

Background: ApoM overexpression generates larger nascent and plasma HDLs.
Results: ApoMQ22A overexpression generates smaller nascent and plasma HDL and enhances apoM and S1P secretion as compared with apoMWT.
Conclusion: ApoM secretion regulates hepatocyte S1P secretion, and its uncleaved signal peptide delays apoM secretion, promoting larger nascent and plasma HDL particle formation.
Significance: ApoM signal peptide is required for large apoM/S1P-enriched HDL formation.
Keywords: Ceramide, Cholesterol, Liver, Sphingolipid, Sphingosine 1-Phosphate (S1P), Apolipoprotein M, Fumonisin B1, High Density Lipoproteins, Myriocin
Abstract
Apolipoprotein M (apoM), a plasma sphingosine 1-phosphate (S1P) carrier, associates with plasma HDL via its uncleaved signal peptide. Hepatocyte-specific apoM overexpression in mice stimulates formation of both larger nascent HDL in hepatocytes and larger mature apoM/S1P-enriched HDL particles in plasma by enhancing hepatic S1P synthesis and secretion. Mutagenesis of apoM glutamine 22 to alanine (apoMQ22A) introduces a functional signal peptidase cleavage site. Expression of apoMQ22A in ABCA1-expressing HEK293 cells resulted in the formation of smaller nascent HDL particles compared with wild type apoM (apoMWT). When apoMQ22A was expressed in vivo, using recombinant adenoviruses, smaller plasma HDL particles and decreased plasma S1P and apoM were observed relative to expression of apoMWT. Hepatocytes isolated from both apoMWT- and apoMQ22A-expressing mice displayed an equivalent increase in cellular levels of S1P, relative to LacZ controls; however, relative to apoMWT, apoMQ22A hepatocytes displayed more rapid apoM and S1P secretion but minimal apoMQ22A bound to nascent lipoproteins. Pharmacologic inhibition of ceramide synthesis increased cellular sphingosine and S1P but not medium S1P in both apoMWT and apoMQ22A hepatocytes. We conclude that apoM secretion is rate-limiting for hepatocyte S1P secretion and that its uncleaved signal peptide delays apoM trafficking out of the cell, promoting formation of larger nascent apoM- and S1P-enriched HDL particles that are probably precursors of larger apoM/S1P-enriched plasma HDL.
Introduction
HDL cholesterol concentrations are inversely correlated with coronary heart disease (1, 2). The atheroprotective nature of HDL is probably due to its ability to promote reverse cholesterol transport (3), inhibit inflammation (4) and oxidation (5), and transport cardioprotective molecules. One likely atheroprotective HDL molecule is apolipoprotein M (apoM),2 which is present on ∼5% of plasma HDL particles (6). ApoM is proposed to be atheroprotective by stimulating pre-β HDL formation (7–9), promoting macrophage cholesterol efflux (9), increasing antioxidative activity of HDL (10), and transporting sphingosine 1-phosphate (S1P) on HDL (11).
ApoM is secreted with an uncleaved signal peptide due to the lack of a functional signal peptidase cleavage site (6). The retained signal peptide acts as an anchor for membrane and lipoprotein binding (12, 13). The substitution of glutamine 22 for alanine (Q22A) creates a signal peptidase cleavage site in apoM (apoMQ22A), leading to signal peptide processing, poor binding to plasma HDL, and rapid clearance of apoM from plasma by the kidney (12, 13). In HEK293 cells transfected with apoMWT versus apoMQ22A, more apoMQ22A was found in 48 h conditioned serum-free medium, suggesting increased secretion of apoMQ22A (14). These results are compatible with the hypothesis that the signal peptide of ApoM regulates its secretion; however, the interrelationship between apoM secretion kinetics and hepatocyte S1P secretion has not been investigated.
Our previous studies demonstrated that apoM overexpression in ABCA1-expressing HEK293 cells (8) and primary hepatocytes (15) from hepatocyte-specific apoM transgenic (apoM Tg) mice generated larger nascent HDL particles. These nascent HDL particles from hepatocytes might recruit lecithin-cholesterol acyltransferase for cholesterol esterification in vivo, resulting in formation of larger, cholesteryl ester-enriched plasma HDL. Primary hepatocytes from apoM Tg mice also displayed increased sphingolipid synthesis and S1P secretion compared with those from wild type (WT) mice, agreeing with published data indicating that apoM overexpression in HepG2 cells stimulates S1P secretion (16). How apoM stimulates nascent HDL formation and S1P synthesis and secretion remains unknown. Because apoM is poorly secreted (8, 14), we speculated that the intracellular retention of apoM might contribute to the formation of larger nascent HDL particles and S1P enrichment.
In the current study, we investigated the role of the apoM signal peptide in hepatic apoM, HDL, and S1P metabolism. Given our previous findings regarding the role of apoM overexpression in formation of large nascent HDL, we explored the role of the apoM signal peptide in (a) nascent HDL particle formation, (b) S1P production and secretion from hepatocytes, and (c) the overall effect on plasma HDL and lipid metabolism. Our results suggest that the apoM signal peptide promotes association of apoM with plasma lipoproteins and retards apoM secretion, allowing assembly of large apoM- and S1P-enriched nascent and mature plasma HDL that can transport S1P to extrahepatic tissues.
MATERIALS AND METHODS
Animals
Ten-week-old male mice (wild type, C57BL/6J; Jackson Laboratories) were housed in the Wake Forest School of Medicine animal facility with a 12-h light/12-h dark cycle and fed ad libitum a commercial chow diet for 2 weeks before experiments were initiated. All procedures were approved by the Institutional Animal Care and Use Committee of Wake Forest School of Medicine.
Generation of Adenovirus-expressing ApoMWT and ApoMQ22A
Wild type human apoM cDNA FLAG-tagged at the carboxyl terminus was cloned into pcDNA3 and amplified as described (8). A Q22A mutation was introduced into pcDNA3-apoM by QuikChange® site-directed mutagenesis (Stratagene). The primer sequence used for mutagenesis was 5′-C TCC ATC TAC GCG TGC CCT GAG CAC AG-3′. The underlined is the mutated amino acid codon (Q22A). DNA sequences were confirmed by sequencing (Genewiz). Adenoviruses expressing apoMWT (Ad-apoM) and apoMQ22A (Ad-Q22A) were generated using the Adeno-XTM expression system (Clontech) and amplified and purified (17). An adenovirus expressing LacZ (Ad-LacZ) was used as a control. After purification by CsCl gradient ultracentrifugation, the adenovirus was dialyzed against 20 mm Tris-HCl, pH 8.0, 270 mm NaCl, 2 mm MgCl2, and 50% (v/v) glycerol. The adenoviral titer was determined using the Adeno-XTM rapid titer kit (Clontech). Adenoviruses were diluted into 150 μl of saline and injected into C57BL/6J mice at 2.9 × 109 pfu/mouse. Three days post-injection, mice were fasted for 4 h before sacrifice, and plasma and liver samples were collected for analysis. Studies with recombinant adenovirus in vivo were repeated at two additional times (n = 3/genotype) with similar results.
Plasma Lipid and Lipoprotein Measurements
Four-hour fasted mouse plasma was harvested by tail bleeding or cardiac puncture of anesthetized mice at sacrifice. Plasma total cholesterol and triglycerides were measured by enzymatic assays (Wako). Plasma samples were fractionated by a high-resolution Superose 6TM FPLC column (10/300GL, Amersham Biosciences; flow rate, 0.5 ml/min) with an online cholesterol analyzer (18). Lipoprotein fractions from FPLC were collected for further analysis.
Nascent HDL Formation
HEK293 cells stably expressing ABCA1 (19, 20) were cultured in DMEM and transfected with empty vector pcDNA3 (control), pcDNA3-apoM (apoMWT), or pcDNA3-Q22A (apoMQ22A) using Lipofectamine 2000TM (Invitrogen); 24 h later, cells were incubated with [125I]apoA-I (10 μg/ml; 105 cpm) for an additional 24 h in serum-free media (8). After incubation, conditioned media were harvested, concentrated using an Amicon Ultra-10 concentrator, and fractionated using three Superdex-200HR FPLC columns (Amersham Biosciences) connected in a series. The particles were eluted (0.9% NaCl and 0.01% EDTA, pH 7.4, column buffer) at a flow rate of 0.3 ml/min; individual fractions were analyzed for 125I radioactivity, and the 125I profile was plotted. Conditioned media from hepatocytes isolated from mice injected with Ad-apoM or Ad-Q22A were harvested, concentrated, and fractionated using one Superdex-200HR FPLC column after the addition of a trace amount of [125I]apoA-I (1.25 × 105 cpm) and incubated at 4 °C for 30 min. Different lipoprotein fractions (VLDL, intermediate fractions, nascent HDL, and lipid-free fraction) were collected based on the 125I profile (21). Proteins from each fraction were precipitated in TCA, dissolved in SDS sample buffer (Invitrogen), and separated by SDS-PAGE, and human apoM expression was measured on Western blots using anti-FLAG monoclonal antibody M2 (Sigma-Aldrich, catalogue no. F3165).
Isolation of Primary Hepatocytes
Primary hepatocytes were isolated as described previously (22) with minor modifications. After isolation, hepatocytes were centrifuged at 50 × g for 5 min in a 50% Percoll-Williams medium E gradient to pellet live cells, which were then washed with Williams medium E before seeding into 35-mm dishes at a density of 3 × 105 cells/dish.
ApoM Secretion from Primary Hepatocytes
Primary hepatocytes were isolated, incubated with Williams medium E for 2 h, washed, and switched to serum-free DMEM for a 2-h equilibration. Cells were then washed and incubated with methionine/cysteine (Met/Cys)-deficient media for 20 min before the addition of [35S]Met/Cys (100 μCi/35-mm dish) in Met/Cys-deficient media. Cells were incubated for 10 min at 37 °C before the addition of DMEM chase medium containing 10% FBS, 10 mm Met, and 3 mm Cys to prevent further incorporation of the radiolabel into proteins. Hepatocytes were then washed and incubated with chase media for 0, 60, and 120 min. At each chase time point, media were collected and centrifuged to pellet cell debris. Hepatocytes were washed with PBS, and cells were harvested in lysis buffer (1% Triton X-100, 25 mm Tris-HCl, pH 7.5, 150 mm NaCl), supplemented with 1× protease inhibitors (Roche Applied Science, catalogue no. 05892791001) and 2.5 mg/ml BSA. Media were adjusted to the concentration of lysis buffer. Cell lysates and media were then subjected to immunoprecipitation using anti-FLAG monoclonal antibody M2 (Sigma-Aldrich).
Immunoprecipitation of ApoM in Hepatocyte Cell Lysates and Medium
Cell lysate and medium samples were rotated overnight at 4 °C with anti-FLAG monoclonal antibody (5 μl). Protein G-Sepharose beads (Sigma-Aldrich, catalogue no. P3296) were added (20 μl), and samples were rotated for another 2 h before the beads were centrifuged and washed three times with lysis buffer, followed by a final wash with buffer containing 0.1% SDS, 10 mm Tris-HCl, pH 7.5, 2.5 mm EDTA. ApoM immunoprecipitates were denatured at 95 °C for 5 min with SDS sample buffer (Invitrogen) containing DTT (100 mm), and proteins were separated by SDS-PAGE. Gels were heated and dried under vacuum, and radiolabeled proteins were visualized using a phosphor imager (Fujifilm). Band intensities were quantified using Multi Gauge software (Fujifilm).
Myriocin and Fumonisin B1 Treatment
Primary hepatocytes were isolated and seeded for 2 h before they were washed and incubated with serum-free media for 2 h. Myriocin (Sigma-Aldrich) dissolved in methanol was added to the culture dishes at a final concentration of 10 μm, and fumonisin B1 (Sigma-Aldrich) dissolved in ethanol was added at a final concentration of 25 μm. After incubation for 6 h, media were collected and supplemented with phosphatase inhibitors (Roche Applied Science, catalogue no. 04906845001). Control dishes were incubated with the appropriate vehicle. Cells were washed and collected in methanol, and both cells and media were subjected to LC-ESI-MS/MS analysis. Replicate dishes in each group were used to determine protein content by a BCA assay (Thermo Scientific).
LC-ESI-MS/MS Analysis of Sphingolipids
Sphingolipid concentrations in plasma, HDL, hepatocytes, and hepatocyte-conditioned medium were determined using liquid chromatography mass spectroscopy (15, 23).
Western Blotting
Proteins were fractionated by SDS-PAGE and transferred to a nitrocellulose membrane (Schleicher & Schuell) (250 mA, 2 h). Membranes were blocked with 5% nonfat dry milk in Tris-based saline containing 0.1% Tween 20; incubated with primary antibodies anti-FLAG M2 (1:1,000 dilution), anti-GAPDH (Sigma-Aldrich; 1:1,000 dilution), and anti-apoA-I (generated in our laboratory; 1:500 dilution) at 4 °C overnight; washed three times; and then incubated with HRP-conjugated anti-mouse and anti-rabbit secondary antibodies (GE Healthcare; 1:10,000 dilution) for 1 h at room temperature. Blots were incubated with SuperSignal West Pico chemiluminescence substrate (Pierce) and visualized with a Fujifilm LAS-3000 camera. Band intensities were quantified using Multi Gauge software.
Gene Expression Analysis
RNA was isolated from liver using TRIzol (Invitrogen) and reverse-transcribed into cDNA using an Omniscript RT kit (Qiagen). Expression levels were analyzed by quantitative real-time PCR (24). Primers used for the indicated gene expression were as follows: GAPDH, TGTGTCCGTCGTGGATCTG (forward) and CCTGCTTCACCACCTTCTTGAT (reverse); human apoM, TGCCCCGGAAATGGATCTA (forward) and CAGGGCGGCCTTCAGTT (reverse).
Statistics
Results are reported as mean ± S.E. One-way ANOVAs were used to analyze results among three or four groups from adenovirus experiments. Tukey's post hoc test was used to identify differences among groups. Two-way ANOVAs and Bonferroni post hoc tests were used to analyze data from myriocin and fumonisin B1 inhibition experiments. A p value of <0.05 was considered statistically significant. Statistical analyses were performed with GraphPad Prism software.
RESULTS
An Uncleaved ApoM Signal Peptide Is Necessary to Generate Larger Nascent HDL Particles
We previously reported that apoM overexpression in ABCA1-expressing HEK293 (8) and hepatocytes from apoM Tg mice generated larger nascent HDL compared with controls (15). ApoM is poorly secreted from HEK293 cells (8), probably due to retention of the uncleaved signal peptide (14). Replacement of glutamine 22 with alanine in apoM generates a cleavable signal peptide, which increases steady-state amounts of apoM in serum-free media of transfected HEK293 cells (14). We reasoned that without its signal peptide as a hydrophobic anchor, apoMQ22A would not promote larger nascent HDL formation. To test this hypothesis, we transfected plasmids encoding apoMWT, apoMQ22A, and an empty vector (pcDNA3; control) into ABCA1-expressing HEK293 cells and initiated nascent HDL formation by incubating cells with [125I]apoA-I for 24 h. Conditioned media were then collected and fractionated by size exclusion FPLC.
ABCA1-expressing HEK293 cells generated distinct sizes of nascent HDL particles (25) (Fig. 1A). Overexpression of both apoMWT and apoMQ22A stimulated formation of nascent HDLs, but only overexpression of apoMWT resulted in larger nascent HDLs versus control transfection. These data suggest that cleavage of the apoM signal peptide prevented the increase in nascent HDL size observed with expression of apoMWT. We also compared apoM protein expression in cells and media. As shown in Fig. 1B, apoMQ22A had slightly faster migration during SDS-PAGE compared with apoMWT due to processing by signal peptidase (processed form), agreeing with previous studies (14). After 24 h of incubation, we observed comparable levels of apoMWT and apoMQ22A in media but less apoMQ22A protein in cells, suggesting that apoMQ22A was secreted more efficiently.
FIGURE 1.
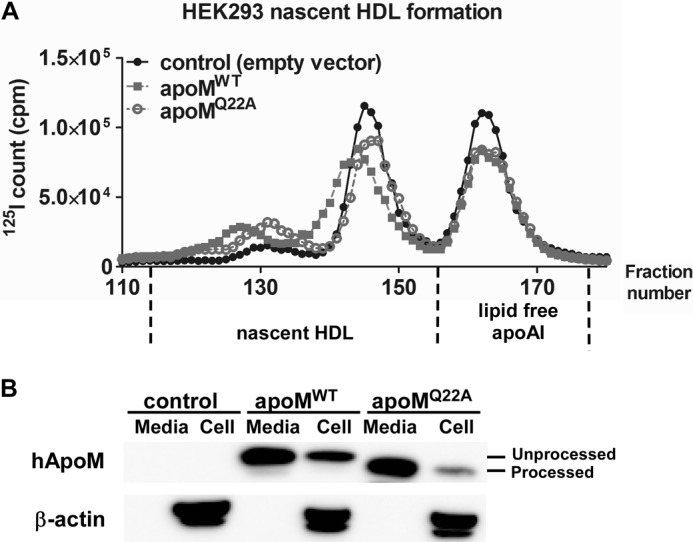
ApoMWT, but not apoMQ22A, overexpression stimulates generation of larger nascent HDL particles. ABCA1-expressing HEK293 cells were transfected with plasmids expressing apoMWT, apoMQ22A, or empty vector (pcDNA3, control) for 24 h before incubation with [125I]apoA-I (105 cpm, 10 μg/ml) in serum-free DMEM for 24 h. Media and cells were then collected for analysis. A, media were fractionated using three high-resolution Superdex-200 FPLC columns in a series to separate nascent HDL particles of varying sizes. Elution was monitored by 125I quantification. B, cells were lysed, and equivalent amounts of media and cells were Western blotted for human apoM (hApoM) using an anti-FLAG monoclonal antibody and β-actin. Processed and unprocessed apoM are indicated.
Adenoviral Overexpression of ApoMQ22A Results in Lower Plasma ApoM Concentrations
We previously found that hepatocyte-specific apoM Tg mice had increased plasma HDL size but not HDL cholesterol (HDL-C) concentrations (15). In this study, we generated adenoviruses expressing apoMWT (Ad-apoM), apoMQ22A (Ad-Q22A), and β-galactosidase (Ad-LacZ) to study the role of apoM and its signal peptide in mature HDL formation in vivo. C57BL/6J wild type mice were injected with 2.9 × 109 pfu/mouse, with saline injection as a control. Three days after injection, mice were fasted for 4 h and sacrificed, and liver and plasma were collected. Liver apoM mRNA expression was undetectable in the Ad-LacZ group, as expected, and comparable levels were observed in the Ad-apoM and Ad-Q22A groups (Fig. 2A), indicating similar levels of overexpression. Next, we analyzed liver and plasma apoM protein levels. Livers of Ad-Q22A-infected mice contained mainly the processed form of apoMQ22A, and its abundance was marginally decreased compared with Ad-apoM (Fig. 2, B and C), as anticipated from our in vitro data (Fig. 1B). There was also a detectable amount of unprocessed apoMQ22A in liver with Ad-Q22A injection, probably due to a small percentage of nascent apoM that escaped processing by signal peptidase. However, in plasma, Ad-Q22A mice exhibited only the unprocessed form of apoMQ22A, which was less abundant than that in Ad-apoM mice (Fig. 2, D and E).
FIGURE 2.

ApoM overexpression in Ad-LacZ, Ad-apoM, and Ad-Q22A mice. C57BL/6J WT mice (n = 6/group) were injected retro-orbitally with adenovirus (2.9 × 109 pfu in 150 μl of saline) expressing LacZ (Ad-LacZ), apoMWT (Ad-apoM), or apoMQ22A (Ad-Q22A); control mice were injected with saline. Mice were sacrificed 3 days later after a 4-h fast, and plasma and liver were collected. A, RNA was isolated from livers of mice, reverse transcribed into cDNA, and measured by quantitative real-time PCR. Expression of human apoM was normalized to GAPDH. ND, not detectable. Human apoM in proteins from liver (60 μg, n = 4, B; quantified in C) and plasma samples (0.5 μl, n = 3, D; quantified in E) from Ad-LacZ, Ad-apoM, and Ad-Q22A mice was measured on Western blots using an anti-FLAG antibody. Liver lysate and plasma were also measured on Western blots for GAPDH and apoA-I, respectively. *, p < 0.05 by one-way ANOVAs and Tukey's post hoc tests. Error bars, S.E.
Overexpression of ApoMQ22A in Vivo Does Not Affect Plasma HDL Particle Size
We next studied the impact of apoMQ22A overexpression in vivo on plasma lipid concentrations and HDL particle size. Plasma lipid concentrations (total cholesterol (TC) and triglyceride (TG); Fig. 3A) were similar among the four groups of mice. However, FPLC fractionation of plasma revealed that Ad-apoM-treated mice had an apparent shift to larger HDL particle sizes compared with the other groups (Fig. 3B), agreeing with previous results demonstrating larger plasma HDL in apoM Tg mice (15). In contrast, Ad-Q22A-injected mice had only a slight increase in HDL size compared with controls. Additionally, Ad-apoM and Ad-Q22A mice showed a trend toward increased plasma VLDL cholesterol (2.7 ± 0.9 and 1.9 ± 0.2 mg/dl, respectively, versus 1.2 ± 0.3 mg/dl for Ad-LacZ group) and LDL cholesterol concentrations (11.6 ± 2.2 and 10.8 ± 0.9 mg/dl, respectively, versus 6.1 ± 0.4 mg/dl for Ad-LacZ group), agreeing with previous findings in apoM Tg mice (15). HDL-C concentrations were similar among all groups (Fig. 3C). Collectively, our data suggest that overexpression of apoMWT, but not apoMQ22A, increases plasma HDL particle size without affecting plasma HDL-C concentrations.
FIGURE 3.
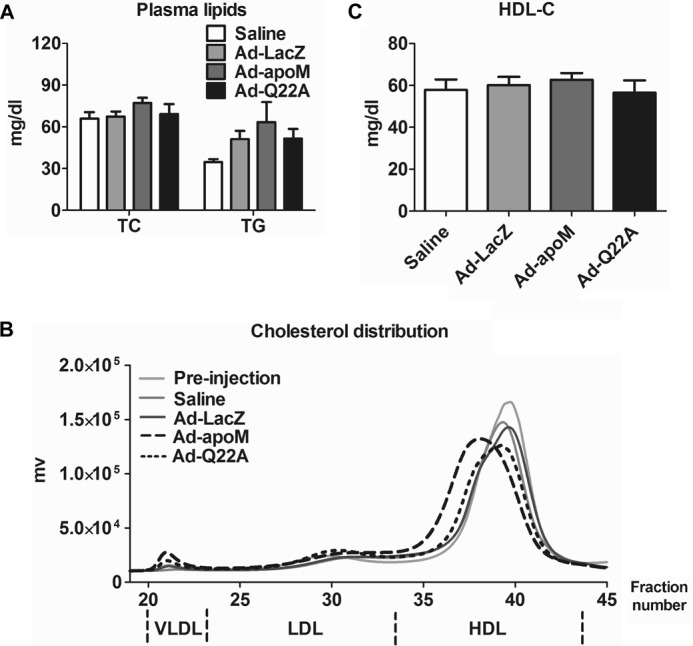
Increased HDL size in Ad-apoM mice but not in Ad-Q22A mice. Mice were treated as described in the legend to Fig. 2, and plasma was collected for analysis. A, plasma total cholesterol (TC) and triglyceride (TG) concentrations were measured by enzymatic assays. B, equivalent volumes of plasma from mice with the indicated treatments were subjected to FPLC size exclusion chromatography to fractionate lipoproteins. A representative FPLC cholesterol profile is shown for each group. C, HDL-C was calculated based on plasma total cholesterol and the fractional HDL cholesterol distribution from FPLC. n = 3/genotype. One-way ANOVA and Turkey's post hoc test were used for statistical analysis among groups. Error bars, S.E.
Ad-apoM-treated Mice Have Higher Plasma S1P Concentrations than Ad-Q22A-treated Mice
We (15) and others (11, 16, 26, 27) have shown that apoM overexpression leads to increased plasma S1P concentrations. Here, we measured plasma S1P concentrations in mice injected with Ad-LacZ, Ad-apoM, and Ad-Q22A. Plasma S1P concentrations were 78% greater in Ad-apoM-injected mice compared with Ad-LacZ-treated mice, whereas Ad-Q22A mice showed only a 10% increase (Fig. 4A), presumably due to less apoM in plasma (Fig. 2D). The results for plasma dihydrosphingosine 1-phosphate (DH-S1P), which lacks the double bond in the sphingoid base, reflected those of plasma S1P. The increases in plasma S1P and DH-S1P concentrations (Fig. 4A) paralleled those in peak HDL fractions (Fig. 4B). Concentrations of sphingosine (Sph) and dihydrosphingosine (DH-Sph), immediate precursors of plasma S1P and DH-S1P, respectively, and ceramide in plasma were similar among groups (data not shown). Collectively, apoMWT overexpression leads to higher plasma and HDL S1P concentrations than apoMQ22A overexpression.
FIGURE 4.
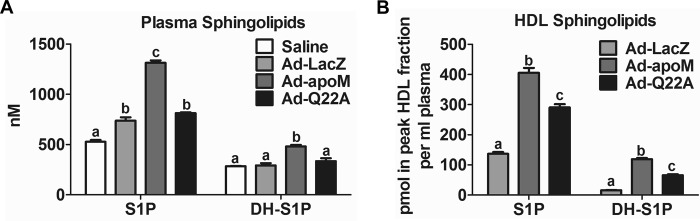
Plasma and HDL S1P and DH-S1P are increased in mice overexpressing apoMWTversus apoMQ22A. Mice were injected with adenoviruses (Fig. 2), and plasma was collected for analysis. Lipids from plasma (A) and peak HDL fractions from FPLC (B) were extracted and subjected to LC-ESI-MS/MS for S1P and DH-S1P analysis. n = 3/genotype. Different letters indicate statistical significance by one-way ANOVAs and Tukey's post hoc tests. Error bars, S.E.
ApoMQ22A Overexpression Results in Increased Hepatocyte S1P Secretion
We previously showed that hepatic apoM overexpression stimulated sphingolipid synthesis, resulting in elevated S1P production and secretion (15). Because plasma S1P concentrations were lower in Ad-Q22A mice than Ad-apoM mice, overexpressed apoMQ22A might be less able to stimulate S1P production and secretion than apoMWT. To test this idea, we first isolated hepatocytes from Ad-LacZ, Ad-apoM, and Ad-Q22A mice and analyzed cellular and medium sphingolipids. Similar to apoM Tg hepatocytes, cellular S1P concentrations increased by ∼2.7-fold in Ad-apoM hepatocytes and ∼2.8-fold in Ad-Q22A hepatocytes versus Ad-LacZ (Fig. 5A). However, in media, S1P concentrations were 4-fold higher for Ad-Q22A versus Ad-apoM and Ad-LacZ cells (Fig. 5B), suggesting that apoMQ22A overexpression enhanced S1P secretion in hepatocytes relative to apoMWT, despite similar S1P production in both types.
FIGURE 5.

Enhanced S1P and apoMQ22A secretion from Ad-Q22A hepatocytes. Mice were injected with adenoviruses (Fig. 2), and hepatocytes were isolated and incubated in 1 ml of serum-free medium for 6 h. Lipids were extracted from cells (A) and media (B), and sphingolipids were analyzed by LC-ESI-MS/MS (n = 4). C, human apoM in cells and media was measured on Western blots with an anti-FLAG antibody. D, conditioned media from hepatocytes isolated from Ad-apoM or Ad-Q22A mice were harvested, concentrated, and fractionated using one Superdex-200HR FPLC column after the addition of a trace amount of [125I]apoA-I (1.25 × 105 cpm) and incubation at 4 °C for 30 min. Different lipoprotein fractions were collected based on 125I elution profile. Inter, intermediate fractions. nHDL, nascent HDL. E, human apoM expression was measured in proteins from each pooled fraction. Samples were precipitated in TCA, dissolved in SDS sample buffer, separated by SDS-PAGE, and analyzed on Western blots using an anti-FLAG antibody. Error bars, S.E.
We also analyzed apoM protein abundance in cultured hepatocytes and media. As shown in Fig. 5C, in both cells and media, hepatocytes from Ad-Q22A-infected mice displayed abundant processed apoM, with minimal residual unprocessed apoM within cells. Hepatocytes from Ad-Q22A mice had less apoM in cells, but more in media, relative to Ad-apoM hepatocytes. These results are consistent with those in transfected HEK293 cells (Fig. 1B), suggesting that apoMQ22A is more efficiently secreted than apoMWT.
To explore apoM distribution among medium lipoproteins, we incubated hepatocyte media with a trace amount of [125I]apoA-I before fractionation by FPLC. Fractions corresponding to VLDL, nascent HDL, and lipid-free apoA-I were collected based on the [125I]apoA-I elution profile (Fig. 5D) (21); lipoproteins with a size range between VLDL and nascent HDL (i.e. intermediate fractions) were also collected. ApoMWT associated mainly with VLDL particles and to a lesser extent with nascent HDL and was undetectable in the lipid-free fractions (Fig. 5E). In contrast, apoMQ22A was distributed mainly in the lipid-free fractions and to a lesser extent in VLDL fractions and was nearly absent in nascent HDL particle fractions. The inability of apoMQ22A to associate with secreted hepatocyte lipoproteins, despite apparent increased apoM and S1P secretion, may be linked to the failure of apoMQ22A to promote formation of larger apoM/S1P-enriched plasma HDL.
ApoMQ22A Is Secreted from Hepatocytes More Rapidly than ApoMWT
To determine the effect of the signal peptide on apoM secretion kinetics, we performed pulse-chase analyses. Primary hepatocytes from Ad-apoM and Ad-Q22A mice were radiolabeled with [35S]Met/Cys for 10 min and chased for 0, 30, 60, and 120 min. At the end of the pulse period (i.e. chase min 0), levels of radiolabeled cellular apoM were similar in Ad-apoM and Ad-Q22A hepatocytes (Fig. 6A), suggesting equivalent levels of synthesis. During the chase, cellular apoM decreased more rapidly in Ad-Q22A versus Ad-apoM hepatocytes (Fig. 6, A and B). Correspondingly, medium apoM increased more rapidly in Ad-Q22A versus Ad-apoM hepatocytes (Fig. 6, A and C). These results suggest that the apoM signal peptide reduces apoM trafficking kinetics, thereby decreasing the rate and perhaps extent of apoM and S1P secretion.
FIGURE 6.

ApoMQ22A secretion is more rapid than apoMWT secretion in hepatocytes. C57BL/6J mice were injected with Ad-apoM or Ad-Q22A; 3 days later, primary hepatocytes were isolated and radiolabeled with [35S]Met/Cys (100 μCi) in Met/Cys-deficient media for 10 min and then chased with Met/Cys complete media for 0, 60, and 120 min. At each time point, cell lysates and media were collected, and human apoM was immunoprecipitated with an anti-FLAG antibody. A, radiolabeled human apoM was separated by SDS-PAGE, visualized by phosphor imager analysis, and quantified using Multi Gauge software. The percentage of apoM at chase time 0 detected in cells (B) and media (C) was calculated and plotted. *, p < 0.05 by Student's t test. This experiment was repeated twice with similar results. Error bars, S.E.
Inhibition of Ceramide Synthase Stimulates Cellular Levels, but Not Secretion, of S1P
Our previous study suggested that apoM limits the rate of hepatocyte S1P secretion (15). Here, we further tested this concept by inhibiting ceramide synthases via fumonisin B1 (FB1) (28) and thereby increase hepatic S1P production and secretion. As anticipated, FB1 treatment decreased hepatocyte ceramide content (Fig. 7A) and increased Sph and DH-Sph (Fig. 7B) for all three groups relative to vehicle control, confirming inhibition of ceramide synthases. FB1-treated hepatocytes also had increased cellular S1P and DH-S1P compared with vehicle control-treated cells (Fig. 7C), with apoMWT and apoMQ22A expression resulting in significantly higher S1P and DH-S1P concentrations relative to LacZ control. Although overexpression of apoMQ22A resulted in increased DH-S1P relative to apoMWT in FB1-treated hepatocytes, hepatocyte S1P content was similar in both groups (Fig. 7C). However, medium S1P concentrations were unaffected (Fig. 7D). Consistent with the results in Fig. 5B, S1P concentrations were considerably higher in Ad-apoMQ22A versus Ad-apoMWT hepatocytes in conditioned media (Fig. 7D). These results suggest that in apoMWT- and apoMQ22A-overexpressing hepatocytes, inhibition of ceramide synthesis via FB1 increases S1P (and DH-S1P) production but not S1P (and DH-S1P) secretion, which is limited by apoMWT and apoMQ22A secretion.
FIGURE 7.

Inhibition of ceramide synthase increases S1P concentrations in Ad-apoM and Ad-Q22A hepatocytes but not media. C57BL/6J mice were injected with Ad-LacZ, Ad-apoM, or Ad-Q22A; 3 days later, primary hepatocytes were isolated and cultured for 3 h and then switched to serum-free media with (+) or without (−) FB1 (25 μm). After further incubation for 6 h, cell lysates and media were collected for lipid extraction and sphingolipid quantification by LC-ESI-MS/MS. A, cellular ceramide (Cer); B, cellular DH-Sph and Sph; C, cellular DH-S1P and S1P; D, medium DH-S1P and S1P (n = 3). p < 0.05 by two-way ANOVAs and Bonferroni post hoc tests considered statistically significant, indicated by different letters. Error bars, S.E.
Inhibition of de Novo Sphingolipid Synthesis Minimally Affects Cellular and Medium S1P in Hepatocytes Expressing ApoMWT and ApoMQ22A
In hepatocytes treated with myriocin, an inhibitor of de novo sphingolipid synthesis, cellular Sph and DH-Sph content was unaltered (Fig. 8A), but ceramide was decreased relative to control for all three groups (Ad-LacZ, Ad-apoM, and Ad-Q22A; Fig. 8B). Myriocin treatment also did not affect hepatocyte S1P levels and resulted in only small reductions in DH-S1P (Fig. 8C), suggesting that acute inhibition of de novo sphingolipid synthesis minimally affects S1P production and secretion, probably due to continued S1P production through the salvage pathway (29). Similar to FB1, myriocin treatment did not affect hepatocyte S1P secretion and minimally increased DH-S1P secretion (Fig. 8D), further supporting the notion that S1P and DH-S1P secretion are determined by levels of apoM secretion.
FIGURE 8.

Inhibition of de novo sphingolipid synthesis minimally affects cellular or medium S1P levels. C57BL/6J mice were injected with Ad-LacZ, Ad-apoM, or Ad-Q22A; 3 days later, primary hepatocytes were isolated and cultured for 3 h and then switched to serum-free media with (+) or without (−) myriocin (10 μm). After further incubation for 6 h, cell lysates and media were collected for lipid extraction and sphingolipid quantification by LC-ESI-MS/MS. A, cellular DH-Sph and Sph; B, cellular Cer; C, cellular DH-S1P and S1P; D, medium DH-S1P and S1P (n = 3). p < 0.05 by two-way ANOVAs and Bonferroni post hoc tests considered statistically significant, indicated by different letters. Error bars, S.E.
DISCUSSION
In this study, we investigated the role of the apoM signal peptide in the formation of large apoM/S1P-enriched plasma HDL particles. Using plasmid- and adenovirus-mediated overexpression and a mutant version of apoM (apoMQ22A) with a signal peptidase cleavage site, the apoMQ22A signal peptide was efficiently cleaved in hepatocytes, resulting in more rapid apoMQ22A secretion compared with apoMWT. However, unlike apoMWT, apoMQ22A failed to 1) bind to hepatocyte-secreted lipoproteins, 2) generate larger nascent HDL in a non-hepatic cell line, 3) generate larger plasma HDL particles, or 4) sustain plasma levels of apoM and S1P, despite unaltered plasma lipid and HDL concentrations. The more rapid secretion of apoMQ22A versus apoMWT mobilized additional hepatocyte-generated S1P, despite comparable production of hepatocyte S1P by Ad-apoM and Ad-Q22A infection. FB1 inhibition of ceramide synthases further increased S1P content in hepatocytes but did not augment S1P secretion by apoMQ22A or apoMWT, lending additional support to the idea that apoM limits the rate of hepatic S1P secretion. Our study is the first to show that the apoM signal peptide slows secretion of apoM and S1P in addition to serving as an anchor for newly secreted apoM-containing lipoproteins (i.e. VLDL and HDL) and facilitates formation of larger nascent and mature plasma HDL particles. These large, apoM-S1P-enriched HDL particles may play a different physiological role in S1P signaling outside the liver, which necessitates a dedicated HDL-mediated pathway for S1P transport in plasma.
ApoMQ22A Is Secreted More Rapidly than ApoMWT in Hepatocytes
ApoM is one of a few hepatic secretory proteins secreted with a retained signal peptide (30, 31). The apoM secretion rate from transfected HEK293 cells is slow relative to an efficiently secreted protein albumin (8). This is presumably due, in part, to retention of the apoM signal peptide, which may alter folding and also anchors apoM to membranes and lipoprotein phospholipid monolayers (6, 14). Indeed, the secretion of apoMQ22A, a mutant version of apoM that contains a signal peptidase cleavage site, is increased in transfected HEK293 cell media compared with apoMWT (14). However, in hepatocytes from apoMWT versus apoMQ22A Tg mice, apoMQ22A concentrations were 60% lower than apoMWT concentrations in conditioned media (13), which paralleled a similar decrease in hepatocyte mRNA, suggesting that apoMQ22A and apoMWT secretion were similar. Both prior studies, however, lacked kinetic data and analyzed only steady-state apoM concentrations in media. Our pulse-chase radiolabeling studies in primary hepatocytes of Ad-Q22A and Ad-apoM mice directly demonstrate more rapid secretion of apoMQ22A than apoMWT (Fig. 6). This result did not stem from differences in synthesis, because amounts of newly synthesized hepatocyte apoMQ22A and apoMWT after the pulse radiolabeling were similar (Fig. 6A). Although apoMQ22A is secreted more rapidly from hepatocytes, its concentration in plasma is low (Fig. 2E) (13), presumably due to poor binding to lipoproteins that results in rapid plasma clearance by the kidney (13). Our results demonstrate that the signal peptide slows apoM secretion from hepatocytes and prolongs its residence time in plasma, both of which probably impact the physiological function of apoM.
ApoM Signal Peptide Is Necessary for Formation of Large Nascent and Plasma HDL Particles
ApoMWT overexpression in HEK293 cells (8) and primary hepatocytes (15) stimulated formation of large nascent HDL particles, whereas apoMQ22A overexpression in HEK293 cells did not (Fig. 1). We speculate that the signal peptide of apoM is essential for its cellular retention, which facilitates enlargement of nascent HDL particles. Our previous study suggested that apoM co-localized with endoplasmic reticulum and Golgi markers in HEK293 cells but not with plasma membrane (8). Endoplasmic reticulum and Golgi compartments are involved in the mobilization of cholesterol by ABCA1 for nascent HDL formation (32). Although nascent HDL particles are primarily assembled at the plasma membrane (33, 34), we previously showed that ∼20% of newly synthesized apoA-I in HepG2 cells is lipidated in the secretory pathway (35). Thus, apoM may mediate transfer of free cholesterol and/or phospholipid to ABCA1 for nascent HDL formation in the endoplasmic reticulum and Golgi compartments. However, there is little evidence for a direct role of apoM in lipid transfer, because it does not directly bind cholesterol (36) or phospholipids unless they are oxidized to end products that resemble lysophosphatidylcholine species (10).
Here, we show that increasing the intracellular secretory trafficking kinetics of apoM by promoting signal peptide processing (i.e. apoMQ22A) negates the increased nascent HDL particle size observed with apoMWT overexpression. This result suggests that apoM may indirectly facilitate lipid addition to nascent HDL particles in the secretory pathway by reducing particle secretion rate via its retained signal peptide. By analogy, apoA-IV overexpression reduces apolipoprotein B secretion and enhances triglyceride and phospholipid transfer to VLDL, resulting in secretion of larger VLDL particles (17, 37, 38). Additional studies will be required to determine whether apoM has an analogous role in the secretory pathway to facilitate increased nascent HDL particle size or whether other mechanisms are involved. It is intriguing that in hepatocyte media, apoMWT is mostly present on VLDL particles, suggesting that it may also play a role in intracellular assembly of VLDL particles.
ApoMQ22A Increases Hepatic S1P Secretion but Does Not Maintain Plasma S1P Concentrations
Using transgenic overexpression and genetic deletion of apoM in mice, Christoffersen et al. (11) defined a novel role for apoM in plasma lipoprotein S1P transport, particularly on HDL particles. We (15) and others (16) have confirmed the importance of apoM in plasma S1P transport using acute adenoviral overexpression and have documented the liver as an important source of plasma S1P (15). Adenoviral overexpression of apoMWT and apoMQ22A increased S1P content in hepatocytes to an equivalent extent (Figs. 5 and 7C). Hence, the increased S1P observed in hepatocyte media can be directly attributable to more rapid secretion of apoMQ22A (Fig. 6), resulting in less cell-associated apoMQ22A (Fig. 5C). Blocking ceramide synthesis with FB1 increased Ad-apoM and Ad-Q22A S1P concentrations in hepatocytes equally but did not further enhance S1P secretion from Ad-Q22A hepatocytes, confirming that apoMQ22A may limit the rate of S1P secretion. However, apoMQ22A overexpression resulted in less, not more, plasma S1P relative to apoMWT. This, however, is probably due to inefficient apoMQ22A binding to nascent hepatocyte and mature plasma lipoproteins and to increased clearance of plasma apoMQ22A by the kidney (13, 27).
Interestingly, apoMQ22A expression also led to greater ceramide content in hepatocytes (Fig. 7A). FB1 inhibition of ceramide synthesis reduced cellular ceramide content in all groups relative to controls (as anticipated) but increased S1P, DH-S1P, Sph, and DH-Sph relative to controls (Fig. 7), suggesting increased sphingosine conversion into S1P due to decreased ceramide production. Although apoMQ22A overexpression did not increase S1P content above that of apoMWT in FB1-treated hepatocytes (Fig. 7C), it did increase ceramide, Sph, DH-Sph, and DH-S1P, suggesting that increased S1P (and DH-S1P) secretion mediated by apoMQ22A (Fig. 7D) stimulates general synthesis of sphingolipids. Why hepatocyte S1P content is not increased under these conditions is unknown.
Conclusions
ApoMWT overexpression stimulates formation of large nascent and apoM/S1P-enriched plasma HDL particles. This phenotype is reversed by overexpression of apoMQ22A, a mutant version of apoM that contains a cleavable signal peptide. Both forms of apoM increase S1P production in hepatocytes, but apoMQ22A enhances S1P secretion because of its more rapid secretion rate. We conclude that apoM is rate-limiting for hepatocyte S1P secretion and that its signal peptide slows secretion, allowing the formation of larger nascent HDLs containing apoM/S1P that are probably precursors of large apoM/S1P-enriched plasma HDL.
Acknowledgments
We gratefully acknowledge Karen Klein (Wake Forest School of Medicine) for editing the manuscript and Wake Forest Cell and Viral Vector Core Laboratory for proliferating adenovirus. Sphingolipid analyses were performed at the Lipidomics Core Facility of the Virginia Commonwealth University Biochemistry and Molecular Biology Department.
This work was supported, in whole or in part, by National Institutes of Health Grants P01HL49373 (to J. S. P. and G. S. S.), R01HL119962 (to J. S. P.), and R37GM043880 (to S. S.). This work was also supported by American Heart Association Predoctoral Fellowship 12PRE12040309 and an RJR-Leon Golberg postdoctoral fellowship (to M. L.). The Lipidomics Core Facility in the Virginia Commonwealth University Biochemistry and Molecular Biology Department is supported in part by funding to the Massey Cancer Center from National Institutes of Health, NCI, Cancer Center Support Grant P30 CA016059.
- apo
- apolipoprotein
- LacZ
- β-galactosidase
- apoM Tg
- hepatocyte-specific apoM transgenic
- FB1
- fumonisin B1
- Ad-LacZ
- adenovirus expressing LacZ
- Ad-apoM
- adenovirus expressing apoMWT
- Ad-Q22A
- adenovirus expressing apoMQ22A
- HDL-C
- HDL cholesterol
- S1P
- sphingosine 1-phosphate
- DH-S1P
- dihydrosphingosine 1-phosphate
- Sph
- sphingosine
- DH-Sph
- dihydrosphingosine
- ESI
- electrospray ionization
- ANOVA
- analysis of variance.
REFERENCES
- 1. Castelli W. P., Anderson K., Wilson P. W., Levy D. (1992) Lipids and risk of coronary heart disease. The Framingham Study. Ann. Epidemiol. 2, 23–28 [DOI] [PubMed] [Google Scholar]
- 2. Gordon D. J., Rifkind B. M. (1989) High-density lipoprotein: the clinical implications of recent studies. N. Engl. J. Med. 321, 1311–1316 [DOI] [PubMed] [Google Scholar]
- 3. Fisher E. A., Feig J. E., Hewing B., Hazen S. L., Smith J. D. (2012) High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 32, 2813–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barter P. J., Nicholls S., Rye K. A., Anantharamaiah G. M., Navab M., Fogelman A. M. (2004) Antiinflammatory properties of HDL. Circ. Res. 95, 764–772 [DOI] [PubMed] [Google Scholar]
- 5. Kontush A., Chapman M. J. (2010) Antiatherogenic function of HDL particle subpopulations: focus on antioxidative activities. Curr. Opin. Lipidol. 21, 312–318 [DOI] [PubMed] [Google Scholar]
- 6. Xu N., Dahlbäck B. (1999) A novel human apolipoprotein (apoM). J. Biol. Chem. 274, 31286–31290 [DOI] [PubMed] [Google Scholar]
- 7. Christoffersen C., Jauhiainen M., Moser M., Porse B., Ehnholm C., Boesl M., Dahlbäck B., Nielsen L. B. (2008) Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J. Biol. Chem. 283, 1839–1847 [DOI] [PubMed] [Google Scholar]
- 8. Mulya A., Seo J., Brown A. L., Gebre A. K., Boudyguina E., Shelness G. S., Parks J. S. (2010) Apolipoprotein M expression increases the size of nascent pre beta HDL formed by ATP binding cassette transporter A1. J. Lipid Res. 51, 514–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wolfrum C., Poy M. N., Stoffel M. (2005) Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat. Med. 11, 418–422 [DOI] [PubMed] [Google Scholar]
- 10. Elsøe S., Ahnström J., Christoffersen C., Hoofnagle A. N., Plomgaard P., Heinecke J. W., Binder C. J., Björkbacka H., Dahlbäck B., Nielsen L. B. (2012) Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis 221, 91–97 [DOI] [PubMed] [Google Scholar]
- 11. Christoffersen C., Obinata H., Kumaraswamy S. B., Galvani S., Ahnström J., Sevvana M., Egerer-Sieber C., Muller Y. A., Hla T., Nielsen L. B., Dahlbäck B. (2011) Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. U.S.A. 108, 9613–9618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Axler O, Ahnström J, Dahlbäck B. (2008) Apolipoprotein M associates to lipoproteins through its retained signal peptide. FEBS Lett. 582, 826–828 [DOI] [PubMed] [Google Scholar]
- 13. Christoffersen C., Ahnström J., Axler O., Christensen E. I., Dahlbäck B., Nielsen L. B. (2008) The signal peptide anchors apolipoprotein M in plasma lipoproteins and prevents rapid clearance of apolipoprotein M from plasma. J. Biol. Chem. 283, 18765–18772 [DOI] [PubMed] [Google Scholar]
- 14. Ahnström J., Axler O., Dahlbäck B. (2010) HDL stimulates apoM secretion. Protein Pept. Lett. 17, 1285–1289 [DOI] [PubMed] [Google Scholar]
- 15. Liu M., Seo J., Allegood J., Bi X., Zhu X., Boudyguina E., Gebre A. K., Avni D., Shah D., Sorci-Thomas M. G., Thomas M. J., Shelness G. S., Spiegel S., Parks J. S. (2014) Hepatic apolipoprotein M (apoM) overexpression stimulates formation of larger apoM/sphingosine 1-phosphate-enriched plasma high density lipoprotein. J. Biol. Chem. 289, 2801–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kurano M., Tsukamoto K., Ohkawa R., Hara M., Iino J., Kageyama Y., Ikeda H., Yatomi Y. (2013) Liver involvement in sphingosine 1-phosphate dynamism revealed by adenoviral hepatic overexpression of apolipoprotein M. Atherosclerosis 229, 102–109 [DOI] [PubMed] [Google Scholar]
- 17. VerHague M. A., Cheng D., Weinberg R. B., Shelness G. S. (2013) Apolipoprotein A-IV expression in mouse liver enhances triglyceride secretion and reduces hepatic lipid content by promoting very low density lipoprotein particle expansion. Arterioscler. Thromb. Vasc. Biol. 33, 2501–2508 [DOI] [PubMed] [Google Scholar]
- 18. Brown J. M., Bell T. A., 3rd, Alger H. M., Sawyer J. K., Smith T. L., Kelley K., Shah R., Wilson M. D., Davis M. A., Lee R. G., Graham M. J., Crooke R. M., Rudel L. L. (2008) Targeted depletion of hepatic ACAT2-driven cholesterol esterification reveals a non-biliary route for fecal neutral sterol loss. J. Biol. Chem. 283, 10522–10534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mulya A., Lee J. Y., Gebre A. K., Boudyguina E. Y., Chung S. K., Smith T. L., Colvin P. L., Jiang X. C., Parks J. S. (2008) Initial interaction of apoA-I with ABCA1 impacts in vivo metabolic fate of nascent HDL. J. Lipid Res. 49, 2390–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. See R. H., Caday-Malcolm R. A., Singaraja R. R., Zhou S., Silverston A., Huber M. T., Moran J., James E. R., Janoo R., Savill J. M., Rigot V., Zhang L. H., Wang M., Chimini G., Wellington C. L., Tafuri S. R., Hayden M. R. (2002) Protein kinase A site-specific phosphorylation regulates ATP-binding cassette A1 (ABCA1)-mediated phospholipid efflux. J. Biol. Chem. 277, 41835–41842 [DOI] [PubMed] [Google Scholar]
- 21. Bi X., Zhu X., Duong M., Boudyguina E. Y., Wilson M. D., Gebre A. K., Parks J. S. (2013) Liver ABCA1 deletion in LDLrKO mice does not impair macrophage reverse cholesterol transport or exacerbate atherogenesis. Arterioscler. Thromb. Vasc. Biol. 33, 2288–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chung S., Timmins J. M., Duong M., Degirolamo C., Rong S., Sawyer J. K., Singaraja R. R., Hayden M. R., Maeda N., Rudel L. L., Shelness G. S., Parks J. S. (2010) Targeted deletion of hepatocyte ABCA1 leads to very low density lipoprotein triglyceride overproduction and low density lipoprotein hypercatabolism. J. Biol. Chem. 285, 12197–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 25. Mulya A., Lee J. Y., Gebre A. K., Thomas M. J., Colvin P. L., Parks J. S. (2007) Minimal lipidation of pre-β HDL by ABCA1 results in reduced ability to interact with ABCA1. Arterioscler. Thromb. Vasc. Biol. 27, 1828–1836 [DOI] [PubMed] [Google Scholar]
- 26. Karuna R., Park R., Othman A., Holleboom A. G., Motazacker M. M., Sutter I., Kuivenhoven J. A., Rohrer L., Matile H., Hornemann T., Stoffel M., Rentsch K. M., von Eckardstein A. (2011) Plasma levels of sphingosine-1-phosphate and apolipoprotein M in patients with monogenic disorders of HDL metabolism. Atherosclerosis 219, 855–863 [DOI] [PubMed] [Google Scholar]
- 27. Sutter I., Park R., Othman A., Rohrer L., Hornemann T., Stoffel M., Devuyst O., von Eckardstein A. (2014) Apolipoprotein M modulates erythrocyte efflux and tubular reabsorption of sphingosine-1-phosphate. J. Lipid Res. 55, 1730–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang E., Norred W. P., Bacon C. W., Riley R. T., Merrill A. H., Jr. (1991) Inhibition of sphingolipid biosynthesis by fumonisins: implications for diseases associated with Fusarium moniliforme. J. Biol. Chem. 266, 14486–14490 [PubMed] [Google Scholar]
- 29. Mullen T. D., Jenkins R. W., Clarke C. J., Bielawski J., Hannun Y. A., Obeid L. M. (2011) Ceramide synthase-dependent ceramide generation and programmed cell death: involvement of salvage pathway in regulating postmitochondrial events. J. Biol. Chem. 286, 15929–15942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sorenson R. C., Bisgaier C. L., Aviram M., Hsu C., Billecke S., La Du B. N. (1999) Human serum paraoxonase/arylesterase's retained hydrophobic N-terminal leader sequence associates with HDLs by binding phospholipids: apolipoprotein A-I stabilizes activity. Arterioscler. Thromb. Vasc. Biol. 19, 2214–2225 [DOI] [PubMed] [Google Scholar]
- 31. Smith A. B., Esko J. D., Hajduk S. L. (1995) Killing of trypanosomes by the human haptoglobin-related protein. Science 268, 284–286 [DOI] [PubMed] [Google Scholar]
- 32. Boadu E., Bilbey N. J., Francis G. A. (2008) Cellular cholesterol substrate pools for adenosine-triphosphate cassette transporter A1-dependent high-density lipoprotein formation. Curr. Opin. Lipidol. 19, 270–276 [DOI] [PubMed] [Google Scholar]
- 33. Gillotte K. L., Davidson W. S., Lund-Katz S., Rothblat G. H., Phillips M. C. (1998) Removal of cellular cholesterol by pre-β-HDL involves plasma membrane microsolubilization. J. Lipid Res. 39, 1918–1928 [PubMed] [Google Scholar]
- 34. Gillotte K. L., Zaiou M., Lund-Katz S., Anantharamaiah G. M., Holvoet P., Dhoest A., Palgunachari M. N., Segrest J. P., Weisgraber K. H., Rothblat G. H., Phillips M. C. (1999) Apolipoprotein-mediated plasma membrane microsolubilization: role of lipid affinity and membrane penetration in the efflux of cellular cholesterol and phospholipid. J. Biol. Chem. 274, 2021–2028 [DOI] [PubMed] [Google Scholar]
- 35. Chisholm J. W., Burleson E. R., Shelness G. S., Parks J. S. (2002) ApoA-I secretion from HepG2 cells: evidence for the secretion of both lipid-poor apoA-I and intracellularly assembled nascent HDL. J. Lipid Res. 43, 36–44 [PubMed] [Google Scholar]
- 36. Ahnström J., Faber K., Axler O., Dahlbäck B. (2007) Hydrophobic ligand binding properties of the human lipocalin apolipoprotein M. J. Lipid Res. 48, 1754–1762 [DOI] [PubMed] [Google Scholar]
- 37. Weinberg R. B., Gallagher J. W., Fabritius M. A., Shelness G. S. (2012) ApoA-IV modulates the secretory trafficking of apoB and the size of triglyceride-rich lipoproteins. J. Lipid Res. 53, 736–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gallagher J. W., Weinberg R. B., Shelness G. S. (2004) ApoA-IV tagged with the ER retention signal KDEL perturbs the intracellular trafficking and secretion of apoB. J. Lipid Res. 45, 1826–1834 [DOI] [PubMed] [Google Scholar]