

Background: Loss of function mutations in TfR2 cause iron overload in the body.
Results: CD81, a scaffold protein, controls the level of TfR2 and links TfR2 to E3 ligase, GRAIL.
Conclusion: CD81 down-regulates TfR2 and increases hepcidin levels in Hep3B cells.
Significance: The association of TfR2 with CD81 controls both TfR2 trafficking and hepcidin mRNA.
Keywords: E3 Ubiquitin Ligase, Iron, Iron Metabolism, Protein Degradation, Tetraspanin
Abstract
Mutations in transferrin receptor 2 (TfR2) cause a rare form of the hereditary hemochromatosis, resulting in iron overload predominantly in the liver. TfR2 is primarily expressed in hepatocytes and is hypothesized to sense iron levels in the blood to positively regulate the expression of hepcidin through activation of the BMP signaling pathway. Hepcidin is a peptide hormone that negatively regulates iron egress from cells and thus limits intestinal iron uptake. In this study, a yeast two-hybrid approach using the cytoplasmic domain of TfR2 identified CD81 as an interacting protein. CD81 is an abundant tetraspanin in the liver. Co-precipitations of CD81 with different TfR2 constructs demonstrated that both the cytoplasmic and ecto-transmembrane domains of TfR2 interact with CD81. Knockdown of CD81 using siRNA significantly increased TfR2 levels by increasing the half-life of TfR2, indicating that CD81 promotes degradation of TfR2. Previous studies showed that CD81 is targeted for degradation by GRAIL, an ubiquitin E3 ligase. Knockdown of GRAIL in Hep3B-TfR2 cells increased TfR2 levels, consistent with inhibition of CD81 ubiquitination. These results suggest that down-regulation of CD81 by GRAIL targets TfR2 for degradation. Surprisingly, knockdown of CD81 decreased hepcidin expression, implying that the TfR2/CD81 complex is involved in the maintenance of hepcidin mRNA. Moreover, knockdown of CD81 did not affect the stimulation of hepcidin expression by BMP6 but increased both the expression of ID1 and SMAD7, direct targets of BMP signaling pathway, and the phosphorylation of ERK1/2, indicating that the CD81 regulates hepcidin expression differently from the BMP and ERK1/2 signaling pathways.
Introduction
Hereditary hemochromatosis is an autosomal recessive iron overload disease caused by mutations in genes encoding the first identified hereditary hemochromatosis protein, HFE (1), hemojuvelin (2), hepcidin (3), transferrin receptor 2 (TfR2)2 (4), and ferroportin (5, 6). Accumulation of excessive iron in the liver, heart, pancreas, and pituitary gland results in hepatic cirrhosis, hepatocellular carcinoma, cardiomyopathy, arrhythmias, diabetes, hypogonadotropic hypogonadism, and arthritis. Disease-causing mutations in TfR2 result in type 3 hereditary hemochromatosis. Type 3 hereditary hemochromatosis patients and mouse models of this disease have lower levels of hepcidin than in either humans or mice with the same degree of iron loading (6–10), suggesting that TfR2 is an upstream regulator of hepcidin (HAMP1) expression. Hepcidin, a master regulator of iron homeostasis, is a peptide hormone predominantly produced by the liver. It modulates serum iron levels, by binding to and causing the down-regulation of the only known ferrous ion exporter, ferroportin (11). In murine studies, hepcidin (Hamp1) expression increases in response to iron loading, thus preventing further iron uptake and decreases with iron deficiency to promote increased iron absorption (12, 13).
In the liver, both HAMP1 and TfR2 are primarily expressed in hepatocytes. TfR2 is proposed to sense iron levels in the blood and positively regulate the expression of hepcidin. Our previous studies using an adeno-associated virus vector (rAAV2/8) to express Tfr2 in hepatocytes of Tfr2-deficient mice demonstrated that Tfr2 expression increases hepcidin expression and reduces hepatic iron overload in the Tfr2-deficient mice (14), directly suggesting that TfR2 regulates hepcidin expression. However, the mechanism and signaling pathway by which TfR2 regulates hepcidin expression remains to be determined.
The bone morphogenetic protein (BMP) signaling pathway plays an important role in the regulation of hepcidin expression (15, 16). Recent studies suggest that the BMP receptor/SMAD1,5,8 (BMPR/SMAD) and ERK1/2 signaling pathways are involved in the TfR2-mediated regulation of hepcidin expression, but these results are somewhat controversial (16–18). In the present study we find that CD81, a tetraspanin scaffold protein, serves to maintain TfR2-sensitive hepcidin expression through BMPR/SMAD- and ERK1/2-independent mechanisms.
TfR2 has a short half-life of 2–8 h that can be stabilized by iron-loaded Tf (holo-Tf) (19–22), supporting the hypothesis that TfR2 could serve as an iron sensor to regulate iron homeostasis by modulating hepcidin expression. It is targeted to the lysosome for degradation through the multivesicular body (MVB) pathway without detectable direct ubiquitination (23). In the present study, a yeast two-hybrid assay using the cytoplasmic domain of TfR2 as the “bait” and a human liver library as the “prey” was employed to identify proteins that are responsible for its trafficking within the cell. CD81 scored as a binding partner of TfR2.
CD81 is a broadly expressed protein and is associated with a wide variety of different biological responses, including cell adhesion, morphology, motility, metastasis, proliferation, differentiation, cell activation, and signal transduction (24). It does so by interacting with a variety of signaling molecules. We were interested in CD81 because of its high level of expression in the liver (25) and its regulation by the ubiquitin E3 ligase, GRAIL (gene related to anergy in lymphocytes, Rnf128) (26). GRAIL mRNA is also abundant in the liver (27). The evidence presented here suggests that CD81 and GRAIL positively regulate TfR2 degradation through GRAIL-mediated degradation of CD81.
EXPERIMENTAL PROCEDURES
Plasmids
The pcDNA3/CD81 plasmid was kindly provided by Dr. Vinicio Carloni (Department of Internal Medicine, University of Florence, Florence, Italy) and subcloned into pcDNA3.1-Myc (Invitrogen). The pcDNA3-TfR2ΔCD encoding TfR2 lacking its cytoplasmic domain was generated from pcDNA3-TfR2 by PCR. The chimera, TfR2CD/TfR1-f, containing the cytoplasmic domain of TfR2 and the transmembrane and ecto-domains of TfR1 followed by a Flag epitope was described previously (21). GRAIL cDNA was amplified from the RNA isolated from Hep3B cells, cloned into the pcDNA3 vector, and verified by sequencing.
Cell Culture, Transfection, and Generation of Stable Cell Lines
Hep3B cells were maintained in Eagle's minimum essential medium (Life Technologies, Inc.) supplemented with 1.0 mm sodium pyruvate, 0.1 mm nonessential amino acids (Life Technologies, Inc.), and 10% FBS. The generation of Hep3B stable cell lines expressing TfR2 (Hep3B-TfR2), Hep3B-TfR2CD/TfR1-f, or Hep3B-TfR2ΔCD cell lines were described previously (21). The stable cell lines were maintained in the minimum essential medium with addition of 400 μg/ml Geneticin (G418; Calbiochem). HEK293 cells were maintained in DMEM with addition of 10% fetal bovine serum. They were transiently transfected with plasmids using Effectene transfection reagent (Qiagen) according to the manufacturer's instructions.
RNA Interference
CD81 siRNA and control siRNA was designed according to the manufacturer's instructions and synthesized by Dharmacon, Thermo Scientific. The CD81 siRNA target sequence was UGAUGUUCGUUGGCUUCCUUU. Human GRAIL (RNF128) siRNA was purchased from Qiagen (Hs_RNF128_5 FlexiTube siRNA (NM_024539 and NM_194463)). All of the siRNAs were transfected into cells using RNAiMax (Invitrogen) and the manufacturer's reverse transfection protocol as described previously (23).
Antibodies
The generation of rabbit anti-human TfR2 (anti-hTfR2, 16637) (19), mouse monoclonal anti-hTfR2 (9F8–1C11), and mouse anti-TfR1 (3B82A1) (28) antibodies were described previously. M2 anti-Flag (Sigma-Aldrich), H68.4 anti-TfR1 (Zymed Laboratories Inc.), anti-actin (Sigma-Aldrich), rabbit anti-Myc beads (Sigma-Aldrich), rabbit anti-GRAIL (Santa Cruz), rabbit anti-Myc (Sigma-Aldrich), and mouse anti-actin (Sigma-Aldrich) antibodies were purchased. Secondary antibodies against rabbit and mouse IgG conjugated to HRP were purchased from Chemicon. Fluorescence-conjugated Alexa Fluor 680 goat anti-rabbit IgG was purchased from Molecular Probes. IRDye 800 donkey anti-mouse IgG secondary antibody was purchased from Rockland Immunochemicals.
Immunoprecipitation
Cells were lysed in IP lysis buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 4% CHAPS, and 5% glycerol) containing 1× Complete mini protease inhibitor mixture (Roche Diagnostic). Cell lysates were cleared by centrifugation at 15,000 × g for 10 min. Protein concentration was measured using the BCA protein assay (Pierce). Lysates (200–500 μg of total proteins) were either precleared with Sepharose-4B/protein A beads for 60 min at 4 °C and then rotated overnight at 4 °C with protein A-coated with antibody or incubated with Myc-beads at 4 °C overnight. After centrifugation, the beads were washed three times with IP lysis buffer, and the proteins were eluted using Laemmli loading buffer (29) without boiling at 95 °C. All samples were subjected to 7.5–12.5% gradient SDS-PAGE, and proteins were transferred to nitrocellulose and immunodetected with mouse monoclonal anti-TfR2 (1:10,000), anti-Myc (1:1,000), M2 anti-Flag (1:5,000), or mouse anti-TfR1 (1:10,000) primary antibodies and fluorescence-conjugated secondary antibodies.
Immunoblots
Cells were lysed on ice in 1% NET-Triton buffer (150 mm NaCl, 5 mm EDTA, 10 mm Tris, 1% Triton X-100, pH 7.4) with Complete mini protease inhibitor mixture (Roche Diagnostic) and cleared by centrifugation at 16,000 × g for 30 min, and the supernatant was collected. Protein concentrations of the cell extracts were measured using the BCA protein assay (Pierce). Cell lysate (50 mg) were reduced and denatured with Laemmli buffer and subjected to SDS-PAGE on 10% or 7.5–12.5% gels. Proteins were transferred to nylon-supported nitrocellulose (Maine Manufacturing, Sanford, ME). Immunoblot analysis was carried out using primary antibodies followed by fluorescently labeled secondary antibodies and quantified using a LI-COR infrared fluorescence detector as described previously (21).
qRT-PCR
Total RNA was isolated from Hep3B cells using the RNAeasy RNA isolation kit (Qiagen) and treated with DNase (Roche Applied Science) to remove any contaminating genomic DNA as previously described (14). Oligo(dT) primers and Superscript II reverse transcriptase were used to synthesize cDNA according to the manufacturer's instructions. Hepcidin, CD81, GRAIL, ID1, SMAD7, and GAPDH mRNA were measured using the primers listed below. The mRNA levels of hepcidin, CD81, and GRAIL were normalized to GAPDH. The results were expressed as the level relative to control siRNA-treated cells. All primers were verified for linearity of amplification (Table 1).
TABLE 1.
Primers used
| Primers | Sequence |
|---|---|
| CD81 forward | 5′-GTG ATC CTG GGT GCC CT-3′ |
| CD81 reverse | 5′-CAT CAT CCA CCA CGG CCT GC-3′ |
| GAPDH forward | 5′-ACC CAC TCC TCC ACC TTT GA-3′ |
| GAPDH reverse | 5′-CTG TTG CTG TAG CCA AAT TCG-3′ |
| Hepcidin forward | 5′-GGC TCT GTT TTC CCA CAA CAG-3′ |
| Hepcidin reverse | 5′-TCC TTC GCC TCT GGA ACA TG-3′ |
| ID1 forward | 5′-ACG ATC GCA TCT TGT GTC GCT-3′ |
| ID1 reverse | 5′-AGA CCC ACA GAG CAC GAA TT-3′ |
| SMAD7 forward | 5′-CAA TGA CCA CGA GTT TAT GCA-3′ |
| SMAD7 reverse | 5′-GTT GAA GAT GAC CTC TAG CCA-3′ |
Half-life Determination
Hep3B-TfR2 cells in a 100-mm plate were transfected with control siRNA (lane C and column C) or CD81 siRNA for 32 h and then split into 35-mm plates for 16 h. Cycloheximide was added into the medium to a final concentration of 150 μg/ml for 0, 2, 4, 8, and 16 h. Cells were solubilized, and the lysates were subjected to SDS-10% PAGE. The TfR2 and actin were detected on immunoblots. The integrated intensity of TfR2 was normalized to that of actin, which did not change detectably over the time course of the experiment. The normalized intensity was expressed as a percentage of the intensity at time 0. Half-life was determined by single exponential decay (Prism, GraphPad, La Jolla, CA). The graph shows the mean of 10 experiments; error bars indicate standard deviation.
Cell Surface Biotinylation
Cells seeded in 35-mm dish for 16 h were transfected with siRNA. After 2 days, they were washed three times with ice-cold PBS+ buffer (PBS with 0.5 mm CaCl2 and 1 mm MgCl2) and incubated with 0.25 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Pierce) in PBS+ buffer for 30 min on ice. The reaction was quenched by washing the cells five-times with ice-cold PBS buffer containing 0.1% glycine (pH 7.6). Cells were solubilized, and cell lysate was cleared by centrifugation at 16,000 × g for 5 min. Biotinylated proteins were isolated with streptavidin-agarose and subjected to SDS-10% PAGE followed by immunodetection with anti-TfR2, anti-TfR1, anti-actin, and anti-Flag antibodies.
Statistical Analysis
The difference between groups was analyzed by the paired two-tailed Student's t test using Prism (Prism; GraphPad).
RESULTS
TfR2 Interacts with CD81
We wanted to determine the proteins that interacted with the cytoplasmic domain of TfR2 because this domain often plays roles both in the trafficking of membrane proteins and in signaling. To identify the interacting partners of TfR2, a yeast two-hybrid screen using the cytoplasmic domain of TfR2 as the “bait” and a human liver library as the “prey” was used. CD81 was identified to interact with TfR2. To confirm the physical interaction between TfR2 and CD81, CD81-Myc and full-length TfR2 or pcDNA3 empty vector were transiently co-transfected into HEK293 cells, an easy to transfect human cell line often used to detect protein-protein interactions. CD81-Myc was immunoprecipitated with anti-Myc antibody. Immunoblot analysis of the immunoprecipitate showed that TfR2 co-precipitated with CD81 (Fig. 1A, left panel). No TfR2 was detected in immunoprecipitates of the mock-transfected samples, indicating that TfR2 does not interact with the Myc antibody. To further confirm the interaction between TfR2 and CD81, a reciprocal immunoprecipitation with anti-TfR2 antibody was performed. CD81 could be detected in TfR2 isolates (Fig. 1A, right panel). Unlike TfR2, TfR1 did not interact detectably with CD81 (Fig. 1B), further suggesting that the interaction between CD81 and TfR2 was specific.
FIGURE 1.
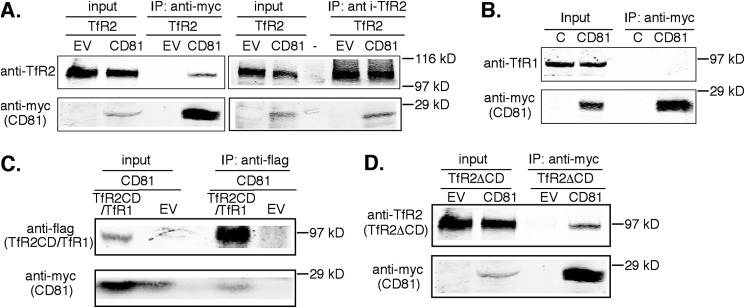
TfR2 interacts with CD81. HEK293 cells were transiently transfected with TfR2 constructs and either CD81-Myc or empty vector (EV). Cells were lysed in IP lysis buffer 48 h post-transfection, 10% of the lysate was reserved (input), and the remaining 90% was subjected to IP with rabbit anti-Myc coupled to beads or anti-TfR2 antibody and protein A-agarose beads. Co-immunoprecipitated proteins were detected with mouse anti-TfR2 and anti-Myc antibodies. A, full-length TfR2 interacts with CD81. TfR2 only co-immunoprecipitates with anti-Myc in the presence of CD81-Myc and the reverse. B, endogenous TfR1 fails to interact with CD81. HEK293 cells were transiently transfected with CD81-Myc. Untransfected HEK293 cells were used as control (lane C). CD81 was immunoprecipitated with anti-Myc beads. Co-immunoprecipitated proteins were detected by Western blot with mouse anti-TfR1 and anti-Myc antibodies. C, the cytoplasmic domain of TfR2 interacts with CD81. TfR2-cyto/TfR1-tm-ecto-Flag (TfR2CD/TfR1-f) or pcDNA3 empty vector and CD81 were transiently co-transfected into HEK293 cells. TfR2CD/TfR1-f was immunoprecipitated with mouse anti-Flag antibody. Co-immunoprecipitated proteins were analyzed by immunoblot with mouse anti-Flag and mouse anti-TfR2 antibodies. D, the truncated TfR2 lacking the cytoplasmic-domain of TfR2 (TfR2ΔCD) interacts with CD81. TfR2ΔCD and CD81-Myc or empty vector were transiently co-transfected into HEK293 cells. CD81 was immunoprecipitated with mouse anti-Myc antibody. Co-immunoprecipitated proteins were analyzed by immunoblot with mouse anti-Myc and mouse monoclonal anti-TfR2 antibodies. All of the immunoprecipitations were repeated at least twice with the same results.
The interaction between TfR2 and CD81 was tested further. Because TfR1 does not interact with CD81, a chimera containing the cytoplasmic domain of TfR2 (TfR2CD) and the transmembrane and ecto-domains of TfR1 with a Flag epitope at the C-terminal (TfR2CD/TfR1-f) was used to confirm the interaction. TfR2CD/TfR1-f and CD81-Myc or empty vector were transiently transfected into HEK293 cells. As predicted from the yeast two-hybrid screen, CD81-Myc interacted with TfR2CD/TfR1-f (Fig. 1C). To determine whether the transmembrane and ecto-domains of TfR2 also interact with CD81, a truncated TfR2 lacking the cytoplasmic domain (TfR2ΔCD) and CD81-Myc were transiently transfected into HEK293 cells. TfR2ΔCD co-precipitated with CD81-Myc (Fig. 1D). Therefore, both the cytoplasmic domain of TfR2 and the transmembrane/ecto-portion of TfR2 interact with CD81.
Increased Expression of CD81 Decreases the Levels of TfR2, TfR2CD/TfR1-f, and TfR2ΔCD
We noticed that increased expression of CD81 resulted in decreased TfR2 in HEK293 cells (Fig. 1, A and D) and suspected that CD81 could be playing a role in the degradation of TfR2. TfR2 is degraded in lysosomes without detectable ubiquitination (23), but those results did not rule out the possibility that a binding partner of TfR2 could be ubiquitinated. CD81 is ubiquitinated by GRAIL (26). To test whether the interaction between CD81 and TfR2 contributed to the down-regulation of TfR2, Hep3B cells were stably transfected with TfR2, TfR2CD/TfR1-f, or TfR2ΔCD. This hepatoma cell line was chosen because it does not express detectable TfR2 but does endogenously express CD81. To test whether CD81 decreased the steady-state level of TfR2, Hep3B-TfR2 cells were transiently transfected with CD81-Myc for 48 h. TfR2 decreased modestly, whereas TfR1 remained the same (Fig. 2A), implying that CD81 was not producing a generalized down-regulation of plasma membrane proteins. Similarly, increased expression of CD81-Myc decreased the levels of TfR2CD/TfR1-f (Fig. 2B) and of TfR2ΔCD (Fig. 2C). Similar to cells expressing TfR2, the level of TfR1 did not change in TfRΔCD-expressing cells. TfR1 level showed a tendency to decline in the TfR2CD/TfR1-f cells, most likely because of the formation of heterodimers between TfR1 and TfR2CD/TfR1-f (Fig. 2B). The ectodomain of TfR1 contains the dimerization motif (30). Together, the results in Figs. 1 and 2 indicate that the level of TfR2 is inversely associated with the expression of CD81.
FIGURE 2.
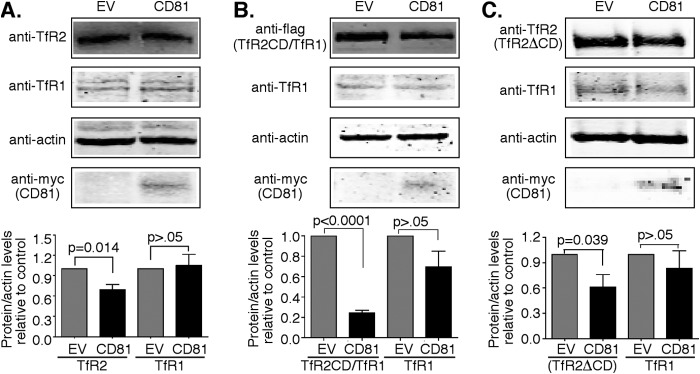
Overexpression of CD81 decreases the levels of TfR2. A, overexpression of CD81 decreases the levels of TfR2 but not TfR1. Hep3B-TfR2 cells were transfected with pcDNA3 empty vector (EV) or pcDNA3-CD81-Myc (CD81) for 48 h. Blots of cell lysates were probed with anti-TfR2, anti-TfR1, and anti-actin antibodies. The levels of TfR2 and TfR1 were normalized to that of actin. The change of TfR2 and TfR1 relative levels and CD81 in response to overexpression of CD81 was normalized to the mock-transfected cells. The graph shows the average results of 16 independent experiments. B, overexpression of CD81 decreases the levels of TfR2CD/TfR1-f. Hep3B-TfR2CD/TfR1-f cells were transfected with empty vector pcDNA3 (empty vector) or pcDNA3-CD81-Myc (CD81) for 48 h. The immunoblot was probed with rabbit-anti-Flag, anti-actin, and H68.4, an antibody to the cytoplasmic domain of TfR1. The changes of TfR2CD/TfR1-f and TfR1 levels were analyzed as described in A. The graphs show the average data from nine experiments. C, overexpression of CD81-Myc decreases the levels of TfR2ΔCD. Hep3B-TfR2ΔCD cells were transfected with control pcDNA3 or pcDNA3-CD81-Myc (CD81) for 48 h. The levels of TfR2ΔCD and TfR1 were immunodetected and analyzed as described in A. The graph in C shows the average data from three experiments.
Knockdown of CD81 Increases the Levels of TfR2, TfR2CD/TfR1-f and TfR2ΔCD
Because increased expression of CD81 decreases the steady-state levels of TfR2, knockdown of CD81 could be expected to increase TfR2 levels. Hep3B-TfR2 cells were transfected with CD81 siRNA to down-regulate endogenous CD81, and TfR2 was detected by immunoblot to test this possibility. TfR2 increased 2.3-fold, whereas TfR1 remained unchanged when CD81 mRNA was decreased by approximately a factor of 2 (Fig. 3A). Similarly, knockdown of CD81 increased TfR2CD/TfR1-f and ΔCDTfR2 (Fig. 3, B and C) when CD81 mRNA decreased by approximately a factor of 2 after treating with CD81 siRNA. A control siRNA (lane C and column C) was used to test for nonspecific siRNA effects in all experiments. Again, the level of TfR1 was higher in the cells expressing TfR2CD/TfR1-f when cells were treated with CD81 siRNA, presumably because of heterodimerization of TfR1 with TfR2CD/TfR1-f. The higher level of TfR2 when CD81 was decreased supports a role for CD81 maintenance of the steady-state level of TfR2.
FIGURE 3.

Knockdown of CD81 increases the levels of TfR2, TfR2CD/TfR1-f, and TfR2ΔCD. A, knockdown of CD81 increases the levels of TfR2 but not TfR1. Hep3B-TfR2 cells were transfected with control siRNA (lane C and column C) or siRNA against CD81 (siRNA CD81) for 48 h. The blot was probed with anti-TfR2, anti-TfR1, and anti-actin antibodies. The protein levels of TfR2 and TfR1 were normalized to that of actin. CD81 mRNA was normalized to GAPDH mRNA. The changes in TfR2 and TfR1 and CD81/GAPDH mRNA in response to knockdown of CD81 were normalized to control siRNA-treated cells. The graphs represent the average data of 20 experiments. B, knockdown of CD81 increases the protein level of the TfR2CD/TfR1-f chimera. Hep3B-TfR2CD/TfR1-f cells were transfected as described in A. The blot of the cell lysates was probed with anti-Flag and anti-actin antibodies. The levels of TfR2CD/TfR1-f and CD81 mRNA were analyzed as described in A. The graphs show the average data from nine experiments. C, knockdown of CD81 increases the levels of TfR2ΔCD but not TfR1. Hep3B-TfR2ΔCD cells were treated and analyzed as described in A. The graphs show the average data from seven experiments.
Knockdown of CD81 Stabilizes TfR2 by Increasing Its Half-life
An increase in the steady-state levels of TfR2 could be caused by a decrease of TfR2 degradation. To test whether this were the case, Hep3B-TfR2 cells were treated with CD81 siRNA or control siRNA. Protein synthesis was inhibited with cycloheximide, and the decay of TfR2 was quantified. The half-life of TfR2 increased from ∼5 h to >16 h when CD81 mRNA was decreased by ∼60% (Fig. 4). These results indicate that knockdown of CD81 stabilizes TfR2 by increasing its half-life and that CD81 promotes the degradation of TfR2.
FIGURE 4.
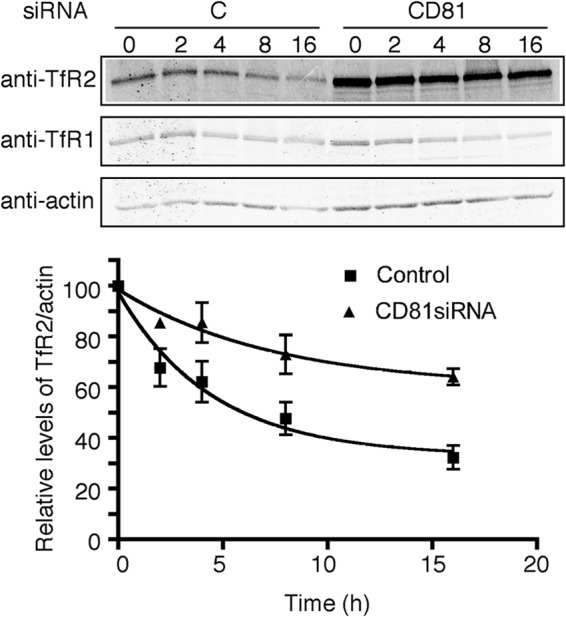
Knockdown of CD81 stabilizes TfR2 by increasing its half-life. The half-life of TfR2 was calculated as described under “Experimental Procedures.” The p value using Student's t test (two-tailed) comparing the two sets of data at each time point is 0.023.
Knockdown of CD81 Decreases the Ratio of Cell Surface to Internal TfR2
The stabilization of TfR2 when CD81 is depleted could result from a number of different mechanisms. CD81 could facilitate the endocytosis of TfR2 from the plasma membrane. Transfection of cells with CD81 siRNA would then increase the ratio of cell surface to total TfR2. Alternately, if CD81 facilitates the trafficking of TfR2 to lysosomes, then its depletion would result in a lower TfR2 cell surface to total ratio. To distinguish between these possibilities, cells were biotinylated at 4 °C, and cell surface proteins in the solubilized lysates were isolated with streptavidin beads. The amount of biotinylated to total TfR2 was quantified on immunoblots (Fig. 5). A decrease of 32% in cell surface TfR2 was seen in cells transfected with CD81 siRNA. No significant change in the amount of TfR1 or its distribution was detected, demonstrating specificity. No biotinylated actin was detected, indicating that the cell remained intact during the labeling procedure. The increase in the total amount of TfR2, as well as the decreased ratio of cell surface to internal TfR2 upon knockdown of CD81, favors the hypothesis that CD81 facilitates the trafficking of TfR2 to lysosomes rather than acting at the plasma membrane to facilitate endocytosis.
FIGURE 5.
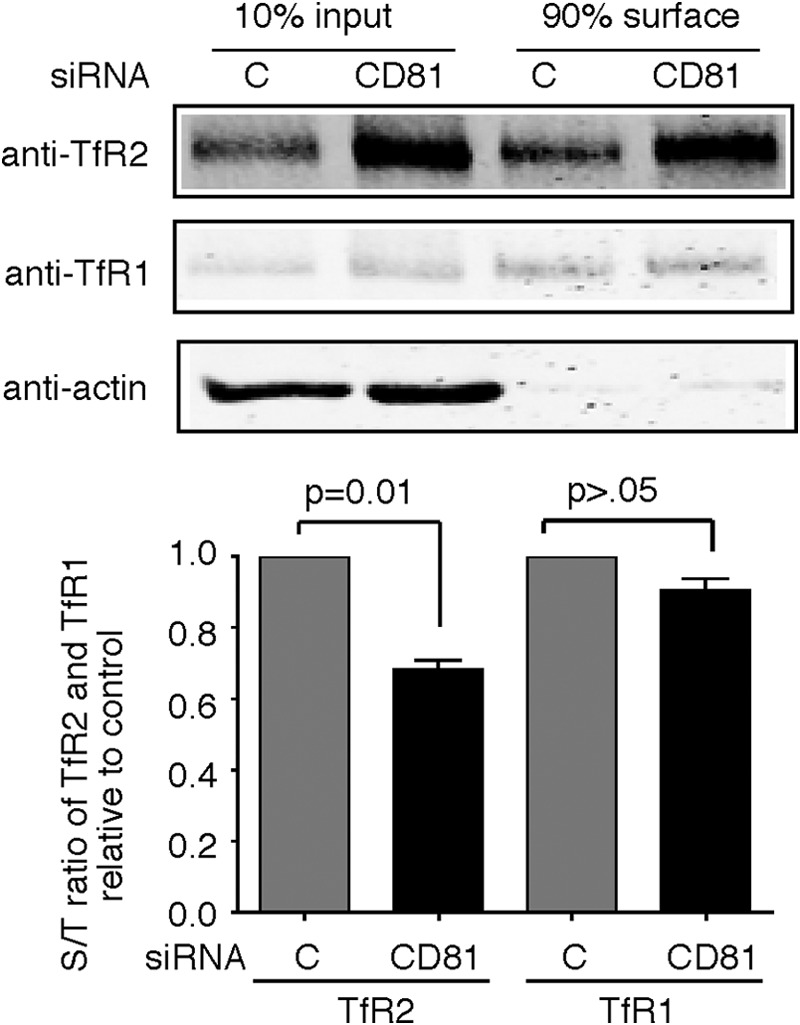
Knockdown of CD81 decreases the surface/total ratio of TfR2. Knockdown of CD81 decreases the surface/total ratio (S/T) of TfR2 but not TfR1. Hep3B-TfR2 cells were transfected with control siRNA (lane C and column C) or CD81 siRNA for 48 h. The cell surface proteins were biotinylated for 30 min on ice. The biotinylated proteins in 90% of the cell lysate were pulled down using streptavidin-agarose beads. 10% lysate was used as input. The samples were subjected to SDS-10% PAGE. The blot was probed with anti-TfR2, anti-TfR1, and anti-actin antibodies. The surface/total ratio of TfR2 and TfR1 was calculated. The fold change of the surface/total ratio of TfR2 and TfR1 in response to knockdown of CD81 was normalized to control siRNA-treated cells. The experiment was repeated four times with similar results.
Steady-state Levels of TfR2 Are Increased by Knockdown of the E3 Ligase, GRAIL
Earlier studies indicated TfR2 is degraded in lysosomes through the MVB pathway without detectable ubiquitination (23). Because CD81 promotes TfR2 degradation and CD81 has been shown to be ubiquitinated by a number of E3 ligases including GRAIL (gene related to anergy in lymphocytes) (26), the association of CD81 with TfR2 could be responsible for the degradation of TfR2. GRAIL is expressed in the liver (31) along with CD81 and TfR2. Therefore, we examined whether GRAIL contributes to the degradation of TfR2 through its interaction with CD81. To test this possibility, siRNA was used to down-regulate endogenous GRAIL in Hep3B-TfR2 cells, and the TfR2 levels were analyzed. TfR2 was significantly increased (Fig. 6A) when GRAIL mRNA was decreased by ∼90% (Fig. 6B), suggesting that knockdown of GRAIL decreases degradation of CD81, thereby increasing TfR2. Consistent with this idea, overexpression of GRAIL in Hep3B-TfR2 cells down-regulated CD81 and did not appreciably down-regulate TfR2 levels (Fig. 6, C and D). These results suggest that in the absence of CD81, TfR2 is not directed to the MVB for degradation.
FIGURE 6.
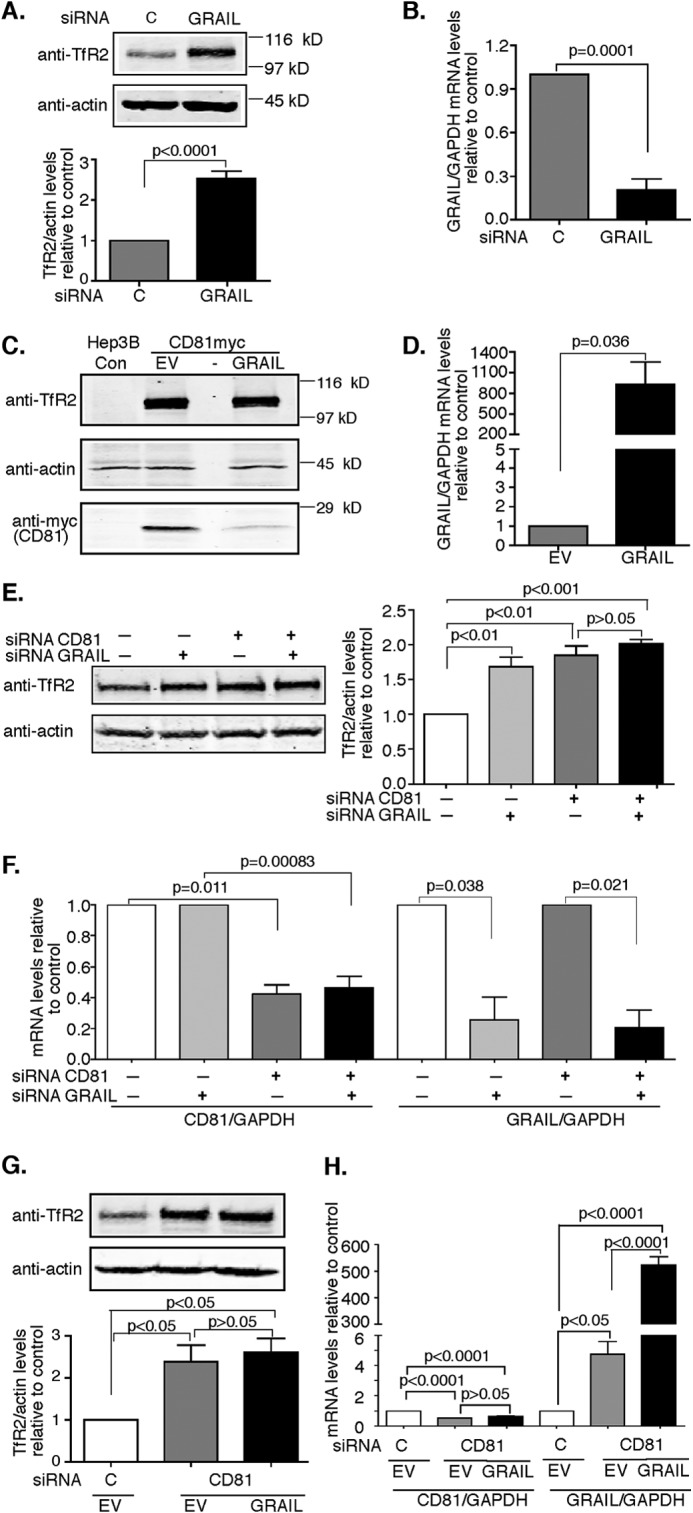
Knockdown of GRAIL increases TfR2 in a CD81-dependent manner. A and B, knockdown of GRAIL increases TfR2. Hep3B-TfR2 cells were treated with control (lane C and column C) or GRAIL siRNA for 48 h. Cell lysates were subjected to 10% SDS-PAGE. The blot was probed with anti-TfR2 and anti-actin antibodies. The levels of TfR2 were normalized to those of actin. The mRNA levels of GRAIL were quantified by qRT-PCR and normalized to GAPDH. The fold change of TfR2 protein levels and GRAIL/GAPDH mRNA levels in response to knockdown of GRAIL was normalized to control siRNA-treated cells. C, overexpression of GRAIL down-regulates CD81 levels in Hep3B-TfR2 cells. Hep3B-TfR2 cells were transient transfected with CD81-Myc and empty vector (EV) or GRAIL for 48 h. Blots of cell lysates were probed with anti-TfR2, anti-actin, and anti-Myc antibodies. Hep3B cells (Hep3B-C) were used to demonstrate the specificity of the Myc and TfR2 antibodies. The mRNA levels of GRAIL were measured by qRT-PCR and analyzed as described in B. E and F, GRAIL does not regulate TfR2 independently of CD81. Hep3B-TfR2 cells were transfected with control siRNA, CD81siRNA, GRAIL siRNA, or both CD81siRNA and GRAIL siRNA for 48 h. TfR2 protein (E) and CD81 and GRAIL (F) mRNA were analyzed as described above. G and H, overexpression of GRAIL does not affect the up-regulation of TfR2 when CD81 is depleted. Hep3B-TfR2 cells were transfected with control siRNA or CD81siRNA for 48 h and then transfected with empty vector or pcDNA-GRAIL. TfR2 protein (G) and CD81 and GRAIL (H) mRNA were analyzed as described above. All of the experiments were repeated twice with similar results. Con, control.
GRAIL could regulate TfR2 degradation independently of CD81. CD81 was depleted using siRNA to test for this possibility. Knockdown of CD81 or GRAIL increased TfR2 by ∼1.8-fold. Knockdown of both CD81 and GRAIL had no additional effect on TfR2 compared with single knockdown of either CD81 or GRAIL (Fig. 6, E and F). These results indicated that GRAIL fails to regulate TfR2 independently of CD81. To further confirm these results, Hep3B-TfR2 cells were transiently transfected with CD81 siRNA for 1 day and then split into two wells followed by transfection with the GRAIL plasmid or empty vector. When CD81 was suppressed by ∼60%, TfR2 increased by ∼2.2-fold whether or not GRAIL was overexpressed (Fig. 6, G and H), suggesting that overexpression of GRAIL has no effect on the TfR2 levels independently of CD81.
Tf also regulates the stability of TfR2 in hepatoma cells and in the liver (19, 22). Down-regulation of GRAIL did not abolish the increase in stability of TfR2 in the presence of Tf.3 Previous studies indicate that Tf increases the recycling of TfR2 from endosomal compartments back to the plasma membrane (21, 23, 32). These results suggest that recycling occurs from an earlier compartment than the CD81/GRAIL-mediated degradation signal occurs.
CD81 Increases Hepcidin Expression in Hep3B Cells in the Presence of TfR2
TfR2 plays an important role in the regulation of hepcidin expression. Lack of functional TfR2 results in low hepcidin levels leading to iron overload. To determine whether the TfR2/CD81 complex regulates hepcidin expression, hepcidin mRNA levels were measured from the cells described in Fig. 3. In Hep3B-TfR2 cells, when CD81 mRNA was suppressed ∼2-fold (Fig. 3B), hepcidin mRNA levels decreased ∼2-fold (Fig. 7A), indicating that CD81 participates in the regulation of hepcidin mRNA in Hep3B cells expressing TfR2. Similar results were obtained in the Hep3B-TfR2CD/TfR1-f and Hep3B-TfR2ΔCD cells (Fig. 7A). These results suggest that both the cytoplasmic domain and transmembrane/ecto-portion of TfR2 interact with CD81 and together with CD81 modulate hepcidin expression.
FIGURE 7.

CD81 and TfR2 are both required for the regulation of hepcidin. A, knockdown of CD81 decreases hepcidin expression in Hep3B-TfR2, Hep3B-TfR2CD/TfR1-f, and Hep3B-ΔCDTfR2 cells. The mRNA levels of hepcidin in the cells transfected with control siRNA (lane C and column C) or CD81 siRNA for 48 h in Fig. 2 were measured by qRT-PCR and normalized to GAPDH. Hepcidin mRNA in response to knockdown of CD81 was normalized to that of control siRNA-treated cells. The experiments were repeated at least four times with similar results. B, knockdown of CD81 does not affect hepcidin expression in Hep3B cells. Hep3B cells were transfected with control siRNA (lane C and column C) or CD81 siRNA for 48 h. The mRNA levels of CD81/GAPDH and hepcidin/GAPDH were analyzed as described in A. The experiments were repeated twice with similar results. C, CD81 does not regulate hepcidin expression through BMP/SMAD signaling pathway. Hep3B-TfR2 cells (100 mm dish) were transfected with control siRNA or CD81 siRNA for 24 h and then split into a 6-well plate and grown overnight. The cells were then treated with or without 10 ng/ml BMP6 for 6 h. The mRNA levels of CD81, hepcidin, ID1, and SMAD7 were measured by qRT-PCR, normalized to GAPDH, and then expressed as the ratio of that of control siRNA-transfected cells (lane C and column C) in the absence of BMP6. The graph shows the average data from four independent experiments. D, knockdown of CD81 increased the levels of phospho-ERK1/2. Hep3B-TfR2 cells transfected control siRNA or CD81 siRNA for 32 h were treated with DMSO or 10 μm U0126 overnight. p-ERK, total ERK1/2, TfR2, and actin were detected by immunoblot analysis. E, U0126 increased hepcidin expression independent of CD81. CD81/GAPDH and hepcidin/GAPDH mRNA levels were normalized to control siRNA-DMSO-treated cells. The experiments in D and E were done in duplicate and repeated twice with similar results. F, U0126 increased hepcidin expression independent of TfR2. Hep3B and Hep3B-TfR2 cells were treated with DMSO (lane C and column C) or 10 μm U0126 for 4 h. The hepcidin/GAPDH mRNA levels were normalized to DMSO-treated cells. F, U0126 increased hepcidin expression independent of TfR2. Hep3B and Hep3B-TfR2 cells were treated with DMSO as a control (lane C and column C) or 10 μm U0126 for 4 h. The hepcidin/GAPDH mRNA levels were normalized to DMSO-treated cells.
We tested whether overexpression of CD81 increases hepcidin expression, because the suppression of CD81 decreased hepcidin expression. Overexpression of CD81 did not reproducibly increase hepcidin expression (data not shown), which indicates that the endogenous level of CD81 is enough to maintain hepcidin levels in Hep3B-TfR2 cells, and CD81 is not a limiting factor in the regulation of hepcidin expression in this cell line. To determine whether CD81 regulates hepcidin expression independently of TfR2, Hep3B cells were used. This cell does not express detectable levels of TfR2. When endogenous CD81 mRNA levels were decreased by 71% using CD81 siRNA, hepcidin mRNA levels were not significantly changed (Fig. 7B), indicating that TfR2 is required for the regulation of hepcidin by CD81.
The BMPR/SMAD signaling pathway plays an important role in the regulation of hepcidin expression (15, 16). Several studies show that a lack of TfR2 results in lower hepcidin and phosphorylated SMAD1,5,8 (pSMAD), which implies that TfR2 may directly regulate the BMPR/SMAD signaling pathway (16, 17). BMP6 is a potent activator of this signaling pathway. BMP6 treatment of cells increased the mRNA levels of hepcidin, to the same extent in control siRNA-treated cells and CD81 siRNA-treated cells (Fig. 7C). If CD81 was directly suppressing hepcidin mRNA through suppressing BMPR/SMAD1,5,8 signaling, then it should suppress the induction of ID1 and SMAD7, two other downstream targets of the BMPR/SMAD signaling pathway. To test whether CD81 increased, the BMP-BMPR pathway cells were treated CD81 siRNA. Knockdown of CD81 increased rather than decreased ID1 and SMAD7 mRNA levels (Fig. 7C). These results suggest that the decrease in hepcidin expression seen in cells depleted of CD81 is not due to a decrease in BMPR/SMAD signaling.
The roles that CD81 and TfR2 play in the control of hepcidin mRNA through the ERK/MAPK signaling pathway was next examined. CD81 sustains T-cell activation through the ERK/MAPK kinase signaling pathway (33, 34). This signaling pathway is reported to be involved in the regulation of hepcidin expression, although the results are somewhat controversial (16, 18, 35). If the CD81/TfR2 complex up-regulates hepcidin expression through the ERK1/2 pathway, then the knockdown of CD81 should decrease the phosphorylation of ERK1/2 (pERK1/2). To determine whether this were the case, Hep3B-TfR2 cells were transfected with control siRNA or CD81 siRNA. p-ERK1/2 was determined by immunoblot. U0126, a specific MAPK/ERK kinase inhibitor, was used as a control for the down-regulation of p-ERK1/2. U0126 decreased the p-ERK1/2 levels in both control siRNA and CD81 siRNA-treated cells (Fig. 7D). Depletion of CD81 expression decreased hepcidin expression as expected (Fig. 7E) but increased the levels of p-ERK1/2 (Fig. 7D). In addition, U0126 increased hepcidin mRNA levels in both control siRNA and CD81 siRNA-treated Hep3B-TfR2 cells (Fig. 7E). To determine whether the increase of hepcidin expression by U0126 is dependent on TfR2, both Hep3B-TfR2 and Hep3B cells were treated with or without U0126 for 4 h. U0126 increased hepcidin mRNA levels to the same extent in both Hep3B-TfR2 and Hep3B cells (Fig. 7F), suggesting that the up-regulation of hepcidin by U0126 is independent of TfR2. Taken together, CD81/TfR2 complex regulates hepcidin expression differently from the predicted BMP/SMAD and ERK1/2 signaling pathways.
DISSCUSSION
In this study, we identified an interaction between CD81 and TfR2 that promoted the CD81-dependent degradation of TfR2 by the E3-ligase, GRAIL. Previous results show that TfR2 is degraded in the lysosome through the MVB pathway without detectable ubiquitination, indicating that TfR2 is degraded either by ubiquitination-independent mechanism or by binding to a ubiquitinated protein. Knockdown of GRAIL expression increased TfR2 levels, suggesting that the silencing of GRAIL blocks TfR2 degradation by blocking CD81 degradation.
CD81 is a scaffold protein involved in signaling and membrane remodeling events (reviewed in Ref. 36). It has been mainly studied for its role in adaptive immunity in B- and T-cell function (reviewed in Ref. 37). In the liver, CD81 is known as the receptor for hepatitis C virus (38). We showed an additional function for CD81 in the liver. Not only did CD81 promote the degradation of TfR2, but it participated in the maintenance of hepcidin mRNA. Signal transduction molecules affect membrane trafficking, and in turn, membrane trafficking also regulates signal transduction events (39). Trafficking of cell surface receptors to the lysosome provide a mechanism for down-regulation of receptor signaling (40). For example, the binding of the EGF to EGF receptor (EGFR) both induces signaling as well as the lysosomal degradation of EGFR through MVB pathway (41–46). In addition, endocytosis of signaling receptors can increase signaling. Endocytic trafficking of EGFR is necessary for full Tyr phosphorylation and activation of MAP kinase of the EGFR (39). Similarly, Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate) promotes degradation of tyrosine kinase receptor through trafficking of the receptor to MVBs to attenuate tyrosine kinase receptor signaling (47). β-Arrestin mediates internalization and degradation of protease-activated receptor 2 and to turn off protease-activated receptor 2 signaling (48). It also facilitates activation of ERK1/2 kinase and acts a as scaffold to bring ERK1/2 and JunK3 together to promote signaling from endocytic vesicles (49). Thus endosomal vesicles, MVB, and lysosome may serve as primary compartments to stimulate and later attenuate signaling. We propose that CD81 acts both to increase TfR2 signaling and degradation.
Interestingly, we found that knockdown of CD81 significantly decreased hepcidin expression in Hep3B-TfR2 but not in Hep3B cells without detectable TfR2 protein, indicating that CD81 and TfR2 are both required for the regulation of hepcidin expression. The BMPR/SMAD1,5,8 signaling pathway plays a central role in the regulation of hepcidin expression (50, 51). BMP binds to BMP receptors, BMP receptor I phosphorylates BMP receptor II, which then phosphorylates SMAD1,5,8. The phospho-SMAD1/5/8 associates with SMAD4. This complex translocates to the nucleus to activate SMAD binding elements of the hepcidin promoter, thereby activating hepcidin expression (37, 52, 53). Tfr2-deficient mice have lower levels of phosphorylated SMAD1,5,8 relative to liver iron status, indicating that TfR2 could regulate hepcidin expression through BMPR/SMAD signaling pathway (17, 54). Reduction of CD81 by siRNA decreased hepcidin expression by ∼50% but increased mRNA levels of ID1 and SMAD7 by ∼3-fold. In addition, CD81 does not affect the stimulation of hepcidin, ID1, and SMAD7 by BMP6. These results suggest that the regulation of CD81 on hepcidin expression is independent of BMPR/SMAD signaling pathway.
Stimulation of ERK1/2 is another possible pathway that could lead to the control of hepcidin expression. Poli et al. (16) found that both p-Smad and p-ERK1/2 were decreased in Tfr2 null mice and that TfR2 and HFE increased holo-Tf-mediated induction of p-ERK1/2 in HepG2 cells. Ramey et al. (18) showed that iron-loaded transferrin (holo-Tf) activates ERK1/2 kinase and increases the levels of p-ERK1/2, p-Smad, and hepcidin in mouse primary hepatocytes, indicating that holo-Tf mediates cross-talk between ERK1/2 and BMP signaling pathway to regulate hepcidin expression. These findings suggest that TfR2 might regulate hepcidin expression through BMP/SMAD and/or ERK1/2 signaling pathway. Calzolari et al. (35) showed that holo-Tf and anti-TfR2 antibody stimulates ERK1/2 kinase in K562 cells but not in a TfR2-negative subclone of K562 cells, indicating that holo-Tf and anti-TfR2 antibody activates ERK1/2 signaling through binding to TfR2. However, our results did not show the involvement of TfR2 in ERK1/2 signaling.
A previous study reported that TfR2 binds CD81 and is internalized in a caveolin-dependent manner (35), which is in contrast to another study showing that TfR2 is internalized through AP-2-dependent clathrin-mediated pathway (23). The basis for these differences is not totally clear.
In conclusion, we have shown that CD81 interacts with TfR2, and CD81 promotes hepcidin expression and TfR2 degradation via a GRAIL-mediated pathway. The increase in hepcidin expression involves neither the BMPR/SMAD nor the ERK1/2 signaling pathway. How TfR2 regulates hepcidin expression will be the subject of future study.
Acknowledgments
We thank Kristina DeMaster and Annie Wentz for the technical support and Dr. An-Sheng Zhang and Dr. Ningning Zhao (Oregon Health & Science University) for careful reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-DK072166 and R37-DK054488 (to C. A. E.). This work was also supported by American Heart Association Grant 11POST5250029 and a Collins Medical Trust grant (to J. C.).
J. Chen and C. A. Enns, unpublished results.
- TfR
- transferrin receptor
- BMP
- bone morphogenetic protein
- BMPR
- BMP receptor
- MVB
- multivesicular body
- IP
- immunoprecipitation
- qRT-PCR
- quantitative RT-PCR
- EGFR
- EGF receptor.
REFERENCES
- 1. Feder J. N., Gnirke A., Thomas W., Tsuchihashi Z., Ruddy D. A., Basava A., Dormishian F., Domingo R., Jr., Ellis M. C., Fullan A., Hinton L. M., Jones N. L., Kimmel B. E., Kronmal G. S., Lauer P., Lee V. K., Loeb D. B., Mapa F. A., McClelland E., Meyer N. C., Mintier G. A., Moeller N., Moore T., Morikang E., Prass C. E., Quintana L., Starnes S. M., Schatzman R. C., Brunke K. J., Drayna D. T., Risch N. J., Bacon B. R., Wolff R. K. (1996) A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 13, 399–408 [DOI] [PubMed] [Google Scholar]
- 2. Papanikolaou G., Samuels M. E., Ludwig E. H., MacDonald M. L., Franchini P. L., Dubé M. P., Andres L., MacFarlane J., Sakellaropoulos N., Politou M., Nemeth E., Thompson J., Risler J. K., Zaborowska C., Babakaiff R., Radomski C. C., Pape T. D., Davidas O., Christakis J., Brissot P., Lockitch G., Ganz T., Hayden M. R., Goldberg Y. P. (2004) Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet. 36, 77–82 [DOI] [PubMed] [Google Scholar]
- 3. Roetto A., Papanikolaou G., Politou M., Alberti F., Girelli D., Christakis J., Loukopoulos D., Camaschella C. (2003) Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 33, 21–22 [DOI] [PubMed] [Google Scholar]
- 4. Camaschella C., Roetto A., Calì A., De Gobbi M., Garozzo G., Carella M., Majorano N., Totaro A., Gasparini P. (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 25, 14–15 [DOI] [PubMed] [Google Scholar]
- 5. Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S. J., Moynihan J., Paw B. H., Drejer A., Barut B., Zapata A., Law T. C., Brugnara C., Lux S. E., Pinkus G. S., Pinkus J. L., Kingsley P. D., Palis J., Fleming M. D., Andrews N. C., Zon L. I. (2000) Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781 [DOI] [PubMed] [Google Scholar]
- 6. Wallace D. F., Pedersen P., Dixon J. L., Stephenson P., Searle J. W., Powell L. W., Subramaniam V. N. (2002) Novel mutation in ferroportin1 is associated with autosomal dominant hemochromatosis. Blood 100, 692–694 [DOI] [PubMed] [Google Scholar]
- 7. Nemeth E., Roetto A., Garozzo G., Ganz T., Camaschella C. (2005) Hepcidin is decreased in TFR2 hemochromatosis. Blood 105, 1803–1806 [DOI] [PubMed] [Google Scholar]
- 8. Ahmad K. A., Ahmann J. R., Migas M. C., Waheed A., Britton R. S., Bacon B. R., Sly W. S., Fleming R. E. (2002) Decreased liver hepcidin expression in the hfe knockout mouse. Blood Cells Mol. Dis. 29, 361–366 [DOI] [PubMed] [Google Scholar]
- 9. Bridle K. R., Frazer D. M., Wilkins S. J., Dixon J. L., Purdie D. M., Crawford D. H., Subramaniam V. N., Powell L. W., Anderson G. J., Ram G. A. (2003) Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 361, 669–673 [DOI] [PubMed] [Google Scholar]
- 10. Wallace D. F., Summerville L., Lusby P. E., Subramaniam V. N. (2005) First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut 54, 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nemeth E., Tuttle M. S., Powelson J., Vaughn M. B., Donovan A., Ward D. M., Ganz T., Kaplan J. (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 [DOI] [PubMed] [Google Scholar]
- 12. Pigeon C., Ilyin G., Courselaud B., Leroyer P., Turlin B., Brissot P., Loréal O. (2001) A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276, 7811–7819 [DOI] [PubMed] [Google Scholar]
- 13. Weinstein D. A., Roy C. N., Fleming M. D., Loda M. F., Wolfsdorf J. I., Andrews N. C. (2002) Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood 100, 3776–3781 [DOI] [PubMed] [Google Scholar]
- 14. Gao J., Chen J., De Domenico I., Koeller D. M., Harding C. O., Fleming R. E., Koeberl D. D., Enns C. A. (2010) Hepatocyte-targeted HFE and TFR2 control hepcidin expression in mice. Blood 115, 3374–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guihard S., Clay D., Cocault L., Saulnier N., Opolon P., Souyri M., Pagès G., Pouysségur J., Porteu F., Gaudry M. (2010) The MAPK ERK1 is a negative regulator of the adult steady-state splenic erythropoiesis. Blood 115, 3686–3694 [DOI] [PubMed] [Google Scholar]
- 16. Poli M., Luscieti S., Gandini V., Maccarinelli F., Finazzi D., Silvestri L., Roetto A., Arosio P. (2010) Transferrin receptor 2 and HFE regulate furin expression via mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/Erk) signaling: implications for transferrin-dependent hepcidin regulation. Haematologica 95, 1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wallace D. F., Summerville L., Crampton E. M., Frazer D. M., Anderson G. J., Subramaniam V. N. (2009) Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 50, 1992–2000 [DOI] [PubMed] [Google Scholar]
- 18. Ramey G., Deschemin J. C., Vaulont S. (2009) Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 94, 765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson M. B., Enns C. A. (2004) Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 104, 4287–4293 [DOI] [PubMed] [Google Scholar]
- 20. Johnson M. B., Chen J., Murchison N., Green F. A., Enns C. A. (2007) Transferrin receptor 2: evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol. Biol. Cell 18, 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen J., Enns C. A. (2007) The cytoplasmic domain of transferrin receptor 2 dictates its stability and response to holo-transferrin in Hep3B cells. J. Biol. Chem. 282, 6201–6209 [DOI] [PubMed] [Google Scholar]
- 22. Robb A., Wessling-Resnick M. (2004) Regulation of transferrin receptor 2 protein levels by transferrin. Blood 104, 4294–4299 [DOI] [PubMed] [Google Scholar]
- 23. Chen J., Wang J., Meyers K. R., Enns C. A. (2009) Transferrin-directed internalization and cycling of transferrin receptor 2. Traffic 10, 1488–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Levy S., Todd S. C., Maecker H. T. (1998) CD81 (TAPA-1): a molecule involved in signal transduction and cell adhesion in the immune system. Annu. Rev. Immunol. 16, 89–109 [DOI] [PubMed] [Google Scholar]
- 25. Reynolds G. M., Harris H. J., Jennings A., Hu K., Grove J., Lalor P. F., Adams D. H., Balfe P., Hübscher S. G., McKeating J. A. (2008) Hepatitis C virus receptor expression in normal and diseased liver tissue. Hepatology 47, 418–427 [DOI] [PubMed] [Google Scholar]
- 26. Lineberry N., Su L., Soares L., Fathman C. G. (2008) The single subunit transmembrane E3 ligase gene related to anergy in lymphocytes (GRAIL) captures and then ubiquitinates transmembrane proteins across the cell membrane. J. Biol. Chem. 283, 28497–28505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anandasabapathy N., Ford G. S., Bloom D., Holness C., Paragas V., Seroogy C., Skrenta H., Hollenhorst M., Fathman C. G., Soares L. (2003) GRAIL: an E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity 18, 535–547 [DOI] [PubMed] [Google Scholar]
- 28. Vogt T. M., Blackwell A. D., Giannetti A. M., Bjorkman P. J., Enns C. A. (2003) Heterotypic interactions between transferrin receptor and transferrin receptor 2. Blood 101, 2008–2014 [DOI] [PubMed] [Google Scholar]
- 29. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 30. Lawrence C. M., Ray S., Babyonyshev M., Galluser R., Borhani D. W., Harrison S. C. (1999) Crystal structure of the ectodomain of human transferrin receptor. Science 286, 779–782 [DOI] [PubMed] [Google Scholar]
- 31. Nakamichi S., Senga Y., Inoue H., Emi A., Matsuki Y., Watanabe E., Hiramatsu R., Ogawa W., Kasuga M. (2009) Role of the E3 ubiquitin ligase gene related to anergy in lymphocytes in glucose and lipid metabolism in the liver. J. Mol. Endocrinol. 42, 161–169 [DOI] [PubMed] [Google Scholar]
- 32. Johnson A. E., Audigier S., Rossi F., Jard S., Tribollet E., Barberis C. (1993) Localization and characterization of vasopressin binding sites in the rat brain using an iodinated linear AVP antagonist. Brain Res. 622, 9–16 [DOI] [PubMed] [Google Scholar]
- 33. Rocha-Perugini V., Zamai M., González-Granado J. M., Barreiro O., Tejera E., Yañez-Mó M., Caiolfa V. R., Sanchez-Madrid F. (2013) CD81 controls sustained T cell activation signaling and defines the maturation stages of cognate immunological synapses. Mol. Cell. Biol. 33, 3644–3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carloni V., Mazzocca A., Ravichandran K. S. (2004) Tetraspanin CD81 is linked to ERK/MAPKinase signaling by Shc in liver tumor cells. Oncogene 23, 1566–1574 [DOI] [PubMed] [Google Scholar]
- 35. Calzolari A., Raggi C., Deaglio S., Sposi N. M., Stafsnes M., Fecchi K., Parolini I., Malavasi F., Peschle C., Sargiacomo M., Testa U. (2006) TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J. Cell Sci. 119, 4486–4498 [DOI] [PubMed] [Google Scholar]
- 36. Levy S., Shoham T. (2005) The tetraspanin web modulates immune-signalling complexes. Nat. Rev. Immunol. 5, 136–148 [DOI] [PubMed] [Google Scholar]
- 37. Truksa J., Lee P., Beutler E. (2009) Two BMP responsive elements, STAT, and bZIP/HNF4/COUP motifs of the hepcidin promoter are critical for BMP, SMAD1, and HJV responsiveness. Blood 113, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zona L., Tawar R. G., Zeisel M. B., Xiao F., Schuster C., Lupberger J., Baumert T. F. (2014) CD81-receptor associations: impact for hepatitis C virus entry and antiviral therapies. Viruses 6, 875–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vieira A. V., Lamaze C., Schmid S. L. (1996) Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 274, 2086–2089 [DOI] [PubMed] [Google Scholar]
- 40. Seaman M. N., Burd C. G., Emr S. D. (1996) Receptor signalling and the regulation of endocytic membrane transport. Curr. Opin. Cell Biol. 8, 549–556 [DOI] [PubMed] [Google Scholar]
- 41. Carpenter G., Cohen S. (1979) Epidermal growth factor. Annu. Rev. Biochem. 48, 193–216 [DOI] [PubMed] [Google Scholar]
- 42. Miller K., Beardmore J., Kanety H., Schlessinger J., Hopkins C. R. (1986) Localization of the epidermal growth factor (EGF) receptor within the endosome of EGF-stimulated epidermoid carcinoma (A431) cells. J. Cell Biol. 102, 500–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dunn W. A., Connolly T. P., Hubbard A. L. (1986) Receptor-mediated endocytosis of epidermal growth factor by rat hepatocytes: receptor pathway. J. Cell Biol. 102, 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lai W. H., Guyda H. J., Bergeron J. J. (1986) Binding and internalization of epidermal growth factor in human term placental cells in culture. Endocrinology 118, 413–423 [DOI] [PubMed] [Google Scholar]
- 45. Carpenter G., Lembach K. J., Morrison M. M., Cohen S. (1975) Characterization of the binding of 125-I-labeled epidermal growth factor to human fibroblasts. J. Biol. Chem. 250, 4297–4304 [PubMed] [Google Scholar]
- 46. Gorden P., Carpentier J. L., Cohen S., Orci L. (1978) Epidermal growth factor: morphological demonstration of binding, internalization, and lysosomal association in human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 75, 5025–5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lloyd T. E., Atkinson R., Wu M. N., Zhou Y., Pennetta G., Bellen H. J. (2002) Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell 108, 261–269 [DOI] [PubMed] [Google Scholar]
- 48. Cottrell G. S., Amadesi S., Schmidlin F., Bunnett N. (2003) Protease-activated receptor 2: activation, signalling and function. Biochem. Soc. Trans. 31, 1191–1197 [DOI] [PubMed] [Google Scholar]
- 49. Soh U. J., Dores M. R., Chen B., Trejo J. (2010) Signal transduction by protease-activated receptors. Br. J. Pharmacol. 160, 191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Babitt J. L., Huang F. W., Wrighting D. M., Xia Y., Sidis Y., Samad T. A., Campagna J. A., Chung R. T., Schneyer A. L., Woolf C. J., Andrews N. C., Lin H. Y. (2006) Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 38, 531–539 [DOI] [PubMed] [Google Scholar]
- 51. Babitt J. L., Huang F. W., Xia Y., Sidis Y., Andrews N. C., Lin H. Y. (2007) Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J. Clin. Invest. 117, 1933–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang R. H., Li C., Xu X., Zheng Y., Xiao C., Zerfas P., Cooperman S., Eckhaus M., Rouault T., Mishra L., Deng C. X. (2005) A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2, 399–409 [DOI] [PubMed] [Google Scholar]
- 53. Verga Falzacappa M. V., Casanovas G., Hentze M. W., Muckenthaler M. U. (2008) A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. 86, 531–540 [DOI] [PubMed] [Google Scholar]
- 54. Corradini E., Rozier M., Meynard D., Odhiambo A., Lin H. Y., Feng Q., Migas M. C., Britton R. S., Babitt J. L., Fleming R. E. (2011) Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology 141, 1907–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]