

Background: Aggregation of α-Syn is associated with PD pathogenesis.
Results: Despite being natively unfolded, a site-specific structure exists in α-Syn that is significantly altered by familial PD-associated E46K, A53T, and A30P mutations.
Conclusion: Altered site-specific structure of the PD-associated mutants may attribute to their different aggregation propensity.
Significance: This study contributes to understanding the relationship between structure and aggregation of α-Syn.
Keywords: α-Synuclein, Amyloid, Fluorescence Anisotropy, Parkinson Disease, Protein Aggregation, Protein Misfolding
Abstract
Human α-synuclein (α-Syn) is a natively unstructured protein whose aggregation into amyloid fibrils is associated with Parkinson disease (PD) pathogenesis. Mutations of α-Syn, E46K, A53T, and A30P, have been linked to the familial form of PD. In vitro aggregation studies suggest that increased propensity to form non-fibrillar oligomers is the shared property of these familial PD-associated mutants. However, the structural basis of the altered aggregation propensities of these PD-associated mutants is not yet clear. To understand this, we studied the site-specific structural dynamics of wild type (WT) α-Syn and its three PD mutants (A53T, E46K, and A30P). Tryptophan (Trp) was substituted at the N terminus, central hydrophobic region, and C terminus of all α-Syns. Using various biophysical techniques including time-resolved fluorescence studies, we show that irrespective of similar secondary structure and early oligomerization propensities, familial PD-associated mutations alter the site-specific microenvironment, solvent exposure, and conformational flexibility of the protein. Our results further show that the common structural feature of the three PD-associated mutants is more compact and rigid sites at their N and C termini compared with WT α-Syn that may facilitate the formation of a partially folded intermediate that eventually leads to their increased oligomerization propensities.
Introduction
α-Synuclein (α-Syn)4 is a 140-amino acid protein abundantly expressed in human brain (1). α-Syn is mostly localized in the nucleus and presynaptic terminals of neurons with some fractions existing in association with the synaptic vesicles (2–4). Although the physiological function of α-Syn has not been fully determined, it has been suggested to play a role in neurotransmitter (dopamine) release and regulation of the reserve pool of synaptic vesicles at nerve terminals (5, 6). Aggregation of α-Syn has been pathogenically linked to Parkinson disease (PD), which is a very common neurodegenerative disease (7–9) in aged populations. The presence of intraneuronal inclusions, Lewy bodies, mainly composed of fibrillar aggregates of α-Syn in the patient's brain is the most important pathological hallmark of PD (7, 8, 10). Several genetic, neuronal cell culture studies and animal models of PD suggest that α-Syn plays a central role in PD pathogenesis (11–15). The protein is intrinsically disordered in vitro under physiological conditions and has the tendency to self-assemble into amyloid fibrils upon incubation for a longer time (16, 17). Several studies have suggested that the oligomeric intermediates in the fibril formation pathway of α-Syn are the most potent neurotoxic species responsible for PD pathogenesis (18, 19). On the contrary, many recent reports have described that α-Syn fibrils are toxic (20), and exogenously added α-Syn fibrils can seed aggregation of endogenous α-Syn into Lewy body-like inclusions in cultured neuronal cells (21, 22), suggesting a critical role for α-Syn fibrils in PD pathogenesis. The mechanism of neurodegeneration in PD and the form of α-Syn (fibril/oligomers) that is responsible for toxicity are still not clear. Hence, it is very important to study and understand the entire aggregation pathway of α-Syn and the factors that affect its aggregation.
The protein α-Syn has been widely studied in terms of its structural aspects (23). The primary structure of α-Syn can be divided into three main regions: the N-terminal region (residues 1–60), middle region (residues 61–95), and C-terminal region (residues 96–140) (1). The N-terminal region has seven loosely repeated motifs (11 residues) that extend to the middle hydrophobic region (1). α-Syn can bind to synthetic phospholipid vesicles through its N terminus, and these loosely repeated motifs are involved in forming α-helices in association with the vesicle membrane (24, 25). It has also been reported that additional N-terminal repeats in the sequence of α-Syn disfavor β-sheet structure formation (26). The middle region is the most hydrophobic part of α-Syn and is known as non-amyloid β-component (NAC) of Alzheimer disease plaques (27). The NAC region is highly amyloidogenic, and this region drives the fibrillation of α-Syn. The 12-residue stretch (71–82) in the middle of NAC is reported to be necessary and sufficient for α-Syn fibrillation (28). A homologue of α-Syn, β-Syn, which lacks this 12-residue stretch, does not have the ability to aggregate into amyloid fibrils (28). The C-terminal region of α-Syn is rich in acidic amino acid residues, making it negatively charged (1). The C terminus of α-Syn has been suggested to regulate its fibrillation as C-terminal truncation results in a faster fibrillation rate of the protein (29). Moreover, the existence of long range intramolecular interactions between the C terminus and stretches of residues in the middle NAC region as well as in the N terminus of α-Syn has been reported (30, 31). α-Syn thus exists as an ensemble of conformers that are on average structurally more compact than expected for a completely unfolded protein (30, 31).
Several genetic studies have revealed missense mutations E46K, A53T, and A30P as well as the recently discovered H50Q, G51D, and A53E and locus duplication and triplication of α-Syn gene linked to rare familial forms of PD (32, 33). The effect of three well known familial PD-associated mutations, E46K, A53T, and A30P, on the structure and aggregation properties of α-Syn has been largely studied (34–37). These studies suggest that none of the three PD mutations (A53T, E46K, and A30P) change the overall conformation of the protein. These mutant proteins are also natively unfolded similar to wild type (WT) α-Syn (34, 35). The in vitro aggregation studies have shown that the mutant A30P has an enhanced rate of non-fibrillar oligomer formation but a slower fibril formation rate compared with WT α-Syn (34, 38), whereas the other two mutants, E46K and A53T, have a faster fibril forming rate compared with WT α-Syn (35–37, 39). It has been suggested that increased propensity to form non-fibrillar oligomers is the shared property of these familial PD-associated mutants of α-Syn (38, 40). How a single missense mutation in an intrinsically disordered protein can drastically affect its aggregation properties is an important question to be addressed. Several studies using various biophysical techniques including nuclear magnetic resonance (NMR) spectroscopy, time-resolved fluorescence energy transfer measurements, single molecule force spectroscopy, and molecular dynamics simulation have suggested that the PD-associated mutations A53T, E46K, and A30P alter the long range intramolecular interactions, structural compactness, and conformational equilibrium of the protein (41–47). However, the impact of these mutations on the structural properties of α-Syn is highly debated in the current literature (41, 42). Despite all these studies, the structural basis for the increased oligomerization propensities of these PD-associated mutants is not yet clearly understood. Two possibilities could give rise to the altered aggregation properties: first, a difference in early stage oligomerization, and second, a site-specific structural difference between WT and PD mutant α-Syns that could eventually lead to their increased oligomerization rate. In the current study, we address both these possibilities. We studied the initial oligomer distribution of different α-Syns using photo-induced cross-linking of unmodified proteins (PICUP) and their site-specific structural dynamics using time-resolved fluorescence. Our results show that although α-Syn and its familial PD-associated mutants have the same overall secondary structure and initial oligomer distribution pattern, the site-specific structural dynamics is significantly altered in the PD-associated mutants, and this difference could be correlated to their increased oligomerization propensities. Our results shed light on the unique structural properties of α-Syn and its PD-associated mutants, which is useful in understanding their different aggregation behavior. Furthermore, results from our current study together with our previously published work on site-specific structural differences of the fibrils of α-Syn and its familial PD-associated mutants (48) show that different site-specific transformation of dynamics and microenvironment occur for α-Syn and its different PD mutants during fibril formation.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Most of the chemicals were procured from Sigma. For all the experiments, the water used was double distilled as well as deionized by a Milli-Q water purification system (Millipore Corp., Bedford, MA).
Site-directed Mutagenesis
Site-directed mutagenesis was performed to create the tryptophan (Trp) mutants of α-Syn using polymerase chain reaction (PCR) and subsequent DpnI digestion for selection of mutants (49). The template DNA used for the PCR reaction was a construct of pRK172 vector containing insert of α-Syn (WT, A30P, E46K, or A53T). The pRK172 construct of α-Syn was a kind gift from Prof. Roland Riek, ETH Zurich, Switzerland (50). The protocol used for performing site-directed mutagenesis was essentially the same as that reported previously (48). The mutations were confirmed by DNA sequencing.
Protein Expression and Purification
All α-Syns were expressed in Escherichia coli BL21(DE3) strain and purified based on the standard protocol (51) with slight modification (52).
Low Molecular Weight Sample Preparation and Fibrillization Assay of All α-Syns
For preparing low molecular weight forms of all α-Syns, the purified proteins (lyophilized) were dissolved in buffer (20 mm Gly-NaOH, pH 7.4, 0.01% sodium azide) at 40 mg/ml concentration by adding a few microliters of 2 mm NaOH. The final pH of each protein solution was adjusted to 7.4 by adding a few microliters of 1 mm HCl. A micro pH meter (S20 SevenEasy, Mettler Toledo, Switzerland) was used for measuring pH of the protein solutions. All the protein solutions were then centrifuged at 13,000 × g for 30 min at 4 °C to remove any insoluble particles. The supernatants were dialyzed in a mini dialysis unit of 10-kDa cutoff (Slide-A-Lyzer MINI Dialysis Devices, Pierce) overnight against the same buffer and then filtered with 100-kDa molecular mass-cutoff filters (Centricon YM-100, Millipore Corp.). The resulting low molecular weight solutions were clear and free of any larger aggregates. To determine the protein concentration in low molecular weight solutions, absorbance at 280 nm was measured using a Jasco (V-650) spectrophotometer. The molar absorptivity (ϵ) used here is 5960 m−1 cm−1 for WT, E46K, A30P, and A53T α-Syn and 11,460 m−1 cm−1 for their Trp-substituted variants. Fibrillation assay was carried out according to protocols published previously (53). Briefly, microcentrifuge tubes (1.5 ml) containing 500 μl of low molecular weight solutions of each α-Syn at 300 μm concentrations were incubated at 37 °C in a rotating mixer (EchoTherm model RT11, Torrey Pines Scientific) at 50 rpm. At regular time intervals during incubation, aliquots of protein solution were diluted to 5 μm in 150 μl of Gly-NaOH buffer, pH 7.4, 0.01% sodium azide. 1.5 μl of 1 mm thioflavin T (ThT) solution (prepared in Tris-HCl buffer, pH 8.0, 0.01% sodium azide) was added to the diluted protein solution. ThT fluorescence was recorded using a Horiba-Jobin Yvon (Fluomax4) spectrofluorometer in a rectangular 10-mm quartz microcuvette (Hellma, Forest Hills, NY) with excitation at 450 nm and emission in the range of 460–500 nm. The slit width for both excitation and emission was kept at 5 nm. ThT fluorescence obtained at 480 nm was plotted for all proteins against incubation time.
Transmission Electron Microscopy
The α-Syn aggregation stocks were diluted in distilled water to a final concentration of ∼40 μm and spotted on a carbon-coated Formvar grid (Electron Microscopy Sciences, Fort Washington, PA). The sample was wiped gently using a filter paper after 5-min incubation, and the grid was given a quick water wash. After this, negative staining was done wherein 1% (w/v) aqueous uranyl formate solution was applied to the grid and incubated for 2 min. The stain was then gently removed by wiping. The grids were air-dried for 5 min and then subjected to transmission electron microscopy. Electron microscopy of the samples was carried out using a JEOL JEM-2100 microscope at 120 kV with 6000× magnification.
Circular Dichroism (CD) Spectroscopy
5 μm low molecular weight solution of each of WT α-Syn and its PD-associated mutants was used for CD spectroscopy. The CD spectra were measured by a CD polarimeter (Jasco 810) by putting the solutions into a quartz cell (Hellma) with 0.1 cm path length. All spectra were recorded at 25 °C in the wavelength range of 190–260 nm. With each protein sample, three independent experiments were performed. Smoothing and buffer spectrum subtraction were used for raw data processing according to the manufacturer's instructions.
Cross-linking of Proteins
A PICUP experiment was carried out according to the protocol established by Bitan et al. (54). Briefly, 18 μl of 20 μm freshly prepared low molecular weight form of α-Syn and its mutants were taken in clear, thin walled 0.2-ml PCR tubes. 1 μl of 3 mm tris(bipyridine)ruthenium(II) chloride and 1 μl of 60 mm ammonium persulfate were added to the low molecular weight solution. The reaction mixture was irradiated with light for 1 s (controlled by a camera shutter). The reaction mixture was quenched immediately by addition of 5 μl of 5× SDS-PAGE loading dye containing 5% β-mercaptoethanol and then boiled in a water bath at 95 °C for 5 min. The cross-linked products were separated and visualized by SDS-PAGE followed by silver staining. 4 μl of reaction products of each protein and a standard protein ladder were added to the wells of a polyacrylamide gel. Silver staining was performed using a silver staining kit from Invitrogen (SilverXpress). The cross-linking experiment was performed in triplicates to ensure the reproducibility of results.
Steady-state Fluorescence Studies
6 μm concentration of each low molecular weight sample was used for steady-state Trp fluorescence studies. The samples were put in a rectangular 10-mm quartz microcuvette (Hellma), and Trp fluorescence spectra were recorded using a Fluoromax4 spectrofluorometer with excitation set at 295 nm and emission monitored in the range of 305–500 nm. The excitation as well as emission slit width was set to 6 nm.
Time-resolved Fluorescence Intensity Decay Kinetics
The time-resolved fluorescence intensity decay experiments were carried out using a rhodamine 6G dye laser (Spectra Physics, Mountain View, CA) pumped by a passively mode-locked frequency-doubled neodymium-doped yttrium aluminum garnet laser (Vanguard, Spectra Physics) and a time-correlated single photon counting setup coupled to a microchannel plate photomultiplier (model R2809u, Hamamatsu Corp). Pulses (1-ps duration) of 590 nm radiation from the rhodamine 6G dye laser were frequency-doubled to 295 nm by using a frequency doubler (Spectra Physics). For obtaining the instrument response function (IRF), a dilute colloidal suspension of dried nondairy coffee whitener was excited at 295 nm, and the emission was collected at the same wavelength (295 nm) with the emission polarizer oriented at the “magic” angle of 54.7° with respect to excitation polarizer. The width (full width at half-maximum) of IRF was ∼40 ps. A 100 μm concentration of each low molecular weight sample was excited at 295 nm at a pulse repetition rate of 4 MHz. The emission for each protein sample was measured at their emission maxima (λmax) (354 nm for V71W and V3W, 356 nm for A124W, and 360 nm for A140W Trp-substituted variants of WT and PD mutant α-Syns) as determined from steady-state fluorescence. Decays with peak counts of 10,000 were collected for analysis for discrete lifetimes, and decays with peak counts of 20,000 were collected for analysis using the maximum entropy method (MEM) (the detailed method is discussed later under “Analysis of Fluorescence Intensity Decay by MEM”) with the emission polarizer oriented at the magic angle (54.7°) with respect to the excitation polarizer. All the decays were deconvoluted with respect to the IRF.
Time-resolved Fluorescence Anisotropy Decay Kinetics
For time-resolved anisotropy measurements, the low molecular weight sample volume, concentration, experimental setup, and parameters used were the same as those used for time-resolved fluorescence intensity decay kinetics experiments. Peak counts of 10,000 were collected for both parallel (emission polarizer oriented at 0° with respect to excitation polarizer) and perpendicular (emission polarizer oriented at 90° with respect to excitation polarizer) fluorescence intensity measurements. I‖ and I⊥ represent parallel and perpendicular fluorescence intensity, respectively. All the decays were deconvoluted with respect to the IRF.
Analysis of Fluorescence Intensity Decay for Discrete Lifetimes
The time-resolved fluorescence intensity decay curves were analyzed for discrete lifetimes using a nonlinear least square iterative deconvolution method based on the Levenberg-Marquardt algorithm (55). The time-resolved intensity decays were expressed as a sum of three exponential function as follows.
where I(t) is the fluorescence intensity collected with the emission polarizer oriented at the magic angle (54.7°) with respect to the excitation polarizer at time t and αi is the amplitude associated with the fluorescence lifetime τi such that ∑αi = 1. The mean fluorescence lifetime was calculated as τm = ∑αiτi.
The parameters obtained from the fitting of time-resolved fluorescence intensity decays with a three-exponential function (Equation 1) are shown in Tables 1 and 2. The goodness of fit was assessed from the reduced χ2 values (0.9–1.2) and from the randomness of the residuals obtained from analysis.
TABLE 1.
Parameters associated with fluorescence intensity decays
The time-resolved fluorescence intensity decays of all low molecular weight (LMW) samples fitted satisfactorily to a three-exponential function (Equation 1). The fluorescence lifetimes (τ) and their amplitudes (α) as well as the mean fluorescence lifetime (τm) values derived from the fitting are shown. The error associated with fluorescence lifetime measurement and its amplitude is ∼5–10%. The error associated with τm is <5%.
| LMW sample | τ1 (ns) (α1) | τ2 (ns) (α2) | τ3 (ns) (α3) | τm (ns) |
|---|---|---|---|---|
| [V3W]WT | 0.63 (0.31) | 1.93 (0.48) | 4.1 (0.21) | 1.98 |
| [V71W]WT | 0.42 (0.26) | 1.29 (0.37) | 3.72 (0.37) | 1.96 |
| [A124W]WT | 0.22 (0.18) | 1.43 (0.4) | 3.7 (0.42) | 2.16 |
| [A140W]WT | 0.47 (0.12) | 1.52 (0.20) | 4.38 (0.68) | 3.34 |
| [V3W]E46K | 0.19 (0.37) | 1.36 (0.23) | 5.39 (0.4) | 2.54 |
| [V71W]E46K | 0.51 (0.32) | 1.84 (0.40) | 4.25 (0.28) | 2.09 |
| [A124W]E46K | 0.3 (0.17) | 1.48 (0.37) | 3.78 (0.46) | 2.34 |
| [A140W]E46K | 0.59 (0.19) | 2.28 (0.27) | 4.98 (0.54) | 3.42 |
| [V3W]A53T | 0.34 (0.20) | 1.62 (0.53) | 3.99 (0.27) | 2.00 |
| [V71W]A53T | 0.39 (0.26) | 1.42 (0.40) | 3.71 (0.34) | 1.93 |
| [A124W]A53T | 0.4 (0.22) | 1.69 (0.39) | 3.85 (0.39) | 2.25 |
| [A140W]A53T | 0.10 (0.17) | 0.88 (0.22) | 4.33 (0.61) | 2.85 |
| [V3W]A30P | 0.30 (0.18) | 1.32 (0.45) | 3.53 (0.37) | 1.95 |
| [V71W]A30P | 0.60 (0.37) | 1.82 (0.35) | 3.92 (0.28) | 1.96 |
| [A124W]A30P | 0.41 (0.18) | 1.51 (0.39) | 3.64 (0.43) | 2.23 |
| [A140W]A30P | 0.42 (0.17) | 1.90 (0.24) | 4.56 (0.59) | 3.22 |
TABLE 2.
Parameters associated with fluorescence intensity decays
The time-resolved fluorescence intensity decays of all the denatured samples fitted satisfactorily to a three-exponential function (Equation 1). The fluorescence lifetimes (τ) and their amplitudes (α) as well as the mean fluorescence lifetime (τm) values derived from the fitting are shown. The error associated with fluorescence lifetime measurement and its amplitude is ∼5–10%. The error associated with τm is <5%.
| Sample | τ1 (ns) (α1) | τ2 (ns) (α2) | τ3 (ns) (α3) | τm (ns) |
|---|---|---|---|---|
| [V3W]WT + 6 m GdnHCl | 0.25 (0.19) | 0.98 (0.28) | 2.48 (0.53) | 1.64 |
| [V71W]WT + 6 m GdnHCl | 0.32 (0.19) | 1.10 (0.36) | 3.53 (0.45) | 2.05 |
| [A124W]WT + 6 m GdnHCl | 0.26 (0.15) | 1.18 (0.36) | 3.17 (0.49) | 2.02 |
| [A140W]WT + 6 m GdnHCl | 0.21 (0.12) | 1.42 (0.19) | 4.99 (0.69) | 3.74 |
| [V3W]E46K + 6 m GdnHCl | 0.21 (0.54) | 1.2 (0.22) | 3.8 (0.24) | 1.29 |
| [V71W]E46K + 6 m GdnHCl | 0.34 (0.33) | 1.54 (0.33) | 4.02 (0.34) | 1.99 |
| [A124W]E46K + 6 m GdnHCl | 0.24 (0.2) | 1.39 (0.36) | 3.33 (0.44) | 2.01 |
| [A140W]E46K + 6 m GdnHCl | 0.65 (0.26) | 3.42 (0.34) | 5.71 (0.4) | 3.62 |
| [V3W]A53T + 6 m GdnHCl | 0.29 (0.29) | 1.49 (0.4) | 3 (0.31) | 1.61 |
| [V71W]A53T + 6 m GdnHCl | 0.34 (0.33) | 1.43 (0.33) | 3.94 (0.34) | 1.92 |
| [A124W]A53T + 6 m GdnHCl | 0.34 (0.21) | 1.39 (0.34) | 3.28 (0.45) | 2.02 |
| [A140W]A53T + 6 m GdnHCl | 0.46 (0.24) | 3.19 (0.28) | 5.59 (0.48) | 3.69 |
| [V3W]A30P + 6 m GdnHCl | 0.21 (0.22) | 1.23 (0.36) | 2.8 (0.42) | 1.67 |
| [V71W]A30P + 6 m GdnHCl | 0.33 (0.33) | 1.41 (0.33) | 3.93 (0.34) | 1.91 |
| [A124W]A30P + 6 m GdnHCl | 0.19 (0.12) | 1.1 (0.37) | 3.19 (0.51) | 2.06 |
| [A140W]A30P + 6 m GdnHCl | 0.48 (0.24) | 3.18 (0.28) | 5.56 (0.48) | 3.67 |
Analysis of Fluorescence Intensity Decay by MEM
The MEM analysis of time-resolved fluorescence intensity decays was performed as described in detail previously (48). Briefly, the MEM analysis considers the fluorescence intensity decay to arise from a distribution of many discrete lifetime values in the considered range (10 ps to 10 ns). Initially, the analysis assigns equal probability (amplitude) to all the lifetime values, and in each successive iteration, this lifetime distribution is modified to minimize the χ2 and maximize the Shannon-Jaynes entropy function (Equation 2):
where S is the entropy and αi is the probability (amplitude) of the ith lifetime. If several lifetime distributions have similar χ2 values, the one having the maximum value of entropy (S) is accepted. For all of our MEM analyses, the χ2 values were in the range of 0.9–1.2.
Analysis of Fluorescence Anisotropy Decay Kinetics
The time-resolved anisotropy decay curves were derived using Equation 3.
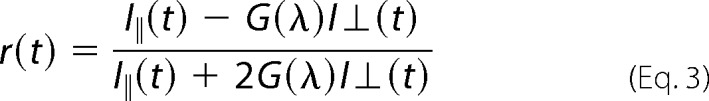 |
where I‖(t) and I⊥(t) are the experimentally obtained parallel and perpendicular fluorescence intensity, respectively, at time t; r(t) is the time-dependent anisotropy; and G(λ) is the geometry factor at the wavelength of emission (λ). The value of G(λ) for the optics for measuring emission of each protein sample was calculated independently using a 50 μm solution of N-acetyltryptophanamide.
Analysis of the time-resolved fluorescence anisotropy decays was done by globally fitting I‖(t) and I⊥(t) as follows.
where r0 is the anisotropy in the absence of any rotational diffusion and βi is the amplitude associated with the ith rotational correlation times φi such that ∑βi = 1. Two rotational correlation times (φ1 and φ2) and the amplitudes associated with each of them (β1 and β2) were derived from fits of the fluorescence anisotropy decays to Equations 4–6 and are represented in Tables 3 and 4. The mean rotational correlation time was calculated as φm = ∑βiφi.
TABLE 3.
Parameters associated with fluorescence anisotropy decays
The time-resolved fluorescence anisotropy decays of all low molecular weight (LMW) samples fitted satisfactorily to a two-exponential function (Equations 4–6). The rotational correlation times (φ) and their amplitudes (β) derived from the fitting are shown. The error associated with rotational correlation time and its amplitude is ∼10–20%.
| LMW sample | φ1 (ns) (β1) | φ2 (ns) (β2) |
|---|---|---|
| [V3W]WT | 0.36 (0.67) | 4.5 (0.33) |
| [V71W]WT | 0.25 (0.56) | 3.12 (0.44) |
| [A124W]WT | 0.21 (0.45) | 2.48 (0.55) |
| [A140W]WT | 0.13 (0.72) | 1.70 (0.28) |
| [V3W]E46K | 1.3 (0.1) | 6.1 (0.9) |
| [V71W]E46K | 0.31 (0.66) | 3.80 (0.34) |
| [A124W]E46K | 1.3 (0.1) | 6.1 (0.9) |
| [A140W]E46K | 0.57 (0.60) | >10 (0.40) |
| [V3W]A53T | 0.36 (0.64) | 5.4 (0.36) |
| [V71W]A53T | 0.29 (0.60) | 3.70 (0.40) |
| [A124W]A53T | 0.5 (0.46) | 6.83 (0.54) |
| [A140W]A53T | 0.20 (0.66) | 3.36 (0.34) |
| [V3W]A30P | 0.36 (0.65) | 4.5 (0.35) |
| [V71W]A30P | 0.27 (0.58) | 3.70 (0.42) |
| [A124W]A30P | 0.35 (0.57) | 3.2 (0.43) |
| [A140W]A30P | 0.35 (0.70) | 4.60 (0.30) |
TABLE 4.
Parameters associated with fluorescence anisotropy decays
The time-resolved fluorescence anisotropy decays of all denatured samples fitted satisfactorily to a two-exponential function (Equations 4–6). The rotational correlation times (φ), their amplitudes (β), and mean rotational correlation times (φm) derived from the fitting are shown. The error associated with rotational correlation time and its amplitude is ∼10–20%.
| Sample | φ1 (ns) (β1) | φ2 (ns) (β2) | φm (ns) |
|---|---|---|---|
| [V3W]WT + 6 m GdnHCl | 0.12 (0.47) | 1.05 (0.53) | 0.613 |
| [V71W]WT + 6 m GdnHCl | 0.15 (0.55) | 1.72 (0.45) | 0.86 |
| [A124W]WT + 6 m GdnHCl | 0.14 (0.38) | 1.825 (0.62) | 1.18 |
| [A140W]WT + 6 m GdnHCl | 0.13 (0.6) | 1.30 (0.4) | 0.6 |
| [V3W]E46K + 6 m GdnHCl | 0.41 (0.45) | 2.2 (0.55) | 1.4 |
| [V71W]E46K + 6 m GdnHCl | 0.16 (0.51) | 1.8 (0.49) | 0.96 |
| [A124W]E46K + 6 m GdnHCl | 0.17 (0.4) | 1.9 (0.6) | 1.21 |
| [A140W]E46K + 6 m GdnHCl | 0.12 (0.55) | 1.3 (0.45) | 0.65 |
| [V3W]A53T + 6 m GdnHCl | 0.14 (0.33) | 1 (0.66) | 0.71 |
| [V71W]A53T + 6 m GdnHCl | 0.12 (0.53) | 1.77 (0.47) | 0.9 |
| [A124W]A53T + 6 m GdnHCl | 0.17 (0.42) | 1.86 (0.58) | 1.15 |
| [A140W]A53T + 6 m GdnHCl | 0.15 (0.54) | 1.3 (0.46) | 0.68 |
| [V3W]A30P + 6 m GdnHCl | 0.17 (0.55) | 1.15 (0.45) | 0.61 |
| [V71W]A30P + 6 m GdnHCl | 0.12 (0.45) | 1.7 (0.55) | 0.99 |
| [A124W]A30P + 6 m GdnHCl | 0.17 (0.4) | 1.9 (0.6) | 1.21 |
| [A140W]A30P + 6 m GdnHCl | 0.12 (0.46) | 1.04 (0.54) | 0.62 |
For some low molecular weight samples, a limiting value of φ2 >10 ns (Table 3) is due to the finite time window offered by the fluorescence lifetime of Trp (τm ∼ 1.2–3.5 ns). The r0 value of 0.25 was calculated by an independent experiment using low molecular weight form of one of the Trp mutant α-Syns in 70% glycerol. The r0 value 0.25 with a window of 0.01 was used during analysis of anisotropy decays. The goodness of fit was assessed from the reduced χ2 values (1.2–1.7) as well as from the randomness of the residuals.
Dynamic Quenching of Fluorescence Using Acrylamide as Quencher
For the dynamic fluorescence quenching experiments, the low molecular weight sample volume, concentration, experimental setup, and parameters used were the same as those used for time-resolved fluorescence intensity decay kinetics experiments. 1 μl of acrylamide (from a 5 m stock solution) was subsequently added to the same low molecular weight solution and mixed thoroughly. After each addition of acrylamide, fluorescence intensity decay with peak counts of 10,000 was collected with the emission polarizer oriented at the magic angle (54.7°) with respect to the excitation polarizer. For each decay, the mean fluorescence lifetime (τm) was calculated by deconvolution with respect to the IRF and fitting to a three-exponential function as described under “Analysis of Fluorescence Intensity Decay for Discrete Lifetimes.” The concentration of acrylamide in the resulting low molecular weight solution after each addition was calculated. τ0/τ was then plotted against the increasing concentrations of acrylamide. The data points were fitted with the Stern-Volmer equation (Equation 7), which gives a straight line (56).
where τ0 is the mean fluorescence lifetime of the low molecular weight sample in the absence of acrylamide, τ is the mean fluorescence lifetime of low molecular weight sample at each acrylamide concentration, kq is the bimolecular rate constant for dynamic quenching, and [Q] is the acrylamide concentration (in m). The extent of solvent exposure was estimated by calculating kq from Equation 7.
Guanidine Hydrochloride (GdnHCl) Denaturation of Trp-substituted α-Syn Proteins
Denaturation was performed by adding GdnHCl from an 8 m stock solution (Pierce, Thermo Scientific) to a final concentration of 6 m in low molecular weight solutions of each of the Trp mutants V3W, V71W, A124W, and A140W of WT α-Syn and its PD-associated mutants. The denatured proteins were then subjected to time-resolved fluorescence studies. The parameters obtained from the fitting of time-resolved fluorescence intensity and anisotropy decays of all the denatured samples are shown in Tables 2 and 4, respectively.
RESULTS
Site-specific Structural Dynamics of α-Syn in Its Soluble Form
In the present work, we focused on the starting point of the α-Syn aggregation process. We studied the site-specific structural dynamics of freshly prepared soluble (low molecular weight) form of WT α-Syn. The fluorescence properties of Trp are extremely sensitive to its microenvironment (57, 58) and are widely used to monitor changes in local structure and dynamics of proteins (48, 59–61). Trp and Tyr fluorescence have been previously used effectively to probe the structural dynamics of several proteins like amyloid β (Aβ) and α-Syn during their aggregation (59, 62, 63). Therefore, single Trp-substituted mutants of α-Syn were studied using multiple time-resolved fluorescence techniques. Trp residues were substituted at specific sites in each of the three regions (N-terminal, C-terminal, and middle NAC region) of WT α-Syn using site-directed mutagenesis. Fig. 1A shows the amino acid sequence of WT α-Syn and its four Trp-substituted mutants designed for studying the site-specific structure of α-Syn. The amino acid residue valine (Val) at positions 3 and 71 of α-Syn was chosen for substitution with Trp because both Val and Trp have bulky and hydrophobic side chains. Therefore, Trp substitution in these positions is not expected to have much effect on the overall hydrophobicity and net charge of the protein. Moreover, substitution of Val with Trp has been previously used effectively to study site-specific conformational changes in Aβ protein (associated with Alzheimer disease) during its aggregation (64). The C-terminal substitution A124W and A140W are also reported not to change the structural properties of α-Syn and have been used previously for studying the structural features of α-Syn oligomers (59). Here, Trp fluorescence was used as a probe to study different aspects of site-specific structural dynamics of α-Syn. The microenvironment experienced by Trp was studied using steady-state fluorescence as well as time-resolved fluorescence intensity decay kinetics. Site-specific solvent accessibility and conformational flexibility were studied using dynamic fluorescence quenching and time-resolved anisotropy decay kinetics, respectively.
FIGURE 1.

Site-specific structural dynamics of α-Syn. A, amino acid sequences of the protein α-Syn and its four Trp-substituted mutants designed for the study. The substituted Trp residues are shown in blue, and the rectangular boxes indicate the mutation site in the sequences. To delineate the site-specific structural dynamics of α-Syn, freshly prepared low molecular weight solutions of Trp3, Trp71, Trp124, and Trp140 α-Syns were studied using different fluorescence methods. B, steady-state fluorescence spectra showing higher fluorescence intensity of Trp140 compared with Trp3, Trp71, and Trp124. C, time-resolved fluorescence intensity decay kinetics fitted to a three-exponential function (smooth lines) showing slower decay of the fluorescence intensity of Trp140 compared with Trp3, Trp71, and Trp124. D, mean fluorescence lifetime (τm = Σαiτi) values calculated by fitting the fluorescence intensity decays using a three-exponential function represented as a bar diagram (mean ± S.D., n = 3) indicating a different microenvironment experienced by Trp140, Trp124, Trp3, and Trp71. E, fluorescence lifetime distributions obtained by analyzing the time-resolved fluorescence intensity decays using MEM showing that Trp3, Trp71, and Trp124 are conformationally more heterogeneous compared with Trp140. F, Stern-Volmer plots derived from dynamic quenching of fluorescence using lifetime measurements showing a higher slope for Trp140 followed by Trp3, Trp71, and Trp124. G, the bimolecular rate constants for dynamic quenching (kq) calculated from the Stern-Volmer plots and represented as a bar diagram (mean ± S.D., n = 2) indicating that Trp140 is more solvent-exposed followed by Trp3, Trp71, and Trp124. I, time-resolved fluorescence anisotropy decay kinetics fitted to a two-exponential function (smooth lines) showing faster decay of fluorescence anisotropy of Trp140 compared with Trp3, Trp71, and Trp124. J, rotational correlation time associated with the local motion of Trp (φ1) calculated by fitting the fluorescence anisotropy decays and represented as a bar diagram (mean ± S.D., n = 2) indicating that Trp140 is conformationally more flexible compared with Trp3 and Trp71. The statistical significance was calculated by one-way analysis of variance followed by Newman-Keuls multiple comparison post hoc test: *, p < 0.05; **, p < 0.01; NS, not significant, p > 0.05. Error bars represent S.D. AU, arbitrary units.
The steady-state Trp fluorescence measurement of different Trp-substituted α-Syns revealed that all four positions in the low molecular weight state of α-Syn experience a polar environment with fluorescence emission maxima (λmax) between 350 and 360 nm (Fig. 1B). Trp3 and Trp71 have similar fluorescence emission spectra where λmax is ∼354 nm and intensity is ∼5 × 105 arbitrary units. Trp124 has a slightly red-shifted fluorescence spectrum (λmax ∼ 356 nm), and intensity is ∼5 × 105 arbitrary units. Interestingly, Trp140 has a fluorescence spectrum where the λmax was further red-shifted to ∼360 nm, suggesting that Trp140 experiences a more polar microenvironment followed by Trp124, Trp3, and Trp71. The fluorescence intensity of Trp140 was also higher (∼1 × 106 arbitrary units) compared with Trp3, Trp71, and Trp124, suggesting less Trp quenching at position 140. Different amino acid sequences and side chains near the different Trp sites may contribute to the different fluorescence intensities observed (Fig. 1B). The site-specific microenvironment was further studied using time-resolved fluorescence intensity decay kinetics. The intensity decay kinetics of Trp3, Trp71, Trp124, and Trp140 α-Syns was fitted satisfactorily to a three-exponential function (Fig. 1C), a behavior typical of Trp-containing proteins (57). Mean lifetime (τm = Σαiτi) values were derived from the fitting of intensity decays (Table 1). Several processes can modulate the fluorescence lifetime of Trp including interaction with solvent molecules, peptide bonds, and fluorescence quenching by the side chains of nearby amino acids in the protein. Thus, the interpretation of changes in fluorescence lifetime is often ambiguous (58, 61, 65). Our data show that the fluorescence intensities of Trp3 and Trp71 decayed faster (with shorter τm) followed by those of Trp124 and Trp140 (Fig. 1, C and D). This could be due to either the different structural dynamics at the four positions and/or different amino acid residues near these sites in the primary sequence of the α-Syn. To extend our understanding on the site-specific microenvironment of WT α-Syn, we analyzed the time-resolved fluorescence intensity decay kinetics using MEM (Fig. 1E). The MEM analysis provides a distribution of fluorescence lifetimes, which gives an idea of the conformational heterogeneity of the fluorescent probe (66). Our MEM results show dual peaks for Trp71, Trp124, and Trp140 α-Syns. Trp3, Trp71, and Trp124 α-Syns have relatively broadened lifetime distributions as compared with that of Trp140 α-Syn (Fig. 1E). This clearly indicates that Trp3, Trp71, and Trp124 are conformationally more heterogeneous as compared with Trp140 in the low molecular weight state of α-Syn. Moreover, the major peak of Trp140 is shifted to longer lifetime values as compared with those of Trp3, Trp71, and Trp124, consistent with our results from discrete analysis of the intensity decays (τm of Trp140 was longer than that of Trp3, Trp71, and Trp124). Therefore, the above results indicate that the structural dynamics and microenvironment of positions 3, 71, 124, and 140 are indeed different in soluble α-Syn.
The solvent accessibility of α-Syn at the four different Trp positions was studied by dynamic quenching of fluorescence using acrylamide as quencher (Q). The quenching experiment done using fluorescence lifetime measurements yielded linear plots of τ0/τ versus [Q] (Stern-Volmer plots) (Fig. 1F). The bimolecular quenching rate constant (kq) derived from the Stern-Volmer plots is an estimate of the solvent accessibility of Trp in the protein (56). A larger kq value indicates a greater extent of solvent exposure and vice versa. The kq values are represented as a bar diagram to compare the solvent accessibility at the four different positions (Trp3, Trp71, Trp124, and Trp140) of α-Syn (Fig. 1G). The data suggest that all four positions are solvent-exposed in the low molecular weight state of α-Syn having kq values of the same order of magnitude as that of N-acetyltryptophanamide, which was used as a standard reference (kq ∼ 11 × 109 m−1 s−1). The kq values represented in Fig. 1G suggest that Trp124 is relatively more solvent-protected followed by Trp71, Trp3, and Trp140. This indicates that α-Syn is not a completely unfolded protein as reported previously (30, 31). The protein has some residual structure, which is responsible for the differential solvent exposure at the different sites of the protein.
The site-specific conformational flexibility of the low molecular weight state of α-Syn was studied using time-resolved fluorescence anisotropy decay kinetics. For all Trp3, Trp71, Trp124, and Trp140 α-Syns, the fluorescence anisotropy decayed in two phases (Fig. 1H). The anisotropy decays of all α-Syns were fitted satisfactorily to a sum of two exponential function. This model assumes a population having uniform fluorescence dynamics properties with each protein molecule associated with two types of motions: the local motion of the Trp residue in the protein and the global tumbling motion of the entire protein molecule (67). Therefore, the two rotational correlation times derived from two exponential fits of the fluorescence anisotropy decays (Table 3) were interpreted to be associated with the local motion of Trp (shorter rotational correlation time; φ1) and the global motion (longer rotational correlation time; φ2) of the protein molecule. The shorter rotational correlation time (φ1) provides information about the site-specific conformational flexibility of the protein (68). The bar diagram compares the φ1 values of the four positions of α-Syn (Fig. 1I). The shorter φ1 value for Trp140 as compared with those of Trp3 and Trp71 indicates that position 140 is conformationally more flexible than positions 3 and 71 of WT α-Syn, which is expected for the last residue of the polypeptide.
The above data suggest that α-Syn in its low molecular weight state has different site-specific structure and dynamics at NAC, N-, and C-terminal regions. To further confirm this observation, we denatured the low molecular weight form of Trp3, Trp71, Trp124, and Trp140 α-Syns by adding 6 m GdnHCl to each of them. If α-Syn in its low molecular weight state has a specific structure, the addition of denaturant (6 m GdnHCl) should disrupt this structure and should alter the site-specific structural dynamics of the protein. We indeed observed a change in the site-specific structural dynamics upon denaturation. The fluorescence intensity decay of Trp3 and Trp140 was affected upon denaturation (Fig. 2A). In contrast, the fluorescence intensity decay of Trp71 and Trp124 was not significantly altered upon denaturation. The τm of Trp3 decreased, whereas that of Trp140 increased upon denaturation (Fig. 2B). Furthermore, for Trp3 and Trp71, the fluorescence anisotropy decayed faster upon denaturation (Fig. 2C). The bar diagram comparing the φ1 values of the four different Trp positions of low molecular weight α-Syn with and without denaturant indicates a significant decrease in the φ1 value of Trp3 and Trp71 upon denaturation (Fig. 2D). Our analysis of the anisotropy decays shows a decrease in rotational correlation times associated with both local motion (φ1) and global motion (φ2) of Trp3 and Trp71 upon denaturation (Table 4). This indicates that positions 3 and 71 are initially structured, and upon denaturation, they become more flexible not only in terms of their local conformation but also with respect to the global conformational properties of the protein. The fluorescence anisotropy decays of Trp124 and Trp140, however, were not much altered upon denaturation. This suggests that because positions 124 and 140 are flexible enough at the low molecular weight state their flexibility is not further enhanced upon denaturation (Fig. 2D). Interestingly, unlike the φ1 values that became nearly the same for all four positions of α-Syn upon denaturation, the τm values remained different (τm of Trp3 < Trp71 = Trp124 < Trp140) (Fig. 2B). This indicates that the amino acid residues in the primary sequence of the protein near the Trp site affect their fluorescence lifetime. Overall, these results imply that all four Trp positions in the low molecular weight state of α-Syn are solvent-accessible due to its random coil structure. However, α-Syn in its low molecular weight state has a definite site-specific structure/dynamics, which gets disrupted upon its denaturation.
FIGURE 2.
Effect of denaturation on the site-specific structure of α-Syn. The freshly prepared low molecular weight solutions of Trp3, Trp71, Trp124, and Trp140 α-Syns were denatured by adding 6 m GdnHCl and subjected to time-resolved fluorescence studies. Black and green represent the low molecular weight state and denatured state, respectively, of each Trp mutant of α-Syn. A, time-resolved fluorescence intensity decay kinetics fitted to a three-exponential function (smooth lines) showing a change in decay kinetics of Trp3, Trp71, Trp124, and Trp140 upon denaturation. B, mean fluorescence lifetime (τm = Σαiτi) values calculated by fitting the fluorescence intensity decays and represented as a bar diagram (mean ± S.D., n = 3) indicating a change in microenvironment experienced by Trp3 and Trp140 upon denaturation. C, time-resolved fluorescence anisotropy decay kinetics fitted to a two-exponential function (smooth lines) showing a faster decay of fluorescence anisotropy of Trp3 and Trp71 upon denaturation. D, rotational correlation times associated with the local motion of Trp (φ1) calculated by fitting the fluorescence anisotropy decays and represented as a bar diagram (mean ± S.D., n = 2) indicating an increase in conformational flexibility of Trp3 and Trp71 upon denaturation. The statistical significance is as follows: *, p < 0.05; **, p < 0.01; NS, not significant, p > 0.05. Error bars represent S.D.
Overall Structure and Early Oligomer Distribution of α-Syn and Its PD-associated Mutants
Little is currently known about the structural basis for the difference in the aggregation properties between WT α-Syn and its three well known familial PD-associated mutants (E46K, A53T, and A30P). Therefore, we studied the secondary structure of WT α-Syn and the PD mutants. The far-UV CD spectra of the freshly prepared low molecular weight samples suggest that none of the PD mutations affect the gross initial structure of α-Syn, having nearly overlapping spectra characteristic of an overall unfolded structure. However, all the α-Syns have CD spectra with a minimum in the vicinity of 198 nm and a positive signal near ∼190–195 nm (Fig. 3A), indicating the possibility of some residual structure in all the proteins. It is possible that the propensity to form higher order oligomers by freshly prepared protein/peptide could be one of the key factors determining the aggregation propensity of amyloidogenic proteins (69). However, these oligomeric species are metastable and exist in a dynamic equilibrium with the monomeric form of the protein. Hence, it is very difficult to biophysically characterize and quantify the relative abundance of each of these oligomeric species. We used the PICUP method for studying the early oligomer distribution of α-Syn (69). PICUP is a powerful technique in which rapid covalent cross-linking of unmodified proteins allows the separation and quantification of small metastable protein oligomers, thereby giving an instant snapshot of the oligomerization pattern of a protein in solution (54). Previously, PICUP has been effectively used to study the early oligomerization event of α-Syn (70) as well as another amyloidogenic protein, Aβ40/42 (69), which is associated with Alzheimer disease. When early oligomerization of α-Syn was studied using PICUP, it was observed that considerable amounts of dimeric, trimeric, and pentameric forms of the protein exist in equilibrium with the monomeric form in freshly prepared low molecular weight α-Syn solution (Fig. 3B). The PD-associated mutants A53T, E46K, and A30P also have an early oligomer distribution comprising monomeric, dimeric, trimeric, and pentameric species, which is qualitatively similar to that of WT protein (Fig. 3B). This implies that the PD-associated mutations do not affect significantly the early oligomer distributions of α-Syn.
FIGURE 3.
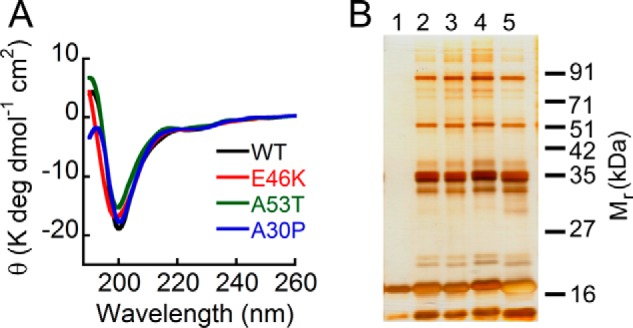
Effect of PD-associated mutations on overall structure and early oligomerization of α-Syn. A, far-UV CD spectra of low molecular weight solutions of WT, E46K, A53T, and A30P α-Syns showing that all the proteins have a minimum in the vicinity of 198–200 nm. B, the early oligomerization was studied by subjecting the freshly prepared low molecular weight solutions of WT α-Syn and its PD-associated mutants to PICUP followed by SDS-PAGE. A silver-stained gel is shown: lane 1, non-cross-linked WT α-Syn; lane 2, cross-linked A30P; lane 3, cross-linked A53T; lane 4, cross-linked E46K; lane 5, cross-linked WT α-Syn. In non-cross-linked WT α-Syn, a band corresponding to monomer (∼17 kDa) was observed, whereas in all cross-linked α-Syns, distinct bands corresponding to monomer (∼17 kDa), dimer (∼35 kDa), trimer (∼51 kDa), and pentamer (∼85 kDa) were observed, indicating that WT α-Syn and its PD-associated mutants have similar early oligomer distributions. deg, degrees.
Effect of Trp Substitution Mutations on the Aggregation Propensities of WT α-Syn and PD-associated Mutants
Because the PD-associated mutations do not affect the early oligomer distributions of α-Syn, we analyzed the effect of E46K, A53T, and A30P PD mutations on the site-specific structural dynamics of α-Syn. For this study, a Trp residue was substituted at four specific sites in each of the three regions of E46K, A53T, and A30P α-Syns similarly as we performed for the WT protein (Fig. 4). We performed the cross-linking as well as aggregation kinetics study using the ThT binding assay with the multiple Trp-substituted variants of WT α-Syn and its PD-associated mutants. The cross-linking studies with the freshly prepared low molecular weight form of all proteins show that all proteins have similar early oligomer distributions, which include monomeric along with dimeric, trimeric, and pentameric species (Fig. 5B). This implies that introduction of a Trp residue at any of the four different positions did not alter the early oligomer distribution of either the WT protein or the PD-associated mutants. Furthermore, our aggregation kinetics study using the ThT binding assay shows that Trp substitution did not largely alter the aggregation behavior of the parent protein (Fig. 5A). The PD mutants E46K and A53T as well as their Trp-substituted variants formed amyloid faster (within ∼48–72 h for E46K and ∼72–85 h for A53T) compared with WT α-synuclein and its corresponding Trp-substituted proteins, which formed amyloid fibrils within ∼96–120 h of incubation (Fig. 5A). The PD mutant A30P and its corresponding Trp-substituted variants had the slowest amyloid forming rate (within ∼175–200 h of incubation). Although there were small variations in the amyloid forming rates across the parent protein and its Trp-substituted variants, none of the Trp mutations significantly altered the amyloid forming propensity of its parent protein. Furthermore, at the end of the aggregation process, all proteins formed amyloids having very similar fibrillar morphology as observed by transmission electron microscope (Fig. 6 and see also Ref. 48). The results suggest that the Trp substitutions do not change the overall aggregation properties of the protein and therefore are suitable probes for our study.
FIGURE 4.

Design of Trp substitution mutations in the PD-associated mutants of α-Syn. Amino acid sequences of the PD-associated mutants E46K, A53T, and A30P of α-Syn and their designed Trp-substituted mutants are shown. The PD mutations are shown in red, and the Trp substitutions are shown in blue. The rectangular boxes indicate the Trp mutation site in the three regions, N terminus (V3W), middle NAC (V71W), and C terminus (A124W and A140W), of the PD-associated mutants.
FIGURE 5.

Effect of Trp substitutions on the aggregation propensities and early oligomerization of the respective parent proteins. A, amyloid formation of WT α-Syn and its three PD-associated mutants (E46K, A53T, and A30P) along with their Trp-substituted variants monitored by ThT fluorescence. ThT fluorescence at 480 nm is plotted as a function of time (in h). Mutants E46K and A53T and their Trp-substituted variants have a faster rate of amyloid formation, whereas A30P and its Trp-substituted variants have a slower amyloid forming rate compared with WT α-Syn and its Trp-substituted variants. B, the early oligomerization was studied for α-Syn and its Trp mutants using PICUP followed by SDS-PAGE. A silver-stained gel shows the cross-linked products of the parent protein (lane 1), V3W variant (lane 2), V71W variant (lane 3), A124W variant (lane 4), and A140W variant (lane 5). In all proteins, distinct bands corresponding to monomer (∼17 kDa), dimer (∼33 kDa), trimer (∼54 kDa), and pentamer (∼71 kDa) were observed, indicating that none of the Trp substitutions affect the early oligomer size distributions of their parent proteins. Error bars represent S.D. AU, arbitrary units. In B, SDS-PAGE gels (from left to right) represent proteins samples of WT, A30P, E46K, and A53T α-synuclein.
FIGURE 6.

Morphological analysis of amyloid fibril formed by the Trp-substituted mutants. Electron micrographs show amyloid fibrils formed by WT α-Syn and its selected Trp-substituted mutants. Scale bars, 200 nm.
Effect of PD-associated Mutations on the Site-specific Structural Dynamics of α-Syn
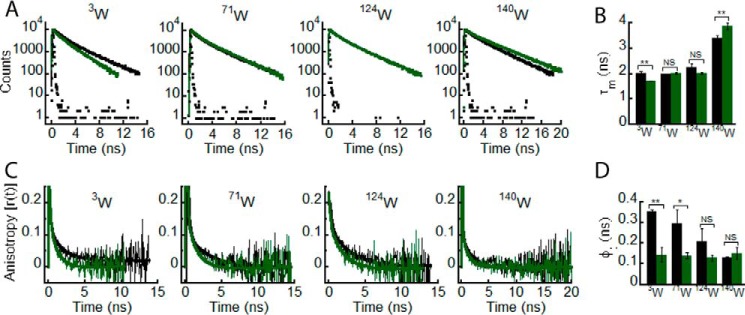
Freshly prepared low molecular weight solutions of all Trp-substituted mutants of WT, E46K, A53T, and A30P α-Syns were subjected to time-resolved fluorescence studies. Similar to WT protein, the site-specific microenvironment at the N terminus of all α-Syns was studied using time-resolved fluorescence intensity decay kinetics, and the data were fitted to a three-exponential function. The intensity decay of Trp3 in E46K mutant was slower as compared with that of Trp3 in WT, A53T, and A30P α-Syns (Fig. 7A). When the τm values for each protein were calculated, we observed that the Trp at the N terminus of E46K had a longer τm (∼2.6 ns) compared with that of Trp at the N terminus of WT, A53T, and A30P α-Syns (τm ∼ 2 ns) (Fig. 7B). The data suggest that the site-specific microenvironment at the N terminus of α-Syn is altered in the PD mutant E46K and remains unaltered in the other two PD mutants A53T and A30P. Furthermore, the site-specific solvent accessibility at the N terminus of WT α-Syn was compared with the other three mutants using dynamic fluorescence quenching method. The kq values were derived from the Stern-Volmer plots (Fig. 7C). The data show that the kq values of Trp at the N terminus of PD-associated mutants are lesser as compared with that of WT α-Syn (Fig. 7D). This indicates that the PD-associated mutations decrease the site-specific solvent accessibility of α-Syn at the N terminus. When the site-specific conformational dynamics at Trp3 was studied using time-resolved fluorescence anisotropy decay kinetics, all proteins showed anisotropy decay at a very similar rate except for E46K α-Syn (Fig. 7E). We also observed a clear increase in the rotational correlation time (φ1) for Trp3 of E46K compared with that of WT, A30P, and A53T (Fig. 7F). The rotational correlation time associated with global motion of the protein (φ2) was also longer for Trp3 of E46K compared with WT, A30P, and A53T (Table 3). This means that the local as well as global conformational rigidity of the N terminus is greater for E46K compared with WT, A30P, and A53T. It has been shown that the residues (1–30) in the N terminus of α-Syn are not part of the fibril core of the protein (71) but are partially buried in oligomers of α-Syn and are involved in binding to membranes (59). The effect of PD-associated mutations on the site-specific structural dynamics at the N terminus of α-Syn further suggests that this region may contribute significantly to the oligomerization of the protein.
FIGURE 7.
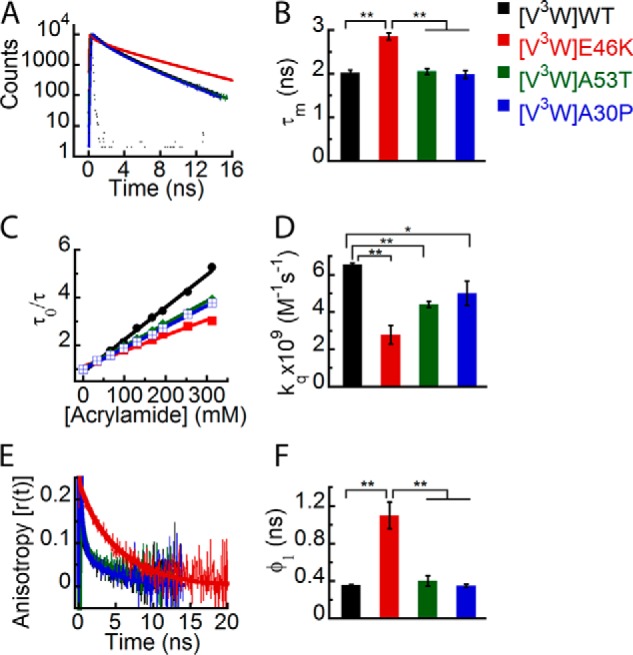
Effect of PD-associated mutations on the site-specific structural dynamics of α-Syn at its N terminus (Trp3). Freshly prepared low molecular weight solutions of Trp3 mutant of WT, E46K, A53T, and A30P α-Syns were studied using different fluorescence methods. A, time-resolved fluorescence intensity decay kinetics showing slower decay of Trp3 in E46K mutant compared with Trp3 in WT, A53T, and A30P α-Syns. B, mean fluorescence lifetimes calculated from the fluorescence intensity decays and represented as a bar diagram (mean ± S.D., n = 3) indicating that Trp3 of the E46K mutant experiences a different microenvironment compared with Trp3 of WT, A53T, and A30P α-Syns. C, Stern-Volmer plots showing a higher slope for Trp3 of WT α-Syn compared with Trp3 of the PD mutants. D, bimolecular rate constants for dynamic quenching (kq) calculated from the Stern-Volmer plots and represented as a bar diagram (mean ± S.D., n = 2) showing that Trp3 of WT α-Syn is more solvent-exposed compared with that of the PD mutants. E, time-resolved fluorescence anisotropy decay kinetics showing slower decay of fluorescence anisotropy of Trp3 of E46K compared with Trp3 of WT and A30P α-Syns. F, rotational correlation times (φ1) derived from the fluorescence anisotropy decays and represented as a bar diagram (mean ± S.D., n = 2) showing that Trp3 of E46K is conformationally more rigid compared with Trp3 of WT, A53T, and A30P α-Syns. The statistical significance is as follows: *, p < 0.05; **, p < 0.01. Error bars represent S.D. The color code shown on the right-hand side (top) in the figure indicates different α-synuclein Trp mutants.
Similar to the N terminus, the structural dynamics of Trp at position 71 in the NAC region of WT α-Syn and its PD-associated mutants was studied. The NAC region of α-Syn is considered to be the most essential part for aggregation and amyloid formation of the protein (28). The time-resolved intensity decay kinetics of Trp71 of all α-Syns is almost overlapping (Fig. 8A). The τm values suggest that there is no significant difference in the microenvironment (similar τm values ∼2 ns) of the Trp residue at position 71 for all α-Syns (Fig. 8B). The site-specific solvent accessibility studied using dynamic fluorescence quenching also showed overlapping Stern-Volmer plots (Fig. 8C) and similar values of kq (Fig. 8D) for all proteins, clearly indicating that Trp at position 71 had a similar extent of solvent accessibility in WT α-Syn and its PD mutants. Moreover, the time-resolved fluorescence anisotropy decay kinetics (Fig. 8E) and the corresponding rotational correlation time (φ1) values (Fig. 8F) were also similar for Trp71 in all α-Syns. The fluorescence data for Trp at position 71 clearly indicate that the PD-associated mutations do not have any major effect on the site-specific structure and dynamics of α-Syn in the NAC region.
FIGURE 8.
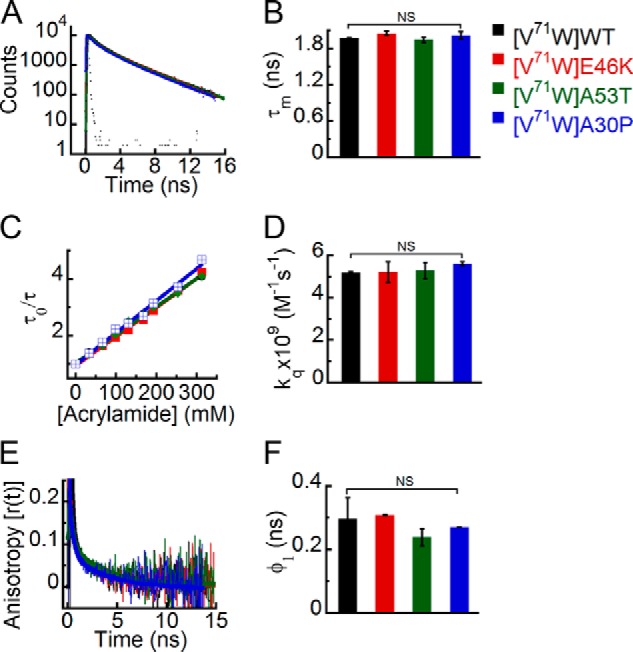
Effect of PD-associated mutations on the site-specific structural dynamics of α-Syn at its middle NAC region (Trp71). Freshly prepared low molecular weight solutions of Trp71 mutant of WT, E46K, A53T, and A30P α-Syns were studied using different fluorescence methods. A, time-resolved fluorescence intensity decay kinetics showing similar decay of fluorescence intensity from Trp71 in WT α-Syn and its PD mutants E46K, A53T, and A30P. B, mean fluorescence lifetime derived from the fluorescence intensity decays kinetics (mean ± S.D., n = 3) showing a similar microenvironment experienced by Trp at position 71 of all α-Syns (WT, E46K, A53T, and A30P). C, Stern-Volmer plots derived from dynamic quenching of fluorescence using lifetime measurements showing that Trp71 in WT α-Syn and its PD mutants E46K, A53T, and A30P have similar slopes. D, the bimolecular rate constants for dynamic quenching (kq) (mean ± S.D., n = 2) showing a similar extent of solvent exposure of Trp71 in WT α-Syn and its PD mutants. E, time-resolved fluorescence anisotropy decay kinetics showing similar anisotropy decays of Trp at position 71 of all α-Syns. F, rotational correlation times (φ1) derived from the fluorescence anisotropy decays (mean ± S.D., n = 2) reflecting similar conformational flexibility of Trp at position 71 of all α-Syns. The statistical significance is as follows: *, p < 0.05; **, p < 0.01; NS, not significant, p > 0.05. Error bars represent S.D. The color code shown on the right-hand side (top) in the figure indicates different α-synuclein Trp mutants.
When we studied the effect of PD-associated mutations on the site-specific conformational dynamics at position 124 in the C terminus, the time-resolved fluorescence intensity decay kinetics of Trp in all α-Syns showed no apparent differences (Fig. 9A). Their corresponding τm values were also similar, suggesting that all proteins experience a similar microenvironment at position 124 (Fig. 9B). The site-specific solvent accessibility and kq values (Fig. 9, C and D) were lower for Trp124 in all the PD mutants compared with WT α-Syn. The data indicate that position 124 is less solvent-accessible in all the PD mutants when compared with WT. However, among the PD mutants, we did not find any significant difference in solvent accessibility. The time-resolved anisotropy decay kinetics revealed that Trp124 of the PD-associated mutants had slower fluorescence anisotropy decay kinetics compared with Trp124 of WT α-Syn (Fig. 9E). The corresponding rotational correlation time (φ1) was longest for Trp124 of E46K and A53T followed by A30P and WT α-Syn (Fig. 9F). Moreover, rotational correlation time associated with global motion of the protein (φ2) was also longer for the PD mutants as compared with WT (Table 3). The data suggest that Trp at position124 of all three PD mutants is more rigid compared with that in WT α-Syn in terms of both local and global dynamics.
FIGURE 9.
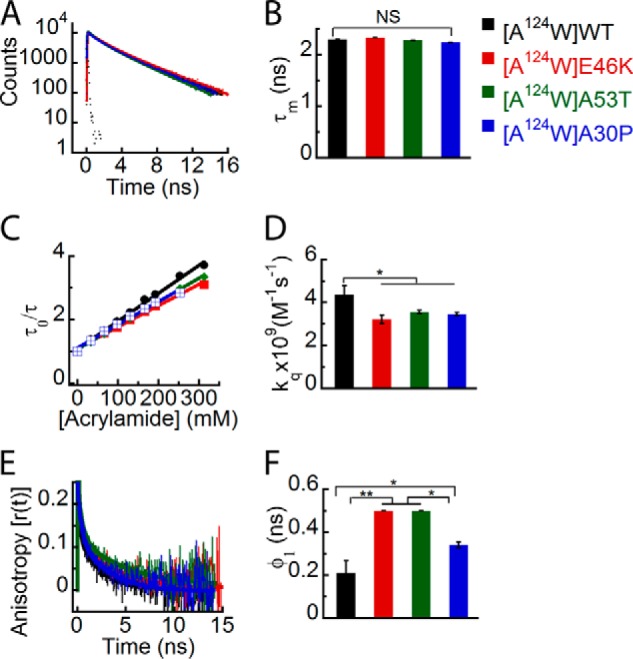
Effect of PD-associated mutations on the site-specific structural dynamics of α-Syn at its C terminus (Trp124). Freshly prepared low molecular weight solutions of Trp124 mutant of WT, E46K, A53T, and A30P α-Syns were studied using different fluorescence methods. A, time-resolved fluorescence intensity decay kinetics showing similar decays of fluorescence intensities by Trp124 in WT α-Syn and its PD mutants. B, mean fluorescence lifetime (mean ± S.D., n = 3) showing a similar microenvironment at position 124 of all α-Syns. C, Stern-Volmer plots derived from dynamic fluorescence quenching showing a higher slope for Trp124 in WT α-Syn compared with the PD mutants. D, the bimolecular rate constant (kq) represented as a bar diagram (mean ± S.D., n = 2) showing that Trp124 in WT α-Syn is more solvent-exposed as compared with Trp124 in the PD mutants. E, time-resolved fluorescence anisotropy decay kinetics showing slower decay of fluorescence anisotropy by Trp124 of all PD mutants compared with that of WT α-Syn. F, rotational correlation time (φ1) as represented by a bar diagram (mean ± S.D., n = 2) showing greater rigidity of Trp at position 124 in all PD mutants. The statistical significance is as follows: *, p < 0.05; **, p < 0.01; NS, not significant, p > 0.05. Error bars represent S.D. The color code in the figure indicates different α-synuclein mutants.
Next, we studied the effect of PD-associated mutations on the site-specific conformational dynamics at position 140 in the C terminus. The time-resolved fluorescence intensity decay kinetics of Trp in all α-Syns showed no apparent differences (Fig. 10A). However, the corresponding τm of Trp140 in A53T was significantly shorter than that of Trp140 in WT, E46K, and A30P α-Syns (Fig. 10B). The data indicate that the PD mutation A53T has an effect on the site-specific microenvironment of α-Syn at position 140. The site-specific solvent accessibility and kq values (Fig. 10, C and D) were lower for Trp140 in all the PD mutants when compared with WT α-Syn. The data indicate that the Trp at position 140 of WT α-Syn is more solvent-accessible than that of the PD-associated mutants. However, among the PD mutants, we did not find any significant difference in solvent accessibility. The time-resolved anisotropy decay kinetics revealed interesting differences between all α-Syn proteins. The Trp140 of E46K had a markedly slower fluorescence anisotropy decay kinetics compared with Trp140 of WT, A53T, and A30P α-Syns. The Trp140 of A53T and A30P also had slower fluorescence anisotropy decay kinetics compared with Trp140 of WT α-Syn (Fig. 10E). The corresponding rotational correlation time (φ1) was longest for Trp140 of E46K followed by A30P, A53T, and WT α-Syns (Fig. 10F). Similar to our observation at position 124, φ2 at position 140 was also longer for all the PD mutants as compared with WT (Table 3). The data suggest that the Trp at the C terminus of all three PD mutants is more rigid compared with WT α-Syn in terms of both local and global conformational dynamics.
FIGURE 10.
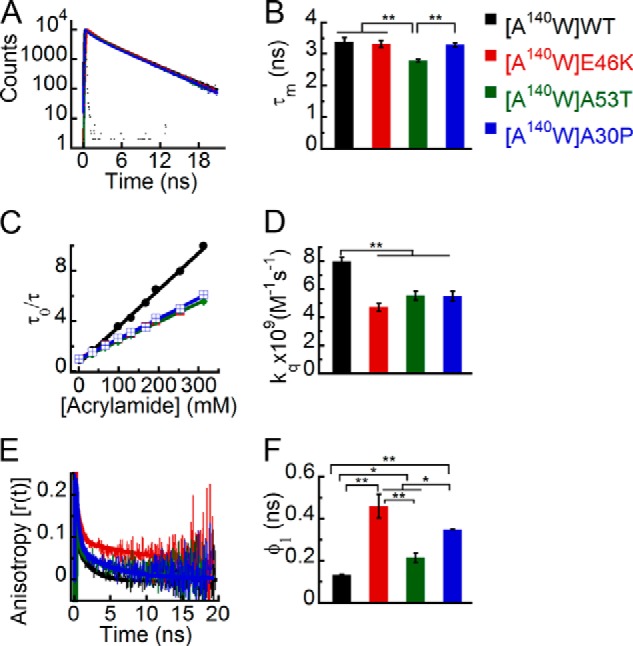
Effect of PD-associated mutations on the site-specific structural dynamics of α-Syn at its C terminus (Trp140). Freshly prepared low molecular weight solutions of Trp140 mutant of WT, E46K, A53T, and A30P α-Syns were studied using different fluorescence methods. A, time-resolved fluorescence intensity decay kinetics showing similar decays of fluorescence intensities by Trp140 in WT α-Syn and its PD mutants. B, mean fluorescence lifetime (mean ± S.D., n = 3) showing the different microenvironment at position 140 by A53T compared with WT, E46K, and A30P α-Syns. C, Stern-Volmer plots derived from dynamic fluorescence quenching showing a higher slope for Trp140 in WT α-Syn compared with the PD mutants. D, the bimolecular rate constant (kq) represented as a bar diagram (mean ± S.D., n = 2) showing that Trp140 in WT α-Syn is more solvent-exposed as compared with Trp140 in the PD mutants. E, time-resolved fluorescence anisotropy decay kinetics showing slower decay of fluorescence anisotropy by Trp140 E46K compared with that of WT, A53T, and A30P α-Syns. The fluorescence anisotropy decay of Trp140 WT α-Syn, however, is faster than that of A53T and A30P mutants. F, rotational correlation time (φ1) as represented by a bar diagram (mean ± S.D., n = 2) showing greater rigidity of Trp at position 140 in all PD mutants. The statistical significance is as follows: *, p < 0.05; **, p < 0.01. Error bars represent S.D. The color code in the figure indicates different α-synuclein mutants.
Effect of PD-associated Mutations on the Sensitivity of α-Syn to Chemical Denaturant (6 m GdnHCl)
We denatured all Trp-substituted variants of WT α-Syn and its PD-associated mutants using 6 m GdnHCl and analyzed them using time-resolved fluorescence studies. These studies could provide important insights in terms of a difference in the degree of sensitivity of different PD mutants and WT protein to chemical denaturant that may be correlated to their altered oligomerization tendency. The time-resolved fluorescence intensity decay kinetics and the corresponding τm values suggest that upon denaturation all the proteins have similar microenvironments at positions 71, 124, and 140 (Figs. 12, A and B; 13, A and B; and 14, A and B). In contrast, at position 3, E46K has a different microenvironment (shorter τm) compared with all the other proteins upon denaturation (Fig. 11, A and B). The time-resolved fluorescence anisotropy decay kinetics showed that upon denaturation all the proteins had similar decays at positions 71, 124, and 140 (Figs. 12C, 13C, and 14C). Because the dynamics of both local and global conformations of the protein changes upon denaturation, we derived a single parameter, the mean rotational correlation time (φm = ∑βiφi), to represent the overall structural dynamics of the denatured proteins. Our analysis revealed that the mean rotational correlation time (φm) was similar for all proteins at positions 71, 124, and 140 (Figs. 12D, 13D, and 14D), suggesting that they have similar overall structural dynamics upon denaturation. Trp at position 3 of E46K, however, had slower anisotropy decay kinetics compared with the other proteins in the denatured state (Fig. 11C). The φm was also longer for Trp3 of E46K compared with all the other proteins (Fig. 11D), indicating that Trp at position 3 of E46K is more rigid compared with all the other proteins. This implies that Trp at position 3 of E46K is relatively less sensitive to GdnHCl denaturation compared with WT, A53T, and A30P.
FIGURE 12.
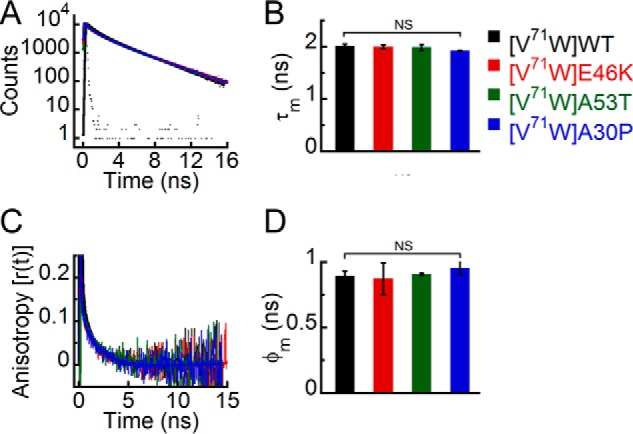
Effect of denaturation on the site-specific (Trp71) structure of PD-associated mutants of α-Syn. The freshly prepared low molecular weight solutions of Trp71 variants of WT α-Syn and the PD-associated mutants were denatured by adding 6 m GdnHCl and subjected to time-resolved fluorescence studies. A, time-resolved fluorescence intensity decay kinetics fitted to a three-exponential function (smooth lines) showing that all proteins have similar decay kinetics in the denatured state. B, mean fluorescence lifetime (τm = Σαiτi) values calculated by fitting the fluorescence intensity decays and represented as a bar diagram (mean ± S.D., n = 3) indicating that all proteins have a similar microenvironment at Trp71 in the denatured state. C, time-resolved fluorescence anisotropy decay kinetics fitted to a two-exponential function (smooth lines) showing similar decay of fluorescence anisotropy at Trp71 of all proteins. D, mean rotational correlation time associated with the motion of Trp (φm = Σβiφi) calculated by fitting the fluorescence anisotropy decays and represented as a bar diagram (mean ± S.D., n = 2) indicating similar conformational rigidity at Trp71 of all proteins. The statistical significance is as follows: NS, not significant, p > 0.05. Error bars represent S.D. The color code in the figure indicates different α-synuclein mutants.
FIGURE 13.
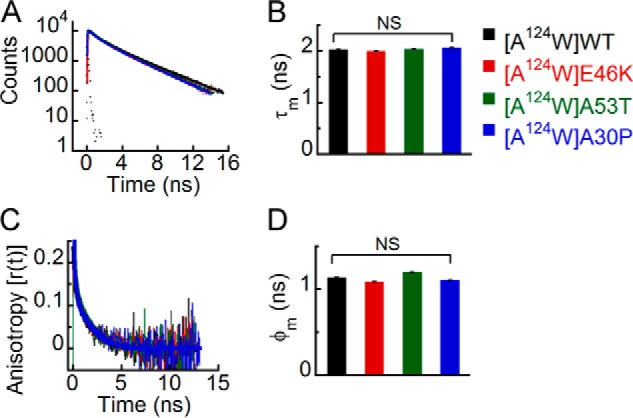
Effect of denaturation on the site-specific (Trp124) structure of PD-associated mutants of α-Syn. The freshly prepared low molecular weight solutions of Trp124 variants of WT α-Syn and the PD-associated mutants were denatured by adding 6 m GdnHCl and subjected to time-resolved fluorescence studies. A, time-resolved fluorescence intensity decay kinetics fitted to a three-exponential function (smooth lines) showing that all proteins have similar decay kinetics in the denatured state. B, mean fluorescence lifetime (τm = Σαiτi) values calculated by fitting the fluorescence intensity decays and represented as a bar diagram (mean ± S.D., n = 3) indicating that all proteins have a similar microenvironment at Trp124 in the denatured state. C, time-resolved fluorescence anisotropy decay kinetics fitted to a two-exponential function (smooth lines) showing similar decay of fluorescence anisotropy at Trp124 of all proteins. D, mean rotational correlation time associated with the motion of Trp (φm = Σβiφi) calculated by fitting the fluorescence anisotropy decays and represented as a bar diagram (mean ± S.D., n = 2) indicating similar conformational rigidity at Trp124 of all proteins. The statistical significance is as follows: NS, not significant, p > 0.05. Error bars represent S.D. The color code shown on the right-hand side (top) in the figure indicates different α-synuclein Trp mutants.
FIGURE 14.
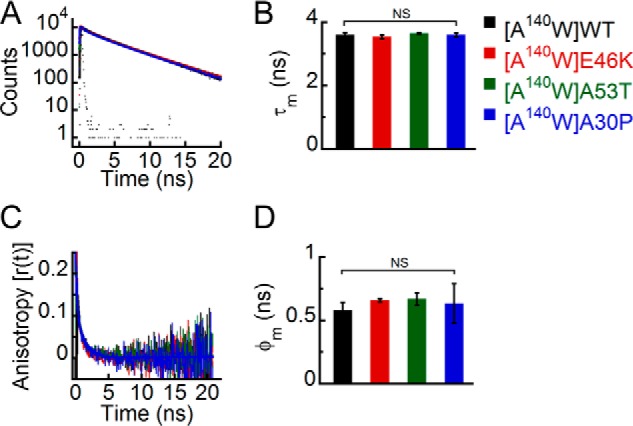
Effect of denaturation on the site-specific (Trp140) structure of PD-associated mutants of α-Syn. The freshly prepared low molecular weight solutions of Trp140 variants of WT α-Syn and the PD-associated mutants were denatured by adding 6 m GdnHCl and subjected to time-resolved fluorescence studies. A, time-resolved fluorescence intensity decay kinetics fitted to a three-exponential function (smooth lines) showing that all proteins have similar decay kinetics in the denatured state. B, mean fluorescence lifetime (τm = Σαiτi) values calculated by fitting the fluorescence intensity decays and represented as a bar diagram (mean ± S.D., n = 3) indicating that all proteins have a similar microenvironment at Trp140 in the denatured state. C, time-resolved fluorescence anisotropy decay kinetics fitted to a two-exponential function (smooth lines) showing similar decay of fluorescence anisotropy at Trp140 of all proteins. D, mean rotational correlation time associated with the motion of Trp (φm = Σβiφi) calculated by fitting the fluorescence anisotropy decays and represented as a bar diagram (mean ± S.D., n = 2) indicating similar conformational rigidity at Trp140 of all proteins. The statistical significance is as follows: NS, not significant, p > 0.05. Error bars represent S.D. The color code in the figure indicates different α-synuclein mutants.
FIGURE 11.
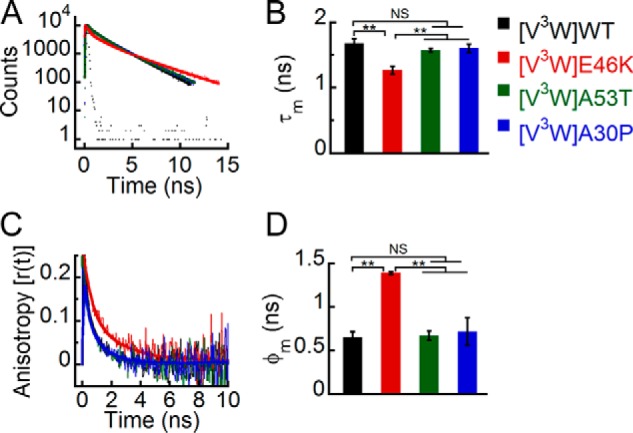
Effect of denaturation on the site-specific (Trp3) structure of PD-associated mutants of α-Syn. The freshly prepared low molecular weight solutions of Trp3 variants of WT α-Syn and the PD-associated mutants were denatured by adding 6 m GdnHCl and subjected to time-resolved fluorescence studies. A, time-resolved fluorescence intensity decay kinetics fitted to a three-exponential function (smooth lines) showing that the mutant E46K has a faster decay kinetics at Trp3 compared with the other proteins even in the denatured state. B, mean fluorescence lifetime (τm = Σαiτi) values calculated by fitting the fluorescence intensity decays and represented as a bar diagram (mean ± S.D., n = 3) indicating that E46K has a different microenvironment at Trp3 compared with the other proteins. C, time-resolved fluorescence anisotropy decay kinetics fitted to a two-exponential function (smooth lines) showing slower decay of fluorescence anisotropy of Trp3 of E46K compared with the other proteins. D, mean rotational correlation time associated with the motion of Trp (φm = Σβiφi) calculated by fitting the fluorescence anisotropy decays and represented as a bar diagram (mean ± S.D., n = 2) indicating higher conformational rigidity of Trp3 in E46K compared with the other proteins. The statistical significance is as follows: *, p < 0.05; **, p < 0.01; NS, not significant, p > 0.05. Error bars represent S.D. The color code shown on the right-hand side (top) in the figure indicates different α-synuclein Trp mutants.
Comparison of Site-specific Structure of the Soluble and Fibrillar States of α-Syn and Its PD-associated Mutants
We compared the site-specific structural dynamics of WT α-Syn and its PD-associated mutants at their low molecular weight (the starting point of aggregation process) and fibrillar states (48) (Table 5). Because in our previous study on α-Syn fibrils (48), [V3W]E46K (the Trp substitution mutations are shown in square brackets, whereas the familial PD-associated mutations are shown outside the square brackets) protein had such a low expression level and yield from which we could not get fibrils, we designed another Trp mutant, F4W, at the N terminus of E46K for the study. Therefore, we have included the comparison of data of fibrils of [F4W]E46K with that of the low molecular weight form of [F4W]E46K in Table 5. The comparison (Table 5) shows that τm values of the N terminus (Trp3/Trp4) remained very similar in the low molecular weight state as well as the fibrillar state of WT, E46K, A53T, and A30P. However, upon fibril formation, the fluorescence of Trp at position 71 was significantly quenched (a decrease in τm values) in all α-Syns. Similar to the NAC region, at the C terminus, the Trp fluorescence intensity was quenched after fibril formation by all proteins except A53T (Table 5). The time-resolved fluorescence intensity decay kinetics thus suggests that a significant change in site-specific microenvironment occurs mostly at the middle NAC region and C terminus for most α-Syns after their aggregation. The dynamic fluorescence quenching studies revealed that there was an overall decrease in the site-specific solvent exposure (a decrease in kq) at all three regions (NAC and N and C termini) of WT as well as PD mutants of α-Syn upon their conversion from the low molecular weight to the fibrillar state (Table 5). The kq value of Trp at the N terminus decreased by ∼2.7 times in WT α-Syn and ∼1.9 times in the PD mutants (Table 5) upon fibril formation. The data indicate that Trp at the N terminus of WT α-Syn undergoes a greater extent of burial than that of the PD-associated mutants upon fibrillation. The Trp at position 71 of WT, A53T, and A30P α-Syns underwent more than a 10-fold decrease in the kq value upon fibril formation. In contrast, Trp71 in the mutant E46K underwent an ∼3.7× decrease in kq value after fibrillation, suggesting a larger extent of burial of position 71 in WT, A53T, and A30P compared with E46K. Furthermore, upon fibril formation, Trp at position 140 in all α-Syns underwent an ∼3.3× decrease in kq value. The data suggest that the C terminus (Trp140) undergoes a similar extent of burial in all proteins upon fibril formation.
TABLE 5.
Comparison of site-specific structural dynamics of low molecular weight and fibrillar forms of WT α-Syn and its PD-associated mutants
The mean fluorescence lifetime (τm), bimolecular quenching rate constant (kq), and rotational correlation time associated with the local motion (φ1) of Trp at the N-terminal, NAC, and C-terminal regions of low molecular weight (LMW) as well as fibrils of WT α-Syn and its PD-associated mutants are shown. The error associated with τm and kq is <5 and <10%, respectively. The error associated with φ1 is ∼10–20%. The symbols beside the values of parameters of fibrils represent their statistical significance in comparison with the values of their respective low molecular weight forms at the particular site. The statistical significance is denoted as follows: *, p < 0.05; **, p < 0.01; NS, not significant, p > 0.05. ND, not done.
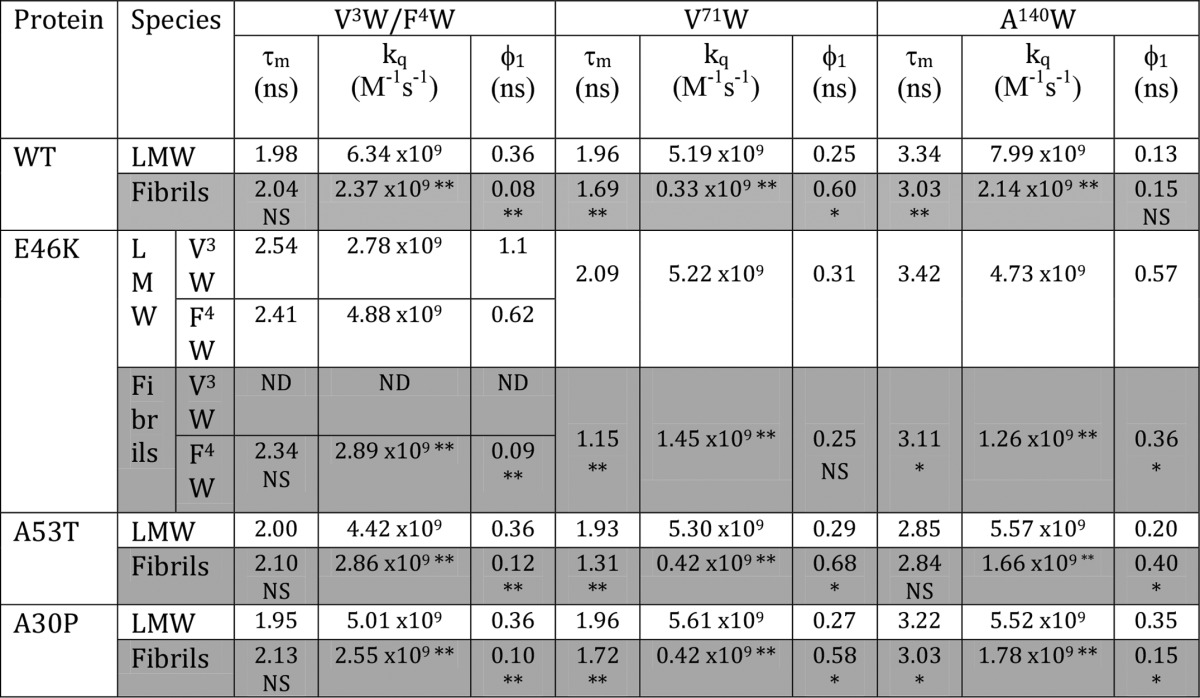
The time-resolved anisotropy decay kinetics data indicate that there was a decrease in φ1 values at the N terminus (Trp3/Trp4) upon conversion of low molecular weight to the fibrillar state for all proteins, suggesting a decrease in site-specific rigidity upon fibril formation (Table 5). Furthermore, the φ1 values increased for the middle NAC region (Trp71) upon fibril formation by WT, A53T, and A30P α-Syns, whereas the φ1 values remained similar for Trp71 of E46K in both low molecular weight and fibrillar states (Table 5). The data suggest that the conformational rigidity at position 71 of all proteins except E46K increases upon fibril formation. At the C terminus (Trp140), φ1 values decreased in E46K and A30P α-Syns upon their fibrillation; however, φ1 increased in the A53T mutant upon its fibril formation. However, the φ1 remained almost the same (φ1 ∼ 0.15 ns) in the low molecular weight and fibrillar states of WT α-Syn. This indicates that at the C terminus site-specific rigidity decreases upon fibril formation of E46K and A30P α-Syns. In WT α-Syn, there was no significant change in rigidity at position 140 upon fibrillation, whereas in A53T mutant, position 140 became more rigid upon conversion into fibrils.
DISCUSSION
A detailed study of the mechanism of aggregation of α-Syn and the factors that affect aggregation has relevance in understanding the etiology of PD (23, 34, 35). α-Syn under physiological conditions is natively unfolded (16) and can aggregate into highly ordered β-sheet-rich amyloid fibrils in vitro upon incubation (17). It is extremely intriguing as to how an essentially disordered protein can self-assemble into a highly ordered structure. Several investigations have been carried out to understand the molecular mechanism of α-Syn fibrillation. These studies revealed that α-Syn fibrillation follows a nucleation-dependent mechanism (72, 73). A model has been proposed to explain the fibrillation mechanism in which the first critical step is the structural transformation of α-Syn from a natively unfolded conformation into an aggregation-competent partially folded conformation (74). This partially folded intermediate further self-assembles into soluble oligomers, which give rise to a fibril nucleus. The fibril nucleus recruits soluble protein and eventually forms fibrillar aggregates. In this model, the conformational transformation of α-Syn from natively unfolded to partially folded intermediate and the formation of a nucleus are the two key rate-limiting steps that determine the kinetics of fibrillation (74).
Consistent with the previous observations, our results also suggest that under physiological conditions α-Syn is substantially unfolded (Fig. 3A) (16). However, several research groups have reported that α-Syn has special and non-random conformational properties. Despite being unstructured overall, α-Syn has been suggested to exist as an ensemble of conformers that are on average structurally more compact than expected for a completely unstructured protein (30, 31). In the current study, we substituted a Trp residue at its four different positions in the middle NAC region (Trp71) and N (Trp3) and C termini (Trp124 and Trp140) to elucidate the site-specific structure of α-Syn. Consistent with previous reports, we observed that a definite site-specific structure exists in α-Syn. The Trp at positions 3, 71, and 124 in WT α-Syn are conformationally more heterogeneous as compared with the Trp at position 140 (Fig. 1E). Furthermore, our dynamic fluorescence quenching studies show that Trp124 is more solvent-protected followed by Trp71, Trp3, and Trp140 (Fig. 1, F and G). The Trp at position 140 is conformationally more flexible compared with Trp at positions 3 and 71, whereas Trp at position 124 had intermediate flexibility (Fig. 1, H and I). To confirm the existence of site-specific structure in α-Syn, we studied the Trp-substituted mutants in completely denatured conditions (in 6 m GdnHCl) (Fig. 2, A–D). Interestingly, the site-specific structure of α-Syn was destroyed upon denaturation, which is evident by the significant increase in conformational flexibility of the Trp at positions 3 and 71 of α-Syn. The site-specific conformational flexibility became nearly the same in all three regions of the protein (middle NAC and N and C termini) in the denatured state, suggesting that α-Syn has a distinct site-specific structure at its three regions.
Previous in vitro studies suggest that the three PD-associated mutations (E46K, A53T, and A30P) in α-Syn lead to an increased propensity of non-fibrillar oligomer formation (34, 36, 37, 39, 40). All three PD mutants of α-Syn are natively unfolded under physiological conditions similar to WT protein (34, 35). Our results also suggest that the overall structure of α-Syn is not affected by the PD-associated mutations as evidenced by their almost overlapping CD spectra (Fig. 3A). The structural basis of this drastic alteration of aggregation kinetics of α-Syn by these PD-associated mutations is thus not clear. α-Syn, which is an aggregation-prone protein, is reported to exist in a dynamic equilibrium with its lower order oligomers (70, 75). Factors affecting aggregation of α-Syn have been reported to change the early oligomer distribution pattern of the protein (75, 76). Because WT α-Syn and its PD mutants have a similar overall structure, another possibility could be that they may possess a different oligomer distribution immediate after solubilization, which could lead to different nucleation rates and eventually result in altered aggregation kinetics. For example, it has been suggested that the more aggregation-prone Aβ42 preferentially forms pentameric/hexameric oligomers (paranuclei) in contrast to Aβ40, which mostly forms dimers, trimers, and tetramers (69). In contrast to Aβ studies, our PICUP data indicate that there is no significant difference in the early oligomer distribution of WT and the PD mutants of α-Syn (Fig. 3B).
We hypothesized that PD-associated mutations may influence the site-specific structure/dynamics of α-Syn, which might lead to their altered propensity to self-associate and eventually result in different oligomerization/fibrillation rates. We studied the effect of PD mutations (E46K, A53T, and A30P) on the site-specific structural dynamics of α-Syn. Significant site-specific structural alterations were observed due to the PD-associated mutations at the N and C termini of α-Syn. The time-resolved fluorescence intensity decay studies suggest that among the PD-associated mutations E46K and A53T alter the site-specific microenvironment at the N terminus and C terminus, respectively (Figs. 7, A and B, and 10, A and B). However, the other mutation, A30P, does not affect the microenvironment at any of the four positions of the protein. The dynamic fluorescence quenching studies reveal that all the PD-associated mutations decrease the site-specific solvent accessibility at both the N (Trp3) and C termini (Trp124 and Trp140) of α-Syn without having any major effect at the middle NAC region (Figs. 7D, 8D, 9D, and 10D). The conformational dynamics studies using time-resolved fluorescence anisotropy decay suggest that the E46K mutation enhances the rigidity at the N terminus and that all the PD-associated mutations enhance the conformational rigidity at the C terminus (positions 124 and 140) (Figs. 7, E and F; 9, E and F; and 10, E and F). None of the mutations, however, have any effect on the conformational dynamics at the middle NAC region (position 71) (Fig. 8, E and F). The data further suggest that the site-specific structural differences are most evident in the E46K mutant. The E46K mutation reduces the site-specific solvent accessibility as well as the conformational flexibility at both the N and C termini of α-Syn. Our denaturation studies showed that Trp at position 3 of E46K is rigid and faces a different microenvironment compared with all the other proteins upon denaturation. This observation of rigid structure being resistant to a high concentration of denaturant (6 m GdnHCl) is very unusual for proteins in general. However, this unique observation is consistent with the previous reports that E46K has a more compact structure with increased long range intramolecular interactions between its N and C termini (35, 42, 45). The higher resistance of Trp at position 3 of E46K to denaturation suggests that these intramolecular interactions are quite strong. Thus, our results suggest that even in the absence of any overall structure in α-Syn, the intramolecular interactions existing in the protein are affected by the PD-associated mutations. This effect may further lead to significant alterations in the structural dynamics in these PD mutants at locations far from the mutation site.
Here, we also correlate the alterations in structural dynamics of α-Syn caused by the PD-associated mutations (E46K, A53T, and A30P) with their shared property of increased propensity to form oligomers. Our results suggest that the common structural property of the three PD-associated mutants (E46K, A53T, and A30P) is reduced solvent accessibility (both at the N terminus and at the C terminus) and reduced conformational flexibility (at the C terminus), which may facilitate the formation of an aggregation-prone partially folded intermediate (the key rate-limiting step in the aggregation pathway) (74). This eventually may lead to their increased rate of oligomerization. It should be noted that oligomerization and fibril formation are two different processes and may follow distinct pathways. Fibril formation of α-Syn requires the folding of residues ∼30–110 of the protein into a β-sheet-rich structure, which forms the core of fibrils (71). However, in the mutant A30P, the β-structure-breaking residue proline at position 30 may hinder this process, thus slowing the fibril forming rate irrespective of its accelerated oligomerization rate. This potentially accounts for the increased oligomerization rate in the PD mutant A30P and increased fibrillation rate in the other two mutants, E46K and A53T.
Comparison of the site-specific structural dynamics of WT α-Syn and its PD-associated mutants at their low molecular weight state (the starting point of the aggregation process) with their fibrillar state (48) (Table 5) reveals that the three regions (middle NAC and N and C termini) of all α-Syns become more buried after fibril formation. Trp at position 71 underwent maximum burial in all proteins upon fibrillation as it resides in the region (residues ∼30–110) that forms the core of α-Syn fibrils (71). We further observed that despite having site-specific structural differences at their N terminus in the low molecular weight state, all the proteins (WT, E46K, A53T, and A30P α-Syns) result in fibrils having similar site-specific structure/dynamics at their N terminus. Because the PD mutants start with a more solvent-protected site at the N terminus (Trp3/Trp4), they undergo a lesser extent of burial at this region during fibril formation as compared with WT. Interestingly, the middle NAC region has similar site-specific microenvironment and structural dynamics in the low molecular weight state of WT and PD mutants. However, after fibril formation, all these proteins end up with a completely different site-specific microenvironment in the NAC region. The data suggest that different PD mutations manifest different alterations in the microenvironment and structure in the NAC region of α-Syn during fibril formation. The structural dynamics of the C terminus is largely altered by all three PD-associated mutations in both the low molecular weight and fibrillar states. All three PD mutations decrease the site-specific solvent accessibility at the C terminus of α-Syn in the low molecular weight state, and this effect persists even in the fibrillar state of the protein. All the PD mutations increase the site-specific rigidity at the C terminus of α-Syn in the low molecular weight state, but in the fibrillar state, only A53T and E46K mutant fibrils have higher rigidity compared with WT fibrils at their C terminus. In contrast, the A30P fibrils have site-specific rigidity similar to that of WT α-Syn fibrils at their C terminus. This indicates that the mutations E46K, A53T, and A30P differentially affect the site-specific conformational changes occurring in the NAC, N-, and C-terminal regions during the aggregation of α-Syn.
CONCLUSION
Our time-resolved fluorescence studies reveal that α-Syn despite being largely unfolded has a definite site-specific structure at its three distinct regions (the N terminus, middle NAC, and C terminus). The low molecular weight form of α-Syn is far from a random coil. The PD-associated mutants E46K, A53T, and A30P have a more compact and rigid structure that eventually may lead to their increased rate of oligomer formation. The comparison of site-specific structure at the soluble and fibrillar states of WT α-Syn and its PD mutants infers that different extents of site-specific conformational transitions take place for different PD mutants. The present study therefore helps to delineate the alteration of structure/aggregation of α-Syn due to the familial PD-associated mutations.
This work was supported in part by Department of Biotechnology, Government of India Grants BT/PR14344Med/30/501/2010 and BT/PR13359/BRB/10/752/2009.
- α-Syn
- α-synuclein
- PD
- Parkinson disease
- NAC
- non-amyloid β-component
- PICUP
- photo-induced chemical cross-linking of unmodified proteins
- ThT
- thioflavin T
- IRF
- instrument response function
- MEM
- maximum entropy method
- Aβ
- amyloid β
- GdnHCl
- guanidine hydrochloride.
REFERENCES
- 1. Clayton D. F., George J. M. (1998) The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21, 249–254 [DOI] [PubMed] [Google Scholar]
- 2. Iwai A., Masliah E., Yoshimoto M., Ge N., Flanagan L., de Silva H. A., Kittel A., Saitoh T. (1995) The precursor protein of non-Aβ component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475 [DOI] [PubMed] [Google Scholar]
- 3. Zhang L., Zhang C., Zhu Y., Cai Q., Chan P., Uéda K., Yu S., Yang H. (2008) Semi-quantitative analysis of α-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 1244, 40–52 [DOI] [PubMed] [Google Scholar]
- 4. Lee S. J., Jeon H., Kandror K. V. (2008) α-Synuclein is localized in a subpopulation of rat brain synaptic vesicles. Acta Neurobiol. Exp. 68, 509–515 [DOI] [PubMed] [Google Scholar]
- 5. Abeliovich A., Schmitz Y., Fariñas I., Choi-Lundberg D., Ho W. H., Castillo P. E., Shinsky N., Verdugo J. M., Armanini M., Ryan A., Hynes M., Phillips H., Sulzer D., Rosenthal A. (2000) Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252 [DOI] [PubMed] [Google Scholar]
- 6. Cabin D. E., Shimazu K., Murphy D., Cole N. B., Gottschalk W., McIlwain K. L., Orrison B., Chen A., Ellis C. E., Paylor R., Lu B., Nussbaum R. L. (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 22, 8797–8807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 8. Baba M., Nakajo S., Tu P. H., Tomita T., Nakaya K., Lee V. M., Trojanowski J. Q., Iwatsubo T. (1998) Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884 [PMC free article] [PubMed] [Google Scholar]
- 9. Parkinson J. (2002) An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 14, 223–236 [DOI] [PubMed] [Google Scholar]
- 10. Spillantini M. G., Crowther R. A., Jakes R., Hasegawa M., Goedert M. (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lansbury P. T., Jr., Brice A. (2002) Genetics of Parkinson's disease and biochemical studies of implicated gene products. Curr. Opin. Genet. Dev. 12, 299–306 [DOI] [PubMed] [Google Scholar]
- 12. Zhou W., Hurlbert M. S., Schaack J., Prasad K. N., Freed C. R. (2000) Overexpression of human α-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 866, 33–43 [DOI] [PubMed] [Google Scholar]
- 13. Xu J., Kao S. Y., Lee F. J., Song W., Jin L. W., Yankner B. A. (2002) Dopamine-dependent neurotoxicity of α-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 8, 600–606 [DOI] [PubMed] [Google Scholar]
- 14. Feany M. B., Bender W. W. (2000) A Drosophila model of Parkinson's disease. Nature 404, 394–398 [DOI] [PubMed] [Google Scholar]
- 15. Recchia A., Debetto P., Negro A., Guidolin D., Skaper S. D., Giusti P. (2004) α-Synuclein and Parkinson's disease. FASEB J. 18, 617–626 [DOI] [PubMed] [Google Scholar]
- 16. Uversky V. N., Li J., Souillac P., Millett I. S., Doniach S., Jakes R., Goedert M., Fink A. L. (2002) Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of α-synuclein assembly by β- and γ-synucleins. J. Biol. Chem. 277, 11970–11978 [DOI] [PubMed] [Google Scholar]
- 17. Conway K. A., Harper J. D., Lansbury P. T., Jr. (2000) Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry 39, 2552–2563 [DOI] [PubMed] [Google Scholar]
- 18. Karpinar D. P., Balija M. B., Kügler S., Opazo F., Rezaei-Ghaleh N., Wender N., Kim H. Y., Taschenberger G., Falkenburger B. H., Heise H., Kumar A., Riedel D., Fichtner L., Voigt A., Braus G. H., Giller K., Becker S., Herzig A., Baldus M., Jäckle H., Eimer S., Schulz J. B., Griesinger C., Zweckstetter M. (2009) Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 28, 3256–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Winner B., Jappelli R., Maji S. K., Desplats P. A., Boyer L., Aigner S., Hetzer C., Loher T., Vilar M., Campioni S., Tzitzilonis C., Soragni A., Jessberger S., Mira H., Consiglio A., Pham E., Masliah E., Gage F. H., Riek R. (2011) In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pieri L., Madiona K., Bousset L., Melki R. (2012) Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 102, 2894–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luk K. C., Song C., O'Brien P., Stieber A., Branch J. R., Brunden K. R., Trojanowski J. Q., Lee V. M. (2009) Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20051–20056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Volpicelli-Daley L. A., Luk K. C., Patel T. P., Tanik S. A., Riddle D. M., Stieber A., Meaney D. F., Trojanowski J. Q., Lee V. M. (2011) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uversky V. N. (2003) A protein-chameleon: conformational plasticity of α-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 21, 211–234 [DOI] [PubMed] [Google Scholar]
- 24. Davidson W. S., Jonas A., Clayton D. F., George J. M. (1998) Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449 [DOI] [PubMed] [Google Scholar]
- 25. Bussell R., Jr., Eliezer D. (2003) A structural and functional role for 11-mer repeats in α-synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 329, 763–778 [DOI] [PubMed] [Google Scholar]
- 26. Kessler J. C., Rochet J. C., Lansbury P. T., Jr. (2003) The N-terminal repeat domain of α-synuclein inhibits β-sheet and amyloid fibril formation. Biochemistry 42, 672–678 [DOI] [PubMed] [Google Scholar]
- 27. Uéda K., Fukushima H., Masliah E., Xia Y., Iwai A., Yoshimoto M., Otero D. A., Kondo J., Ihara Y., Saitoh T. (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 11282–11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giasson B. I., Murray I. V., Trojanowski J. Q., Lee V. M. (2001) A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 276, 2380–2386 [DOI] [PubMed] [Google Scholar]
- 29. Murray I. V., Giasson B. I., Quinn S. M., Koppaka V., Axelsen P. H., Ischiropoulos H., Trojanowski J. Q., Lee V. M. (2003) Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42, 8530–8540 [DOI] [PubMed] [Google Scholar]
- 30. Dedmon M. M., Lindorff-Larsen K., Christodoulou J., Vendruscolo M., Dobson C. M. (2005) Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 127, 476–477 [DOI] [PubMed] [Google Scholar]
- 31. Bertoncini C. W., Jung Y. S., Fernandez C. O., Hoyer W., Griesinger C., Jovin T. M., Zweckstetter M. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 1430–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fujioka S., Ogaki K., Tacik P. M., Uitti R. J., Ross O. A., Wszolek Z. K. (2014) Update on novel familial forms of Parkinson's disease and multiple system atrophy. Parkinsonism Relat. Disord. 20, Suppl. 1, S29–S34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pasanen P., Myllykangas L., Siitonen M., Raunio A., Kaakkola S., Lyytinen J., Tienari P. J., Pöyhönen M., Paetau A. (2014) A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol. Aging 35, 2180.e1–5 [DOI] [PubMed] [Google Scholar]
- 34. Li J., Uversky V. N., Fink A. L. (2001) Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 40, 11604–11613 [DOI] [PubMed] [Google Scholar]
- 35. Fredenburg R. A., Rospigliosi C., Meray R. K., Kessler J. C., Lashuel H. A., Eliezer D., Lansbury P. T., Jr. (2007) The impact of the E46K mutation on the properties of α-synuclein in its monomeric and oligomeric states. Biochemistry 46, 7107–7118 [DOI] [PubMed] [Google Scholar]
- 36. Conway K. A., Harper J. D., Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 37. Greenbaum E. A., Graves C. L., Mishizen-Eberz A. J., Lupoli M. A., Lynch D. R., Englander S. W., Axelsen P. H., Giasson B. I. (2005) The E46K mutation in α-synuclein increases amyloid fibril formation. J. Biol. Chem. 280, 7800–7807 [DOI] [PubMed] [Google Scholar]
- 38. Li J., Uversky V. N., Fink A. L. (2002) Conformational behavior of human α-synuclein is modulated by familial Parkinson's disease point mutations A30P and A53T. Neurotoxicology 23, 553–567 [DOI] [PubMed] [Google Scholar]
- 39. Choi W., Zibaee S., Jakes R., Serpell L. C., Davletov B., Crowther R. A., Goedert M. (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 576, 363–368 [DOI] [PubMed] [Google Scholar]
- 40. Conway K. A., Lee S. J., Rochet J. C., Ding T. T., Williamson R. E., Lansbury P. T., Jr. (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 571–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bertoncini C. W., Fernandez C. O., Griesinger C., Jovin T. M., Zweckstetter M. (2005) Familial mutants of α-synuclein with increased neurotoxicity have a destabilized conformation. J. Biol. Chem. 280, 30649–30652 [DOI] [PubMed] [Google Scholar]
- 42. Rospigliosi C. C., McClendon S., Schmid A. W., Ramlall T. F., Barré P., Lashuel H. A., Eliezer D. (2009) E46K Parkinson's-linked mutation enhances C-terminal-to-N-terminal contacts in α-synuclein. J. Mol. Biol. 388, 1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J. C., Langen R., Hummel P. A., Gray H. B., Winkler J. R. (2004) α-Synuclein structures from fluorescence energy-transfer kinetics: implications for the role of the protein in Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 16466–16471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brucale M., Sandal M., Di Maio S., Rampioni A., Tessari I., Tosatto L., Bisaglia M., Bubacco L., Samorì B. (2009) Pathogenic mutations shift the equilibria of α-synuclein single molecules towards structured conformers. Chembiochem 10, 176–183 [DOI] [PubMed] [Google Scholar]
- 45. Wise-Scira O., Dunn A., Aloglu A. K., Sakallioglu I. T., Coskuner O. (2013) Structures of the E46K mutant-type α-synuclein protein and impact of E46K mutation on the structures of the wild-type α-synuclein protein. ACS Chem. Neurosci. 4, 498–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coskuner O., Wise-Scira O. (2013) Structures and free energy landscapes of the A53T mutant-type α-synuclein protein and impact of A53T mutation on the structures of the wild-type α-synuclein protein with dynamics. ACS Chem. Neurosci. 4, 1101–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wise-Scira O., Aloglu A. K., Dunn A., Sakallioglu I. T., Coskuner O. (2013) Structures and free energy landscapes of the wild-type and A30P mutant-type α-synuclein proteins with dynamics. ACS Chem. Neurosci. 4, 486–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sahay S., Anoop A., Krishnamoorthy G., Maji S. K. (2014) Site-specific fluorescence dynamics of α-synuclein fibrils using time-resolved fluorescence studies: effect of familial Parkinson's disease-associated mutations. Biochemistry 53, 807–809 [DOI] [PubMed] [Google Scholar]
- 49. Sambrook J. F., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 50. Jakes R., Spillantini M. G., Goedert M. (1994) Identification of two distinct synucleins from human brain. FEBS Lett. 345, 27–32 [DOI] [PubMed] [Google Scholar]
- 51. Volles M. J., Lansbury P. T., Jr. (2007) Relationships between the sequence of α-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol. 366, 1510–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Singh P. K., Kotia V., Ghosh D., Mohite G. M., Kumar A., Maji S. K. (2013) Curcumin modulates α-synuclein aggregation and toxicity. ACS Chem. Neurosci. 4, 393–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ghosh D., Mondal M., Mohite G. M., Singh P. K., Ranjan P., Anoop A., Ghosh S., Jha N. N., Kumar A., Maji S. K. (2013) The Parkinson's disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry 52, 6925–6927 [DOI] [PubMed] [Google Scholar]
- 54. Bitan G., Lomakin A., Teplow D. B. (2001) Amyloid β-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J. Biol. Chem. 276, 35176–35184 [DOI] [PubMed] [Google Scholar]
- 55. Bevington P. R. (1969) Data Reduction and Error Analysis for the Physical Sciences, McGraw Hill, Inc., New York, NY [Google Scholar]
- 56. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., Springer, New York, NY [Google Scholar]
- 57. Royer C. A. (2006) Probing protein folding and conformational transitions with fluorescence. Chem. Rev. 106, 1769–1784 [DOI] [PubMed] [Google Scholar]
- 58. Beechem J. M., Brand L. (1985) Time-resolved fluorescence of proteins. Annu. Rev. Biochem. 54, 43–71 [DOI] [PubMed] [Google Scholar]
- 59. van Rooijen B. D., van Leijenhorst-Groener K. A., Claessens M. M., Subramaniam V. (2009) Tryptophan fluorescence reveals structural features of α-synuclein oligomers. J. Mol. Biol. 394, 826–833 [DOI] [PubMed] [Google Scholar]
- 60. Anoop A., Ranganathan S., Das Dhaked B., Jha N. N., Pratihar S., Ghosh S., Sahay S., Kumar S., Das S., Kombrabail M., Agarwal K., Jacob R. S., Singru P., Bhaumik P., Padinhateeri R., Kumar A., Maji S. K. (2014) Elucidating the role of disulfide bond on amyloid formation and fibril reversibility of somatostatin-14: relevance to its storage and secretion. J. Biol. Chem. 289, 16884–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Krishnamoorthy G. (2012) Motional dynamics in proteins and nucleic acids control their function: revelation by time-domain fluorescence. Curr. Sci. 102, 266–276 [Google Scholar]
- 62. Maji S. K., Amsden J. J., Rothschild K. J., Condron M. M., Teplow D. B. (2005) Conformational dynamics of amyloid β-protein assembly probed using intrinsic fluorescence. Biochemistry 44, 13365–13376 [DOI] [PubMed] [Google Scholar]
- 63. Dusa A., Kaylor J., Edridge S., Bodner N., Hong D. P., Fink A. L. (2006) Characterization of oligomers during α-synuclein aggregation using intrinsic tryptophan fluorescence. Biochemistry 45, 2752–2760 [DOI] [PubMed] [Google Scholar]
- 64. Garzon-Rodriguez W., Vega A., Sepulveda-Becerra M., Milton S., Johnson D. A., Yatsimirsky A. K., Glabe C. G. (2000) A conformation change in the carboxyl terminus of Alzheimer's Aβ (1–40) accompanies the transition from dimer to fibril as revealed by fluorescence quenching analysis. J. Biol. Chem. 275, 22645–22649 [DOI] [PubMed] [Google Scholar]
- 65. Chen Y., Barkley M. D. (1998) Toward understanding tryptophan fluorescence in proteins. Biochemistry 37, 9976–9982 [DOI] [PubMed] [Google Scholar]
- 66. Lakshmikanth G. S., Sridevi K., Krishnamoorthy G., Udgaonkar J. B. (2001) Structure is lost incrementally during the unfolding of barstar. Nat. Struct. Biol. 8, 799–804 [DOI] [PubMed] [Google Scholar]
- 67. Jha A., Udgaonkar J. B., Krishnamoorthy G. (2009) Characterization of the heterogeneity and specificity of interpolypeptide interactions in amyloid protofibrils by measurement of site-specific fluorescence anisotropy decay kinetics. J. Mol. Biol. 393, 735–752 [DOI] [PubMed] [Google Scholar]
- 68. Singh T. S., Rao B. J., Krishnamoorthy G. (2012) GTP binding leads to narrowing of the conformer population while preserving the structure of the RNA aptamer: a site-specific time-resolved fluorescence dynamics study. Biochemistry 51, 9260–9269 [DOI] [PubMed] [Google Scholar]
- 69. Bitan G., Kirkitadze M. D., Lomakin A., Vollers S. S., Benedek G. B., Teplow D. B. (2003) Amyloid β-protein (Aβ) assembly: Aβ 40 and Aβ 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. U.S.A. 100, 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li H. T., Lin X. J., Xie Y. Y., Hu H. Y. (2006) The early events of α-synuclein oligomerization revealed by photo-induced cross-linking. Protein Pept. Lett. 13, 385–390 [DOI] [PubMed] [Google Scholar]
- 71. Vilar M., Chou H. T., Lührs T., Maji S. K., Riek-Loher D., Verel R., Manning G., Stahlberg H., Riek R. (2008) The fold of α-synuclein fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 8637–8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wood S. J., Wypych J., Steavenson S., Louis J. C., Citron M., Biere A. L. (1999) α-Synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J. Biol. Chem. 274, 19509–19512 [DOI] [PubMed] [Google Scholar]
- 73. Fink A. L. (2006) The aggregation and fibrillation of α-synuclein. Acc. Chem. Res. 39, 628–634 [DOI] [PubMed] [Google Scholar]
- 74. Uversky V. N., Li J., Fink A. L. (2001) Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 276, 10737–10744 [DOI] [PubMed] [Google Scholar]
- 75. Ariesandi W., Chang C. F., Chen T. E., Chen Y. R. (2013) Temperature-dependent structural changes of Parkinson's α-synuclein reveal the role of pre-existing oligomers in α-synuclein fibrillization. PLoS One 8, e53487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ono K., Mochizuki H., Ikeda T., Nihira T., Takasaki J., Teplow D. B., Yamada M. (2012) Effect of melatonin on α-synuclein self-assembly and cytotoxicity. Neurobiol. Aging 33, 2172–2185 [DOI] [PubMed] [Google Scholar]