

Background: Physiological signals that negatively regulate CYP450s are not well understood.
Results: Loss of Abcb6 in mice results in suppression of CYP450 activity.
Conclusion: Suppression of P450 activity in Abcb6 deficiency may result from altered endogenous metabolites involved in maintaining homeostasis.
Significance: Understanding metabolite alterations in Abcb6 deficiency should help understand the physiological signals and the mechanisms involved in negative regulation of P450s.
Keywords: ABC Transporter, Cytochrome P450, Gene Regulation, Membrane Transport, Metabolic Regulation
Abstract
Although endogenous mechanisms that negatively regulate cytochrome P450 (P450) monooxygenases in response to physiological and pathophysiological signals are not well understood, they are thought to result from alterations in the level of endogenous metabolites, involved in maintaining homeostasis. Here we show that homeostatic changes in hepatic metabolite profile in Abcb6 (mitochondrial ATP-binding cassette transporter B6) deficiency results in suppression of a specific subset of hepatic P450 activity. Abcb6 null mice are more susceptible to pentobarbital-induced sleep and zoxazolamine-induced paralysis, secondary to decreased expression and activity of Cyp3a11 and Cyp2b10. The knock-out mice also show decrease in both basal and xeno-inducible expression and activity of a subset of hepatic P450s that appear to be related to changes in hepatic metabolite profile. These data, together with the observation that liver extracts from Abcb6-deficient mice suppress P450 expression in human primary hepatocytes, suggest that this mouse model may provide an opportunity to understand the physiological signals and the mechanisms involved in negative regulation of P450s.
Introduction
The cytochrome P450s constitute a superfamily of monooxygenases that play key roles in the biotransformation of endogenous and exogenous compounds (1–4). Modification of P450 enzymatic activity by inhibition, induction, or activation is linked to altered biological activity of endogenous substrates and therapeutic drugs, resulting in human diseases and harmful drug-drug interactions (4–6). Many CYP genes are inducible in response to a variety of chemical signals, and considerable insight has been gained into the molecular mechanisms that regulate the transcriptional induction of P450s (5, 7–9). Evidence suggests that CYP expression can also be decreased in response to physiological and pathophysiological signals (10–12). However, compared with our knowledge of P450 induction, our understanding of the fundamental mechanisms involved in P450 suppression is still incomplete.
ABCB6 (the ATP-binding cassette transporter B6) belongs to a superfamily of integral membrane proteins that play important roles in many cellular processes and in the therapeutic response to medications (13–16). ABCB6 was initially characterized as a mitochondrial transporter capable of regulating porphyrin synthesis (17). However, recent studies suggest that loss of Abcb6 expression in mice does not affect basal porphyrin synthesis (18). Consistent with this observation, in humans, loss of function mutations in the ABCB6 gene results in a variety of complex human diseases whose pathogenesis is independent of porphyrin synthesis (19–23). Thus, the physiological role of Abcb6 in vivo remains elusive.
Here we show that Abcb6 deficiency suppresses CYP450 expression in mice and human hepatocytes. We report that ablation of Abcb6 reprograms hepatic metabolite profile that negatively regulates hepatic CYP expression, probably as a homeostatic response to promote survival.
EXPERIMENTAL PROCEDURES
Chemicals
All reagents were purchased from Sigma-Aldrich unless stated otherwise.
Abcb6 Null Mice
Abcb6 knock-out mice were generated on C57BL6/N background using ES cells developed by the trans-NIH Knock-Out Mouse Project. The ablation cassette (velocigene cassette [bacterial β-galactosidase-polyadenylation signal-loxP (locus of X over P1) site-human ubiquitin C gene promoter-neomycin phosphotransferase-polyadenylation signal-loxP site]) used to generate Abcb6 knock-out mice replaces the Abcb6 ORF containing exons 3–5 with the β-galactosidase-hUBC/em7-neomycin-poly(A) cassette, where the neomycin expression cassette is flanked by loxP sites (24). Microinjection of ES cells and generation of Abcb6 heterozygous mice were done according to standard procedures. Mice were genotyped using appropriate primers. The first primer pair anchors to exon 1 (WT-F; see Fig. 1a) and exon 3 (WT-R; see Fig. 1a) and amplifies 470 bp of the WT allele. The sequences of the primers are as follows: WT-F, 5′-GCCCCCAGTCTTATACTCTACACG-3′, and WT-R, 5′-CCCATGCCTCTCTCTGCTTTCC-3′. The second primer pair anchors to exon 1 (KO-F; see Fig. 1a) and the neomycin cassette (KO-R; see Fig. 1a) and amplifies 620 bp of the mutant allele. The primer sequences are as follows: KO-F, 5′-CACACCTCCCCCTGAACCTGAAA-3′, and KO-R, 5′-CGTGACCCCTCTCAGAGTTAGGAAAG-3′).
FIGURE 1.

Targeted disruption of Abcb6 results in a pleiotropic phenotype. a, schematic presentation of the wild-type locus (Abcb6 genomic fragment), targeted construct (targeting construct), and deleted locus (disrupted allele). Boxes represent exons. Arrows represent genotyping primer priming site. F, forward; R, reverse primers. b, PCR analysis of genomic DNA from tails. c, expression of Abcb6 mRNA in liver as determined by real time RT-PCR. Mean ± S.E. data from three independent measurements are shown. *, significantly different from Abcb6+/+ mice; p < 0.001. #, significantly different from Abcb6+/− mice; p < 0.001. d, immunostaining for Abcb6 in liver. Top panel Abcb6 immunostaining; bottom panel actin immunostaining (loading control). e, HPLC analysis of hepatic heme levels. Mean ± S.E. data from three independent measurements are shown. Heme concentration was calculated from a heme standard curve. f, expression of hepatic enzymes involved in heme synthesis as determined by real time RT-PCR in Abcb6+/+ (WT) and Abcb6−/− (KO) mouse. Mean ± S.E. data from three independent measurements are shown. g, Mendelian inheritance pattern of Abcb6 allele. h, representative photo of age-matched (6 weeks) and sex-matched (males) Abcb6−/− short stature mice and mice with no gross abnormal phenotype presented alongside a ruler for reference. ALAS1, aminolevulinic acid synthase; ALAD, aminolevulinate dehydratase; UROS, uroporphyrinogen III synthase; UROD, uroporphyrinogen III decarboxylase; CPOX, coproporphyrinogen oxidase; FECH, ferrochelatase.
Animal Studies
All animal experiments were approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee. Mice were housed in polycarbonate cages (four per cage), provided normal diet and water ad libitum, and maintained on a 12-h/12-h light-dark cycle at 22 ± 5 °C and 50 ± 20% relative humidity. All Abcb6 knock-out mice used in these studies were mice that escaped stunted growth (those that appear normal in Fig. 1h). The reason for this is because most of these small/stunted mice do not survive beyond 5–6 weeks after weaning.
Cell Culture, Transduction, and Treatment
Human liver-derived cell lines Hep3B and Huh7 were from the American Type Culture Collection (Manassas, VA). Hep3B and Huh7 cells were cultured in modified Eagle's medium supplemented with 10% FBS, 2 mm l-glutamine, and 100 units/ml penicillin. Knockdown of endogenous ABCB6 expression in Hep3B and Huh7 cells were achieved, as previously described (21), using ABCB6-specific shRNA lentiviral particles. ABCB6-shRNA viral particles were obtained from Sigma-Aldrich. Stable cell lines harboring either ABCB6-shRNA or the scrambled shRNA were cultured in selection medium containing 0.6 μg/ml puromycin. ABCB6 knockdown in these cell lines was confirmed by RT-PCR and Western blot analysis using gene-specific primers and protein-specific antibodies (21).
Human liver specimens were obtained from The University of Kansas hospital in accordance with all hospital policies and an approved IRB protocol. Human hepatocytes were isolated and cultured as previously described (25).
Mitochondria Isolation, Liposome Reconstitution, and Transport Studies
Mitochondria were isolated as previously described (17). Briefly, liver tissue was homogenized using a Dounce homogenizer in MS buffer (210 mmol/liter mannitol, 70 mmol/liter sucrose, 5 mmol/liter Tris, pH 7.4, and 1 mmol/liter EDTA) containing protease inhibitor mixture (Roche Applied Science). The supernatant was collected after centrifugation at 1,500 × g for 10 min. The supernatant was centrifuged at 9,000 × g for 15 min to pellet mitochondria. Crude mitochondria were purified from the endoplasmic reticulum as previously described (26). Liposome preparation and liposome transport studies were conducted as previously described (27).
Preparation of Microsomes
Microsomes were prepared from Abcb6+/+, Abcb6+/−, and Abcb6−/− mice liver tissues as described (28). Briefly, 0.3–0.5 g of liver tissue was homogenized in homogenization buffer (0.154 m KCl, 0.25 m sucrose in 0.05 m phosphate buffer, pH 7.5) and centrifuged at 17,000 × g for 30 min at 4 °C. The supernatant was subjected to centrifugation at 100,000 × g for 90 min at 4 °C. The resulting microsomal pellet was resuspended in resuspension buffer (20% glycerol in 0.1 m phosphate buffer pH 7.5). Microsomal protein concentrations were determined using the Bio-Rad protein assay reagent. Microsomes were stored at −80 °C until used for Western blot analysis and/or P450 activity assays.
Immunoblotting
Western blot analysis of mitochondrial and microsomal proteins was carried out as previously described (28, 29). Polyclonal primary antibodies were used to detect P450 oxidoreductase (catalog no. ab13513; Abcam, Cambridge, MA); Cyp2e1 (catalog no. ab19140; Abcam) Cyp2b10 (catalog no. AB9916; Millipore, Billerica, MA), Gapdh (catalog no. 2118; Cell Signaling, Danvers, MA), Abcb6 (21), and Cyp3a and Cyp1a (kind gift from Dr. Xiaochao Ma, School of Pharmacy, University of Pittsburgh, PA). Immunoreactive proteins were detected using polyclonal goat anti-rabbit horseradish peroxidase IgG secondary antibodies (Thermo Scientific, Waltham, MA) and visualized using SupersignalTM chemiluminescent horseradish peroxidase substrate (Thermo Scientific). Densitometric analysis was performed using ImageJ analysis software (National Institutes of Health).
RNA Isolation, Reverse Transcription, and Real Time PCR Analysis
RNA isolation from liver tissue was done using TRIzol® reagent (Invitrogen). 1 μg of RNA was used for reverse complementation using iScriptTM cDNA synthesis kit, following the manufacturer's protocol (Bio-Rad). Real time PCR was performed using the CFX384TM real time PCR system (Bio-Rad), as described previously by using primer sets specific for the mouse genes (29).
Microarray Analysis
Microarray and Microarray data analysis was performed as described previously (30).
Mass Spectrometry-based P450 Assay
The CYP activities, Cyp3a11 (midazolam to hydroxymidazolam), Cyp2b6 (bupropion to hydroxybupropion), Cyp2e1 (chlorzoxazone to hydroxychlorzoxazone), and Cyp1a2 (melatonin to hydroxymelatonin), were determined in microsomes isolated from mouse liver, using probe substrate metabolism assays as described (28). Briefly, the incubation reaction consisted of 3 μm midazolam, 5 μm chlorozoxazone, 50 μm bupropion, or 30 μm melatonin with 0.03 mg of mouse liver microsomes, in a final volume of 200 μl of 1× phosphate-buffered saline (pH 7.4). Following 5 min of preincubation at 37 °C, the reaction was initiated by adding NADPH (final concentration, 1.0 mm) and incubated for an additional 10 min for midazolam, 15 min for chlorozoxazone, 15 min for melatonin, and 20 min for bupropion, with shaking at 37 °C. Incubations were terminated by adding 200 μl of ice-cold acetonitrile, and the incubation reaction was centrifuged at 18,000 × g for 10 min. Supernatants from the centrifugation were transferred to an auto sampler vial, and 5 μl was injected into the ultra performance liquid chromatography-quadrupole time of flight mass spectrometry (UPLC-QTOFMS; SYNAPT-G1 Waters, Milford, MA)3 system for metabolite analysis.
Heme Measurement
Heme measurement was done using liver tissue from Abcb6+/+, Abcb6+/−, and Abcb6−/− mice as described (26). Briefly, 25 mg of liver tissue was homogenized in 100 μl of 1× phosphate-buffered saline, pH 7.4. Heme extraction was performed using 2× volumes of ethyl acetate and acetic acid mixture (4:1). The sample was vortexed briefly and centrifuged at 20,000 × g for 5 min. Each supernatant was diluted 5× with 50% acetonitrile and transferred to an auto sampler vial, of which 5 μl was injected into the UPLC-QTOFMS system for heme analysis.
UPLC-QTOFMS Analysis
UPLC-QTOFMS analyses were performed as described previously (31). Briefly, a 100 mm × 2.1 mm (Acquity 1.7 μm) UPLC BEH C-18 column (Waters) was used to separate metabolites. The flow rate of the mobile phase was 0.3 ml·min−1, with a gradient ranging from 2 to 98% aqueous acetonitrile, containing 0.1% formic acid in a 10-min run. QTOFMS was operated in a positive mode with electrospray ionization. The source temperature and desolvation temperature were set at 120 and 350 °C, respectively. Nitrogen was applied as the cone gas (10 liter·h−1) and desolvation gas (700 liter·h−1), and argon as the collision gas. QTOFMS was calibrated with sodium formate and monitored by the intermittent injection of lock mass leucine enkephalin, in real time, generating a reference ion at m/z 556.2771. The capillary voltage and cone voltage were set at 3.5 kV and 35 V in positive ion mode. Mass chromatograms and mass spectra were acquired by MassLynx software (Waters) in centroid format from m/z 50 to 1000. Identification of major metabolites was performed using MakerLynx software (Waters), based on accurate mass measurement (mass errors less than 10 ppm). All incubations were performed in duplicate.
Pentobarbital-induced Sleeping Time
Pentobarbital-induced sedation of Abcb6+/+ and Abcb6−/− mice was determined, after animals were injected with a single intraperitoneal dose of sodium pentobarbital (40 mg·kg−1 body weight) in corn oil. Sleep latency was determined by measuring the duration of time it took for the animals to completely lose their righting reflex, following pentobarbital injection. Sedation was determined by measuring the duration of sleep time, defined as the period from the moment of complete loss of righting reflex to the time the animals regained their reflexes.
Zoxazolamine-induced Paralysis
Zoxazolamine-induced paralysis of Abcb6+/+ and Abcb6−/− mice was determined after animals were injected with a single intraperitoneal dose of zoxazolamine (300 mg·kg−1) in corn oil. Mice were then placed on their backs. Paralysis time was determined by measuring the duration of inactivity, defined as the period of time from the moment of complete loss of reflex to the time the animals regained enough consciousness to right themselves. In experiments where TCPOBOP was used to induce Cyp2b10 expression, mice were given a single intraperitoneal dose of TCPOBOP (3 mg·kg−1) in corn oil or corn oil alone. All animals received intraperitoneal zoxazolamine (425 mg·kg−1) in corn oil 24 h following TCPOBOP treatment. Paralysis time was measured as described above.
Hemin Treatment Mice
All hemin solutions were freshly prepared as described (32). Briefly, hemin (Sigma-Aldrich) was dissolved together with Trizma base in 0.1 m NaOH solution and diluted in saline. Next, pH 11–12 of the hemin solution was adjusted to pH 8 with HCl. The solution was then filter sterilized, protected from light, and directly used. The dose and frequency of hemin administration to mice, were based on previous reports (33), and our own preliminary studies, which demonstrated that 5 mg·kg−1 heme, given intraperitoneally every 12 h for 3 days, increased hepatic heme levels and induced HO activity, with no evidence of liver toxicity. The main study included three mice per group (Abcb6+/+ and Abcb6−/−) per experiment, which were treated with either vehicle alone or hemin (1.25, 2.5, or 5 mg·kg−1), administered intraperitoneally once every 12 h for 3 days. At the end of treatment, mice were sacrificed, and livers were harvested and frozen in liquid nitrogen, for subsequent biochemical analysis and RNA preparation.
P450 Promoter-LUC Reporter Assays in HepG2 Cells
Promoter-reporter luciferase activity assays were performed as previously described (29). Briefly, HepG2 cells at ∼60% confluency were transfected with either (a) 200 ng of CYP3A4-luc-reporter plasmid (34) (pGL3-CYP3A4-luc), along with 50 ng of PXR expression plasmid (34) (pcDNA3-hPXR), a generous gift from Dr. Taosheng Chen (St. Jude Children's Research Hospital, Memphis, TN); (b) 200 ng of Cyp2b10-luc-reporter plasmid (35) (pGL3–2b10-luc), along with 50 ng of CAR expression plasmid (35) (pCMX-mCAR), a generous gift from Dr. Ronald M. Evans (Salk Institute for Biological Studies, La Jolla, CA); or (c) 250 ng of CYP1A-luc reporter plasmid (p1A1-FL) a generous gift from Dr. Robert Barouki (INSERM, Paris, France). All transfections were performed using Lipofectamine reagent following the manufacturer's protocol (Invitrogen). All transfections also included Renilla luciferase (100 ng/well) as an internal transfection control. Following overnight transfection, the transfected cells were treated with either (a) rifampicin (5 μm), (b) TCPOBOP (500 nm), or (c) 3-methylcholanthrene (3-MC; 10 μm) and the indicated amount of either Abcb6+/+ or Abcb6−/− liver extract. The relative activity of the promoter constructs was determined, after subtraction of the values obtained for the corresponding luciferase control vectors, and the results were expressed in relative terms. All experiments were performed at least three times, with a minimum of four replicates per experiment.
Preparation of Mouse Liver Extracts
Metabolite extraction from liver samples was done as described (36). Briefly, 100 mg of liver tissue was ground and suspended in 4 ml of a 1:1 water/methanol mixture. The homogenate was cooled on ice, and precipitated material was removed by centrifugation at 20,000 × g for 15 min. The supernatant was removed and evaporated in a SpeedVac (Labconco Inc., Kansas City, MO). The residue was resuspended in 300 μl of water/methanol (1:1), filtered through a 0.2 μm ultracentrifuge filter (Millipore), and used for cell treatment.
mRNA Stability Assay
In these studies, HepG2 cells grown to subconfluence were treated with actinomycin D (Sigma), at a final concentration of 2.5 μm, to arrest de novo RNA synthesis. To determine the half-life of P450 transcript, actinomycin D-treated cells were harvested at 0, 2, 4, 6, 8, 16, 20, and 24 h, and P450 mRNA was quantified by RT-PCR as described above. GAPDH mRNA levels were also monitored as controls. In experiments designed to determine the effect of Abcb6+/+ and Abcb6−/− mouse liver extract on P450 mRNA stability, cells were pretreated with actinomycin D (2.5 μm) for 4 h before exposing them to either Abcb6+/+ or Abcb6−/− liver extract. The cells were cultured in the presence of liver extract for 16 h before harvesting the cells for mRNA analysis. Throughout the liver extract treatment period, the cells were exposed to actinomycin D to prevent transcription of nascent mRNA. Total RNA isolation and RT-PCR of analysis of transcripts following actinomycin D and liver extract treatment were performed as described above.
Statistical Analysis
Statistical analysis of the observed values was performed using the Student's t test. All calculations were performed with SPSS statistical software package (SPSS Inc., Chicago, IL). All values are expressed as either means ± S.D. or ± S.E. Significant differences between the groups were determined with SPSS 10.0 software (SPSS Inc.). A difference was considered significant at the p < 0.05 level.
RESULTS
Abcb6 Knock-out Mice Show Pleiotropic Phenotype
To disrupt the Abcb6 gene, the region extending from exons 3 to 5 of Abcb6 was replaced with a neomycin-containing cassette by homologous recombination (Fig. 1a). We confirmed successful ablation, by the virtual loss of Abcb6 gene (Fig. 1, b and c) and protein expression (Fig. 1d). Elimination of Abcb6 in mice resulted in a pleiotropic phenotype. Pups lacking Abcb6 show a non-Mendelian inheritance, with nearly 40% fewer mice of the Abcb6−/− genotype than expected (Fig. 1g). Interestingly, we also observed relatively small Abcb6−/− mice among the littermates that escaped embryonic lethality (Fig. 1h; frequency of ∼20%). The remaining Abcb6−/− mice that escaped both embryonic lethality and short stature appeared normal without any observable gross phenotype.
Abcb6 has been suggested to play a role in porphyrin synthesis (17). Thus, the significance of Abcb6 expression to hepatic heme synthesis was evaluated in Abcb6−/− mice. As shown in Fig. 1e, hepatic heme levels decreased in Abcb6+/− and Abcb6−/− mice, in a manner that was consistent with a gene dose effect. However, the values did not reach statistical significance. Further, we did not see any compensatory changes in the hepatic heme synthetic pathway enzymes in Abcb6−/− mice (Fig. 1f). These results suggest that although Abcb6 is capable of transporting heme precursors into the mitochondria (18, 27) and plays a role in regulating porphyrin synthesis in vitro (17, 18, 26), Abcb6 deficiency in vivo does not have a significant impact on basal hepatic porphyrin or heme levels. These findings are consistent with recent reports that demonstrate a lack of functional association between Abcb6 and basal porphyrin synthesis in erythroid cells in vivo (18).
Abcb6 Knock-out Mice Demonstrate Altered Xenobiotic Response
During the course of a surgical procedure, we observed that the Abcb6−/− mice (which appeared normal) given pentobarbital as an anesthetic were more susceptible to pentobarbital-induced sleep compared with Abcb6+/+ mice. To confirm this further, we repeated the pentobarbital-induced sleep experiment in the absence of any surgical procedure. After a single dose of pentobarbital (40 mg/kg), Abcb6−/− mice demonstrated significantly increased sleeping time (Fig. 2a, right panel) compared with Abcb6+/+ mice, despite both groups showing similar sleep latency (Fig. 2a, left panel).
FIGURE 2.

Abcb6 deficiency suppresses xenobiotic response. a, right panel, pentobarbital-induced sleeping time but not (left panel) sleep latency is augmented in Abcb6−/− mice given a single injection (intraperitoneal) of pentobarbital (40 mg/kg). Mean ± S.E. data representative of three independent experiments with six mice per group per experiment. *, significantly different from Abcb6+/+ mice; p < 0.01. b, isoflurane-induced sleeping time (right panel) and sleep latency (left panel) are not altered Abcb6−/− mice. Mean ± S.E. data representative of three independent experiments with six mice per group per experiment. c, ATP-dependent pentobarbital transport is not different between (left panel) mitochondria isolated from Abcb6 WT and Abcb6 KO mice or in (right panel) Abcb6 reconstituted liposomes. The data are representative of three independent experiments. d–g, Cyp3a11 (d), Cyp1a2 (e), and Cyp2b10 (f) enzyme activities but not Cyp2e1 (g) enzyme activity is attenuated in Abcb6−/− mice. P450 enzyme activity was measured in liver microsomes as described under “Experimental Procedures.” Enzyme activity data presented as means ± S.E. are representative of three independent experiments with four mice per group per experiment. *, significantly different from Abcb6+/+ mice; p < 0.01. #, significantly different from Abcb6+/− mice; p < 0.01.
Increased susceptibility of Abcb6−/− mice to pentobarbital could occur, if Abcb6−/− mice were sensitive to anesthetics in general. To test this, we repeated the sleep experiment, substituting isoflurane in place of pentobarbital. Following exposure to isoflurane, all mice in the Abcb6+/+ and Abcb6−/− groups had similar sleep latency (Fig. 2b, left panel) and similar sleeping time (Fig. 2b, right panel). These results suggest that loss of Abcb6 expression in vivo does not affect the general anesthetic response but has a profound consequence on the sedative effect of pentobarbital.
The length of sedation following treatment with pentobarbital, is directly proportional to hepatic metabolism and/or hepatic clearance of the parent compound (37). Given that Abcb6 is a transporter, increased sensitivity of Abcb6−/− mice to pentobarbital could result from increased accumulation of the parent compound in the absence of Abcb6. However, such a hypothesis would require pentobarbital to be a transport substrate of Abcb6. To test this hypothesis, we measured Abcb6 -mediated pentobarbital transport using (a) mitochondria isolated from Abcb6+/+ and Abcb6−/− mouse liver and (b) liposomes reconstituted with either a transport-competent Abcb6 (Abcb6-WT) or a transport-incompetent Abcb6 (Abcb6-MT). As shown in Fig. 2c, we found no difference in energy-dependent pentobarbital transport kinetics between Abcb6+/+ and Abcb6−/− mitochondria (Fig. 2c, left panel) or liposomes (Fig. 2c, right panel). These results suggest that pentobarbital is not a transport substrate of Abcb6.
Decreased P450 Activity in Abcb6 Knock-out Mice
Pentobarbital is a classic CYP substrate and is metabolized to an inactive compound by hepatic CYP enzyme activity, predominantly by Cyp3a11 (37). Thus, increased sensitivity of Abcb6−/− mice to pentobarbital could also result from altered Cyp3a11 activity. To test this, we measured Cyp3a11 activity in Abcb6−/− liver microsomes, by measuring the biotransformation of midazolam, a commonly used in vitro probe for the prediction of Cyp3a11 activity (38). As shown in Fig. 2d, Abcb6−/− liver microsomes showed significant decrease in midazolam biotransformation, suggesting decreased Cyp3a11 activity in Abcb6−/− mice.
We next evaluated whether the altered P450 activity in Abcb6 deficiency was specific to Cyp3a11. In addition to Cyp3a, mammalian hepatic CYPs involved in the metabolism of endogenous compounds and xenobiotics include members of the Cyp1a, Cyp2a, Cyp2b, Cyp2c, Cyp2d, and Cyp2e subfamilies (9, 10). Among these, Cyp3a, Cyp2e, Cyp2b, and Cyp1a forms account for majority of liver microsomal total CYP content and activity (39, 40). Thus, to understand the extent of change in P450 activity in Abcb6−/− mice, we measured the biotransformation of melatonin, bupropion, and chlorozoxazone, commonly used in vitro probes for predicting Cyp1a2, Cyp2b10, and Cyp2e1 activities, respectively (41–44). Interestingly, we found a significant decrease in the biotransformation of melatonin and bupropion but not chlorozoxazone in Abcb6−/− liver microsomes (Fig. 2, e–g, respectively). These results suggest that Abcb6 deficiency in mice modifies the activity of a specific set of hepatic P450s.
Results presented in Fig. 2d demonstrating significant decrease in Cyp2b10 activity in Abcb6−/− mice prompted us to test whether Abcb6−/− mice were more susceptible to zoxazolamine-induced paralysis. Zoxazolamine, a muscle relaxant, is inactivated by CYP enzyme activity with a preference for Cyp2b10 and like pentobarbital is traditionally used as an in vivo measure of altered P450 activity (45). Therefore, the length of paralysis following treatment with zoxazolamine is indicative of Cyp2b10 activity. As shown in Table 1, Abcb6+/+ mice treated with zoxazolamine recovered from paralysis no later than 6 h. In contrast, Abcb6−/− mice were paralyzed for more than 12 h. As with pentobarbital, zoxazolamine did not appear to be a transport substrate of Abcb6 (data not shown). Taken together, results from the pentobarbital and the zoxazolamine studies suggest that loss of Abcb6 function modifies xenobiotic response in mice.
TABLE 1.
Zoxazolamine paralysis test
Zoxazolamine-induced paralysis is augmented in Abcb6−/− mice given a single injection (intraperitoneally) of 300 mg/kg zoxazolamine. The data grouped together are from two independent experiments with six mice per group per experiment.
| Abcb6+/+ |
| 1/12 > 1 h paralysis; recovered |
| 2/12 > 2 h paralysis; recovered |
| 8/12 > 4 h paralysis; recovered |
| 1/12 > 6 h paralysis; recovered |
| Abcb−/− |
| 12/12 > 12 h paralysis; recovered |
Absence of Abcb6 Suppresses Hepatic P450 Transcription
Studies investigating the mechanisms responsible for variability in CYP function suggest a role for transcriptional gene activation/repression to be particularly important (9, 11, 40). Given the lack of association between hepatic heme levels and loss of Abcb6 in Abcb6−/− mice, we hypothesized that decreased CYP activity in Abcb6−/− mice might be a result of decreased expression of CYPs. To test this hypothesis, we measured Cyp3a11, Cyp2b10, Cyp1a2, and Cyp2e1 expression in Abcb6+/+ and Abcb6−/− mice liver. As presented in Fig. 3a, we found significant decrease in mRNA levels of Cyp3a11, Cyp2b10, and Cyp1a2 but not Cyp2e1 in Abcb6−/− mice. Further, the decreased mRNA levels of Cyp2b10, Cyp3a11, and Cyp1a2 correlated well with the respective protein expression in Abcb6+/+ and Abcb6−/− mice (Fig. 3b, left and right panels). These results are consistent with the P450 activity assays described in Fig. 2d, suggesting that altered P450 activity in Abcb6−/− mice is due to decreased expression of P450s.
FIGURE 3.

Abcb6 deficiency attenuates constitutive expression of hepatic P450s. a, Cyp3a11, Cyp2b10, and Cyp1a2 but not Cyp2e1 mRNA expression was down-regulated in Abcb6−/− mice. mRNA expression was determined by real time RT-PCR. The data are presented as means ± S.E. from four independent measurements with three mice per group per experiment. *, significantly different from Abcb6+/+ mice; p < 0.01. #, significantly different from Abcb6+/− mice; p < 0.01. b, left panel, expression of hepatic Cyp3a11, Cyp2b10, and Cyp1a2 but not Cyp2e1 or P450 oxidoreductase (POR) protein is attenuated in Abcb6−/− mice. Protein expression was determined by immunostaining using protein specific antibodies as described under “Experimental Procedures.” The data are representative of three independent experiments with three mice per group per experiment. Right panel, cumulative (average of all mice in the three independent experiments) ImageJ analysis of Cyp3a11, Cyp2b10, Cyp1a2, and Cyp2e1 immunostained protein bands in Abcb6+/+ and Abcb6−/− mice liver. The data are normalized to Gapdh and presented as means ± S.E. *, significantly different from Abcb6+/+ mice; p < 0.01. c, microarray analysis of hepatic P450 mRNA expression in Abcb6−/− mice shows a pattern that is similar to the one observed using real time RT-PCR analysis. d–f, real time RT-PCR analysis of hepatic P450 mRNA in Abcb6 deficiency shows Abcb6 allele-specific (d), non-allele-specific (e), and Abcb6-independent (f) expression pattern. The data are presented as means ± S.E. from three independent measurements with three mice per group per experiment. *, significantly different from Abcb6+/+ mice; p < 0.01. #, significantly different from Abcb6+/− mice; p < 0.01.
The attenuation of Cyp3a11, Cyp2b10, and Cyp1a2 but not Cyp2e1 expression in Abcb6 deficiency suggests that Abcb6-mediated effect on hepatic P450 expression might not be identical. Of the 102 putatively functional full-length mouse P450 genes, nearly 40% of them (∼42 P450s) are expressed in liver (39, 40). However, mechanisms regulating these hepatic P450 enzymes, their pattern, and the level of expression vary. Therefore, to gain a broad understanding and to evaluate the extent of alteration, in hepatic P450 expression in Abcb6−/− mice, we performed a global hepatic microarray analysis (Fig. 3c) followed by a targeted P450 real time RT- PCR analysis, for the 42 P450s using gene specific primers (Fig. 3, d–f). We found three different patterns of hepatic P450 expression in Abcb6−/− mice; 10 of the 42 P450s analyzed showed a direct correlation with loss of Abcb6, in a manner that was consistent with allele-specific effects (gene dose effect; Fig. 3d); 17 of the 42 P450s showed a direct correlation with loss of both Abcb6 alleles, but no significant difference was seen with loss of a single allele (Abcb6+/+ versus Abcb6−/− but not Abcb6+/+ versus Abcb6+/− mice; Fig. 3e), whereas the remaining 12 P450s showed no association with Abcb6 genotype (Fig. 3f). Together, these results suggest that loss of Abcb6 expression in mice modifies the expression of a specific subset of hepatic P450s.
Xeno-inducible Expression of CYP450 Is Compromised in Abcb6 Deficiency
In addition to basal expression, the extent to which xenobiotics can induce CYP enzyme expression is of pharmacological importance because in principle this activation will affect not only the metabolism of the specific xenobiotic but also the metabolism of any compound processed by cytochromes (46, 47). Thus, to investigate the extent to which the inducible expression of CYPs is altered in Abcb6 deficiency, we assessed the expression and activity of Cyp3a11 and Cyp2b10, in response to prototypical rodent specific inducers of Cyp3a11 (pregnenolone 16 α-carbonitrile) and Cyp2b10 (1,4-bis[2-(3,5-dichloropyridyloxy)] benzene; TCPOBOP). As shown in Fig. 4, the inducible mRNA expression of both Cyp3a11 (Fig. 4a) and Cyp2b10 (Fig. 4b) was attenuated in Abcb6 null mice. Further, consistent with decreased mRNA expression, Cyp2b10 protein expression, enzyme activity, and zoxazolamine sensitivity were altered in Abcb6 null mice (Fig. 4, c–e, respectively). These results suggest that loss of Abcb6 expression modifies P450 xeno-regulation in vivo.
FIGURE 4.

Abcb6 deficiency attenuates xeno-inducible expression of Cyp3a11 and Cyp2b10. a and b, expression of hepatic Cyp3a11 mRNA in response to pregnenolone 16 α-carbonitrile (a) and expression of hepatic Cyp2b10 mRNA in response to TCPOBOP (b) are attenuated in Abcb6−/− mice. mRNA expression was determined by real time RT-PCR. The data are representative of three independent experiments with three mice per group per experiment. The data are presented as means ± S.E. *, significantly different from Abcb6+/+ mice with or without pregnenolone 16 α-carbonitrile; p < 0.01. #, significantly different from Abcb6+/+ mice plus or minus TCPOBOP; p < 0.01. c, left panel, TCPOBOP-inducible expression of hepatic Cyp2b10 protein is attenuated in Abcb6−/− mice. Protein expression was determined by immunostaining for Cyp2b10. The data representative of two independent experiments with four mice per group in each experiment. Right panel, cumulative (average of all mice in the two independent experiments) ImageJ analysis of immunostained protein bands. The data are normalized to tubulin and presented as means ± S.E. *, significantly different from Abcb6+/+ mice; p < 0.01. d, TCPOBOP-inducible Cyp2b10 enzyme activity is attenuated in Abcb6−/− mice. Cyp2b10 enzyme activity was measured in TCPOBOP-treated and untreated liver microsomes as described under “Experimental Procedures.” *, significantly different from Abcb6+/+ mice; p < 0.01. e, TCPOBOP-mediated reduction in zoxazolamine-induced paralysis is attenuated in Abcb6−/− mice. The data presented as means ± S.E. are representative of two independent experiments with six mice per group per experiment. Results are significantly different from those for Abcb6+/+ mice; p < 0.01.
Decreased P450 Expression in Abcb6 Knock-out Mice Is Not Heme-dependent
Although hepatic heme concentration in the Abcb6−/− mice was not statistically different from that of Abcb6+/+ mice (Fig. 1e), the concentrations were lower in the Abcb6−/− mice compared with Abcb6+/+ mice. Given that heme is an important molecule and is thought to regulate the expression of certain P450s (33, 48), we wanted to confirm that these minor decreases in hepatic heme levels in Abcb6−/− mice were not responsible for the differential P450 phenotypes. To test this, we supplemented Abcb6−/− mice with heme, as described under “Experimental Procedures,” until the hepatic heme levels in the Abcb6−/− matched the values that were seen in the Abcb6+/+ mice. Hematin at the concentration used in these studies did not show any hepatic or blood toxicity (Fig. 5b) but showed increased hepatic heme levels in Abcb6−/− mice to approximately the same levels as seen in Abcb6+/+ mice (Fig. 5a). Further, consistent with previous observations, Hemeoxygenase expression was induced in response to heme treatment (33, 49) (Fig. 5c, right panel), whereas ALAS, the rate-limiting enzyme in heme synthesis that is negatively regulated by heme levels (33) was reduced (Fig. 5C, left panel). Analysis of Cyp3a11, Cyp2b10, and Cyp1a2 expression in heme-supplemented Abcb6−/− mouse livers continued to demonstrate decreased mRNA (Fig. 5d) and protein expression (Fig. 5, e and f), suggesting that decreased P450 expression in Abcb6−/− mice is not associated with heme availability. We also ruled out the possibility that P450 suppression in Abcb6−/− mice was associated with liver injury, because Abcb6−/− mice did not show any observable gross or histological alteration in liver (data not shown), nor did they demonstrate any increase in serum biomarkers of liver injury (data not shown).
FIGURE 5.

CYP450 expression continues to decrease in Abcb6−/− mice supplemented with exogenous heme. a, hepatic heme levels in Abcb6−/− mice given exogenous heme as described under “Experimental Procedures.” b, exogenous heme supplementation in mice (at the concentration used in these studies) does not cause liver injury as measured by serum alanine aminotransferase levels. c, left panel, ALAS mRNA expression is negatively regulated following exogenous heme treatment in both Abcb6+/+ and Abcb6−/− mice. Right panel, hemeoxygenase 1 (HO1) is induced in response to heme treatment in both Abcb6+/+ and Abcb6−/−. d, mRNA expression of Cyp1a2, Cyp3a11, and Cyp2b10 continues to decrease in mice supplemented with exogenous heme. e and f, top panels, Cyp1a2 protein levels (e) and Cyp3a11 protein levels (f) continue to decrease in Abcb6−/− mice supplemented with exogenous heme (data shown for 5 mg/kg heme). Bottom panels, ImageJ analysis of Cyp1a2 (e) and Cyp3a11 (f) protein bands normalized to Gapdh expression. In all panels, the data represent means ± S.E. *, significant differences from untreated control mice; p < 0.01. #, significant differences from 1.25 mg/kg hemin-treated mice; p < 0.01. Results are representative of three independent experiments with three mice per treatment group per experiment.
ABCB6 Deficiency Suppresses P450 Expression in Human Hepatocytes
Considerable homology exists between many mouse and human P450s, and it is believed that the knowledge of mouse P450 expression and function can provide insight into P450 expression and function in humans (50). Thus, to understand the association between loss of ABCB6 expression and the pattern of P450 expression in humans, we evaluated P450 expression in human hepatoma cells (Hep3B and Huh7) deficient in endogenous ABCB6. Loss of ABCB6 expression in these cells was achieved by knockdown of ABCB6 expression using shRNA as previously described (21). Consistent with the mouse studies, loss of ABCB6 expression in the hepatomas resulted in a significant decrease in CYP1A2, CYP2B6, and CYP3A4 transcript but not CYP2E1 (Fig. 6, a and c). Further, as in the mouse studies, loss of ABCB6 expression in the hepatomas did not alter cellular heme levels (Fig. 6, b and d). These results suggest that P450 suppression in ABCB6 deficiency might be similar between mice and humans.
FIGURE 6.

Abcb6 deficiency attenuates P450 expression in human hepatoma Huh7 and Hep3B cells. a and c, expression of CYP1A2, CYP2B6, and CYP3A4 but not CYP2E1 mRNA is repressed in ABCB6 knockdown Huh7 (a) and Hep3B (c) cells (bottom panels in a and c shows Abcb6 protein expression in knockdown cells). b and d, loss of endogenous ABCB6 expression does not affect heme levels in Huh7 (b) and Hep3B (d) cells. The data are presented as means ± S.E. from three independent measurements. *, significantly different from ABCB6 scrambled shRNA cells; p < 0.01. Heme levels were measured by HPLC as described under “Experimental Procedures.” mRNA expression was determined by real time RT-PCR.
Liver Extracts from Abcb6 Knock-out Mice Suppress P450 Expression in Primary Human Hepatocytes
Suppression of CYP genes in vivo are thought to result from alterations in the level of endogenous metabolites involved in maintaining homeostasis that prevent transmission of activating signals, thus preventing transcription initiation (12). Given that Abcb6 has been postulated to serve as a metabolic transmembrane transporter, we hypothesized that Abcb6 deficiency in vivo might lead to alteration in the levels of critical hepatic metabolites that could lead to transcriptional repression. To test this hypothesis, we cultured immortalized cells (HepG2) and primary human hepatocytes in media supplemented with liver extracts derived from either Abcb6+/+ or Abcb6−/− mice and measured P450 expression. We found that liver extracts from Abcb6−/− mice, dose-dependently repressed CYP3A4, CYP2B6, and CYP1A2 expression both in HepG2 cells (Fig. 7a) and in primary human hepatocytes (Fig. 7b). In contrast, no reduction in CYP expression was seen in hepatocytes supplemented with liver extracts from Abcb6+/+ mice. These results suggest that metabolite differences in Abcb6−/− liver prevent transcriptional initiation of P450 gene or stability of the P450 transcript. A comprehensive metabolomics study is currently in progress to identify the metabolite differences in Abcb6 deficiency.
FIGURE 7.

Liver extract from Abcb6−/− mice suppresses P450 expression in human hepatocytes. HepG2 (a) and primary human hepatocytes (b) cultured in the presence of Abcb6−/− liver extracts show decreased CYP1A2, CYP3A4, and CYP2B6 expression. Results in a and b representative of three independent experiments for HepG2 and two independent primary human hepatocyte cultures. Cultures were treated with liver extracts isolated from three mice in each group for each experiment. *, significantly different from Abcb6+/+ mice liver extract treated HepG2 or primary human hepatocytes; p < 0.05. #, significantly different from Abcb6+/+ mice liver extract treated HepG2 cells; p < 0.01.
P450 Suppression in Abcb6 Deficiency Is Not Mediated by Decreased Stability of P450 mRNA Transcript
As mentioned in the last paragraph, processes that regulate the abundance of mRNA transcript in cells could involve both transcriptional and post-transcriptional mechanisms. Although the transcriptional mechanisms are thought to be mediated by regulation of the promoter activity, the post-transcriptional mechanisms are thought to be mediated by regulation of mRNA transcript stability. Thus, as a first step in exploring the molecular mechanism(s) by which the altered hepatic metabolite in Abcb6−/− mice repress CYP expression, we first evaluated P450 mRNA stability. For this purpose, we first established the half-life of endogenous P450 mRNAs in HepG2 cells, by measuring the nascent mRNA transcript levels, following inhibition of transcriptional initiation, using actinomycin D as described under “Experimental Procedures.” We found that the endogenous half-life of all the P450 mRNA transcripts tested in this study was ∼ 20 h (Fig. 8a). We next evaluated the half-life of each of these P450 mRNAs (CYP3A4, CYP1A2, CYP2B6, and CYP2E1) in HepG2 cells cultured in the presence of either Abcb6+/+ or Abcb6−/− liver extract. In these studies, HepG2 cells were preincubated with actinomycin D for 4 h, before the addition of Abcb6+/+ or Abcb6−/− mouse liver extracts, to prevent transcription initiation. As seen in Fig. 8 (b–d, left panels), we found no difference in the stability of any of the P450 mRNAs tested, when cultured in the presence of either Abcb6+/+ or Abcb6−/− mouse liver extracts. In contrast, parallel studies that were conducted to assess P450 mRNA expression, in the absence of actinomycin D, continued to demonstrate decreased mRNA transcripts (Fig. 8, b–d, right panels). Taken together, these results suggest that P450 suppression in Abcb6 deficiency is not a result of decreased stability of P450 transcript but might involve transcriptional repression.
FIGURE 8.
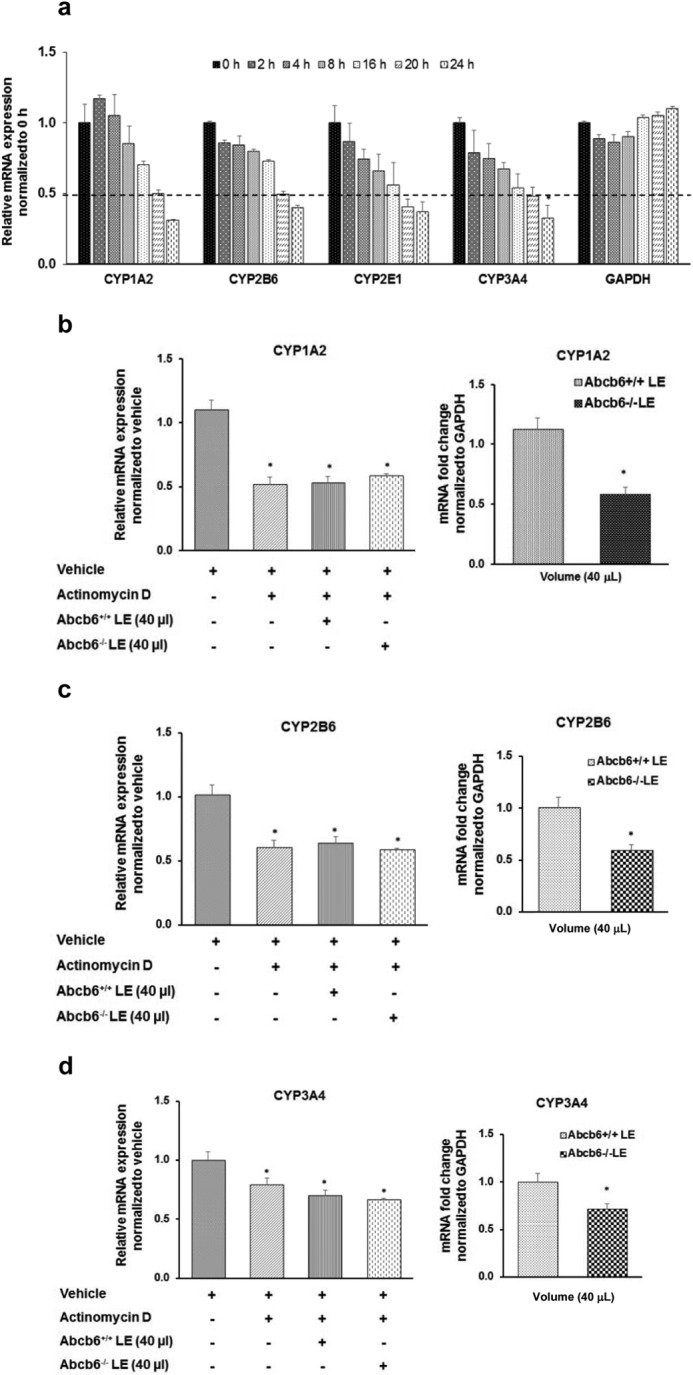
Liver extract from Abcb6−/− mice does not affect P450 mRNA stability. Nascent RNA synthesis was inhibited with actinomycin D (2.5 μm) as described under “Experimental Procedures.” a, RT-PCR analysis of CYP1A2, CYP2B6, CYP2E1, and CYP3A4 mRNA carried out to trace the remaining amount of respective CYP mRNAs with time showed an mRNA half-life of ∼ 20 h for all mRNAs. GAPDH mRNA levels were also monitored as control for stable mRNA. b–d, Abcb6−/− liver extract (LE) did not affect the stability of either CYP1A2 (b), CYP2B6 (c), or CYP3A4 (d) mRNA as measured by RT-PCR. The data shown (a–d) represent means ± S.E. for three independent experiments. *, significantly decreased mRNA expression in the presence of actinomycin D. Right panels in b–d show decreased mRNA expression of CYP1A2 (b), CYP2B6 (c), and CYP3A4 (d) in parallel experiments conducted in the absence of actinomycin D.
P450 Suppression in Abcb6 Deficiency Is Mediated via Repression of the P450 Promoter
Mechanistic studies of transcriptional repression of genes in general suggest that this process involves a complex cascade of transcription factors and other regulatory proteins that interact with specific DNA sequences generally located in the 5′-flanking region of genes to promote or inhibit transcription (9, 12). Further, as shown in Fig. 4, Abcb6−/− mice treated with either TCPOBOP (a predominantly CAR activator) or pregnenolone 16 α-carbonitrile (a predominantly PXR activator) demonstrate attenuated CYP induction. Based on these observations, we hypothesized that P450 suppression in Abcb6 deficiency might be mediated by CAR and/or PXR. Thus, as a first step in exploring the molecular mechanism of transcriptional repression, by which the altered hepatic metabolites in Abcb6−/− mice repress CYP expression, we conducted experiments using gene specific promoter-reporter plasmids, in transactivation assays to test whether suppression of CYPs in the absence of Abcb6 involved the minimal xeno-inducible 5′-flanking sequence of CYP genes regulated by CAR and/or PXR. For this purpose, we used a combination of human (CYP3A4 and CYP1A) and mouse (Cyp2b10) P450 promoter-reporter plasmids cloned upstream of firefly luciferase in transactivation assays to test whether transactivation of CYP3A4, CYP1A and Cyp2b10 promoters were suppressed in HepG2 cells treated with Abcb6−/− liver extracts. In the initial set of experiments, we used 40 μl of liver extract for promoter transactivation studies, because this volume was able to decrease endogenous mRNA levels in these HepG2 cells. Interestingly, we did not see suppression of the xeno-inducible regions of the promoter-reporter plasmids in response to 40 μl of Abcb6−/− liver extracts (Fig. 9, a–c, left panels), despite decreased endogenous P450 expression in these transfected cells (Fig. 9, a–c, right panels). On closer examination, it was realized that given the robust activation of the CYP promoter in the presence of inducers, the increased stability of luciferase, and the lack of knowledge of altered metabolite concentrations in liver extract, the assumption that 40 μl of liver extract would have the same effect on CYP promoter activity as it did with the endogenous mRNA expression in the absence of inducers was probably not correct. It is quite possible that the metabolite concentrations in liver extract were probably insufficient to generate significant differences in the activity of the promoter above the signal noise in the presence of the inducers. To explore this further, we redesigned the experiments to ask the following questions: (a) Does increasing the concentration of liver extract in the promoter transactivation studies show any difference, and are these differences dose-dependent? (b) Can stable knockdown of Abcb6 alter P450 promoter activity in the absence of exogenous treatment with liver extract. As shown in Fig. 9, we found that increasing the concentration of liver extract decreased both Cyp2b10 (Fig. 9d) and CYP3A4 (Fig. 9e) promoter activity in a dose-dependent manner. Further, we found that rifampicin-inducible CYP3A4 promoter activity was suppressed in ABCB6-deficient hepatoma cells in the absence of exogenous liver extract (Fig. 9f). Together, these results demonstrate that one potential mechanism of decreased P450 expression in Abcb6 deficiency could involve repression of P450 promoter activity. A comprehensive study is currently under progress to identify the transcription factors that may be involved in transcriptional suppression of P450 in Abcb6 deficiency.
FIGURE 9.

CYP3A4 promoter transactivation is suppressed in Abcb6 deficiency. a–c, effect of Abcb6−/− liver extract on CYP1A, Cyp2b10, and CYP3A4 promoter activity. HepG2 cells were transfected with xeno-inducible region of the CYP1A promoter (a), mCAR expression plasmid and the xeno-inducible region of Cyp2b10 promoter (b), and hPXR expression plasmid and the xeno-inducible region of CYP3A4 promoter (c). The transfected cells were cultured in the presence of either Abcb6+/+ or Abcb6−/− liver extract and then treated with prototypical inducers 3-MC (CYP1A) (a), TCPOBOP (Cyp2b10) (b), or rifampicin (CYP3A4) (c). After 24 h, luciferase activities were determined. The data are means ± S.E. of three independent experiments. *, significantly different from luciferase activity in the absence of inducers; p < 0.01. Right panels show decreased mRNA expression of CYP1A (a), CYP2B6 (b), and CYP3A4 (c) in the promoter transfected HepG2 cells treated with Abcb6+/+ or Abcb6−/− liver extracts in the absence of inducers. d and e, increasing concentrations of Abcb6−/− liver extract suppresses Cyp2b10 (d) and CYP3A4 (e) promoter activity but not their empty vector controls (right panels). HepG2 cells were cotransfected with mCAR expression plasmid and the xeno-inducible region of Cyp2b10 promoter (d) and hPXR expression plasmid and the CYP3A4 promoter (e). The cotransfected cells were then cultured in the presence of either Abcb6+/+ or Abcb6−/− liver extract followed by treatment with the prototypical inducer of Cyp2b10 (TCPOBOP) (d) or CYP3A4 (rifampicin) (e). After 24 h, luciferase activity was determined. The data are means ± S.E. of three independent experiments. *, significantly different from luciferase activity in the absence of inducers; p < 0.01. #, significantly reduced luciferase activity compared with cells cultured in the presence of Abcb6+/+ liver extract. $, significantly reduced luciferase activity compared with cells cultured in the absence of any liver extract. f, CYP3A4 promoter activity is suppressed in HepG2 cells stably transfected with Abcb6-specific shRNA. HepG2 cells stably transfected with either scrambled (control) shRNA or Abcb6-specific shRNA were cotransfected with hPXR expression plasmid and CYP3A4 promoter-reporter plasmid. The transfected cells were then treated with prototypical inducer of CYP3A4 (rifampicin). After 24 h, luciferase activity was determined. The data are means ± S.E. of three independent experiments. *, significantly different from luciferase activity in the absence of inducers; p < 0.01. $, significantly different from luciferase activity from cells treated with 5 μm rifampicin. &, significantly different from cells stably transfected with scrambled shRNA. Scr ShRNA, scramble shRNA; B6 ShRNA, Abcb6-specific shRNA; Rif, rifampicin.
DISCUSSION
The expression of cytochrome P450 genes in many species is highly regulated during development, by nutritional status, and by hormonal factors, including sex steroids, glucocorticoids, growth hormone, insulin, and inflammatory cytokines (7, 40). Although the mechanisms and consequences of regulation of P450s by drugs and chemicals have been intensively studied, relatively little is known about the mechanisms by which P450s are regulated by physiological factors (12). We show here that endogenous homeostatic responses in Abcb6 deficiency negatively regulate CYP450 expression leading to altered xenobiotic response in both mouse and human hepatocytes. These findings indicate that Abcb6 plays an unexpectedly broad role in maintaining cellular homeostasis.
Abcb6 deficiency in mice results in a pleiotropic phenotype, suggesting incomplete penetrance of the Abcb6 genotype. This observation is relatively consistent with recent reports of growth and proliferation defects associated with loss of Abcb6 function in humans and in zebrafish (22). Although Abcb6 is capable of transporting heme precursors into the mitochondria (17, 18) and plays a role in regulating porphyrin synthesis in vitro (17, 26), Abcb6 deficiency in vivo did not have a significant impact on hepatic porphyrin or heme levels. This suggests that in vivo Abcb6 might not be important for basal porphyrin synthesis. These findings are consistent with recent reports that demonstrate a lack of functional association between Abcb6 and basal porphyrin synthesis in erythroid cells in vivo (18).
In the human population, there are many incompletely understood incidences of a functional association between ABCB6 and human disease (19, 22, 51). However, the mechanistic association between loss of ABCB6 function and the pathogenesis of the disease is not clear. In light of our data, it will be of great interest to investigate whether some of these disease pathologies could be explained by loss of expression and activity of CYP450s. For example, retinal defects leading to ocular disease in humans has a strong association with loss of CYP1B1 expression (52, 53). A similar association in retinal defects is also seen with loss of ABCB6 expression in humans (22). Although these results are correlative at present, they suggest a potential functional interaction between Abcb6 and Cyp1b1 in ocular disease. Studies currently underway in a zebrafish model of Abcb6 deficiency, which show strong association between Abcb6 function and ocular defects similar to those seen in humans, should help explain Cyp1b1 and Abcb6 functional association in ocular disease.
In summary, the results presented in this manuscript provide evidence suggesting a complex physiological function for Abcb6 in vivo. Although the results presented in these studies do not define the homeostatic changes in Abcb6−/− mice that suppress Cyp450 expression, they provide compelling evidence suggesting an imbalance in endogenous metabolite homeostasis in vivo. This observation is consistent with the hypothesis that endogenous metabolites can activate or deactivate regulatory networks that govern homeostatic processes in vivo (7, 40). Our current ongoing studies using Abcb6 conditional knock-out mouse and the Abcb6 transgenic mice in combination with the Abcb6 knock-out mice described here should help us define the complex physiological function(s) of Abcb6 in vivo. These studies should help us understand whether the Abcb6−/− mouse phenotype is due to (a) a pathophysiological response to stress signals, (b) an adaptive homeostatic response, or (c) part of a tightly regulated physiological pathway mediated by Abcb6. In addition, these mouse models should provide an opportunity to understand the homeostatic mechanisms that regulate CYP450 expression in vivo.
Acknowledgments
We thank Dr. Robert Barouki (Université Paris Descartes, Paris, France) for CYP1A-luciferase plasmid, Dr. Taosheng Chen (St. Jude Children's Research Hospital, Memphis, TN) for CYP3A4-promoter luciferase and PXR expression plasmid, and Dr. Ronald Evans (Salk Institute, La Jolla, CA) for Cyp2b10-promoter luciferase and CAR expression plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grants U01HG004085 (to Velocigene at Regeneron Inc.) and U01HG004080 (to the CSD Consortium). This work was also supported by Grant U42RR024244 from Children's Hospital Oakland Research Institute, Grant 5P20RR021940-07 from the National Center for Research Resources, Grant 8P20GM103549-07 from the National Institute of General Medical Sciences, and the Biomedical Research Training Grant for the University of Kansas Medical Center (to H. C.).
- UPLC-QTOFMS
- ultra performance liquid chromatography-quadrupole time of flight mass spectrometry
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)] benzene
- PXR
- pregnane x receptor.
REFERENCES
- 1. Gonzalez F. J., Nebert D. W. (1990) Evolution of the P450 gene superfamily: animal-plant 'warfare', molecular drive and human genetic differences in drug oxidation. Trends Genet. 6, 182–186 [DOI] [PubMed] [Google Scholar]
- 2. Miller M. S., Juchau M. R., Guengerich F. P., Nebert D. W., Raucy J. L. (1996) Drug metabolic enzymes in developmental toxicology. Fundam. Appl. Toxicol. 34, 165–175 [DOI] [PubMed] [Google Scholar]
- 3. Nebert D. W., McKinnon R. A. (1994) Cytochrome P450: evolution and functional diversity. Prog. Liver Dis. 12, 63–97 [PubMed] [Google Scholar]
- 4. Nebert D. W., Russell D. W. (2002) Clinical importance of the cytochromes P450. Lancet 360, 1155–1162 [DOI] [PubMed] [Google Scholar]
- 5. Alsaad A. M., Zordoky B. N., Tse M. M., El-Kadi A. O. (2013) Role of cytochrome P450-mediated arachidonic acid metabolites in the pathogenesis of cardiac hypertrophy. Drug Metab. Rev. 45, 173–195 [DOI] [PubMed] [Google Scholar]
- 6. Nebert D. W., Dalton T. P. (2006) The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat. Rev. Cancer 6, 947–960 [DOI] [PubMed] [Google Scholar]
- 7. Nebert D. W., Gonzalez F. J. (1987) P450 genes: structure, evolution, and regulation. Annu. Rev. Biochem. 56, 945–993 [DOI] [PubMed] [Google Scholar]
- 8. Prough R. A., Linder M. W., Pinaire J. A., Xiao G. H., Falkner K. C. (1996) Hormonal regulation of hepatic enzymes involved in foreign compound metabolism. FASEB J. 10, 1369–1377 [DOI] [PubMed] [Google Scholar]
- 9. Ramana K. V., Kohli K. K. (1998) Gene regulation of cytochrome P450–an overview. Indian J. Exp. Biol. 36, 437–446 [PubMed] [Google Scholar]
- 10. Cheng P. Y., Wang M., Morgan E. T. (2003) Rapid transcriptional suppression of rat cytochrome P450 genes by endotoxin treatment and its inhibition by curcumin. J. Pharmacol. Exp. Ther. 307, 1205–1212 [DOI] [PubMed] [Google Scholar]
- 11. Morgan E. T. (1997) Regulation of cytochromes P450 during inflammation and infection. Drug Metab. Rev. 29, 1129–1188 [DOI] [PubMed] [Google Scholar]
- 12. Riddick D. S., Lee C., Bhathena A., Timsit Y. E., Cheng P. Y., Morgan E. T., Prough R. A., Ripp S. L., Miller K. K., Jahan A., Chiang J. Y. (2004) Transcriptional suppression of cytochrome P450 genes by endogenous and exogenous chemicals. Drug Metab. Dispos. 32, 367–375 [DOI] [PubMed] [Google Scholar]
- 13. Abraham A., Karathedath S., Varatharajan S., Markose P., Chendamarai E., Jayavelu A. K., George B., Srivastava A., Mathews V., Balasubramanian P. (2014) ABCB6 RNA expression in leukemias: expression is low in acute promyelocytic leukemia and FLT3-ITD-positive acute myeloid leukemia. Ann. Hematol. 93, 509–512 [DOI] [PubMed] [Google Scholar]
- 14. Dean M. (2009) ABC transporters, drug resistance, and cancer stem cells. J. Mammary Gland Biol. Neoplasia 14, 3–9 [DOI] [PubMed] [Google Scholar]
- 15. Huls M., Russel F. G., Masereeuw R. (2009) The role of ATP binding cassette transporters in tissue defense and organ regeneration. J. Pharmacol. Exp. Ther. 328, 3–9 [DOI] [PubMed] [Google Scholar]
- 16. Moitra K., Dean M. (2011) Evolution of ABC transporters by gene duplication and their role in human disease. Biol. Chem. 392, 29–37 [DOI] [PubMed] [Google Scholar]
- 17. Krishnamurthy P. C., Du G., Fukuda Y., Sun D., Sampath J., Mercer K. E., Wang J., Sosa-Pineda B., Murti K. G., Schuetz J. D. (2006) Identification of a mammalian mitochondrial porphyrin transporter. Nature 443, 586–589 [DOI] [PubMed] [Google Scholar]
- 18. Ulrich D. L., Lynch J., Wang Y., Fukuda Y., Nachagari D., Du G., Sun D., Fan Y., Tsurkan L., Potter P. M., Rehg J. E., Schuetz J. D. (2012) ATP-dependent mitochondrial porphyrin importer ABCB6 protects against phenylhydrazine toxicity. J. Biol. Chem. 287, 12679–12690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cui Y. X., Xia X. Y., Zhou Y., Gao L., Shang X. J., Ni T., Wang W. P., Fan X. B., Yin H. L., Jiang S. J., Yao B., Hu Y. A., Wang G., Li X. J. (2013) Novel mutations of ABCB6 associated with autosomal dominant dyschromatosis universalis hereditaria. PLoS One 8, e79808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Helias V., Saison C., Ballif B. A., Peyrard T., Takahashi J., Takahashi H., Tanaka M., Deybach J. C., Puy H., Le Gall M., Sureau C., Pham B. N., Le Pennec P. Y., Tani Y., Cartron J. P., Arnaud L. (2012) ABCB6 is dispensable for erythropoiesis and specifies the new blood group system Langereis. Nat. Genet. 44, 170–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Polireddy K., Chavan H., Abdulkarim B. A., Krishnamurthy P. (2011) Functional significance of the ATP-binding cassette transporter B6 in hepatocellular carcinoma. Mol. Oncol. 5, 410–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang L., He F., Bu J., Zhen Y., Liu X., Du W., Dong J., Cooney J. D., Dubey S. K., Shi Y., Gong B., Li J., McBride P. F., Jia Y., Lu F., Soltis K. A., Lin Y., Namburi P., Liang C., Sundaresan P., Paw B. H., Li W., Li D. Y., Phillips J. D., Yang Z. (2012) ABCB6 mutations cause ocular coloboma. Am. J. Hum. Genet. 90, 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang C., Li D., Zhang J., Chen X., Huang M., Archacki S., Tian Y., Ren W., Mei A., Zhang Q., Fang M., Su Z., Yin Y., Liu D., Chen Y., Cui X., Li C., Yang H., Wang Q., Wang J., Liu M., Deng Y. (2013) Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J. Invest. Dermatol. 133, 2221–2228 [DOI] [PubMed] [Google Scholar]
- 24. Skarnes W. C., Rosen B., West A. P., Koutsourakis M., Bushell W., Iyer V., Mujica A. O., Thomas M., Harrow J., Cox T., Jackson D., Severin J., Biggs P., Fu J., Nefedov M., de Jong P. J., Stewart A. F., Bradley A. (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xie Y., McGill M. R., Dorko K., Kumer S. C., Schmitt T. M., Forster J., Jaeschke H. (2014) Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol. Appl. pharmacol. 279, 266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lynch J., Fukuda Y., Krishnamurthy P., Du G., Schuetz J. D. (2009) Cell survival under stress is enhanced by a mitochondrial ATP-binding cassette transporter that regulates hemoproteins. Cancer Res. 69, 5560–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chavan H., Khan M. M., Tegos G., Krishnamurthy P. (2013) Efficient purification and reconstitution of ATP binding cassette transporter B6 (ABCB6) for functional and structural studies. J. Biol. Chem. 288, 22658–22669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang X., Zhang B., Molony C., Chudin E., Hao K., Zhu J., Gaedigk A., Suver C., Zhong H., Leeder J. S., Guengerich F. P., Strom S. C., Schuetz E., Rushmore T. H., Ulrich R. G., Slatter J. G., Schadt E. E., Kasarskis A., Lum P. Y. (2010) Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 20, 1020–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chavan H., Krishnamurthy P. (2012) Polycyclic aromatic hydrocarbons (PAHs) mediate transcriptional activation of the ATP binding cassette transporter ABCB6 gene via the aryl hydrocarbon receptor (AhR). J. Biol. Chem. 287, 32054–32068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manzardo A. M., Gunewardena S., Wang K., Butler M. G. (2014) Exon microarray analysis of human dorsolateral prefrontal cortex in alcoholism. Alcoholism Clin. Exp. Res. 38, 1594–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li F., Lu J., Ma X. (2011) Profiling the reactive metabolites of xenobiotics using metabolomic technologies. Chem. Res. Toxicol. 24, 744–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wagener F. A., Eggert A., Boerman O. C., Oyen W. J., Verhofstad A., Abraham N. G., Adema G., van Kooyk Y., de Witte T., Figdor C. G. (2001) Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 98, 1802–1811 [DOI] [PubMed] [Google Scholar]
- 33. Jover R., Hoffmann F., Scheffler-Koch V., Lindberg R. L. (2000) Limited heme synthesis in porphobilinogen deaminase-deficient mice impairs transcriptional activation of specific cytochrome P450 genes by phenobarbital. Eur. J. Biochem. 267, 7128–7137 [DOI] [PubMed] [Google Scholar]
- 34. Lin W., Wu J., Dong H., Bouck D., Zeng F. Y., Chen T. (2008) Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J. Biol. Chem. 283, 30650–30657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie W., Barwick J. L., Simon C. M., Pierce A. M., Safe S., Blumberg B., Guzelian P. S., Evans R. M. (2000) Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 14, 3014–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gaikwad N. W. (2013) Ultra performance liquid chromatography-tandem mass spectrometry method for profiling of steroid metabolome in human tissue. Anal. Chem. 85, 4951–4960 [DOI] [PubMed] [Google Scholar]
- 37. Roth L. J., Leifer E. (1949) The metabolism of radioactive pentobarbital in mice. J. Biol. Chem. 178, 963–965 [PubMed] [Google Scholar]
- 38. Watanabe M., Tateishi T., Asoh M., Nakura H., Tanaka M., Kumai T., Kobayashi S. (1998) Effects of glucocorticoids on pharmacokinetics and pharmacodynamics of midazolam in rats. Life Sci. 63, 1685–1692 [DOI] [PubMed] [Google Scholar]
- 39. Wrighton S. A., Stevens J. C. (1992) The human hepatic cytochromes P450 involved in drug metabolism. Crit. Rev. Toxicol. 22, 1–21 [DOI] [PubMed] [Google Scholar]
- 40. Gonzalez F. J., Lee Y. H. (1996) Constitutive expression of hepatic cytochrome P450 genes. FASEB J. 10, 1112–1117 [DOI] [PubMed] [Google Scholar]
- 41. Facciolá G., Hidestrand M., von Bahr C., Tybring G. (2001) Cytochrome P450 isoforms involved in melatonin metabolism in human liver microsomes. Eur. J. Clin. Pharmacol. 56, 881–888 [DOI] [PubMed] [Google Scholar]
- 42. Faucette S. R., Hawke R. L., Lecluyse E. L., Shord S. S., Yan B., Laethem R. M., Lindley C. M. (2000) Validation of bupropion hydroxylation as a selective marker of human cytochrome P450 2B6 catalytic activity. Drug Metab. Dispos. 28, 1222–1230 [PubMed] [Google Scholar]
- 43. Härtter S., Ursing C., Morita S., Tybring G., von Bahr C., Christensen M., Röjdmark S., Bertilsson L. (2001) Orally given melatonin may serve as a probe drug for cytochrome P450 1A2 activity in vivo: a pilot study. Clin. Pharmacol. Ther. 70, 10–16 [DOI] [PubMed] [Google Scholar]
- 44. Easterbrook J., Fackett D., Li A. P. (2001) A comparison of aroclor 1254-induced and uninduced rat liver microsomes to human liver microsomes in phenytoin O-deethylation, coumarin 7-hydroxylation, tolbutamide 4-hydroxylation, S-mephenytoin 4′-hydroxylation, chloroxazone 6-hydroxylation and testosterone 6β-hydroxylation. Chem. Biol. Interact. 134, 243–249 [DOI] [PubMed] [Google Scholar]
- 45. Wei P., Zhang J., Egan-Hafley M., Liang S., Moore D. D. (2000) The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 407, 920–923 [DOI] [PubMed] [Google Scholar]
- 46. Tralau T., Luch A. (2013) The evolution of our understanding of endo-xenobiotic crosstalk and cytochrome P450 regulation and the therapeutic implications. Expert Opin. Drug Metab. Toxicol. 9, 1541–1554 [DOI] [PubMed] [Google Scholar]
- 47. Xu M., Ju W., Hao H., Wang G., Li P. (2013) Cytochrome P450 2J2: distribution, function, regulation, genetic polymorphisms and clinical significance. Drug Metab. Rev. 45, 311–352 [DOI] [PubMed] [Google Scholar]
- 48. Srivastava G., Bawden M. J., Hansen A. J., May B. K. (1989) Heme may not be a positive regulator of cytochrome-P450 gene expression. Eur. J. Biochem. 178, 689–692 [DOI] [PubMed] [Google Scholar]
- 49. Jover R., Lindberg R. L., Meyer U. A. (1996) Role of heme in cytochrome P450 transcription and function in mice treated with lead acetate. Mol. Pharmacol. 50, 474–481 [PubMed] [Google Scholar]
- 50. Nelson D. R., Zeldin D. C., Hoffman S. M., Maltais L. J., Wain H. M., Nebert D. W. (2004) Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14, 1–18 [DOI] [PubMed] [Google Scholar]
- 51. Andolfo I., Alper S. L., Delaunay J., Auriemma C., Russo R., Asci R., Esposito M. R., Sharma A. K., Shmukler B. E., Brugnara C., De Franceschi L., Iolascon A. (2013) Missense mutations in the ABCB6 transporter cause dominant familial pseudohyperkalemia. Am. J. Hematol. 88, 66–72 [DOI] [PubMed] [Google Scholar]
- 52. Mookherjee S., Acharya M., Banerjee D., Bhattacharjee A., Ray K. (2012) Molecular basis for involvement of CYP1B1 in MYOC upregulation and its potential implication in glaucoma pathogenesis. PLoS One 7, e45077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vasiliou V., Gonzalez F. J. (2008) Role of CYP1B1 in glaucoma. Annu. Rev. Pharmacol. Toxicol. 48, 333–358 [DOI] [PubMed] [Google Scholar]