline oxidation pathway as an efficient mechanism for supplying energy to the pathogenic protozoa T. cruzi. The potential for using this enzyme as a drug target is also discussed.
EXPERIMENTAL PROCEDURES
Ethics Statement
Animals were used to obtain a polyclonal monospecific serum against a recombinant protein. The protocol was approved by the ethical committee for animal use in research of the Institute of Biomedical Sciences at University of São Paulo. All procedures followed Brazilian regulations and were approved under protocol number 017/2008.
Microorganisms and Culture
Parasites
The epimastigote (E) forms of T. cruzi (CL strain, clone 14) were cultured at 28 °C by successive passages in undefined medium (LIT) supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS) (Vitrocell®, Campinas, SP, Brazil) (36). Metacyclic trypomastigotes (M) were obtained from 5 × 108 epimastigote cells in the stationary growth phase (4th day), washed once with saline phosphate buffer 1× (PBS), and transferred to Grace's medium, pH 6.0 (Gibco®, Life Technologies) for in vitro differentiation over 9 days at 28 °C. The metacyclic trypomastigotes were further purified using ion-exchange chromatography in diethylaminoethylcellulose columns as described previously (37). Intracellular forms were collected from mammalian tissue culture cell-derived trypomastigotes (TCTs) in Chinese hamster ovary cells (CHO-K1) cultivated in RPMI 1640 medium supplemented with 10% FCS, 0.15% NaHCO3 (w/v), 100 units ml−1 penicillin, and 100 mg ml−1 of streptomycin in a humid atmosphere containing 5% CO2. A parasite/cell ratio of 50 trypomastigotes per CHO-K1 cell was used for infection assays. Amastigotes (A), intracellular epimastigotes (IE), and TCT forms were isolated on the 2nd, 4th, and 7th days post-infection, respectively (13). Cell numbers were counted using a Neubauer chamber.
Bacteria
The Escherichia coli strain DH5-α was used for all cloning procedures. The cells were grown at 37 °C in Luria-Bertani (LB) medium supplemented with 100 μg ml−1 ampicillin and/or 2% agar base (w/v).
Yeast Manipulations
The S. cerevisiae strain S288c was used as the wild-type strain for functional complementation assays. The yeast mutant YHR037w (harboring a disruption within the PUT2 gene) is a homozygous haploid derived from the BY4741 strain (Euroscarf, Germany). This latter strain was used in a systematic deletion project and is derived from the parental S288c strain (38). YHR037w has a kanamycin resistance cassette (KANMX4) flanking the PUT2 gene (YHR07w::KANMX4), resulting in a deficient strain for P5CDH activity. Aiming to restore the auxotrophies for histidine and leucine in the mutant YHR037w, their intrinsic deletions were reverted by introducing the LEU2 and HIS3 ORFs. All of the transformation procedures were performed as described previously (39). The yeast strains were grown either in rich or selective media. The rich media (YPD-2×) contained 20 g liter−1 yeast extract, 40 g liter−1 peptone, 40 g liter−1 d-glucose, and 2% agar base (w/v) when necessary. The ΔPUT2, control, and TcP5CDH strains were maintained in the presence of 300 μg·ml−1 geneticin (G418). After transformations, the cells were grown in synthetic complete dropout medium (SC-gal), which contained 0.17% yeast nitrogen bases without amino acids or ammonium sulfate (w/v), 0.5% ammonium sulfate (w/v), 2% galactose (w/v), and dropout powder containing all amino acids except for histidine, leucine, methionine, and uracil, as described previously (40). Functional analysis was performed in synthetic depleted (SD-gal) media, which contained 0.17% yeast nitrogen bases without amino acids or ammonium sulfate (w/v), 2% galactose, 40 μg·ml−1 methionine, and either 10 mm proline or 0.5% urea (w/v) as the only nitrogen source.
In Silico Analysis
Amino acid sequences for P5CDHs from distinct species were aligned using the ClustalW tool under default parameters and were manually checked (41). Sequences were retrieved from UniProtKB database, and the access code numbers are indicated within parentheses as follows: Homo sapiens (P30038), Mus musculus (Q8CHT0), T. cruzi (Q4DRT8), Trypanosoma brucei (Q38BS5), Leishmania major (Q9NKR5), S. cerevisiae (P07275), E. coli (P5CDH domain between residues 623 and 1106 of P09546), Thermus thermophilus (Q72IB9), and Arabidopsis thaliana (Q8VZC3). Signal peptides, transmembrane domains, and α-helix predictions were performed using the phobius and heliQuest tools (, respectively, under default parameters. The identification of transmembrane domains was performed with the InterProScan database using the region between residues Trp-180 and Ser-300 of the TcP5CDH sequence.
Cloning Procedures
The full-length ORF encoding for Δ1-pyrroline-5-carboxylate dehydrogenase (identification number in the TriTryp database, Tc00.1047053510943.50) was amplified by PCR using 10 ng of total epimastigote genomic DNA as a template, 1 unit of Taq-DNA polymerase (Fermentas®), 1.5 mm MgCl2, 0.25 mm dNTPs mix, 1× Taq buffer with KCl, and 20 pmol of each oligonucleotide. The gene-specific primers were designed with restriction sites for the enzymes BamHI and XhoI, which are underlined, as follows: Tcp5cdhFw 5′-GGATCCATGTTACGTCGCACACTGC-3′ and Tcp5cdhRv 5′-CTCGAGCTAAACAAACAGGCGGTC-3′. The PCR conditions were as follows: temperature of 95 °C (2 min) and 35 cycles of an initial denaturation at 92 °C (1 min), annealing at 61 °C (30 s), and elongation at 72 °C (2 min), with a final extension at 72 °C (10 min). A single fragment of expected size (≈1.7 kb) was amplified and cloned into the pCR-2.1 TOPO vector (Invitrogen®) according to the manufacturer's instructions. To express recombinant TcP5CDH in the bacteria, the ORF TcP5CDH was amplified using a reverse primer in which the stop codon was removed. The PCR product was digested using BamHI and XhoI enzymes and ligated into the pET28a (+) expression vector (Novagen®), which was previously digested using the same restriction enzymes. This construct allows for the recombinant expression of a C-terminal His6-tagged fusion protein.
For functional assays in the yeast model, the circularized plasmid pYEp351 harboring the LEU2 gene was introduced into the ΔPUT2 strain, resulting in ΔPUT2-LEU2+ (42). The ORF encoding for HIS3 was released from the pMA210 vector (43) by endonuclease digestion with BamHI and subsequently inserted into the same site of the pYES2 polylinker region (Invitrogen®). The resulting pYES-HIS3 construct was then inserted into the ΔPUT2-LEU2+, producing the ΔPUT2-LEU2+HIS3+URA3+ strain (herein referred to as the “control strain”). The TcP5CDH ORF was excised from the pCR-2.1 TOPO vector and cloned into the BamHI and XhoI sites of pYES2 to produce the pYES-TcP5CDH construct. Next, the control strain was transformed with pYES-TcP5CDH, resulting in the ΔPUT2-LEU2+ HIS3+ URA3+ TcP5CDH+ (TcP5CDH) strain. All of the constructs were confirmed by DNA sequencing.
Analysis of Recombinant TcP5CDH
Bacterial Expression of TcP5CDH
A plasmid construct containing TcP5CDH (pET28-TcP5CDH) was used to transform E. coli BL21 (DE3) cells (Stratagene®) that were previously transformed with the pGro7 plasmid (Takara®). This vector contains a chaperone system that is expressed upon the addition of arabinose. Bacterial cells were grown in LB medium containing chloramphenicol 20 μg·ml−1 and kanamycin 30 μg·ml−1 at 37 °C. When an absorbance (A) λ600 nm = 0.4 was reached, 0.5 mg·ml−1 l-arabinose was added to induce chaperone expression. Subsequently, when an A λ600 nm of 0.6 was reached, the expression of TcP5CDH-His6 was induced upon the addition of 200 μm isopropyl α-d-thiogalactopyranoside, and the cultures were incubated for 4 h at 28 °C under constant agitation (200 rpm). The recombinant TcP5CDH-His6 enzyme was purified by metal-chelate affinity chromatography using a Ni2+-nitriloacetic acid (column (Qiagen). The TcP5CDH-His6 was eluted using 250 mm imidazole and dialyzed against P5CDH buffer (50 mm HEPES-NaOH, 150 mm NaCl, 0.5 mm EDTA, 0.5 mm tris(2-carboxyethyl)phosphine, 5% glycerol (v/v), 0.02% n-dodecyl β-d-maltoside (DDM) (w/v), pH 8.1). The protein was further concentrated within a range of 0.3–2.5 mg ml−1 using the Centricon (molecular mass cutoff of 10 kDa) centrifugation system (Millipore). Protein homogeneity was assessed by SDS-PAGE (44), and protein concentrations were determined with the Bradford method using bovine serum albumin as a standard (45).
Polyclonal Serum Production
Tyrosine aminotransferase (TcTAT) and aspartate aminotransferase (TcASATm) were kindly provided by Dr. Cristina Nowicki (Faculty of Pharmacy and Biochemistry, University of Buenos Aires, Argentina) (46–48). Recombinant glyceraldehyde-3-phosphate dehydrogenase (TcGAPDH) was obtained as reported previously (49). All of these proteins, as well as the recombinant TcP5CDH, were used as immunogens to produce polyclonal antibodies in mice according to standard protocols (50).
Oligomeric State Determination of TcP5CDH
To determine the molecular mass of TcP5CDH under nondenaturing conditions, the recombinant protein was dialyzed against P5CDH-buffer and fractionated using a Superdex 200TM 16/60 GL column (Amersham Biosciences) coupled to an ÄKTA purifier system (GE Healthcare). The column was previously equilibrated with P5CDH buffer and run at a flow rate of 0.8 ml·min−1. Fractions in which protein content was detected (by absorbance at λ280 nm) were pooled (1 ml) and further analyzed by SDS-PAGE. Stokes radius and elution volumes were used for the determination of molecular size and compared with dynamic light scattering (DLS) data. DLS measurements were performed on a Zetasizer μV (Malvern®) reader at 10 °C in a 10-μl cuvette.
Small angle x-ray scattering (SAXS) data were collected on the SAXS2 beamline at the Brazilian Synchrotron Light Laboratory. Scattering was recorded within the range of the momentum transfer 0.014 < q < 0.34 Å−1, where q = 4πsinθ/λ; 2θ is the scattering angle, and λ = 1.48 Å is the x-ray wavelength. The measurements were performed under vacuum with exposure times of 5 min to decrease parasitic scattering. TcP5CDH scattering was measured at a concentration of 2.5 mg ml−1 in P5CDH buffer (without surfactant) in the absence of ligand. SAXS analysis was performed using the ATSAS program package (51). The distance distribution function p(r) and the radius of gyration (Rg) were evaluated using the indirect Fourier transform program GNOM (52). Molecular weight was evaluated from SAXS curves using SAXSmoW (53). The low resolution dummy residues models of TcP5CDH were restored from experimental data using ab initio modeling implemented in the GASBOR program (54). CRYSOL (55) was used to evaluate the simulated SAXS curve and crystallographic structure parameters (Protein Data Bank code 1UZB). The ab initio low resolution models and the crystallographic structure were superimposed using the SUPCOMB program (56).
Subcellular Localization of TcP5CDH
Immunofluorescence Microscopy
Immunofluorescence assays were performed to assess the predominant T. cruzi life cycles (epimastigotes, metacyclic trypomastigotes, amastigotes, and trypomastigotes derived from cell infections) by assessing the co-localization of TcP5CDH with the mitochondrial MitoTracker-CMXROS probe (Invitrogen®). Briefly, parasite forms (5 × 106 cells) were preincubated at 28 °C for stages E and M, 33 °C for stages A and IE, and 37 °C for TCT (30 min) in the presence of 50 nm MitoTracker, which was dissolved in the corresponding culture medium. An additional in situ assay using CHO-K1 cells infected with TCTs was also performed. Thus, CHO-K1 cells (4 × 105 cells/well) were layered in a sterile glass slip embedded into 24-well plates for 24 h at 37 °C under a 5% CO2 atmosphere. Next, the cells were infected with the TCT forms (2 × 107 per well) and cultured for 48 h at 33 °C for preincubation with MitoTracker as described above. The cells were harvested, washed twice with PBS, and seeded onto poly-l-lysine-coated coverslips. The cells were fixed (20 min) with 4% p-formaldehyde (w/v) in PBS, permeabilized (5 min) by the addition of 0.1% Triton X-100, and blocked (30 min) with 2% BSA dissolved in PBS (PBS/BSA). The preparations were incubated for 1 h at room temperature with anti-TcP5CDH (diluted 1:150 in PBS/BSA), washed five times with PBS, and further incubated (50 min, light-protected) with AlexaFluor488-coupled goat anti-mouse immunoglobulin G (Invitrogen®) secondary antibody (1:400). DNA was stained by a further incubation (1 min) with Hoechst 33258 probe (1:2500 in PBS) (Invitrogen®). The coverslips were visualized using an Axio Imager motorized M.2 (Carl Zeiss®) epifluorescence microscope. Microscope Z-sections were processed and merged using ImageJ version 1.4p (National Institutes of Health) software for a 64-bit iOS system.
Digitonin Titration
Selective digitonin permeabilization of the T. cruzi epimastigote forms was performed as described previously (46). Briefly, cell pellets (60 mg) were harvested and resuspended in 1 ml of TSB buffer (25 mm Tris-HCl, pH 7.6, 0.25 m sucrose, 1 mm EDTA, 1 mm PMSF, and 50 μl of protease/inhibitor mixture from Sigma®) in the presence of increasing concentrations of digitonin (ranging from 0 to 5 mg·ml−1) (Sigma®). The cells were incubated with digitonin for 5 min at 25 °C and centrifuged (2 min, 18,000 × g at 25 °C). The resulting supernatants were gently transferred to new tubes, and the pellets were resuspended in 1 ml of TSB and subjected to further sonication. The enzymatic activities corresponding to pyruvate kinase, hexokinase, citrate synthase (cytosolic, glycosomal, and mitochondrial markers, respectively), and TcP5CDH were measured in all of the supernatant and pellet fractions as described elsewhere. All of the fractions were also subjected to Western blot analysis using antibodies against TcTAT, TcGAPDH, and TcProDH (cytosolic, glycosomal, and mitochondrial markers, respectively) as well as an antibody against TcP5CDH.
Detection of TcP5CDH in Mitochondrial Fractions
The isolation of mitochondrial vesicles from T. cruzi epimastigotes was performed as described recently (57). The solubilization of mitochondrial membrane proteins was performed in the presence of 0.1% Triton X-100 (v/v) (5 min at 4 °C), and clarification was performed by centrifugation (16,000 × g for 20 min at 4 °C). The supernatant was separated to confirm the presence of the proteins by enzymatic activity measurements (total protein mass, 100 μg) and by Western blotting analysis (total protein mass, 40 μg per lane). For the latter analysis, an additional total cell-free extract from epimastigotes was used as a control (58).
Native Gel Electrophoresis (NGE)
Recombinant TcP5CDH and mitochondrial fractions from T. cruzi epimastigotes were resolved in NGE as described previously (59). Briefly, 4 μg of purified TcP5CDH was mixed with 3× NGE sample buffer (5% Coomassie Blue G-250 (w/v) and 0.75 m aminocaproic acid) and resolved in nondenaturing conditions followed by a comparison with the migration profile of the NativeMarkTM protein standard (Novex®) or high molecular weight native marker (GE Healthcare). Mitochondrial fractions were prepared and solubilized using either 10% DDM (w/v) dissolved in water or 10% digitonin (DIG) (w/v) dissolved in a buffer containing 50 mm NaCl, 50 mm imidazole, 5 mm aminocaproic acid, and 4 mm PMSF. Detergent (in grams) ratios of 2:1 and 4:1 per g of total protein content were used for DDM and digitonin treatments, respectively. Thus, samples were incubated for 5 min on ice, and the solubilized material was separated by centrifugation (16,000 × g for 20 min at 4 °C). The samples were mixed with 3× NGE sample buffer and loaded (50 μg of mitochondrial protein) onto native gels.
Protein Identification by Liquid Chromatography Coupled to Tandem Mass Spectrometry
NGE slices containing the protein bands of interest were excised. After reduction with DTT (10 mm) and alkylation of the Cys groups using iodoacetamide (50 mm), modified porcine trypsin (Promega®) was added at a final mass ratio of 1:50 (trypsin/protein). The samples were digested overnight at 37 °C, after which they were vacuum-dried and dissolved in 1% acetic acid (v/v). To identify proteins, the resulting tryptic peptide mixtures were analyzed by nano-liquid chromatography coupled to mass spectrometry. The peptides were injected into a C-18 reverse-phase nano-column (100 μm inner diameter and 15 cm; TK mediterraneaTM Sea, Teknokroma) and analyzed on a continuous acetonitrile gradient consisting of 0–43% B for 90 min and 50–90% B for 1 min (B = 95% acetonitrile, 0.5% acetic acid). The peptides were eluted from the reverse-phase nano-column at a flow rate of ∼300 nl/min into an emitter nanospray needle for real time ionization and peptide fragmentation on an LTQ-Orbitrap mass spectrometer (Thermo Fisher, San José, CA). An enhanced FT-resolution spectrum (resolution = 6000) and the MS/MS spectra of the five most intense parent ions were analyzed during the chromatographic run (130 min). Dynamic exclusion was set at 0.5 min. For protein identification, tandem mass spectra were extracted, and the charge-states were deconvoluted using Proteome Discoverer 1.2.0.207 (Thermo Fisher Scientific). All of the MS/MS samples were analyzed by SEQUESTTM (Thermo Fisher Scientific, version 1.0.43.2), MASCOTTM (Matrixscience, version 2.1) and X! Tandem (The GPM, version 2007.01.01.1). All searches were performed assuming complete trypsin digestion. Two mixed cleavages were allowed, and errors of 15 ppm or 0.8 Da were set for full MS and MS/MS spectra searches, respectively. Oxidation on Met, phosphorylation on Ser or Thr and deamidation on Gln or Asn were selected as dynamic modifications. Identification was performed by querying the Decoy database for FDR analysis. Scaffold (version Scaffold_3_00_03, Proteome Software Inc., Portland, OR) was used to validate the identified MS/MS peptides and proteins. Protein probabilities were assigned by the Protein Prophet algorithm. Proteins that contained similar peptides and that could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Expression Levels of TcP5CDH in the Parasite Developmental Stages
Quantification of mRNA Levels
Total RNA was extracted using the TRIzol reagent (Invitrogen®) from all parasitic stages and CHO-K1 cells (as a control). Each RNA preparation was performed in triplicate according to the manufacturer's protocols. Yeast RNA preparations were performed according to a standardized method (40). After extraction, the RNA samples were treated with RNase-free DNase (Fermentas®), which was controlled under denaturing conditions and quantified spectrophotometrically following standard procedures (60). First strand cDNA was synthesized from total RNA (5 μg) using a random primer set and the Superscript III® reverse transcriptase mix according to the manufacturer's protocols (Invitrogen®). Based on the T. cruzi nucleotide coding sequences TcP5CDH and TcGAPDH as housekeeping gene GenBankTM accession numbers are as follows: TcP5CDH, XM811985.1; TcGAPDH, AI007393), specific primers for quantitative real time PCR (qRT-PCR) analysis were designed. TcP5CDH forward, 5′-ATGCTTGGTGTGCACGAACA-3′, and TcP5CDH reverse, 5′-CATCGATAACGGCGCACATA-3′ (product size 76 bp), and TcGAPDH forward, 5′-GTGGCAGCACCGGTAACG-3′, and TcGAPDH reverse, 5′-CAGGTCTTTCTTTTGCGAAT-3′ (product size 110 bp) were separately prepared in a mixture containing 1.6 pmol of each primer for each corresponding gene. qRT-PCR was performed in a Mastercycler ep RealPlex (Eppendorf, Germany) using the SYBR Green fluorescence quantification system (Fermentas, Life Sciences). The PCR conditions were as follows: 95 °C (10 min) and then 40 cycles of 94 °C (1 min), 57 °C (1 min), and 72 °C (2 min) followed by a denaturation curve. The data obtained were analyzed using RealPlex version 1.5 software. Fold changes in the expression of each transcript were determined by applying the comparative method (2−ΔΔCt) using epimastigote stage data as a calibrator (61). Differences among samples were analyzed with a one-way analysis of variance followed by Tukey's post test using GraphPad Prism version 5.00 software for Windows (GraphPad software). The significance level (p value) was determined with a confidence interval of 95% in a two-tail distribution.
Western Blot Analysis
Total protein extracts from the different stages of T. cruzi and CHO-K1 cells were prepared for Western blot assays. The cells were harvested, washed twice with PBS, and resuspended in 20 μl of extraction buffer (50 mm Tris-HCl, pH 7.4, 50 mm NaCl, 2 mm EGTA, 2 mm EDTA, 20% glycerol (v/v), 1% Triton X-100 (v/v), 1 mm PMSF, and protease inhibitor mixture (Sigma®). The cells were chilled on ice (30 min) and clarified by centrifugation (16,000 × g for 15 min at 4 °C). Supernatants corresponding to 2 × 107 T. cruzi cells or 5 × 106 CHO-K1 cells were mixed with 2× SDS-PAGE Laemmli buffer (44), boiled (5 min at 95 °C), and further subjected to SDS-PAGE protein electrophoresis. Protein extracts from the yeast cells were extracted as described previously (62) and used in both enzymatic activity assays and Western blotting analysis. Then the proteins were electrotransferred (1.2 mA × cm2 of gel during 75 min) onto 45-μm Hybond-C extra nitrocellulose membranes (Amersham Biosciences) and stained with 0.5% Ponceau Red solution (p/v) dissolved in 10% acetic acid (v/v). Polyclonal mouse antisera produced against TcP5CDH (1:2000), TcGAPDH (1:2500), or TcASATm (1:3000) (46) were diluted in PBS containing 0.1% Tween 20 (PBS-T) (v/v) and used to probe for the blotted proteins (1 h at room temperature). The membranes were washed three times with PBS-T and then incubated in the presence of horseradish peroxidase-coupled goat anti-mouse IgG (Sigma®) secondary antibody (1:2500). Protein signals were developed using the SuperSignal® West Pico chemiluminescent substrate (Thermo Fisher Scientific) following the manufacturer's protocols.
Biochemical Characterization of TcP5CDH
Chemical Synthesis and Analysis of P5C
P5C was synthesized via the meta-peroxidation of dl-5-hydroxylysine (Sigma®) and purified by ion-exchange chromatography as described previously (63). Eluted P5C was maintained in acidic medium (1 m HCl) and quantified using a colorimetric method with o-aminobenzaldehyde (OAB) (16). Absorbance (λ443 nm) of the hydroquinone complex (P5C-OAB) was measured in 1-cm cuvettes using 0.25 m HCl as a reference instead of eluted samples. The molar extinction coefficient (ϵ) of the complex was 2,590 m−1·cm−1 (16, 63). The intracellular P5C levels in the yeast cells were determined according to a method described previously (32). The purity and mass of synthetic P5C were analyzed by isocratic HPLC and mass spectrometry, respectively.
Enzymatic Activity Determinations
The enzymatic activity of TcP5CDH was determined spectrophotometrically (λ443 nm) by monitoring the production of the reduced form of NADH as the co-product of oxidized γGS. Steady-state activities were determined at 28 °C under saturating concentrations in a mixture containing 100 mm HEPES-NaOH, pH 7.2, 0.6 mm synthetic P5C (freshly prepared), 2 mm of either NAD+ or NADP+ (NAD(P)+), brought to a volume of 3 ml with deionized water. Enzymatic reactions were initiated upon the addition of either 10 μg of recombinant TcP5CDH-His6 or 100 μg of cell-free total extract, depending on the assay. Measurements were performed in 1-cm quartz cuvettes at 28 °C with magnetic stirring. Initial velocities were determined in the linear range of the time course reaction using the ϵ value for NADH (6200 m−1·cm−1). The kinetic data (Km and kcat) were determined for the three substrates (l-γGS and NAD(P)+). Substrate parameters were assayed by varying the P5C concentrations (10–1000 μm) with a fixed concentration of 2 mm NAD+. Cofactor parameters were separately assayed by varying the concentrations of each NAD(P)+ (10–2500 μm) with a fixed concentration of 0.3 mm P5C. The effect of temperature on the specific activity of TcP5CDH was assayed by raising the reaction temperature within a range of 15–85 °C. The effect of pH variation on TcP5CDH activity was assayed in 100 mm reaction buffer adjusted to different pH values within a range of 5–9. To determine the activation energy at a steady state of TcP5CDH, the Arrhenius equation was applied. Values were plotted, and the resulting curves were adjusted to the Michaelis-Menten function using OriginPro version 8.0 software (OriginLab). Substrate specificity was assessed separately in the presence of 0.4 mm distinct aldehydes structurally related to γGS (succinate semi-aldehyde, acetaldehyde, malonic acid, glutaric acid, and glutamic acid). The activities of citrate synthase (EC number 4.1.3.7), hexokinase (2.7.1.1), and pyruvate kinase (2.7.1.40) were measured using standard procedures. The activity of fumarate reductase was measured as described previously (64).
Physiological Assays with P5C
Nutritional Starvation Assays
The ability of T. cruzi cells to use P5C during nutritional starvation was evaluated. Briefly, epimastigote forms (2 × 107 cells) were exposed to nutritional depletion for 48 h at 28 °C in either PBS or PBS supplemented with 1 mm of each substrate analyzed (d-glucose, l-proline, l-glutamate, dl-P5C, and HCl as a control for the P5C treatment). Next, cell viability was determined in the presence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for 4 h (58, 65).
ATP Measurements
To determine whether the parasite produces ATP from distinct sources after nutritional starvation, an enzymatic light assay was used. Briefly, epimastigote forms (5 × 106 cells) were starved by incubation for 24 h at 28 °C in PBS. During this period, intracellular ATP levels are depleted without affecting cell viability, as confirmed by microscopy. Next, the parasites were incubated (1 h at 28 °C) with 1 mm of the corresponding substrates to restore the energy requirements, and the intracellular ATP contents were measured using a bioluminescence cell kit (Sigma®) according to the manufacturer's protocols.
Infection Assay during P5C Supplementation
To evaluate the effect of P5C on the intracellular cycle of T. cruzi, CHO-K1 cells (5 × 104 cells per well) were seeded into 24-well plates and incubated for 24 h at 37 °C under a 5% CO2 atmosphere. Next, the cells were washed with PBS, and 2 × 107 TCT forms were added to each well in either RPMI 1640 media supplemented with 10% FCS, PBS supplemented with 10% FCS, or PBS supplemented with increasing concentrations of P5C (20, 50, and 100 μm) to a final volume of 500 μl and incubated for 3 h as described above. After the incubation, the cells were washed three times with PBS and fresh RPMI 1640 medium 2% plus 10% FCS. After an additional 24 h of incubation, the infections were further incubated at 33 °C for 5 days. After this period, the number of parasites released into the culture media was determined by counting using a hemocytometer.
RESULTS
Identification and in Silico Analysis of TcP5CDH
A 1686-bp open reading frame encoding a putative 62.4-kDa TcP5CDH was found in the genome database. This sequence is henceforth referred to as TcP5CDH1. Two additional sequences were also found. TcP5CDH2 (Tc00.1047053509351.10) exhibits 98% amino acid sequence identity with the N-terminal portion of TcP5CDH1, whereas TcP5CDH3 (Tc00.1047053503577.9) shares a 73% amino acid sequence identity with the C terminus of TcP5CDH1. TcP5CDH1 was sequenced (EBA database access code number HG326611) and further used for functional characterization experiments as described in this work.
Analysis of the TcP5CDH1 amino acid sequence revealed high sequence identity with other hemoflagellate putative orthologs, such as T. brucei (82%) and L. major (68%), followed by orthologs from eukaryote organisms such as S. cerevisiae PUT2 protein (48%), Drosophila melanogaster P5CDh1 (45%), H. sapiens ALDH4A1 (42%), the E. coli putA domain (29%), and T. thermophilus TtP5CDH (32%) from bacteria and archaea (Fig. 1A). A query of the InterProscan database revealed the presence of conserved domains that characterize proteins belonging to the aldehyde dehydrogenases family, NAD+-binding (Glu302, Phe387, and Leu441) and glutamate-binding (Cys336 and Ser340) enzymes, in which the specific residues are likely to be involved in catalytic processes, as reported previously in archaea and human orthologs (Fig. 1A) (25, 66).
FIGURE 1.

In silico analysis of the deduced amino acid sequence for TcP5CDH. A, alignment of primary sequences for P5CDHs from different species. The aligned sequences correspond to orthologous proteins from H. sapiens (ALDH4A1), D. melanogaster (P5CDh1), T. cruzi (TcP5CDH), T. brucei (TbP5CDH), L. major (LmP5CDH), S. cerevisiae (PUT2), E. coli (putA), and T. thermophilus (TtP5CDH). Accession code numbers are detailed under “Experimental Procedures.” The alignment was performed using the ClustalW method (default parameters) (41). Protein signatures found in the mTP (Met1–Ser18) indicating the MLRR (†††) and alanine-rich FAFAYA (¥¥¥) domains and the putative cleavable site (arrow) of TcP5CDH. The predicted trans-membrane region (Phe200–Trp221) is highly conserved among trypanosomatid sequences (- - -), as are the conserved catalytic residues Glu302, Phe387, Leu441 (stars), and Cys336, Ser340 (circles), as shown by previous structural data. B, α-helical structure presented in mTP region using the heliQuest software. The resulting model depicts the positive (+) (Arg3, Arg4, and Lys16) and hydrophobic (H) (Leu2, Leu6, Ala9, Ala11, and Ala13) residues and the putative Thr17 (arrow) cleavable site. C, bioinformatic analysis for the transmembrane region found for TcP5CDH. The complete amino acid sequence for TcP5CDH (561 residues) was used as the input sequence for the Phobius predictor tool. The graphic layout shows the probability values (0–1) for the transmembrane (TM) domains in gray, cytoplasmic (C) in green, noncytoplasmic (NC) in blue and signal peptide (SP) in red.
Currently, all reported eukaryotic forms of P5CDH are mitochondrial enzymes. An attempt to identify mitochondrial targeting sequences by means of available bioinformatics resources (SignalP, MitoProt, and TargetP) did not reliably support the presence of this targeting sequence. However, visual analysis of the N-terminal portion (Met1–Val20) revealed protein fingerprints common in mitochondrial targeting sequences from kinetoplastid organisms (67). The presence of three positively charged residues (Arg3, Arg4, and Lys16), a conserved-motif MLRR, an alanine-repeat track (FAFAYA), and a putative α-helix suggested a mitochondrial location (Fig. 1, A and B). In addition, we identified a hydrophobic region (Phe198-Gly215) with characteristics of a putative transmembrane α-helix spanning domain (Fig. 1C).
TcP5CDH Can Complement the Activity of a PUT-deficient Yeast Strain (ΔPUT2)
To confirm that TcP5CDH encodes a protein with P5CDH activity, a ΔPUT2 yeast mutant strain lacking P5CDH activity, which is unable to use proline as the single nitrogen source, was used for functional complementation. As the designated yeast strain exhibited intrinsic auxotrophy for histidine and leucine, it was initially modified to be able to synthesize these metabolites by the insertion of the genes HIS3 and LEU2 (Table 1). These modifications allowed for the use of a selective medium without these amino acids, which was crucial for the assay, as an excess of these amino acids might facilitate their use as alternative nitrogen sources in S. cerevisiae (68). Thus, yeasts that are able to grow in selective media containing only a carbon, nitrogen, and a sulfur source were obtained. TcP5CDH1 was cloned into the pYES vector, and the construct was inserted into the modified yeast ΔPUT2. Phenotypic analysis of the transformed cells in defined media revealed that proline was a suitable nitrogen source, as was ammonium sulfate (used as control), for both the wild-type (S288c) and the complemented TcP5CDH strain (Fig. 2A). In addition, the sensitivity of S288c to G418, the plasmid selection marker, was verified. Furthermore, the presence of the transcript for TcP5CDH was detected only in the TcP5CDH-transformed clones (Fig. 2B), and TcP5CDH (which shares 44% identity with S. cerevisiae orthologs) (Fig. 1A) was detected with an anti-TcP5CDH serum, indicating the presence of the T. cruzi protein in the TcP5CDH-transformed clones (Fig. 2C). Finally, both the intracellular P5C content and functional activity of TcP5CDH was assessed. The specific activity of P5CDH in the TcP5CDH complemented strain was compatible with that obtained for the wild-type cells (Fig. 2D). This result was consistent with the similar levels of intracellular pools of free P5C. As expected, this activity was evidenced by the accumulation of P5C in both ΔPUT2 and control cells (Fig. 2D). These data demonstrate that the protein product of TcP5CDH functionally reverts the phenotype of the ΔPUT2 mutant in terms of both the increased levels of the intermediate P5C and its inability to metabolize proline as a single nitrogen source in a defined medium, indicating its P5CDH enzymatic activity.
TABLE 1.
List summarizing the yeast strains used in this study
The abbreviations used are as follows: ΔPUT2, knockout strain for the proline utilization 2 gene (PUT2); control, control strain; TcP5CDH, complemented strain harboring cds for P5CDH in T. cruzi. The WT strain lacks auxotrophy, whereas the ΔPUT2 strain exhibits auxotrophy for HIS, MET, LEU, and URA and is also deficient in P5CDH activity. Both control and TcP5CDH strains exhibit auxotrophy only for MET, whereas the control strain is unable to grow when proline is the only nitrogen source.
| Strain | Name | Plasmids inserted | Resultant genotype |
|---|---|---|---|
| S288c | WT | None | MATα; SUC2; GAL2; MAL; MEL; FLO1; FLO8-1; HAP1; HO; BIO1; BIO6 |
| BY4741 | ΔPUT2 | None | MATa; HIS3Δ1; LEU2Δ0; MET15Δ0; URA3Δ0; YHR037w::KANMX4 |
| Control | pYEp351, pYES-HIS3 | MATa; MET15Δ0; YHR037w::KANMX4 | |
| TcP5CDH | pYEp351, pYES-HIS3, pYES-TcP5CDH | MATa; MET15Δ0; YHR037w::KANMX4; TcP5CDH |
FIGURE 2.

Yeast functional complementation assay. PUT2 activity-deficient mutant strain was transformed by the episomal insertion of the TcP5CDH gene. A, drop tests in selective media. For phenotypic analysis, yeast cells were grown in liquid medium (until A600 nm = 1) and then serially diluted (1–10−5) in sterile water prior to spotting. Synthetic complete plates (SC) contained a dropout mix with all amino acids, including uracil, ammonium sulfate, and galactose as nitrogen and carbon sources (SC1). SC without uracil or ammonium sulfate but with proline and G418 added are referred to as SC2. Synthetic depleted plates (SD) contained only methionine, ammonium sulfate, and galactose (SD1) or methionine, proline, and galactose (SD2). Final concentrations are described under “Experimental Procedures.” Cells were incubated at 28 °C for 2 or 4 days for the SC and SD tests, respectively. B, transcriptional levels of TcP5CDH were assayed by RT-PCR with specific primers. Amplified products were resolved in 1% agarose gel (w/v) and stained with 0.5 μg·ml−1 ethidium bromide. Gel samples were loaded as follows: 1st lane, 1-kb DNA ladder (Fermentas®); 2nd lane, products amplified using either total genomic DNA from epimastigote forms; 3rd lane, total cDNA from wild-type yeast; 4th lane, cDNA from the ΔPUT2 mutant; or 5th lane, cDNA from the TcP5CDH mutant as templates in PCR. C, Western blot analysis with protein extracts was performed as indicated. Membranes were probed against anti-TcP5CDH produced in mice (1:2000) and developed as described elsewhere. D, biochemical analysis of yeast cells. Cell-free extracts (100 μg) from 500 ml of yeast cells, grown in either SC1 or SC2 media, were used as an enzyme source in enzymatic determinations (left axis) or reacted with OAB for the quantification of free P5C levels. Intracellular P5C levels are expressed as the concentration (μm) ratio with respect to the biomass of dried yeast cells (grams) (right axis). The data are representative of three independent measurements.
Expression, Purification, and Determination of the Oligomeric State of TcP5CDH
To confirm the functionality of the TcP5CDH protein product, its DNA sequence was cloned and expressed in E. coli as a protein fused with a six histidine tag in the C-terminal region. The TcP5CDH-His6 was purified by affinity chromatography using a Ni2+-nitriloacetic resin followed by size-exclusion chromatography (SEC) and enzymatically active in the presence of detergent (DDM). In addition, a recombinant TcP5CDH product, lacking the N-terminal targeting pre-sequence (first 22 residues), showed no alterations in the enzymatic rates (data not shown).
The chromatogram (λ280 nm) obtained from the SEC of recombinant TcP5CDH-His6 exhibited two major peaks (P1 and P2) with different elution times, suggesting different oligomerization states (Fig. 3A and Table 2). These fractions were also resolved by SDS-PAGE, which revealed a single protein of the expected size (≈64 kDa) only in the P2 samples (Fig. 3A). The estimation of molecular size for P1 using SEC, along with the fact that SDS-PAGE did not resolve the proteins in the samples, suggested that the protein recovered in P1 was aggregated. Consistently, P1 did not show enzymatic activity, whereas P2 presented a P5CDH activity (Table 2). Next, to confirm that P1 consisted of aggregated protein and to determine the oligomeric state of active TcP5CDH, we analyzed the data from SEC, DLS, and SAXS measurements and NGE. SEC measurements, DLS, and NGE for P2 revealed a high molecular mass oligomer of 715, 649, and ≈700 kDa, respectively (Fig. 3A and Table 2). The molecular size of TcP5CDH within the P2 fraction is consistent with the DLS data, collected at different concentrations. In addition, the DLS profile showed that the homogeneous (not aggregated) state of TcP5CDH is consistent at different concentrations (tested: 0.3, 0.75, and 1.5 mg/ml). Taken together, these data suggest that TcP5CDH adopts an oligomeric conformation with 10 to 12 subunits. Next, SAXS experiments with the TcP5CDH apoenzyme in solution (from P2) were performed to confirm the absence of aggregates and the oligomeric state of the enzyme in this fraction. The linear region observed at the beginning of the curve (values of q between 0.015 and 0.048 Å−1 or q2 between 0.00025 and 0.00225 Å as expressed in Fig. 3 D, and D, inset) was considered to obtain Rg values by using the method of Guinier. The linearity of the curve in this range showed that the protein solution was homogeneous, meaning that no aggregates were evidenced. To estimate the size of the complex (number of subunits) as well as its molecular weight, the obtained curve was compared with a theoretical one generated for a crystallized hexameric P5CDH as described under “Experimental Procedures.” As can be seen, the theoretical curve obtained from a hexameric crystallographic structure of P5CDH overlapped the experimental curve obtained with TcP5CDH (P2 fraction) (Fig. 3D). This behavior strongly supported that TcP5CDH is present predominantly as a 364.6-kDa hexamer at a concentration of 2.5 mg ml−1. The typical scattering curve and distance distribution function p(r) obtained with TcP5CDH at 2.5 mg ml−1 are presented in Fig. 3, D and E, respectively. All model curves yielded optimal fits with the experimental data. The experimental values of Dmax and Rg, 200 and 65.21 ± 0.53 Å, respectively, suggest that TcP5CDH is approximately in a globular configuration (Fig. 3F).
FIGURE 3.

Analysis of purified TcP5CDH-His6 in solution. A, recombinant TcP5CDH-His6 was produced in E. coli and purified by affinity chromatography and SEC. After SEC separation, two major peaks (λ280 nm) were observed (P1 and P2), and the protein sample was subjected to electrophoresis in a 10% SDS-polyacrylamide gel stained with Coomassie Blue (right inset). Size determinations were determined from calibration curve used in SEC separation (left inset). B, BNGE of the pooled fractions from SEC. Fractions 44–47 (corresponding to P1) and 51–54 (corresponding to P2) were pooled, concentrated, and resuspended at 0.5 μg/μl in P5CDH buffer in the presence or absence of 0.02% DDM. Molecular mass determinations were determined by comparing the samples migration against protein standards. C, DLS of P2 was performed at three different concentrations. Fractions 51–54 (corresponding to P2) were pooled, concentrated, and resuspended at 0.3, 0.75, and 1.5 μg/μl in P5CDH buffer in the presence of 0.02% DDM. Hydrodynamic radius (RH) and molecular mass (MW) were similar for the tested concentrations. D, distance distribution function of TcP5CDH from the experimental x-ray scattering data. The determined value of maximum distance (Dmax) is indicated. E, experimental solution scattering curves of TcP5CDH-His6 (log I versus q) and the results of the fitting procedures. Fraction P2 was resuspended in buffer containing 90 mm HEPES-NaOH, pH 7.2, and 5% glycerol (v/v) and was concentrated up to 2.5 mg ml−1. Scattering curves obtained from experimental data for TcP5CDH from the high resolution model (Protein Data Bank code 1UZB) and scattering patterns computed from the Gasbor model were plotted as indicated. The inset displays the correspondent Guinier plot (log I versus q2). F, low resolution structure of the TcP5CDH in solution as obtained by Gasbor (spheres) with superposition of the high resolution model (1UZB) (ribbons). The models were rotated 90° with respect to the y axis. a.u., absorbance units.
TABLE 2.
Comparative analysis of the molecular mass for TcP5CDH
Recombinant TcP5CDH dissolved in fraction P2 (Fig. 3A) was subjected to gel filtration (GF), which facilitated the determination of the Stokes radius and the molecular mass from the resulting equation (Fig. 3, inset). P2 fraction was used to compare the molecular mass of TcP5CDH by either DLS or SAXS measurements. Native gel analysis of P2 revealed an estimation of the molecular mass based on its electrophoretic mobility, whereas the Stokes radius could not be detected (ND).
| Technique | Stokes radius | Molecular mass |
|---|---|---|
| Å | kDa | |
| GF | 81.1 | 714.9 |
| DLS | 97.1a | 649.3 |
| SAXS | 65.21b | 364.6 |
| BNGE | ND | ≈480/≈720 |
a DLS assays revealed a Stokes radius with a mono-dispersal peak.
b The SAXS measurements of TcP5CDH in solution resulted in a gyration radius of 65.21 ± 0.53 Å and a maximal diameter of 200 Å based on a distance distribution function calculated using Fit2D and SAXS MoW software (53).
Enzymatic Conversion of dl-P5C in Glutamate
The biochemical characterization of TcP5CDH involves the synthesis of dl-P5C, which is not commercially available. dl-P5C was prepared by oxidizing dl-5-hydroxylysine with periodate in acidic media (63). The concentration of dl-P5C was determined using the OAB colorimetric assay (Fig. 4A). MS analysis (Fig. 4B) indicated the presence of dl-P5C (m/z 114), the corresponding hydrolyzed ring-opened form (γGS, m/z 132), and the chlorinated derivative (m/z 162). After a purification step, the produced dl-P5C was immediately used to investigate the enzymatic activity of both native and recombinant TcP5CDH (Fig. 4C). Although quantum chemical calculations indicate that the equilibrium in water is greatly shifted to P5C rather than to γGS (ΔG0 = 41 kJ mol−1, K = 2.2 × 107; Fig. 4D), crystallographic evidence suggests that γGS is the substrate of TcP5CDH (Inagaki et al. (25)). Therefore, the product of periodate oxidation of l-proline will be referred to as P5C throughout the text, whereas γGS will be assumed to be enzymatically converted into l-glutamate (see under “Discussion”).
FIGURE 4.

Analytical test of chemically synthesized P5C. A, wavelength scans of synthesized P5C. The DHQ yellow-colored complex absorbs light within the visible light range (λ443 nm), enabling its quantification as ϵ = 2590 m−1·cm−1. B, mass spectrum of synthetic P5C. After synthesis, the product was dissolved in two distinct solutions (pH 4.5 and 7.0) using a mixture of H3PO4/K+ phosphate buffer. Next, the sample was dissolved (1:2) in formic acid 1% (v/v) and further injected in a Finnigan Surveyor Mass Spectrometer Quadrupole Plus-MSQ (Thermo Fisher Scientific). Chromatograms from each injection were recorded and merged to analyze the differences among peaks of interest. Peaks corresponding to P5C, γGS, and Cl-γGS were detected, as indicated. A single peak with a mass-to-charge ratio of 114.15 m/z was detected for P5C. C, wavelength scans for reactants involved in the enzymatic assay for TcP5CDH. The reaction assay was prepared as described under “Experimental Procedures,” and the reaction was initiated with 200 μg of T. cruzi total lysates. Next, wavelength scans were performed (260 nm min−1), and the UV-visible light spectra were recorded over the reaction time. The graph indicates the three main peaks for P5C/γGS, NAD+, and NADH obtained at 220, 280, and 340 nm, respectively. D, structural representation of the spontaneous conversion between ring (P5C) and opened (γGS) forms in aqueous medium. Optimized structures of SMD-B3LYP/6–311++G (d,p) and values for the free-cyclization energy and the equilibrium constant (Keq) are depicted. Lowest unoccupied energy molecular orbital (LUMO) depicts the sites for potential interaction with nucleophilic species.
The effects of the reaction conditions were investigated. The Km value for the oxidation of γGS to l-Glu in the presence of TcP5CDH-His6 and saturating NAD+ was found to be 72 ± 21 μmol liter−1. When mitochondrial lysates were used as a cell-free source of the enzyme, a Km of 43 ± 0.5 μmol liter−1 was observed (Table 3). Both recombinant and mitochondrial lysate-derived enzymes exhibited increased affinities for γGS in the presence of the cofactor NAD+ (Km = 72.3 μmol liter−1) compared with NADP+ (Km = 342 μmol liter−1) (Table 3). The enzymatic conversion was pH-dependent, and an optimal pH of 7.2 for either mitochondrial lysates or recombinant TcP5CDH-His6 was determined (Fig. 5A). Although TcP5CDH is inactivated at temperatures above 45 °C, activation parameters were determined within a temperature range between 20 and 45 °C (Fig. 5B). The resulting curved Arrhenius plot depicts two distinct barriers, one exhibiting a lower value of 5.8 kJ mol−1 and the other with a higher value of 32.2 kJ·mol−1 (Fig. 5B). Finally, as P5CDHs are aldehyde dehydrogenases, aldehydes other than γGS might serve as substrates. Thus, we evaluated the specificity of TcP5CDH. We observed that the enzyme showed negligible specific activity in the presence of other structurally related metabolites such as succinate semialdehyde, acetaldehyde, malonate, glutarate, and adipic acid. Based on genomic and experimental data, a biochemical scheme of the proline-glutamate interconversion pathway is represented for T. cruzi (Fig. 6).
TABLE 3.
Kinetic parameters for the natural substrate (γGS) and cofactors estimated in both the recombinant form (TcP5CDH-His6) (left) and mitochondrial vesicles from T. cruzi (right)
Measurements were performed on saturated concentrations of NAD+ (2 mm) for γGS determinations and γGS-saturated (1 mm) NAD(P)+ determinations as described previously. To determine kcat values, 10 μg of recombinant TcP5CDH or 100 μg of mitochondrial proteins were used in a 3-ml reaction volume. Enzymatic activities were initiated upon the addition of the enzyme, and changes in absorbance were measured over 5 or 15 min for recombinant TcP5CDH or mitochondrial extracts, respectively (at 28 °C with constant stirring).
| Substrate | Mitochondrial lysate |
TcP5CDH-His6 |
||||
|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | |
| μm | s−1 | μm−1 s−1 | μm | s−1 | μm−1 s−1 | |
| γGS | 43 ± 0.5 | 130.7 ± 10 | 3 ± 0.1 | 72 ± 21 | 7 ± 0.5 | 0.097 ± 0.011 |
| NAD+ | 39 ± 3,8 | 89.2 ± 1.6 | 2.3 ± 0.26 | 72.3 ± 14.1 | 7.5 ± 0.4 | 0.103 ± 0.028 |
| NADP+ | 302 ± 66 | 116.8 ± 7.1 | 0.39 ± 0.1 | 342 ± 140 | 1.2 ± 0.14 | 0.003 ± 0.001 |
FIGURE 5.
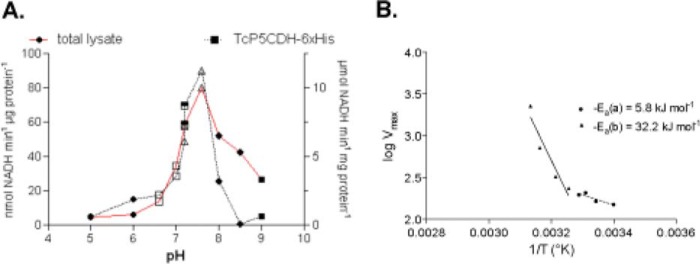
Effect of pH and temperature variations on the TcP5CDH activity. A, pH of the media in the reaction catalyzed by TcP5CDH was modified using different buffer systems. Enzymatic activity was determined in the presence of 2 mm NAD+ disodium salt, 0.3 mm γGS, and 100 mm of reaction buffer as follows: MES-NaOH (pH 5, 6) (filled circles), MOPS-NaOH (pH 6.5, 7) (open squares), HEPES-NaOH, pH 7.2, 7.6 (open triangles), potassium phosphate, pH 7.2, 7.6 (inverted triangles), Tris-HCl, pH 8, 8.5 (diamonds), and CHES (9, 9.5) (filled squares). The reaction was initiated by the addition of the enzyme, and initial velocities were calculated as linear rates at 5 or 15 min (at 30 °C with constant stirring) for the TcP5CDH-His6 or total lysates, respectively. B, effect of temperature variation in reactions catalyzed by TcP5CDH. Enzymatic activity was determined by progressively increasing the reaction temperature (from 20 to 75 °C). Arrhenius plot of the specific activity of TcP5CDH and the temperatures were assayed. y axis, log of Vmax according to temperature values used; x axis, temperature values−1 (Kelvin degrees) tested. The resulting plot was adjusted to a linear function to determine the energy of activation derived from the respective equation (slope = −Ea) (CI = 95%).
FIGURE 6.
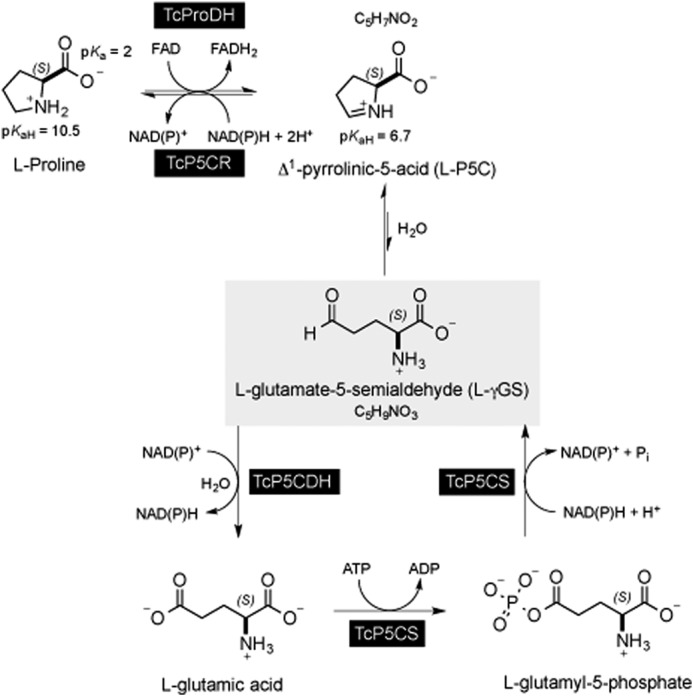
Biochemical steps involving P5C/γGS intermediates. The proline-glutamate interconversion pathway in T. cruzi involves two enzymatic steps in either the catabolic or biosynthetic pathway. l-Proline is oxidized to P5C by the FAD-dependent proline dehydrogenase (TcPRODH) (1.5.99.8) (UniProtKB code number F2WVH3). The reduction of P5C in proline is mediated by the enzyme P5C-reductase (TcP5CDR) (EC 1.5.1.2) (K4EAX2) and is coupled to NADPH oxidation. The reduction of glutamic acid into P5C requires two steps as follows. (i) Glutamic acid is first activated by an ATP-dependent kinase to produce glutamyl 1-phosphate, which is in turn converted to γGS by the NADPH-dependent P5C synthetase (TcP5CS) (EC 2.7.2.11) (K4E0F1). γGS is the equilibrium form of P5C in aqueous medium. (ii) carbonyl moiety of γGS is further oxidized to glutamic acid concomitantly with reduction of NAD(P)H by P5C-dehydrogenase (EC 1.5.1.1) (4) (Q4DRT8), which is the enzyme investigated in this study.
TcP5CDH Activity Is Associated with Mitochondrial Membranes
Kinetic data showed that TcP5CDH was enzymatically active in the presence of either NAD+ or NADP+, and no remarkable differences were observed when saturating concentrations of each cofactor were tested (Fig. 7A). Because mitochondrial membranes are not permeable to these cofactors, we assayed the activity of TcP5CDH using both solubilized and intact mitochondrial preparations from epimastigote forms. The values obtained revealed a basal activity of 4.7 ± 0.5 μmol min−1 mg of protein−1 for TcP5CDH in intact mitochondria (Fig. 7B). This activity increased 5-fold upon the addition of Triton X-100 to the preparations (21 ± 2 μmol min−1 mg of protein−1) (Fig. 7B) and decreased when NADP+ was used as a cofactor (11 ± 1.2 μmol min−1 mg of protein−1), confirming that TcP5CDH activity can be dependent on either cofactor. To determine the mitochondrial compartment in which TcP5CDH is located, the previously obtained data were compared with specific activities for citrate synthase (CS) (mitochondrial matrix enzyme) and fumarate reductase (mitochondrial inner membrane enzyme). Because the mitochondrial matrix content from vesicles is released during extraction, likely during a re-sealing process (10), no CS activity was detected in the mitochondrial vesicles. However, a higher specific activity for fumarate reductase was observed, suggesting that the preparations were enriched with mitochondrial membrane content (data not shown). The enzymatic activities detected in the mitochondrial fractions were also confirmed by Western blot analysis. The mitochondrial isoform of TcASATm was previously reported as a mitochondrial matrix protein (46). Thus, the polyclonal serum anti-TcASATm (expected size 45 kDa) was reactive in T. cruzi total extracts (Te), whereas in mitochondrial (Mc) preparations, the signal was not apparent (Fig. 7C). Conversely, TcP5CDH was detected in both Mc and Te preparations (Fig. 7C), with the Mc preparation exhibiting a slight difference in its band migration compared with the Te preparation (Fig. 7C). Taken together, our biochemical data indicate that TcP5CDH activity occurs in association with mitochondrial membranes in T. cruzi.
FIGURE 7.

Cofactor dependence and membrane-bound activity of TcP5CDH. A, evaluation of cofactor specificity for TcP5CDH. Specific activity was individually determined in the presence of γGS (0.3 mm) and either NAD+ or NADP+ (2 mm). Analysis of TcP5CDH present in mitochondrial vesicles of T. cruzi. B, enzymatic activity was measured in mitochondrial preparations from replicative E forms that were solubilized (+) or not (−) with detergent (Triton X-100). The composition of the reaction mixture was detailed previously, and each reaction was initiated upon the addition of mitochondrial extract as an enzyme source. When Triton X-100 was added, the activity of TcP5CDH significantly increased. When NADP+ was used as a cofactor, a similar increase was observed. All of the measurements were conducted by monitoring the change in absorbance (λ340 nm) at a linear rate (30 °C by 15 min under constant agitation). C, Western blot analysis of TcP5CDH in mitochondrial (Mito) vesicles. Polyclonal serum (diluted 1:3000 in PBS-T plus skim milk 0.3% w/v) against the TcASATm, used as mitochondrial marker, was probed against both mitochondrial and total extracts (Te) from E forms. Similarly, anti-TcP5CDH (1:2000) was used to probe both mitochondria and total extracts. The assay detected TcP5CDH in protein extracts from both total extracts and Mc.
TcP5CDH Oligomer Is Bound to Mitochondrial Membranes
As noted above, an N-terminal region with characteristics of a mitochondrial targeting pre-sequence (Fig. 1, A and B) and a putative trans-membrane spanner were found in TcP5CDH (Fig. 1C). The above-described biochemical data obtained from mitochondrion-derived vesicles support the functionality of both the mitochondrial targeting and the membrane-spanning sequences. Considering that our mitochondrial vesicle preparations resulted in enzymatically active proteins and that TcP5CDH is a membrane-bound protein, we addressed whether the quaternary structure of its oligomer might be present in T. cruzi mitochondria.
To address this question, we analyzed recombinant TcP5CDH by BNGE followed by Western blot analysis. A predominant band with an apparent molecular mass of 720 kDa and a weaker band of ∼440 kDa was detected (Fig. 8A). Mitochondrial proteins in BNGE exhibit distinct migration profiles depending on the type of detergent used (Fig. 8B). When those samples were solubilized with digitonin, electrophoresed in BNGE, transferred, and probed with anti-TcP5CDH, a single reactive band with an apparent molecular mass of 400 kDa was detected (Fig. 8B). These data are consistent with the SAXS estimations of TcP5CDH, further supporting the hexameric conformation of this protein.
FIGURE 8.

TcP5CDH native gel electrophoretic analysis. A, recombinant TcP5CDH-His6 (fraction P2, Fig. 3A) was resolved by NGE using a 4–16% acrylamide/bisacrylamide gradient gel. Lane 1, molecular mass marker for nondenaturing gels (GE Healthcare); lane 2, recombinant TcP5CDH-His6 (4 μg). The gel was run at 4 °C and further stained with Coomassie Blue R-250 solution as described under “Experimental Procedures.” Right panel, Western blot analysis from the NGE sample using purified anti-TcP5CDH (1:500). B, analysis of the TcP5CDH present in the mitochondrial fractions under native conditions. Samples were prepared from epimastigote forms, and the mitochondrial content (MitoC) was solubilized with either DIG or DDM. The mitochondrial fractions were first resolved by BNGE. Both of the samples (DIG and DDM) were electrotransferred to nitrocellulose membranes and probed with polyclonal anti-TcP5CDH; only DIG samples exhibited immunoreactivity. A single band of high molecular mass (≈450 kDa), presumably mitochondrial membrane-bound, was detected (note: the bands marked 1–5 correspond to the gel areas selected for proteomic analysis by MS/MS as summarized in Table 3). C, two-dimensional (2-D) analysis was performed under denaturing conditions (SDS-PAGE 12%). Samples from the first dimension (1D) native gel were excised (dotted region) and embedded in a gel cast for separation using conventional protein SDS-electrophoresis. After electrophoresis (two-dimensional), the proteins were electrotransferred to nitrocellulose membranes and probed with polyclonal anti-TcP5CDH. The boxes indicate proteins that were distinguished under solubilization conditions, showing two and one reactive bands for the DIG and DDM treatments, respectively. In both cases, the proteins exhibited a molecular mass compatible with that expected for TcP5CDH (∼63 kDa).
To verify the molecular mass of native TcP5CDH, mitochondrial membrane fractions were analyzed by BNGE, and the bands migrating within the regions corresponding to those of recombinant TcP5CDH were sampled for MS analysis. As expected, those samples were predominantly enriched in subunits of mitochondrial respiratory complexes such as complex II (succinate dehydrogenase) and complex IV (cytochrome c oxidase) of T. cruzi, as evidenced by in-gel activity assays (data not shown) and MS data (Table 4). Mitochondrial protein samples were solubilized either by using digitonin or DDM and further resolved by BNGE (Fig. 8C). The analyzed bands (numbered 1–5) revealed the presence of different subunits for complex II (bands 3 and 5) and complex IV (bands 1, 4 and 5), with molecular sizes ranging between 550–600 and 350–400 kDa, respectively (Table 4). TcP5CDH was detected in all of the protein samples (1–5; Fig. 8B), with up to 17 peptides and 25% coverage (Table 4). The differences in the migration profiles in which TcP5CDH was detected revealed two types of associations between the oligomeric forms of TcP5CDH and the mitochondrial membranes, exhibiting molecular masses of ∼600 and 350–400 kDa. This range is similar to what can be observed for certain respiratory complexes from T. cruzi (Table 4).
TABLE 4.
Proteins identified from the mitochondrial samples after BNGE
Bands corresponding to the regions where recombinant TcP5CDH was detected on native gels (Fig. 6B) were excised from BN gels (numbered 1–5 in Fig. 6B), treated as described under “Experimental Procedures,” and analyzed by tandem MS/MS. For simplicity, only some of the identified proteins are shown.
| Sample from BNGE | Protein name | GI no. | Peptides no. |
|---|---|---|---|
| 1 | Cytochrome c oxidase subunit IV | 71667854 | 51 |
| 1 | Cytochrome c oxidase subunit V | 71412456 | 32 |
| 1 | Cytochrome c oxidase subunit VI | 71412400 | 30 |
| 5 | Cytochrome c oxidase subunit VII | 71661625 | 17 |
| 4 | Cytochrome c oxidase subunit IV | 71667854 | 94 |
| 4 | Cytochrome c oxidase subunit V | 71412456 | 14 |
| 4 | Cytochrome c oxidase subunit VI | 71412400 | 74 |
| 5 | Cytochrome c oxidase subunit IV | 71667854 | 53 |
| 5 | Cytochrome c oxidase subunit V | 71412456 | 55 |
| 3 | Succinate dehydrogenase | 5931575 | 22 |
| 3 | Succinate dehydrogenase | 71401706 | 10 |
| 5 | Succinate dehydrogenase | 5931575 | 203 |
| 1 | Δ1-Pyrroline-5-carboxylate dehydrogenase | 71657117 | 7 |
| 2 | Δ1-Pyrroline-5-carboxylate dehydrogenase | 71657117 | 4 |
| 3 | Δ1-Pyrroline-5-carboxylate dehydrogenase | 71657117 | 4 |
| 4 | Δ1-Pyrroline-5-carboxylate dehydrogenase | 71657117 | 5 |
| 5 | Δ1-Pyrroline-5-carboxylate dehydrogenase | 71657117 | 17 |
| 1 | β-Subunit of mitochondrial processing peptidase | 71655600 | 12 |
| 1 | β-Subunit ATPase | 71661631 | 47 |
| 3 | α-Subunit of mitochondrial processing peptidase | 71420779 | 6 |
| 2 | Precursor of Reiske iron-sulfate protein | 71655052 | 30 |
To confirm whether the anti-TcP5CDH signal from mitochondrial protein resolved in BNGE corresponds to TcP5CDH, we performed a second-dimensional run under SDS-PAGE conditions, blotted the protein into a membrane, and probed it with the same antibody. Interestingly, digitonin treatment of the mitochondrial fractions released two anti-TcP5CDH-reactive proteins with distinct molecular weights, as observed in the first-dimensional run (Fig. 8C). The higher molecular mass band was more reactive than the lower one; both bands exhibited a molecular weight that was equivalent to that of the monomeric form of TcP5CDH (63 kDa). The higher band showed more reactivity to anti-TcP5CDH, and a slight difference in its migration profile under the second dimension was also observed (Fig. 8C, left side). In contrast, treatment with dodecyl maltoside, a detergent that preserves intact individual mitochondrial complexes, released a single protein of the same molecular mass (Fig. 8C, right side). These results are consistent with our previous observations by SAXS measurements and enzymatic assays in mitochondrial vesicles, supporting a quaternary conformation for TcP5CDH that is characteristic of a homohexamer. Nevertheless, the assumption that TcP5CDH might interact with other mitochondrion-forming supra-molecular complexes and components in T. cruzi cannot be excluded.
Subcellular Localization of TcP5CDH Is Conserved in the Distinct Parasite Stages
The co-localization profile of TcP5CDH with mitochondrial probes in distinct developmental forms of the parasite was used to evaluate its mitochondrial localization. TcP5CDH staining inside mammalian iCHO cells is isoform-specific for T. cruzi and correlates with intracellular parasitic load (Fig. 9A, merged panel). Detection of TcP5CDH with polyclonal serum shows a labeling pattern comparable with that found using the MitoTracker® probe. In addition, marked staining was observed in a central region that co-localizes with the region highlighted by kDNA staining (Fig. 9A, merged panel). The co-localization of TcP5CDH with the MitoTracker® probe suggests the presence of the protein within parasite mitochondria.
FIGURE 9.

Subcellular localization of TcP5CDH. A, TcP5CDH immunolabeling in the predominant life stages of T. cruzi. Axenic forms from E and M forms, A, Ie, and TCT derived from infected CHO-K1 cells (iCHO) were analyzed by microscopy. The cells were preincubated with 50 nm MitoTracker-CMXROS® (MitoT) (red) and fixed on polylysine-coated coverslips for subsequent immunostaining with anti-TcP5CDH (1:100), followed by secondary staining with AlexaFluor®-488 goat anti-mouse IgG (H + L) (Invitrogen®) (1:400). The DNA was stained using Hoechst-33258 (1:5000) (blue). For the iCHO assay, CHO-K1 cells were cultured on 24-well plates containing an embedded glass coverslip, which were subsequently used for TCT infection. After the 3rd day of infection, the cells were washed twice with PBS/BSA (2% w/v) and incubated with MitoTracker, followed by immunostaining with anti-TcP5CDH and Hoechst as detailed above. The resulting images were merged using ImageJ software. The arrows indicate the co-localization of all the three probes within a region proximal to the kinetoplastid DNA. B, Western blot analysis from digitonin-titrated fractions. Epimastigotes were selectively permeabilized with increased digitonin concentrations (0–5 mg ml−1), and the resulting supernatants (S) and pellets (P) were analyzed. All of the samples (40 μg per lane) were subjected to SDS-PAGE (10%), transferred to nitrocellulose membranes, and probed with polyclonal antibodies raised against TcTAT (mass = 45 kDa, cytosolic marker), glyceraldehyde-3-phosphate dehydrogenase (TcGAPDH) (mass = 39 kDa, glycosomal marker), proline dehydrogenase (TcProDH) (mass = 65 kDa, mitochondrial marker), and TcP5CDH as indicated. C, biochemical assays for the subcellular localization of TcP5CDH. Resulting samples from digitonized epimastigotes were also used as enzymatic sources in biochemical assays. The enzymatic activities of pyruvate kinase (○) (cytosol marker), hexokinase (*) (glycosomal marker), citrate synthase (▴) (mitochondrial marker), and TcP5CDH (■) were determined for all of the resulting fractions (S and P). Values plotted on the graph correspond to the ratio between activities on S and P + S and are expressed as a percentage of the enzyme.
Sequential digitonin permeabilization in epimastigote forms was also employed to confirm the mitochondrial localization of TcP5CDH. This permeabilization technique allows for the gradual release of the cellular contents from different compartments, such as the cytosol, glycosomes, reservosomes, and mitochondria, upon treatment with increasing concentrations of digitonin. Samples were evaluated for TcP5CDH and markers by both Western blotting and biochemical activity assays. Western blot analysis showed that TcTAT, a cytosolic marker (48), was released at digitonin concentrations ranging from 0.1 to 5 mg ml−1 (Fig. 9B), whereas TcGAPDH, a glycosomal marker, was released at intermediate digitonin concentrations (0.5–3 mg ml−1). Finally, TcProDH, a marker corresponding to the mitochondrial content, was released at higher digitonin concentrations (1.5–5 mg ml−1) (Fig. 9B). TcP5CDH was detected at intermediate digitonin concentrations (1.5–2.5 mg ml−1), which were compatible with either glycosomal or mitochondrial content (Fig. 9B). To confirm this result, the same samples were used to detect TcP5CDH and subcellular compartment markers by measuring biochemical activities. For this purpose, enzymatic activities for PK (cytosolic), hexokinase (glycosomal), and CS (mitochondrial marker) were also used (Fig. 9C). Thus, the percentage of TcP5CDH activity was predominantly detected at concentrations of 1–5 mg ml−1 of digitonin. This range was higher than that presented in the Western blot analysis. The release of TcP5CDH was slightly different from that of CS (Fig. 9C), which was observed at digitonin concentrations above 2.5 mg ml−1. Glycosomal content was released at intermediate digitonin concentrations (1.5–2.5 mg ml−1), similar to what was observed for hexokinase percentages (Fig. 9C). Hence, the data suggest that CS release requires higher digitonin concentrations compared with that required for TcP5CDH release. This result is consistent with either the outer or inner mitochondrial membrane localization of TcP5CDH.
TcP5CDH Is Up-regulated in the Infective Stages of the Parasite
TcP5CDH expression levels were analyzed in all of the developmental stages of T. cruzi. The mRNA levels were quantified in relative terms using the epimastigote stage as a reference (fold change = 1). Both M and TCT-infected stages exhibited increased mRNA levels (Fig. 10A) (p value < 0.05 in both cases), whereas neither intracellular stage (amastigotes and intracellular epimastigotes) derived from CHO-K1 cells exhibited significant differences compared with the reference stage (Fig. 10A). The enzymatic activities of TcP5CDH were also assayed for all of the life cycle stages. Interestingly, the activity profile correlated with its mRNA profile, with notable differences between trypomastigotes and epimastigotes, amastigotes and intracellular epimastigotes, and metacyclic trypomastigotes and amastigotes (Fig. 10A) (p value < 0.05). In all three of these comparisons, both mRNA and enzymatic levels were increased in the infective stage in comparison with the epimastigote reference stage (6-fold). Although the differences in specific activities were not significant between epimastigotes and metacyclic trypomastigotes, our data showed a relation between transcriptional and activity levels in all the stages analyzed (Fig. 10A). Notably, when the protein levels of TcP5CDH were analyzed by Western blotting, significant differences were not observed (Fig. 10B) with the exception of the intracellular stages (A and IE) compared with the other stages tested (Fig. 10B). Taken together, these data indicate a strong regulation of TcP5CDH throughout the life cycle at least at the post-transcriptional and post-translational levels, consistent with the participation of an enzyme in a nodal metabolic pathway.
FIGURE 10.
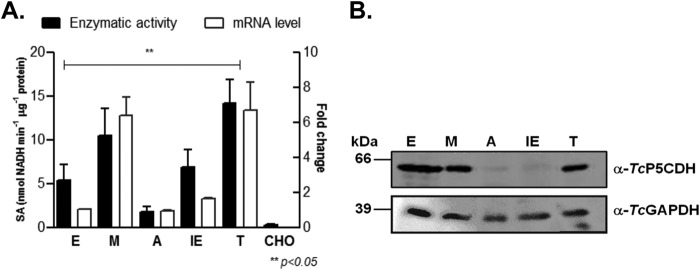
Analysis of TcP5CDH over distinct parasite life stages. A, expression levels of TcP5CDH in the predominant life stages of T. cruzi. Noninfected CHO-K1 cells served as controls. Total cell-free extracts from each parasitic stage were used as enzymatic sources in the TcP5CDH activity assay (left axis). RNA preparations from the stages noted above were used in quantitative real time PCR assays (quantitative RT-PCR). Comparisons were performed using the 2−ΔΔCt method and normalized relative to TcGAPDH expression levels. Values plotted correspond to the levels of fold change compared with the indicated epimastigote stage (right axis). The differences among the samples were calculated using one-way analysis of variance and Tukey's post test considering a p value < 0.05 as significant. B, Western blot analysis for TcP5CDH levels in the above-listed parasitic life stages. Protein sample preparations are described under “Experimental Procedures,” and equivalent amounts (40 μg) of each preparation were loaded per lane. The samples were subjected to SDS-PAGE (10%), transferred to nitrocellulose membranes, and probed with polyclonal antibodies raised against TcP5CDH (1:2000) and TcGADPH (1:2500). The latter did not react with GAPDH isoforms from the CHO-K1 cells used in our assay conditions.
Physiological Role for P5C
Having confirmed that TcP5CDH activity can be NAD(P)+-dependent and that it is associated with mitochondrial membranes, we addressed the ability of parasite cells to use P5C as a nutritional source. To this end, we subjected epimastigote forms to metabolic starvation by incubating the cells in PBS supplemented or not with P5C to evaluate their ability to survive during metabolic stress. Parallel incubations with other metabolites, such as d-glucose and l-proline, which are known to extend parasite survival under such conditions, were run as controls. Notably, P5C synthesis yields a stable product in acidic medium (1 m HCl); thus, the pH must be adjusted to values ≤7.0 prior to the assay; otherwise, the cells are unable to uptake P5C, which results in cell death (data not shown). The fact that parasites can extend their life spans during the conditions described above (Fig. 11A) indicates that P5C is not only metabolized but can also be taken up from the extracellular medium. Therefore, we tested whether P5C participates in ATP production. We starved the cells (in PBS) for 24 h to deplete intracellular ATP stocks and further incubated them in PBS in the presence and absence of P5C. PBS supplemented with proline was used as a control. The intracellular ATP levels were restored in cells incubated with l-proline or P5C (Fig. 11B), indicating the participation of P5C in energy production. This activity was impaired in the presence of a mitochondrial complex IV inhibitor (antimycin A), suggesting that P5C-dependent ATP production was driven by oxidative phosphorylation.
FIGURE 11.

Functional test of P5C in replicative and infective forms of T. cruzi. A, viability test in metabolically stressed cells (over 24 h) in the presence of single nutritional sources (3 mm) as indicated. After the incubation period, the cells were washed once with PBS and incubated (4 h) with MTT reagent in viability assays. Because the P5C purification method was performed in acid medium (HCl), the pH of the medium was adjusted (7.2) with KOH prior to use with the cells. An additional control of P5C-eluting agent (HCl) was also used. Absorbance ratio (590–695 nm) values were converted into percentages of cell viability using LIT as a control. B, determination of ATP levels in epimastigote forms after metabolic starvation. Intracellular ATP was depleted by incubation (30 h at 28 °C) in PBS buffer, followed by a recovery time (1 h) with 1 mm of single catabolic substrates. An additional treatment in the presence of P5C plus 0.5 μm antimycin-A (AA) (a respiratory chain complex IV inhibitor) was also performed. Next, the cells were lysed, and ATP content was determined using a luminescence-based assay following the manufacturer's protocols. The ATP content is expressed relative to cells under normal conditions (as 100%, grown in LIT medium). C, effect of P5C as a catabolic substrate was also tested for the infective TCT forms. iCHO cells were incubated (3 h) under normal (RPMI 1640 medium) and conditional media (PBS + FCS or PBS + P5C), and the number of parasites released into the supernatant (after the 6th day) was determined by hemocytometer counting. No significant (ns) differences were detected when RPMI 1640 medium was compared with PBS + 100 μm P5C, as indicated by one-way analysis of variance followed by Bonferroni's multiple comparison test (*, p < 0.05). Bars in the graph represent the results for three independent replicates (n = 3).
After observing that T. cruzi incorporates P5C from the external media, and because TcP5CDH is up-regulated during infective stages, we evaluated the P5C in the T. cruzi intracellular cycle. To this end, we performed a similar experiment to that described above by incubating metabolically stressed TCT forms under increased concentrations of P5C. Treated parasites were used to infect CHO-K1 cells, and the number of TCT forms released after the completion of their infective cycle (6th day) was determined. As expected, the number of parasites released from infected cells incubated in minimum media (PBS + SFB) was diminished by 50% (p < 0.05) compared with those released from infected cells incubated in control media (RPMI 1640 medium) (Fig. 11C). When the parasites were incubated with the host cells (3 h at 37 °C) under different concentrations of P5C, a dose-response effect on the number of released parasites was observed (Fig. 11C). At 100 μm P5C, no statistically significant differences were observed in the trypomastigotes release between RPMI 1640 medium and P5C-treated parasites, indicating that P5C can efficiently revert the effect of metabolic stress on the parasite invasion and infection. These data provide evidence of a physiological role for TcP5CDH, likely associated with its mitochondrial functions, and its natural substrate in either proliferative or infective forms of T. cruzi.
DISCUSSION
l-Proline is a nonessential amino acid, with different enzymes involved in its oxidation or biosynthetic routes depending on the organisms. In eukaryotes, these enzymes are located in the mitochondria and may participate in other redox processes that occur inside this organelle (70). Although the TcP5CDH N terminus could not be accurately predicted as an mTP using conventional bioinformatic tools, a detailed analysis revealed an N-terminal region (MLRR and FAFAYA motifs and an α-helix spanning the mitochondrial membrane) with protein fingerprints that are characteristic of an mTP for mitochondrial proteins in trypanosomes (67). Once imported, this mTP can be removed by mitochondrial peptidases (Gi: 71655600, 71420779 in Table 4), as indicated by MS data from our mitochondrial preparations. However, because migration of TcP5CDH in mitochondrial samples differs (upper band in Fig. 8C) from that observed in total lysates, either the cleavage of mTP does not occur or TcP5CDH undergoes a post-translational modification when associated with the mitochondria. Consistent with the subcellular localization of TcP5CDH, a proteomic analysis performed on mitochondrial fractions of T. brucei reportedly detected TbP5CDH exclusively in protein samples derived from inner mitochondrial membranes (71). In this compartment, an interaction with mitochondrial complex IV has been suggested (72), and a thioacylation event (with the addition of a palmitic acid) in the cysteine residues of TbP5CDH has also been reported (73). Similarly, we observed a co-localization of P5CDH and complex IV in our proteomic analysis, which, although not demonstrative, may be indicative of an interaction between these two complexes within high molecular weight fractions (Fig. 8B and Table 4).
Because TcP5CDH activity was stimulated (5-fold) upon the addition of Triton X-100 in mitochondrial vesicles and NAD+ diffuses freely through the MOM but not in the MIM (74), we can assume that TcP5CDH is exposed on either the inner face of the MOM or the external face of the MIM. Basal TcP5CDH activity was detected in our mitochondrial preparations in the absence of detergent. Therefore, we propose that TcP5CDH interacts with MIM, and its catalytic site is likely exposed to the mitochondrial matrix (Fig. 12). This latter possibility is consistent with data analyzed from the T. brucei mitochondrial proteome (75). Along with our data for T. cruzi, this evidence supports the existence of P5CDH as a component of mitochondrial membranes, which can be involved in redox processes within the mitochondrion of this unique parasite.
FIGURE 12.
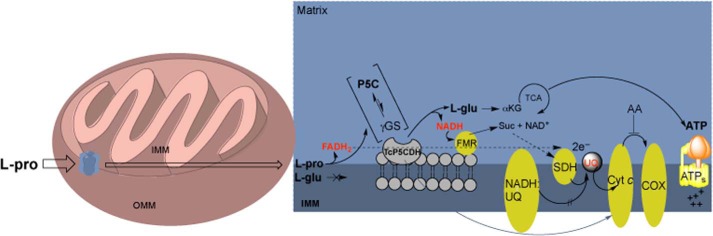
Schematic of the mitochondrial proline metabolic pathway in T. cruzi and its role in bioenergetics. Proline must be taken up from extracellular medium into mitochondria, where it is further oxidized into P5C by FAD-dependent TcProDH concomitantly with the production of FADH2. Next, the P5C is spontaneously converted into γGS, which is enzymatically converted to l-Glu. The TcP5CDH localizes within the inner mitochondrial membrane (IMM) and faces the matrix space, where the conversion of γGS into glutamate occurs at neutral pH. The glutamate produced can be deaminated into α-ketoglutarate (α-KG) and further oxidized in the tricarboxylic acid cycle (TCA), where substrate level phosphorylation can occur at a sufficient succinyl-CoA synthetase level (110). Because the function of the NADH:ubiquinone reductase complex appears limited in T. cruzi (69, 78), NADH can be reoxidized by fumarate reductase (FMR) to produce succinate. Next, e− contained in FADH2 and NADH are transferred (dashed arrows) to the ubiquinone (UQ) pool to a similar degree as succinate dehydrogenase (SDH), promoting the synthesis of ATP by proton F0F1-ATP synthase via the oxidative phosphorylation process. P5C-dependent ATP production is susceptible to inhibition by antimycin-A (AA) at the level of the cytochrome c reductase complex level, thus supporting the above described assumption. However, subsequent protein associations within the mitochondria of this parasite cannot be excluded (gray arrow).
As described above, TcP5CDH interacts with mitochondrial membranes. This interaction is most likely mediated by an α-helix domain (Phe198–Gly215), the structure of which is disrupted by the Pro211 residue, as observed in the deduced amino acid sequence. The detection of TcP5CDH under native conditions in mitochondrial samples solubilized with different detergents suggests that the active form is present within the parasite as an oligomer. The treatment of mitochondrial lysates with DIG (a mild detergent) (76) followed by Western blot analysis revealed a single band (with an apparent size of 380 kDa) when probed using anti-TcP5CDH. However, when this sample was further analyzed by conventional SDS-PAGE (two-dimensional), two reactive bands with molecular masses that were equivalent to the monomeric form of TcP5CDH were observed. The higher band was not detected by BNGE Western blot analysis, which may be explained by the following factors: (i) TcP5CDH forms protein complexes with other mitochondrial components that interfere with epitope exposure and are recognized by anti-TcP5CDH, or (ii) the number of blotted proteins with this molecular mass was below the sensitivity threshold of detection for the antibody. Nevertheless, we confirmed the presence of TcP5CDH in samples compatible with this higher molecular size through MS data analysis. This co-migration with respiratory chain complexes further implicates TcP5CDH as a mitochondrial membrane-bound component and may be an indication of other protein associations. This particular TcP5CDH migration under nondenaturing conditions was also confirmed when the recombinant isoform was resolved by BNGE. The multimeric structure of functional TcP5CDH was also confirmed by SEC, DLS, BNGE, and SAXS analysis. These results indicated a predominantly hexameric arrangement. This is consistent with previous structural data describing a hexameric conformation (25), suggesting that TcP5CDH exists as an oligomer. The enzyme bound to mitochondrial membranes exhibited either a hexameric conformation or a protein super-complex arrangement compatible with a dimer of hexamers. These data were also supported by molecular masses obtained for the recombinant protein from SEC, DLS, and BNGE, which were compatible with this assumption.
Structural studies have reported different oligomeric states for P5CDHs (25–27). In archaea, P5CDH forms a homohexamer in its ligand-free form (25). In contrast, a ring-shaped tetramer was reported for bacteria (27). In the latter group, proline dehydrogenase and P5CDH are not separate enzymes but, rather, exist as a single enzyme that catalyzes the entire conversion of proline into glutamate. Both catalytic domains are arranged in a single bifunctional flavoenzyme in which proline oxidation to glutamate occurs through a P5C-channeling mechanism (27). Thus, the separation of these domains into two distinct enzymes might represent an early evolutionary process.
The affinity of TcP5CDH for γGS (produced from synthetic P5C), as reflected by low Km values (at a scale of micromolars), reveals the ability of T. cruzi cells to efficiently convert P5C into glutamate, the only metabolic fate of P5C in this organism. This finding precludes the accumulation of P5C within the cell, which could otherwise result in an oxidative imbalance and lethality for the parasite. Indeed, yeast cells that are unable to express a functional P5CDH (defective for the PUT2 gene) exhibited an intracellular accumulation of P5C and were unable to grow in selective media in which proline was the only nitrogen source. It has been well described that reactive oxygen species are produced in ΔPUT2 yeasts under these conditions, dramatically affecting cell viability (32). We confirmed this characteristic in our functional complementation assay using ΔPUT2 mutant yeast in which P5C accumulates intracellularly, and cell growth is impaired under minimal proline medium conditions. Although we have not addressed the issue of TcP5CDH trafficking inside yeast cells, we assumed that TcP5CDH was processed via a mechanism that was as efficient as that used for T. cruzi cells.
Proline breakdown in T. cruzi occurs through two oxidation steps (7). First, a dimeric mitochondrial enzyme is catalyzed by a proline dehydrogenase and oxidizes the proline in P5C coupled with a reduction in FAD. The electrons are channeled into the electron transport chain through coenzyme Q and transferred downstream via an identical route as that used in succinate oxidation by succinate dehydrogenase at complex II. Next, TcP5CDH oxidizes γGS to glutamic acid, and NAD+/NADP+ is concomitantly reduced. This process also occurs in the mitochondria, where the entire proline oxidation process is known to occur during ATP production (9, 10).
In water, P5C exists in equilibrium with γGS, and NMR data indicate that the imino-form of P5C predominates at pH > 5.8 (pKaHimino = 6.7; pKaCOOH = 1.8) (19, 63, 79, 80). DFT calculations also indicate that the imino-protonated cyclic form (P5C) is thermodynamically preferred over the zwitterionic form of γGS. Accordingly, crystallographic data indicate that γGS is the substrate of T. thermophilus Δ1-pyrroline-5-carboxylate dehydrogenase and that the nucleophilic attack of the thiol portion of Cys336 to the γGS carbonyl yields an intermediate hemithioacetal, which is subsequently converted to glutamate via NAD+-mediated hydrolysis (25, 66). Assuming that the active site of the T. cruzi P5CDH is similar to that of T. thermophilus, the deprotonation of the –SH group of the cysteine residue (whose pKa is unknown but can be assumed to be ∼6–8) (81) might be crucial to the oxidation of γGS because the thiolate is a more efficient nucleophile than its corresponding protonated form. This hypothesis is corroborated by the fact that the optimal pH values reported for P5CDHs from other species are similar (25, 27, 82) and can explain the lack of activation for the nucleophilic attack of the thiol group reported by Inagaki et al. (25). In other words, although the cyclic form (P5C) is thermodynamically more favorable, this characteristic is irrelevant for the overall oxidation mechanism because the enzymatic oxidation of γGS to glutamate is assumed to be rapid and irreversible (23, 82). As a consequence, γGS is not expected to accumulate. Furthermore, the intracellular pH in the amastigote and TCT forms of T. cruzi is 7.3, a condition that favors the enzymatic conversion of γGS into glutamate (83).
The reaction catalyzed by TcP5CDH was dependent on NAD+ or NADP+ reduction, and the affinity constants (Km values) measured in mitochondrial lysates or TcP5CDH-His6 were similar. However, the Km value for NAD+ in TcP5CDH-His6 was higher (72.3 ± 14.1 μm) than that calculated for NADP+ (342 ± 140 μm). Furthermore, the catalytic efficiency (kcat/Km) exhibited for NADP+ was reduced in both cases, indicating that in the mitochondria of the parasite, TcP5CDH, produces reduced equivalents (NADH and NADPH) that can be used in other biochemical processes (discussed below). The affinity for γGS under saturating concentrations of NAD+ was comparable in both enzymatic preparations, but the differences found in terms of catalytic efficiency can be interpreted as either a consequence of additional NAD+-dependent enzymes or TcP5CS, which interconverts glutamate to P5C via γGS. The Km values determined herein for γGS were consistent with those reported from orthologs in other organisms (25, 84).
The activity of TcP5CDH was dependent on the temperature of the reaction and was optimal at 45 °C, which is higher than the temperatures experienced by the parasite during its life cycle through different hosts (which range from environmental temperatures in the insect vector up to 37 °C). However, similar results have been described for enzymes that are insensitive to thermal inactivation (85). Nonlinear Arrhenius plots can be observed even if the T range is not large, as has been observed for the enzymatic oxidation of γGS (86, 87). This pattern is particularly evident for many enzymatic biochemical reactions occurring in membranes. Thus, different steps of the complex reaction may become rate-limiting at different temperatures and medium pH. Alternatively, there are T-induced phase transitions within the membrane (86, 87). The result reported here is consistent with the complex reaction mechanism involving the presence of the cofactor NAD+ (85).
Our observations regarding TcP5CDH expression show that this enzyme is developmentally expressed throughout the parasite life cycle. An integrated analysis of mRNA, protein levels, and enzymatic rates allowed us to conclude TcP5CDH is up-regulated during both M- and T-infective stages. The apparent discordance between mRNA and protein levels might be attributed to post-transcriptional regulation, a mechanism that has been well described for trypanosomatids (88). Such differential expression of TcP5CDH is likely associated with the metabolite availability faced by T. cruzi in different hosts. In kissing bugs, l-proline was reported to be an abundant amino acid in tissue fluids and urine (89, 90). The predominance of proline in the intracellular pool of amino acids in T. cruzi epimastigotes has also been reported (91). In a comprehensive proteomic analysis of the T. cruzi secretome, proline-related enzymes have been described (92). Proline racemase converts d-proline into l-proline and acts as a host B-cell mitogen (93). The prolyl oligopeptidase hydrolyzes large substrates such as fibronectin and native collagen, which can fulfill proline requirements within the extracellular matrix during the mammal host cell invasion process (94). This redundancy of proline as an energy source has been associated with physiological processes such as in vitro metacyclogenesis (95) and differentiation during the intracellular stages (13). Indeed, proline is the only amino acid that is sufficient for T. cruzi to generate the ATP levels required for the invasion of mammal host cells (12). These previously reported data, along with our observations, indicate that proline metabolism is a crucial part of the T. cruzi infection process, although the role of proline metabolism does not appear to be limited to the generation of ATP. In this regard, we propose that components of proline metabolism might be involved as regulatory or signaling factors during T. cruzi infection. As these factors must be occasionally secreted or exposed on the cell surface when host interactions occur and as we observed that the TcP5CDH immunolabeling profile surrounds the cells in the TCT forms and that P5C can supply the energy required for the infectivity process in CHO-K1, it is conceivable that TcP5CDH functions as a metabolic enzyme that interacts with components of the mammal host cell during the infection process. In Cryptococcus neoformans, a pathogenic yeast, PUT2 is required for the optimal production of major cryptococcal virulence factors, and in mice, infection with a ΔPUT2 mutant was determined to be nonvirulent (96). Another study has shown that the immune response underlying pneumococcal colonization affects Staphylococcus aureus colonization and identified S. aureus P5CDH as a target in a cross-reactive antibody response (97). In trypanosomatids, the participation of enzymes with pivotal roles beyond their canonical metabolic functions has been reported. T. brucei responds to citrate exposure at the initiation of its life cycle during transmission to the tsetse fly (98). Furthermore, in Leishmania parasites, enolase and arginase represent examples of enzymes that participate in the infection process or pathogenesis (99, 100).
Structural data on TcP5CDH, together with its mitochondrial membrane localization and up-regulated activity during infective stages, led us to hypothesize additional functions for this enzyme beyond the production of glutamate. We have observed that TcP5CDH oxidizes P5C in a process that is coupled with NAD(P)H generation, both of which are necessary for other biological processes. For example, NADH might promote oxidative phosphorylation, generating the proton-driven force in the form of an electrochemical potential that can drive the synthesis of ATP (101). Moreover, NADPH serves as a cofactor for trypanothione reductase (102). Indeed, we have observed that P5C can serve as a viable energy source in the ATP production, which was supported by our data showing that antimycin A inhibits this process. The energy can be generated from two possible routes. (i) NADH serves as a substrate for components of the respiratory chain, and therefore, ATP is produced through the electron respiratory chain. Despite the presence of mitochondrial complex I that is limited in functionality in T. cruzi, NADH can be further reoxidized by fumarate reductase (103). (ii) Alternatively, TcP5CDH produces glutamate that can be further deaminated to α-KG, which can be oxidized, producing CO2 and three molecules of NADH, thus enabling extra ATP synthesis. Next, the two-step oxidation of proline produces FADH2 and NADH that can feed the oxidative phosphorylation driving ATP synthesis (Fig. 12). P5C can be produced and secreted to the extracellular medium by mammalian cells (104, 105). Thus, P5C could be used as an energy source by other cells naturally exposed to these metabolites.
Proline synthesis in T. cruzi is operative.3 T. cruzi is able to synthesize proline from glutamate by mean of a bifunctional glutamyl kinase P5C synthetase (UniprotKB access code K4E0F1). P5C is further reduced to proline by P5C reductase (K4EAX2). Hence, the tight regulation of intracellular proline and intermediate pools and related enzymes must be present in T. cruzi throughout its life cycle to avoid futile conversions. As proline is oxidized within the mitochondria of T. cruzi, a transport system likely functions to uptake this amino acid into the organelle. Glutamic acid at physiological pH (7.2) exists in a deprotonated form (106). Interestingly, this glutamic acid is actively taken up from the extracellular medium via a single transporter (107). However, several intracellular sources of the intramitochondrial synthesis of glutamate by enzymatic action have been demonstrated (46, 47, 108). In addition to the oxidation of γGS (catalyzed by TcP5CDH), glutamate can be enzymatically generated from the transamination of l-alanine or l-aspartate, the conversion of l-histidine, or the reductive amination of α-ketoglutarate (α-KG). The latter step is reversible, where α-ketoglutarate acts as an intermediate metabolite of the Krebs cycle (109). These glutamate-related enzymes have been detected in both the mitochondria and the cytosol of T. cruzi (46, 47). Moreover, glutamate might also be converted to glutamine, which is a relevant precursor for amino sugar metabolism and conversion into N-acetylglucosamine. We have also observed that the overexpression of TcP5CDH can stimulate the enzymatic activity of glutamine synthetase.3 Therefore, the glutamate generated in T. cruzi represents an example of an amino acid that exhibits multiple roles beyond protein synthesis. Taken together with the up-regulation of TcP5CDH in the infective forms and with its participation in the respiratory chain to produce ATP, our data highlight this enzymatic step as crucial for T. cruzi viability in both replicative and infective forms.
Conclusions
Here, we demonstrate that the pathogenic species T. cruzi expresses a functional TcP5CDH enzyme. Our enzymatic assays for TcP5CDH demonstrate that glutamate is irreversibly produced from its P5C-opened form γGS, which is coupled to the reduction of either NAD+ or NADP+ within the mitochondria of T. cruzi. Functional assays using mitochondrial preparations from T. cruzi revealed that TcP5CDH is catalytically active as a membrane-bound enzyme. The kinetic parameters for TcP5CDH and its cofactors are consistent with those observed within the mitochondrial fractions of this parasite, suggesting a high catalytic efficiency to oxidize γGS. Moreover, TcP5CDH is present in a hexameric state, and its interaction with mitochondrial membranes is presumably facilitated by an α-helix domain.
Mitochondrial TcP5CDH produces glutamate, concomitantly providing reducing equivalents, which are further used to synthesize ATP via the electron transport chain. In particular, TcP5CDH is differentially up-regulated during infective stages. Overall, we postulate that proline metabolism in T. cruzi represents an efficient mechanism that fulfills energy requirements throughout the life cycle of this parasite. This strategy is correlated with substrate availability in the different hosts of the parasite. Because of the high substrate specificity that TcP5CDH exhibits for γGS, as well as its developmental and mitochondrial functions, we propose that this enzymatic step is crucial for the persistence of this parasite during infectious stages.
Acknowledgments
We acknowledge the following: Dr. C. Nowicki (Universidad de Buenos Aires, Argentina) for providing bacterial clones to express TcASAT and TcTAT; Dr. M. H. Barros (Instituto de Ciências Biomédicas-Universidade de São Paulo) and Dr. L. E. S. Netto (Instituto de Biociências-Universidade de São Paulo) for providing yeast plasmids pYEp351/pMA210 and yeast mutant (ΔPUT2), respectively; J. D. Ferraz for helpful yeast manipulation; Dr. R. Lopes (Center for Applied Technology/Butantã) for MS analysis of P5C; C. Marinho's laboratory (Instituto de Ciências Biomédicas-Universidade de São Paulo) for microscopic assays; and Dr. C. Wrenger for critical reading of the manuscript.
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo Grant 13/18970-6 (to A. M. S.), Instituto Nacional de Biologia Estrutural e Química Medicinal em Doenças Infecciosas, and Conselho Nacional de Desenvolvimento Científico e Tecnológico Grant 475741/2013-7 (to A. M. S.).
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) XM811985.1
L. Marchese, K. Olavarria, B. S. Mantilla, and A. M. Silber, unpublished data.
- P5C
- Δ1-pyrroline-5-carboxylate
- P5CDH
- Δ1-pyrroline-5-carboxylate dehydrogenase
- SAXS
- small angle x-ray scattering
- TcP5CDH
- Δ1-pyrroline-5-carboxylate dehydrogenase from T. cruzi; proline dehydrogenase from T. cruzi
- NGE
- native gel electrophoresis
- γGS
- glutamate-γ-semialdehyde
- SEC
- size-exclusion chromatography
- DDM
- dodecyl maltoside
- OAB
- o-aminobenzaldehyde
- TCT
- tissue culture cell-derived trypomastigote
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- BNGE
- blue native gel electrophoresis
- DLS
- dynamic light scattering
- γGS
- glutamate γ-semialdehyde
- DIG
- digitonin
- E
- epimastigote
- M
- metacyclic trypomastigote
- A
- amastigote
- Ie
- intracellular epimastigote
- TCT
- trypomastigote
- TcTAT
- tyrosine aminotransferase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- TcASATm
- mitochondrial aspartate aminotransferase
- Mc
- mitochondrial
- Te
- total extract
- CS
- citrate synthase
- mTP
- mitochondria target peptide.
REFERENCES
- 1. Chagas C. (1909) Nova tripanozomiaze humana. Estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Mem. Inst. Oswaldo Cruz. 1, 159–218 [Google Scholar]
- 2. Brener Z. (1973) Biology of Trypanosoma cruzi. Annu. Rev. Microbiol. 27, 347–382 [DOI] [PubMed] [Google Scholar]
- 3. World Health Organization (2012) in Epidemiology and Burden of Disease (www.who.int/iris/bitstream/10665/77472/1/WHO_TRS_975_eng.pdf)
- 4. Brener Z. (1971) Life cycle of Trypanosoma cruzi. Rev. Inst. Med. Trop Sao. Paulo. 13, 171–178 [PubMed] [Google Scholar]
- 5. Bastos C. J., Aras R., Mota G., Reis F., Dias J. P., de Jesus R. S., Freire M. S., de Araújo E. G., Prazeres J., Grassi M. F. (2010) Clinical outcomes of thirteen patients with acute Chagas disease acquired through oral transmission from two urban outbreaks in northeastern Brazil. PLoS Negl. Trop. Dis. 4, e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Covarrubias C., Cortez M., Ferreira D., Yoshida N. (2007) Interaction with host factors exacerbates Trypanosoma cruzi cell invasion capacity upon oral infection. Int. J. Parasitol. 37, 1609–1616 [DOI] [PubMed] [Google Scholar]
- 7. Lisvane Silva P., Mantilla B. S., Barisón M. J., Wrenger C., Silber A. M. (2011) The uniqueness of the Trypanosoma cruzi mitochondrion: opportunities to identify new drug target for the treatment of Chagas disease. Curr. Pharm. Des. 17, 2074–2099 [DOI] [PubMed] [Google Scholar]
- 8. Silber A. M., Colli W., Ulrich H., Alves M. J., Pereira C. A. (2005) Amino acid metabolic routes in Trypanosoma cruzi: possible therapeutic targets against Chagas' disease. Curr. Drug. Targets Infect. Disord. 5, 53–64 [DOI] [PubMed] [Google Scholar]
- 9. Sylvester D., Krassner S. M. (1976) Proline metabolism in Trypanosoma cruzi epimastigotes. Comp. Biochem. Physiol. B. 55, 443–447 [DOI] [PubMed] [Google Scholar]
- 10. Paes L. S., Suárez Mantilla B., Zimbres F. M., Pral E. M., Diogo de Melo P., Tahara E. B., Kowaltowski A. J., Elias M. C., Silber A. M. (2013) Proline dehydrogenase regulates redox state and respiratory metabolism in Trypanosoma cruzi. PLoS One 8, e69419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sayé M., Miranda M. R., di Girolamo F., de los Milagros Cámara M., Pereira C. A. (2014) Proline modulates the Trypanosoma cruzi resistance to reactive oxygen species and drugs through a novel dl-proline transporter. PLoS One 9, e92028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martins R. M., Covarrubias C., Rojas R. G., Silber A. M., Yoshida N. (2009) Use of l-proline and ATP production by Trypanosoma cruzi metacyclic forms as requirements for host cell invasion. Infect. Immun. 77, 3023–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tonelli R. R., Silber A. M., Almeida-de-Faria M., Hirata I. Y., Colli W., Alves M. J. (2004) l-Proline is essential for the intracellular differentiation of Trypanosoma cruzi. Cell Microbiol. 6, 733–741 [DOI] [PubMed] [Google Scholar]
- 14. Contreras V. T., Salles J. M., Thomas N., Morel C. M., Goldenberg S. (1985) In vitro differentiation of Trypanosoma cruzi under chemically defined conditions. Mol. Biochem. Parasitol. 16, 315–327 [DOI] [PubMed] [Google Scholar]
- 15. Silber A. M., Tonelli R. R., Lopes C. G., Cunha-e-Silva N., Torrecilhas A. C., Schumacher R. I., Colli W., Alves M. J. (2009) Glucose uptake in the mammalian stages of Trypanosoma cruzi. Mol. Biochem. Parasitol. 168, 102–108 [DOI] [PubMed] [Google Scholar]
- 16. Strecker H. J. (1960) The interconversion of glutamic acid and proline. II. The preparation and properties of Δ1-pyrroline-5-carboxylic acid. J. Biol. Chem. 235, 2045–2050 [PubMed] [Google Scholar]
- 17. Adams E., Frank L. (1980) Metabolism of proline and the hydroxyprolines. Annu. Rev. Biochem. 49, 1005–1061 [DOI] [PubMed] [Google Scholar]
- 18. Vogel H. J., Davis B. D. (1952) Glutamic γ-semialdehyde and Δ1-pyrroline-5-carboxylic acid, intermediates in the biosynthesis of proline. J. Am. Chem. Soc. 74, 109–112 [Google Scholar]
- 19. Williams I., Frank L. (1975) Improved chemical synthesis and enzymic assay of Δ1-pyrroline-5-carboxylic acid. Anal. Biochem. 64, 85–97 [DOI] [PubMed] [Google Scholar]
- 20. Deuschle K., Funck D., Hellmann H., Däschner K., Binder S., Frommer W. B. (2001) A nuclear gene encoding mitochondrial Δ-pyrroline-5-carboxylate dehydrogenase and its potential role in protection from proline toxicity. Plant J. 27, 345–356 [DOI] [PubMed] [Google Scholar]
- 21. Elthon T. E., Stewart C. R. (1981) Submitochondrial location and electron transport characteristics of enzymes involved in proline oxidation. Plant Physiol. 67, 780–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brandriss M. C., Krzywicki K. A. (1986) Amino-terminal fragments of Δ1-pyrroline-5-carboxylate dehydrogenase direct β-galactosidase to the mitochondrial matrix in Saccharomyces cerevisiae. Mol. Cell. Biol. 6, 3502–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haslett M. R., Pink D., Walters B., Brosnan M. E. (2004) Assay and subcellular localization of pyrroline-5-carboxylate dehydrogenase in rat liver. Biochim. Biophys. Acta 1675, 81–86 [DOI] [PubMed] [Google Scholar]
- 24. He F., DiMario P. J. (2011) Drosophila Δ1-pyrroline-5-carboxylate dehydrogenase (P5CDh) is required for proline breakdown and mitochondrial integrity–Establishing a fly model for human type II hyperprolinemia. Mitochondrion 11, 397–404 [DOI] [PubMed] [Google Scholar]
- 25. Inagaki E., Ohshima N., Takahashi H., Kuroishi C., Yokoyama S., Tahirov T. H. (2006) Crystal structure of Thermus thermophilus Δ1-pyrroline-5-carboxylate dehydrogenase. J. Mol. Biol. 362, 490–501 [DOI] [PubMed] [Google Scholar]
- 26. Luo M., Singh R. K., Tanner J. J. (2013) Structural determinants of oligomerization of Δ(1)-pyrroline-5-carboxylate dehydrogenase: identification of a hexamerization hot spot. J. Mol. Biol. 425, 3106–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Srivastava D., Schuermann J. P., White T. A., Krishnan N., Sanyal N., Hura G. L., Tan A., Henzl M. T., Becker D. F., Tanner J. J. (2010) Crystal structure of the bifunctional proline utilization A flavoenzyme from Bradyrhizobium japonicum. Proc. Natl. Acad. Sci. U.S.A. 107, 2878–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Souza W. (2002) Basic cell biology of Trypanosoma cruzi. Curr. Pharm. Des. 8, 269–285 [DOI] [PubMed] [Google Scholar]
- 29. de Souza W., Attias M., Rodrigues J. C. (2009) Particularities of mitochondrial structure in parasitic protists (Apicomplexa and Kinetoplastida). Int. J. Biochem. Cell Biol. 41, 2069–2080 [DOI] [PubMed] [Google Scholar]
- 30. Monzote L., Gille L. (2010) Mitochondria as a promising antiparasitic target. Curr. Clin. Pharmacol. 5, 55–60 [DOI] [PubMed] [Google Scholar]
- 31. Lamour N., Rivière L., Coustou V., Coombs G. H., Barrett M. P., Bringaud F. (2005) Proline metabolism in procyclic Trypanosoma brucei is down-regulated in the presence of glucose. J. Biol. Chem. 280, 11902–11910 [DOI] [PubMed] [Google Scholar]
- 32. Nomura M., Takagi H. (2004) Role of the yeast acetyltransferase Mpr1 in oxidative stress: regulation of oxygen reactive species caused by a toxic proline catabolism intermediate. Proc. Natl. Acad. Sci. U.S.A. 101, 12616–12621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mixson A. J., Phang J. M. (1988) The uptake of pyrroline 5-carboxylate. Group translocation mediating the transfer of reducing-oxidizing potential. J. Biol. Chem. 263, 10720–10724 [PubMed] [Google Scholar]
- 34. Geraghty M. T., Vaughn D., Nicholson A. J., Lin W. W., Jimenez-Sanchez G., Obie C., Flynn M. P., Valle D., Hu C. A. (1998) Mutations in the Δ1-pyrroline 5-carboxylate dehydrogenase gene cause type II hyperprolinemia. Hum. Mol. Genet. 7, 1411–1415 [DOI] [PubMed] [Google Scholar]
- 35. Farrant R. D., Walker V., Mills G. A., Mellor J. M., Langley G. J. (2001) Pyridoxal phosphate de-activation by pyrroline-5-carboxylic acid. Increased risk of vitamin B6 deficiency and seizures in hyperprolinemia type II. J. Biol. Chem. 276, 15107–15116 [DOI] [PubMed] [Google Scholar]
- 36. Camargo E. P. (1964) Growth and differentiation in Trypanosoma cruzi. I. Origin of metacyclic trypanosomes in liquid media. Rev. Inst. Med. Trop Sao. Paulo 12, 93–100 [PubMed] [Google Scholar]
- 37. de Sousa M. A. (1983) A simple method to purify biologically and antigenically preserved bloodstream trypomastigotes of Trypanosoma cruzi using DEAE-cellulose columns. Mem. Inst. Oswaldo Cruz. 78, 317–333 [DOI] [PubMed] [Google Scholar]
- 38. Baudin A., Ozier-Kalogeropoulos O., Denouel A., Lacroute F., Cullin C. (1993) A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 21, 3329–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gietz R. D., Schiestl R. H. (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 [DOI] [PubMed] [Google Scholar]
- 40. Lundblad V., Struhl K. (2003) in Current protocols in Molecular Biology (Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K., eds) pp. 13.10.11–13.10.13, John Wiley & Sons, Inc., Cambridge, UK [Google Scholar]
- 41. Thompson J. D., Higgins D. G., Gibson T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hill J. E., Myers A. M., Koerner T. J., Tzagoloff A. (1986) Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast 2, 163–167 [DOI] [PubMed] [Google Scholar]
- 43. Ma J., Ptashne M. (1987) Deletion analysis of GAL4 defines two transcriptional activating segments. Cell 48, 847–853 [DOI] [PubMed] [Google Scholar]
- 44. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 45. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 46. Marciano D., Llorente C., Maugeri D. A., de la Fuente C., Opperdoes F., Cazzulo J. J., Nowicki C. (2008) Biochemical characterization of stage-specific isoforms of aspartate aminotransferases from Trypanosoma cruzi and Trypanosoma brucei. Mol. Biochem. Parasitol. 161, 12–20 [DOI] [PubMed] [Google Scholar]
- 47. Marciano D., Maugeri D. A., Cazzulo J. J., Nowicki C. (2009) Functional characterization of stage-specific aminotransferases from trypanosomatids. Mol. Biochem. Parasitol. 166, 172–182 [DOI] [PubMed] [Google Scholar]
- 48. Montemartini M., Santomé J. A., Cazzulo J. J., Nowicki C. (1993) Purification and partial structural and kinetic characterization of tyrosine aminotransferase from epimastigotes of Trypanosoma cruzi. Biochem. J. 292, 901–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Souza D. H., Garratt R. C., Araújo A. P., Guimarães B. G., Jesus W. D., Michels P. A., Hannaert V., Oliva G. (1998) Trypanosoma cruzi glycosomal glyceraldehyde-3-phosphate dehydrogenase: structure, catalytic mechanism and targeted inhibitor design. FEBS Lett. 424, 131–135 [DOI] [PubMed] [Google Scholar]
- 50. Cooper H. M., Paterson Y. (2000) Current Protocols in Molecular Biology, pp. 11.12.11–11.12.19, John Wiley & Sons, Inc., New York [Google Scholar]
- 51. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 53. Fischer H., de Oliver Neto M., Napolitano H., Polikarpov I., Craievich A. (2010) Determination of the molecular weight of proteins in solution from a single small-angle x-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43, 101–109 [Google Scholar]
- 54. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Svergun D. I. (1995) CRYSOL–a Program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 56. Kozin M. B., Svergun D. I. (2001) Automated matching of high and low resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 57. Fernandes M. P., Inada N. M., Chiaratti M. R., Araújo F. F., Meirelles F. V., Correia M. T., Coelho L. C., Alves M. J., Gadelha F. R., Vercesi A. E. (2010) Mechanism of Trypanosoma cruzi death induced by Cratylia mollis seed lectin. J. Bioenerg. Biomembr. 42, 69–78 [DOI] [PubMed] [Google Scholar]
- 58. Magdaleno A., Suárez Mantilla B., Rocha S. C., Pral E. M., Silber A. M. (2011) The involvement of glutamate metabolism in the resistance to thermal, nutritional, and oxidative stress in Trypanosoma cruzin. Enzyme Res. 2011, 486928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schägger H., von Jagow G. (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199, 223–231 [DOI] [PubMed] [Google Scholar]
- 60. Sambrook P. M., Russell D. (2001) Molecular Cloning: A Laboratory Manual. 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 61. Bookout A. L., Cummins C. L., Mangelsdorf D. J. (2003) in Current Protocols in Molecular Biology (Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K., eds) pp. 15.18.11–15.18.21, John Wiley & Sons, Inc, Cambridge, UK [Google Scholar]
- 62. Diekert K., de Kroon A. I., Kispal G., Lill R. (2001) Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol. 65, 37–51 [DOI] [PubMed] [Google Scholar]
- 63. Mezl V. A., Knox W. E. (1976) Properties and analysis of a stable derivative of pyrroline-5-carboxylic acid for use in metabolic studies. Anal. Biochem. 74, 430–440 [DOI] [PubMed] [Google Scholar]
- 64. Turrens J. F. (1989) The role of succinate in the respiratory chain of Trypanosoma brucei procyclic trypomastigotes. Biochem. J. 259, 363–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Magdaleno A., Ahn I. Y., Paes L. S., Silber A. M. (2009) Actions of a proline analogue, l- thiazolidine-4-carboxylic acid (T4C), on Trypanosoma cruzi. PLoS One 4, e4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Srivastava D., Singh R. K., Moxley M. A., Henzl M. T., Becker D. F., Tanner J. J. (2012) The three-dimensional structural basis of type II hyperprolinemia. J. Mol. Biol. 420, 176–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Krnáčová K., Vesteg M., Hampl V., Vlček Č., Horváth A. (2012) Euglena gracilis and trypanosomatids possess common patterns in predicted mitochondrial targeting presequences. J. Mol. Evol. 75, 119–129 [DOI] [PubMed] [Google Scholar]
- 68. Godard P., Urrestarazu A., Vissers S., Kontos K., Bontempi G., van Helden J., André B. (2007) Effect of 21 different nitrogen sources on global gene expression in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 3065–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carranza J. C., Kowaltowski A. J., Mendonça M. A., de Oliveira T. C., Gadelha F. R., Zingales B. (2009) Mitochondrial bioenergetics and redox state are unaltered in Trypanosoma cruzi isolates with compromised mitochondrial complex I subunit genes. J. Bioenerg. Biomembr. 41, 299–308 [DOI] [PubMed] [Google Scholar]
- 70. Tanner J. J. (2008) Structural biology of proline catabolism. Amino Acids 35, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Acestor N., Panigrahi A. K., Ogata Y., Anupama A., Stuart K. D. (2009) Protein composition of Trypanosoma brucei mitochondrial membranes. Proteomics 9, 5497–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Acestor N., Ziková A., Dalley R. A., Anupama A., Panigrahi A. K., Stuart K. D. (2011) Trypanosoma brucei mitochondrial respiratome: composition and organization in procyclic form. Mol. Cell. Proteomics 10, M110.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Emmer B. T., Nakayasu E. S., Souther C., Choi H., Sobreira T. J., Epting C. L., Nesvizhskii A. I., Almeida I. C., Engman D. M. (2011) Global analysis of protein palmitoylation in African trypanosomes. Eukaryot. Cell 10, 455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ernster L., Kuylenstierna B. (1970) in Membranes of Mitochondria and Choroplasts (Racker R., ed) pp. 175–197, Van Nostrand Reinhold Co, New York [Google Scholar]
- 75. Panigrahi A. K., Ogata Y., Zíková A., Anupama A., Dalley R. A., Acestor N., Myler P. J., Stuart K. D. (2009) A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics 9, 434–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schägger H. (2002) Respiratory chain supercomplexes of mitochondria and bacteria. Biochim. Biophys. Acta 1555, 154–159 [DOI] [PubMed] [Google Scholar]
- 77. Deleted in proof. [Google Scholar]
- 78. Denicola-Seoane A., Rubbo H., Prodanov E., Turrens J. F. (1992) Succinate-dependent metabolism in Trypanosoma cruzi epimastigotes. Mol. Biochem. Parasitol. 54, 43–50 [DOI] [PubMed] [Google Scholar]
- 79. Bearne S. L., Wolfenden R. (1995) Glutamate γ-semialdehyde as a natural transition state analogue inhibitor of Escherichia coli glucosamine-6-phosphate synthase. Biochemistry 34, 11515–11520 [DOI] [PubMed] [Google Scholar]
- 80. Lewis M. L., Rowe C. J., Sutherland J. D., Wilson E. J., Wright M. C., Sewald N. (1993) The effect of pH on the solution structure of Δ1-pyrroline-2-carboxylic acid as revealed by NMR and electrospray mass spectroscopy. Bioorg. Med. Chem. Lett. 1193–1196 [Google Scholar]
- 81. Harris T. K., Turner G. J. (2002) Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 53, 85–98 [DOI] [PubMed] [Google Scholar]
- 82. Forte-McRobbie C. M., Pietruszko R. (1986) Purification and characterization of human liver “high Km” aldehyde dehydrogenase and its identification as glutamic γ-semialdehyde dehydrogenase. J. Biol. Chem. 261, 2154–2163 [PubMed] [Google Scholar]
- 83. Van Der Heyden N., Docampo R. (2000) Intracellular pH in mammalian stages of Trypanosoma cruzi is K+-dependent and regulated by H+-ATPases. Mol. Biochem. Parasitol. 105, 237–251 [DOI] [PubMed] [Google Scholar]
- 84. Forte-McRobbie C., Pietruszko R. (1989) Human glutamic-γ-semialdehyde dehydrogenase. Kinetic mechanism. Biochem. J. 261, 935–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kohen A., Cannio R., Bartolucci S., Klinman J. P. (1999) Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature 399, 496–499 [DOI] [PubMed] [Google Scholar]
- 86. Silvius J. R., McElhaney R. N. (1981) Nonlinear Arrhenius plots and the analysis of reaction and motional rates in biological membranes. J. Theor. Biol. 88, 135–152 [DOI] [PubMed] [Google Scholar]
- 87. Truhlar D., Kohen A. (2001) Convex Arrhenius plots and their interpretation. Proc. Natl. Acad. Sci. U.S.A. 98, 848–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tschudi C., Young A. S., Ruben L., Patton C. L., Richards F. F. (1985) Calmodulin genes in trypanosomes are tandemly repeated and produce multiple mRNAs with a common 5′ leader sequence. Proc. Natl. Acad. Sci. U.S.A. 82, 3998–4002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Barrett F. M., Friend W. G. (1975) Differences in the concentration of free amino acids in the haemolymph of adult male and female Rhodnius prolixus. Comp. Biochem. Physiol. B. 52, 427–431 [DOI] [PubMed] [Google Scholar]
- 90. Kollien A. H., Schaub G. A. (2000) The development of Trypanosoma cruzi in triatominae. Parasitol. Today 16, 381–387 [DOI] [PubMed] [Google Scholar]
- 91. O'Daly J. A., Serrano L. E., Rodríguez M. B. (1983) Free amino acid pool and proteolytic enzymes in Trypanosoma cruzi cultured in vitro. Int. J. Parasitol. 13, 433–440 [DOI] [PubMed] [Google Scholar]
- 92. Bayer-Santos E., Aguilar-Bonavides C., Rodrigues S. P., Cordero E. M., Marques A. F., Varela-Ramirez A., Choi H., Yoshida N., da Silveira J. F., Almeida I. C. (2013) Proteomic analysis of Trypanosoma cruzi secretome: characterization of two populations of extracellular vesicles and soluble proteins. J. Proteome Res. 12, 883–897 [DOI] [PubMed] [Google Scholar]
- 93. Chamond N., Grégoire C., Coatnoan N., Rougeot C., Freitas-Junior L. H., da Silveira J. F., Degrave W. M., Minoprio P. (2003) Biochemical characterization of proline racemases from the human protozoan parasite Trypanosoma cruzi and definition of putative protein signatures. J. Biol. Chem. 278, 15484–15494 [DOI] [PubMed] [Google Scholar]
- 94. Bastos I. M., Grellier P., Martins N. F., Cadavid-Restrepo G., de Souza-Ault M. R., Augustyns K., Teixeira A. R., Schrével J., Maigret B., da Silveira J. F., Santana J. M. (2005) Molecular, functional and structural properties of the prolyl oligopeptidase of Trypanosoma cruzi (POP Tc80), which is required for parasite entry into mammalian cells. Biochem. J. 388, 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Homsy J. J., Granger B., Krassner S. M. (1989) Some factors inducing formation of metacyclic stages of Trypanosoma cruzi. J. Protozool. 36, 150–153 [DOI] [PubMed] [Google Scholar]
- 96. Lee I. R., Lui E. Y., Chow E. W., Arras S. D., Morrow C. A., Fraser J. A. (2013) Reactive oxygen species homeostasis and virulence of the fungal pathogen Cryptococcus neoformans requires an intact proline catabolism pathway. Genetics 194, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lijek R. S., Luque S. L., Liu Q., Parker D., Bae T., Weiser J. N. (2012) Protection from the acquisition of Staphylococcus aureus nasal carriage by cross-reactive antibody to a pneumococcal dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 109, 13823–13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dean S., Marchetti R., Kirk K., Matthews K. R. (2009) A surface transporter family conveys the trypanosome differentiation signal. Nature 459, 213–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Balaña-Fouce R., Calvo-Álvarez E., Álvarez-Velilla R., Prada C. F., Pérez-Pertejo Y., Reguera R. M. (2012) Role of trypanosomatid's arginase in polyamine biosynthesis and pathogenesis. Mol. Biochem. Parasitol. 181, 85–93 [DOI] [PubMed] [Google Scholar]
- 100. Avilán L., Gualdrón-López M., Quiñones W., González-González L., Hannaert V., Michels P. A., Concepción J. L. (2011) Enolase: a key player in the metabolism and a probable virulence factor of trypanosomatid parasites-perspectives for its use as a therapeutic target. Enzyme Res. 2011, 932549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schneider A., Bouzaidi-Taili N., Chanez A. L., Bulliard L. (2007) in Methods in Molecular Biology (Leister D., Herrmann J. M., eds) pp. 379–387, Humana Press Inc., Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 102. Jockers-Scherübl M. C., Schirmer R. H., Krauth-Siegel R. L. (1989) Trypanothione reductase from Trypanosoma cruzi. Catalytic properties of the enzyme and inhibition studies with trypanocidal compounds. Eur. J. Biochem. 180, 267–272 [DOI] [PubMed] [Google Scholar]
- 103. Christmas P. B., Turrens J. F. (2000) Separation of NADH-fumarate reductase and succinate dehydrogenase activities in Trypanosoma cruzi. FEMS Microbiol. Lett. 183, 225–228 [DOI] [PubMed] [Google Scholar]
- 104. Fleming G. A., Hagedorn C. H., Granger A. S., Phang J. M. (1984) Pyrroline-5-carboxylate in human plasma. Metabolism 33, 739–742 [DOI] [PubMed] [Google Scholar]
- 105. Semon B. A., Phang J. M. (1991) Accumulation of pyrroline 5-carboxylic acid in conditioned medium of cultured fibroblast: stimulatory effects of serum, insulin, and IGF-1. In Vitro Cell Dev. Biol. 27, 665–669 [DOI] [PubMed] [Google Scholar]
- 106. Danbolt N. C. (2001) Glutamate uptake. Prog. Neurobiol. 65, 1–105 [DOI] [PubMed] [Google Scholar]
- 107. Silber A. M., Rojas R. L., Urias U., Colli W., Alves M. J. (2006) Biochemical characterization of the glutamate transport in Trypanosoma cruzi. Int. J. Parasitol. 36, 157–163 [DOI] [PubMed] [Google Scholar]
- 108. Juan S. M., Segura E. L., Cazzulo J. J. (1978) Purification and some properties of the NADP-linked glutamate dehydrogenase from Trypanosoma cruzi. Int. J. Biochem. 9, 395–400 [DOI] [PubMed] [Google Scholar]
- 109. Cazzulo J. J. (1994) Intermediate metabolism in Trypanosoma cruzi. J. Bioenerg. Biomembr. 26, 157–165 [DOI] [PubMed] [Google Scholar]
- 110. Bochud-Allemann N., Schneider A. (2002) Mitochondrial substrate level phosphorylation is essential for growth of procyclic Trypanosoma brucei. J. Biol. Chem. 277, 32849–32854 [DOI] [PubMed] [Google Scholar]