

Abstract
Understanding the mechanism accompanying functional conformational changes associated with protein activation has important implications for drug design. Here, we describe a powerful method, CDSiL-MS (conformational changes and dynamics using stable-isotope labeling and mass-spectrometry), which involves chemical-labeling by isotope-coded forms of N-ethylmaleimide or succinic anhydride to site-specifically label the side-chains of cysteines or lysines, respectively, in native proteins. Subsequent MS-analysis allows the quantitative monitoring of reactivity of residues as a function of time, providing a measurement of the labeling kinetics, thereby enabling elucidation of conformational changes of proteins. We demonstrate the utility of this method using a model G-protein coupled receptor, the β2-adrenergic receptor including experiments that characterize the functional conformational-changes associated with activation of distinct signaling pathways induced by different β-adrenoceptor ligands. The procedure requires five days and can easily be adapted to systems where soluble and detergent-solubilized membrane protein targets, which undergo function-dependent conformational-changes, can be interrogated structurally to allow drug screening.
Keywords: Protein structure, Structural biology, Structure determination, Protein analysis, Stable isotope labeling, N-ethylmaleimide labeling, succinic anhydride labeling, mass spectrometry, reactivity, labeling kinetics, β2-adrenergic receptor (β2AR), G protein-coupled receptors (GPCRs), Drug discovery
INTRODUCTION
Characterization of the conformational changes associated with protein function is a central goal of structural biology, as such information can lead to an understanding of how to modify and regulate a protein's activity. These dynamic changes are difficult to capture using most conventional approaches. X-ray crystallography provides a high-resolution static snapshot of a protein, but is usually unable to reveal structural information in flexible, highly dynamic and functionally important regions. Techniques such as those using cell-based fluorescence resonance energy transfer (FRET) biosensors1-3, site-specific chemical labeling coupled with fluorescent molecules4-6, nuclear magnetic resonance (NMR)7-10, and electron paramagnetic resonance (EPR)11,12 analysis have been utilized previously to infer conformational changes associated with protein activity. Although these biophysical techniques provide insight into conformational changes of a protein, they require a modified or engineered protein construct. For example, FRET based conformation change studies of membrane proteins require large fluorescent reporters (e.g., GFP or its variants fused through genetic approaches) that may perturb a protein's structure and function, while small-molecule fluorescent, NMR, or EPR-based approaches require engineering a series of single residue mutants to incorporate active reporter probes.
Modern mass spectrometry (MS)-based proteomic techniques have progressed rapidly over the past decade particularly with respect to sensitivity, mass accuracy, and faster data analysis time. MS techniques now provide simultaneous detection of multiple peptides from different locations of a native protein in a single experiment and provide global structural information of the protein quickly and with less stringent sample requirements13-18. In particular, with the advent of residue-specific, stable-isotope covalent labeling chemical agents, modern mass spectrometry has emerged as a powerful structural biology tool in measuring reactivity kinetics of surface accessible amino acid side chains associated with different functional protein conformations. This is based on the concept that the chemical reactivity of such amino acid side chains is a function of the local protein microenvironment, and thus would indicate their role in conformational changes within the protein, which could be induced under different conditions, e.g., via ligand binding to the protein of interest resulting in complex formation.
Mass spectrometry experiments using stable-isotope labeling
MS-based techniques to characterize protein conformation and dynamics have been described in several recent reviews, and include amide hydrogen/deuterium exchange (HDX/MS)19-22, which has been widely used in probing soluble protein conformational dynamics as well as the dynamics for technically demanding membrane proteins such as the human β2-adrenergic receptor (β2AR)23. However, HDX studies may be limited by back-exchange and scrambling that frequently preclude precise measurements24-29. Such limitations are minor in other methods such as site-specific covalent stable isotope labeling strategies that are coupled to MS-based quantitative analysis. Strategies for covalent stable isotope labeling include in vivo metabolic incorporation and in vitro chemical tagging30. Among in vivo metabolic incorporation, the 14N/15N labeling approach31-33 and Stable Isotope Labeling by Amino acids in Cell culture (SILAC) are common; the latter being the most popular, developed for use in mammalian cell culture34,35. Similarly, within the in vitro chemical tagging, a number of stable-isotope labeling strategies have been developed in recent years including cysteine specific isotope-coded affinity tags (ICAT)15, amine specific isobaric tag for relative and absolute quantitation (iTRAQ)36 or tandem mass tag (TMT)37-39, enzyme catalyzed 18O-containing water labeling40, and formaldehyde-based reductive methylation of amines41. Methods of accurate quantification strategies of stable isotope labeled peptides from multiplexed proteome samples have also been evolving recently42-44. Most of these labeling technologies have been used however, mainly for the purpose of MS-based quantitative proteomics studies to compare the extents of protein expression or protein post-transcriptional modifications under different biological conditions and not on site-specific conformational changes studies. The general principle, advantages and limitations of each of these labeling technologies, including the ones described here, chemical labeling of N-ethylmaleimide (NEM) and succinic anhydride (SA) are summarized in Table 1.
TABLE 1.
Examples of stable-isotope labeling strategies for MS-based quantitative proteomics
| Labeling | Principle | Advantages | Disadvantages |
|---|---|---|---|
| Metabolic | |||
| SILAC* | Incorporates heavy-labeled selected amino acid counterparts into proteins | Label incorporation to cultures and small organisms like C. elegans and Drosophila. Less variability between samples and incorporations | Not applicability to complex organisms. Labeled amino acids are expensive. Metabolic derivatization of Arg to Pro |
| 14N/15N | Incorporates 14N/15N counterparts into the proteins | Incorporation into many microorganisms | Complexity during data analysis due to incorporation of label at backbone and side-chains. Expected mass difference is unknown before peptide identification |
| Chemical | |||
| ICAT* | Labels cysteine residues at the protein/peptide level | Simplified sample mixture and analysis as it excludes cysteine lacking peptides. Incorporation at the protein level | Only for analysis to proteins/peptides containing cysteines. Expensive reagent. Potential variability as it relies on affinity beads. Increased sample analysis, and time on MS |
| iTRAQ* or TMT* | Labels lysine and N-terminal amines with tags of varying masses | Enables multiplexed sample analysis per MS experiment | Requires tandem MS acquisition; increased sample analysis, and time on MS. High variation |
| NEM* | Labels cysteine residues at the protein/peptide level | Very mild reaction. Cheap reagent. Simple. Analysis at MS level. Applicable to any sample | Only cysteine containing peptides can be used for quantitation |
| SA* | Labels lysine and N-terminal amines primarily | Cheap reagent. Applicable to broader sample nature. Simple | Very fast reaction, which can cause variability and a demand for MS2 analysis |
| Dimethyl labeling | Dimethyl-labeling of lysine and N-terminal amines | Fast reaction. Cheap reagent. Simple. Applicable to any sample | Isotope effect in LC separation. Variability during modifications |
| 16O/18O | Enzyme-facilitated 18O | Cheap reagent. Simple. Applicable to any sample | Incomplete labeling or slow back exchange of 16O and 18O. Incorporation at peptide level |
Stable Isotope Labeling with Amino acids in Cell culture (SILAC); Isotope coded affinity tag (ICAT); Isotope Tags for Relative and Absolute Quantitation (iTRAQ); Tandem Mass Tag (TMT); N-Ethylmaleimide (NEM); and Succinic anhydride (SA).
Challenges in structural analysis of G protein-coupled receptors
G protein-coupled receptors (GPCRs), also known as seven-transmembrane receptors (7TMRs), constitute the largest family of membrane-associated receptors in the mammalian genome and are the targets of nearly half of all clinical drugs on the market45-47. They elicit various distinct physiological outcomes by their response to a diverse array of sensory and chemical stimuli. Upon ligand binding, they undergo conformational changes, and can signal not only through G proteins pathways, but also through G protein independent mechanisms by signaling proteins including β-arrestins48, the multifunctional adapter proteins that also regulate receptor desensitization and trafficking49-51. A number of ligands, referred to as “biased ligands”, have been demonstrated to selectively activate only one or a subset of these pathways (G proteins and β-arrestins, among others)52,53. Several lines of evidence have now demonstrated that such ligands with varied efficacies stabilize distinct receptor conformations54-56. Understanding the structural mechanism(s) of GPCR activation upon binding to different ligands has significant implications in facilitating the design of safer and more efficacious therapeutic agents53.
Recent advances in solving high-resolution, three-dimensional X-ray crystallographic snapshots of different ligand-bound GPCRs57-63, including the crystal structure of the β2AR in complex with heterotrimeric G proteins63, have substantially advanced our understanding in terms of atomic level structural details of these receptors. Despite this progress, GPCRs still face significant challenges to structural biology tools such as X-ray crystallography, mainly due to their intrinsic conformational flexibility, and dynamic character. For example, to overcome the flexibility problem and obtain diffractable crystals, special strategies have been used including insertion of T4 lysozyme to replace the highly dynamic third intracellular loop (ICL3)58,59,61, thermostabilization mutations62, and co-crystallization with antibodies57,61 to stabilize specific ligand-bound receptor conformations57,61. In fact, the highly dynamic structural organizations, including intracellular loops and C-termini of these receptors, remain largely unresolved by X-ray crystallography. Importantly, these structural elements are sites of interactions of activated receptor with effectors (e.g., G-proteins, β-arrestins). Therefore, it is unlikely that X-ray crystallography alone will be able to capture the full complexity and dynamics of GPCRs in which structural flexibility appears to be important to assume different conformations. Hence, complementary approaches that allow characterizing conformational changes and dynamics of these membrane protein GPCRs will be crucial to unraveling novel insights into their mechanism of activation and signaling.
Development of protein CDSiL-MS strategy
Here, we report the development of a general method (CDSiL-MS) for the examination of site-specific protein conformational changes involving in vitro covalent incorporation of stable-isotope reagents at selective sites followed by MS-based quantitative analysis. This procedure was used to a model GPCR, the human β2-adrenergic receptor to characterize various ligand-specific-conformations associated with distinct signaling mechanisms55. The method specifically employs stable-isotope coded forms, incorporating protium atoms (the most common hydrogen isotope; “light”) or deuterium atoms (“heavy”)) into N-ethylmaleimide (NEM) and succinic anhydride (SA) reagents to selectively label thiol groups of cysteines and primary ε-amine groups of lysine side chains, respectively, in a protein or protein complex. N-ethylmaleimide (NEM-H5 or NEMD5) covalently modifies thiol groups of cysteines via a Michael-type addition reaction at the α, β-unsaturated bond and such reactions occur optimally in the pH range between 6.5 and 7.5 (Fig. 1a, top). While, succinic anhydride (SA-H4 or SA-D4) reacts with primary ε-amino groups of lysine side chains and the N-terminal α-amino group of proteins, in their nonprotonated forms, via one of its chemically equivalent electrophilic carbonyl carbons (Fig. 1b, top). Labeling a cysteine residue in a peptide with NEM-H5 or NEM-D5 generates a mass increase of 125.05 Da or 130.08 Da over the corresponding unmodified peptide, due to the mass difference between NEM-H5 and NEM-D5 (Fig. 1a, bottom). Similarly, labeling a lysine residue in a peptide with SA-H4 or SA-D4 generates a mass increase of 100.02 Da or 104.04 Da, over the corresponding unmodified peptide, due to the mass difference between SA-H4 and SA-D4 (Fig. 1b, bottom). The corresponding protiated- and deuterated-labeled peptide ion peaks are then clearly distinguished by these mass differences (i.e., 5 Daltons for NEM and 4 Daltons for SA) during MS analysis using high-resolution matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer (MS).
Figure 1. Reaction scheme for stable-isotope labeling of cysteines and lysines in proteins.
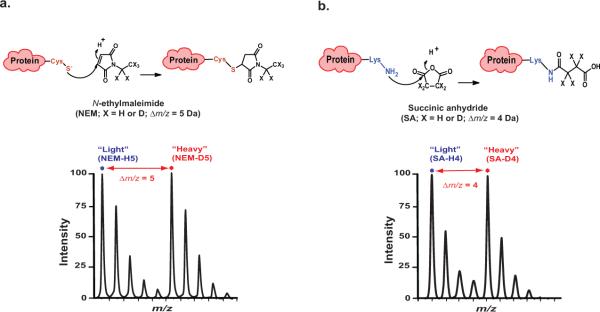
(a) Top, chemical structure of N-ethylmaleimide (NEM-H5 or D5) and the resulting side chain produced by reaction with thiol group of cysteine. Bottom, sample isotope-peak pair (doublet) corresponding to peptide modified at cysteine residue by light and heavy NEM with Δm/z = 5. (b) Top, chemical structure of succinic anhydride (SA-H4 or D4) and the resulting side chain produced by reaction with ε-NH2 group of lysine. Bottom, sample isotope-peak pair (doublet) corresponding to peptide modified at lysine residue by light and heavy SA with Δm/z = 4. Figure adapted from reference55.
The approach involves monitoring the extent of labeling of specific cysteine or lysine residues in protein, quenching at various time points, proteolytically cleaving and quantitatively analyzing using MALDI-TOF MS. Quantification of reactivity of residues as a function of time, typically in minutes, is achieved by comparing signal intensity ratios between the experimental (“heavy”) and corresponding reference (“light”) labeled fragment ion pairs. In general, the advantage of such site-specific stable-isotope chemical labeling techniques is their utilization in MS-based quantitative proteomics to allow characterization of conformational changes of a wide spectrum of proteins and protein complexes both in vitro with purified proteins and in cell systems. A brief overview of the CDSiL-MS approach, materials required, a step-by-step procedures and troubleshooting guidelines are provided below.
Experimental design
Procedure overview
An overview of the experimental workflow of protein CDSiL-MS process is shown in Figure 2. The strategy has four main steps: 1) the reference labeling reaction; 2) experimental time-point labeling reaction; 3) sample processing (reduction, alkylation and proteolytic digestion) and MS analysis; and 4) quantification of site-specific labeling and kinetic analysis.
Figure 2. Overview of CDSiL-MS strategy designed to monitor conformational changes in proteins.

The procedure uses a functionally active, purified protein to quantitatively measure changes in the reactivity of residues to specific stable isotope reagents. (a-b) Labeling of cysteines is initiated in two purified protein pools (2.5 μM; bound to the same ligand; 30 min at 25 °C) by adding 2 mM of either NEM-H5 as in (a) or NEM-D5 as in (b). (a) Reference labeling reaction: reactions are performed for an hour at 25 °C (terminated by adding DTT) and equal sized aliquots are prepared. (b) Experimental time-point labeling reaction: exactly equal sized aliquots are withdrawn at different time points and quenched. (c) Sample processing (reduction, alkylation and proteolysis), and MS analysis: equal amounts of the two pools are mixed, reduced, alkylated, subjected to proteolysis with appropriate enzyme, and MS analyzed to determine peptide fragments that have been modified. The sample time course spectra of singly charged ion ([M+H]+) peak pairs for a peptide modified at cysteine by NEM (H5 and D5) is shown. (d) Sample plot (black circles) of change in the intensity ratios (%R) between peptide signal intensities containing heavy and light reagents over time. Each percent intensity ratio (%R) data point is corrected by a specific site reactivity ratio (Rr) to obtain percent site labeled (%F) as represented by the curve (black squares) for the percent intensity ratios (%R) plot.
Differential labeling and MS analysis
The CDSiL-MS approach can technically be applied to different soluble and membrane proteins. We described it here using a model GPCR, an integral membrane protein, β2AR. To shed a light on mechanism of activation of GPCRs from the perspective of ligand specific conformational rearrangments, we performed CDSiL-MS experiments on β2AR in complex with three distinct β-adrenoceptor ligands (test ligands) exhibiting different pharmacological properties: the full agonist isoproterenol, the inverse agonist ICI-118,551 (ICI), and a biased ligand carvedilol (carv; a β antagonist for G protein activation with selective stimulation of β-arrestin–mediated signaling64). Before experiments, it is important to confirm the activity of the protein being analyzed by performing functional assays55,65. In this regard, for the β2AR, after its expression in Spodoptera frugiperda (Sf9) cells, using baculovirus system, and purification by affinity chromatographic steps, its activity is confirmed using radio-ligand binding assays55,65. When performing labeling reactions, high molar excesses of these test ligands are required relative to receptor, which typically is present at low micromolar concentrations (~2.5 μM). The saturating ligand concentration guarantees locking the receptor to the specific functional conformation that the ligand prefers, thus allowing homogeneous receptor:ligand complex sample preparations suitable for stable-isotope labeling, and MS based quantitative analysis. Obtaining optimal signal intensity of labeled peptide is also important factor for reproducible quantification. Therefore, it is advisable to first perform experiments to determine the relative reactivity condition of each residue to its respective stable-isotope labeled reagent. Such pilot experiments can also be designed to include information in identifying the timescale of the labeling reaction for different residues and distribution between time points for the protein in study (Box 1). Along similar lines, choice of proteolytic enzymes for digestion of protein of interest is also important as it directly ensures accurate quantification of peptides. Therefore, it is important to test initially a group of proteases so that to select appropriate proteases for accurate quantification of peptide of interest by MS. In the current study we used chymotrypsin (which cleaves C-terminally at tryptophan, tyrosine, phenylalanine, leucine residues) to cleave NEM or SA-labeled protein samples. While, trypsin (cleaves C-terminally at lysine and arginine) is only suitable to cleave NEM-labeled samples as it partially cleaves SA-labeled ones (only to those after arginine).
To perform differential stable isotope chemical labeling (“light”/“heavy”) and precisely measure the changes in labeling of reactive residues as a function of time, an accurately measured receptor/protein amount to be labeled is prepared. This protein sample is then split into two equal parts that will serve as the reference (Ref Reaction) and experimental time-point labeling reaction (Exp Reaction). The labeling experiments are designed in a form that the protiated-labeling reagent (NEM-H5 or SA-H4) is used for the Ref Reaction and the deuterated-labeling reagent (NEM-D5 or SA-D4) for the Exp Reaction, although technically can also be designed otherwise vice versa. In the first step of the CDSiL-MS approach, the Ref Reaction is allowed to take place and quenched at a fixed time point, typically the time point of the Exp Reaction at which the labeling reaction has completed is chosen, and snap frozen in liquid N2 (Fig. 2a). While, the Exp Reaction is allowed to take place under identical conditions except with aliquots removed at different time points, followed by quenching and snap freezing of each aliquot (Fig. 2b). Typically, dithiothreitol (DTT) and free L-lysine are used to quench the NEM- and SA-labeling reactions, respectively. Each of the above reactions can be done in parallel or separately depending on the sample sizes and can be stored in −80°C until further use. After completion of labeling, equal aliquots from the Ref and Exp Reactions are mixed, precipitated by chloroform/methanol66, and subjected to in-solution enzymatic digestion using suitable proteases, in the current study chymotrypsin. The resulting peptide digests are desalted on reversed-phase C18 adsorbent micro column and analyzed by MALDI-TOF mass spectrometry (Fig. 2c). Spectral acquisition on MALDI-TOF MS is normally set over a mass range of 700 – 4000 mass-to- charge-ratio (m/z) to analyze suitably sized, reproducible proteolytic peptides fragments away from matrix interferences range, i.e., below 700 m/z.
Quantification is achieved by comparing ion peak signal intensities within the differentially stable-isotope labeled peptide pairs (i.e., the “light”/“heavy” peak pairs or “doublets”) at the initial MS level. Since the mass differences between labeled peptides are 5 and 4 Daltons, overlap between isotopic clusters of light and heavy-labeled peptide peaks is usually negligible. However, even with similar labeling reagents that allow large mass differences, overlapping of isotopic clusters may occur for labeled peptides larger than 3,000 m/z; in this case, depending on the significance and quality of ion signals, peak signal correction may be required. This in turn is dependent on the sequence coverage of the protein interest and the proteolytic enzymes used. Therefore, stringent thresholds need to be applied such as a signal-to-noise ratio greater than 5 and a lack of interfering signals around peptide of interest. In this regard, performing control inverse labeling experiments (i.e., swap labeling in which the Exp Reaction uses light isotope and the Ref Reaction uses heavy isotope in scheme shown in Fig. 2a,b) is recommended in order to quickly identify irrelevant signals and focus on signal of interest ones. Optimal signal ion intensity (i.e., high abundance and signal-to-noise ratio) of labeled peptide ion pairs under study obtained using MS-platform of choice is also important for a reproducible and accurate quantification. As we describe below, to ensure the peptide quantification performance of the MS platform used over a wide dynamic range, the linearity of the response is estimated by comparing the measured peptide ion pair peak intensity ratios to the known amount of the stable isotope labeled peptide added in the experiment55. Such labeling experiments, that validate the technical reproducibility and accuracy of peptide quantification of the MS platform can also be performed in both conventional and inverse stable-isotope chemical labeling strategies.
Generation of site-specific labeling profiles
The reactivity of each site in the different receptor:ligand complexes is obtained by measuring the relative signal intensity ratios of the monoisotopic peaks of the deuterated (“heavy”) to the protiated (“light”) reagent-modified peptides to yield percent intensity ratio, % R(t), for each time point and site. The calculated curves in the form of percent intensity ratios (%R(t)) can then be directly fit with a suitable exponential kinetic model (equations 2-4) or normalized (Fig. 2d). Normalization of percent intensity ratios allows a comparison of reactivities between different residues within a given protein. A normalization factor, termed the reactivity factor, Rr, can be determined experimentally, and relates %R(t) to the percentage of site labeled, %F(t):
| (1) |
where %F(t) is the fraction of the site labeled, %R(t) is the ratio of the heavy-to-light signal intensity for time point t, and Rr is the reactivity factor. An alternative approach to by-pass any normalization of initial percent signal intensity (%R) data is to use a synthetic stable isotope-labeled internal standard of identical sequence to replace “Ref Reaction” in the experimental scheme shown in Figure 2a. The relative reactivity of a given amino acid residue can then be determined from the ion abundance ratio between the standard and the experimental version of the modified peptide (i.e., “light” and “heavy”) at each labeling reaction time point. Hence, the resulting percent intensity ratios (%R) are plotted as a function of time to obtain time-course labeling curves for kinetic analysis. Conceptually the application of a stable isotope-labeled internal standard in CDSiL-MS is similar to that used by the method of absolute quantification (AQUA) which requires internal peptide standard for the quantification of levels post-translational modifications of proteins in complex biological samples67,68.
Methods of analysis of labeling data
In general, the model used to fit the data should be the simplest possible that provides a similar goodness of fit as assessed statistically, e.g., by R squared values. To determine the appropriate fitting model, the reactivity data (in the form of %R or %F) as a function of time in minutes are plotted. These curves (e.g., each site/ligand pair) are tested for their best fit to different exponential models (Fig. 3). If the labeling of a given site in a protein occurs through a simple kinetic mechanism, the labeling data can be fit by simple exponential kinetics behavior. It is likely, however, that some labeling may occur very rapidly in a burst phase, before the first time point of the experiment (Fig. 3a). A simple curve that fits such behavior is:
| (2) |
where t is the labeling time; τ1 is the relaxation time constant; A is the amplitude response of the phase; and the burst phase amplitude is (100 – A).
Figure 3. Different functions that can be used to fit the behavior of labeling kinetics.
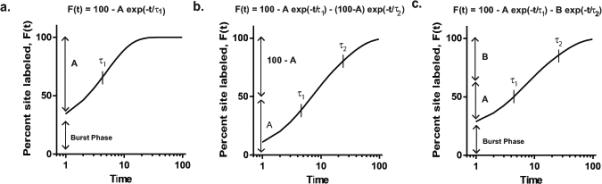
(a) A simple exponential with a burst phase accounting for labeling that occurs faster than the timescale of observation (t1 = 5 min, A = 80). (b) Two exponentials without a burst phase (t1 = 5 min, t2 = 25 min, A = 50). (c) Two exponentials with a burst phase. (t1 = 5 min, t2 = 25 min, A = 40, B = 40).
If there is no burst phase, but rather two relaxations that occur over the timescale of the experiment (Fig. 3b), the data can be fit to:
| (3) |
where τ1 and τ2 are the relaxation time constants of each phase; and A is the fractional amplitude of the τ1 phase. If a burst phase is present in the data as well as two other relaxations (Fig. 3c), the data can be fit to:
| (4) |
where A and B are the amplitudes of the phases with relaxation times τ1 and τ2, respectively and the burst phase amplitude is (100 - A - B). All three models force F(∞)=100, which is expected from the experimental design. Such curve fitting allows a quantitative comparison of the labeling kinetics at different sites in a given conformation of a protein. Moreover, these relaxations are related to the different underlying conformational states and their inter-conversions, and define the minimum number of conformational states in the system.
In our experience, we have found that relaxation times are the most informative of the kinetic parameters used for interpretation and comparison of the labeling kinetics of different amino acid residues on various conformations of a protein. The relaxation times are transformed to a labeling reactivity factor (L-factor), which is equal to the negative logarithm of the relaxation time (-log τ1). The L factor is defined as (-log τ1), so it is proportional to the activation energy associated with residue labeling.
Reactivity measurements are sensitive to the conformational state of the receptor/protein, thus changes in L-factor reflect ligand-dependent conformational rearrangements in the vicinity of the site (residues that are labeled relatively quickly would have high L-factors, and vice versa). The relative reactivity of a given amino acid side chain in a protein with its respective stable-isotope reagent upon conformational changes is also a function of the pKa and protonation state of the residue, which can be modified by changes in the residue's microenvironment within the protein three dimensional structure. Thus, it should be kept in mind that a change in a residue's labeling factor could be due to a number of different changes in the protein microenvironment and not just increased solvent accessibility of a residue.
Determining the linearity range of labeling experiment
The accuracy and sensitivity of peptide quantification over a wide dynamic range is confirmed by performing dilution curves over a series of mixtures of light:heavy-labeled peptides. This experiment is prepared in two separate stable isotope-labeling reaction pools of β2AR (2.5 μM), one with NEM-H5 (2 mM) and the other with NEM-D5 (2 mM), each incubated for 1 hr at room temperature and quenched. The NEM-D5 labeled samples are spiked into NEM-H5 labeled reference samples in seven different dilutions as a dilution series (two-fold concentration dilutions ranging from 1:1 to 1:128) then concurrently digested and MS-analyzed. Quantitative comparison between measured peptide intensity ratios (NEMH5/NEM-D5) and dilution series of expected ratios (1:1 to 1:128) of labeled β2AR allows then to determine linearity range of measurements of isotopically labeled peptide using the instrument. To ensure reliability of the data, we also performed this experiment in an inverse labeling mode (i.e., label swap where NEM-H5 labeled samples are spiked into NEM-D5 labeled samples). A typical example is illustrated with the quantification of one of the longest peptide isotope peak pairs (327CRSPDFRIAF336 modified at Cys327 by NEM-H5 or NEM-D5) shown in Figures 4a and 4b. We observed a strong linear relationship between signal intensities and molar concentration ratios of the labeled protein (slope = 0.9785 ± 0.13; R2 = 0.9996; p < 0.0001). Figure 4c illustrates linearity of response observed in this experimental design i.e., good correlation between intensities and absolute concentrations. Thus, we find that the MALDI-TOF MS analytical platform used here is robust and quantitative with a linear dynamic range of over 2 orders of magnitude.
Figure 4. Quantification of stable-isotope labeled peptides for CDSiL-MS strategy.

Shown are comparison between measured intensity ratios (NEM-H5 /NEM-D5) and expected ratios (seven serial two-fold concentration dilutions, 1:1 to 1:128) of the deuterated NEM-labeled β2AR, titrated against a fixed quantity of the protiated NEM-labeled β2AR performed in both forward (a) and inverse (b) stable-isotope labeling experiment of the β2AR. Percent ratios of signal intensities between deuterated and protiated NEM-labeled peptide peaks are plotted as shown in (c) for a singly charged ion ([M+H]+) peak pair at m/z 1336.6 and 1341.7 corresponding to peptide 327CRSPDFRIAF336 modified at C327 by NEMH5 and NEM-D5, for the forward and inverse labeling strategies, respectively. The linear relation between signal intensities and dilution of labeled-β2AR protein concentrations (slope = 0.9785 ± 0.13; R2 = 0.9996; p < 0.0001) demonstrates the quantitative nature of the MS platform with a linear dynamic range of more than 2 orders of magnitude. Data are means ± s.e.m. of four independent experiments. Figure adapted from reference55.
In addition to the robust nature of its protein quantification, MALDI-TOF MS, has other practical features that include its ability to: (i) rapidly analyze a large number of samples in a short period of time allowing for automation and high-throughput formatting; (ii) generate singly charged precursor ions, which simplifies ion selection from MS spectra; (iii) prevent potential uncertainties that could otherwise occur during chromatographic separation of the deuterated- and protiated-labeled peptide pairs when using LC-MS analysis; (iv) be compatible with sample storage and re-analysis; and (v) provide high quality data when working with purified proteins and protein complexes formed in vitro.
Limitations of this approach
There are potential limitations to the CDSiL-MS approach. For example, the protein of interest may have cysteine or lysine residues that are critical for its activity. Similarly, the protein may have labile disulfide bonds (as Cys residues are assumed to be unreactive in disulfide form) that are important for its structural integrity. In such instances, reactions with NEM or SA would inactivate or structurally compromise the protein. Alternative approaches such as hydrogen/deuterium exchange coupled with mass spectrometry (HDX/MS)21,24, can provide region-specific functional conformational dynamics of proteins in such cases. Also, while one of the advantages of CDSiL-MS is its ability to simultaneously detect reactivities of multiple residues from different locations of a native protein, some proteins may lack native-reactive cysteines or lysines at desirable conformational sensitive regions. In such instances, applications of CDSiL-MS would therefore require the generations of mutant proteins, which is more labor intensive. Finally, when studying dynamics and structural mapping of protein complexes with any covalent stable isotope labeling such as CDSiL-MS, it can be difficult to discern whether changes in labeling are a result of intra- or inter-molecular conformational changes. Again, in such circumstances, complementary approaches may be required.
MATERIALS
REAGENTS
Acetonitrile (ACN) (Sigma-Aldrich, cat. no. 38451) Caution: Chloroform is toxic and flammable. Handle it in a fume hood.
Chloroform (>99.8%; Sigma-Aldrich, cat. no. 366919) Caution: Chloroform is toxic and flammable. Handle it in a fume hood.
Methanol (>99.9%; Sigma-Aldrich, cat. no. 34860) Caution: Methanol is toxic and flammable. Handle it in a fume hood.
Isoproterenol (Sigma-Aldrich, cat. no. I2760)
ICI 118,551 (Sigma-Aldrich, cat. no. I127)
Carvedilol (Sigma-Aldrich, cat. no. C3993)
N-ethylmaleimide-H5 (NEM-H5) (Sigma-Aldrich, cat. no. E3876)
Succinic anhydride-H4 (SA-H4) (Sigma-Aldrich, cat. no. 239690)
N-ethylmaleimide-D5 (NEM-D5, 99% atomic D) (Cambridge Isotopes Laboratories, cat. no. DLM-6711-10)
Succinic anhydride-D4 (SA-D4, 99% atomic D) (Cambridge Isotopes Laboratories, cat. no. DLM-833-5)
Empore C18 (3M, cat. no. 2215)
n-Dodecyl-β-D-maltoside (Sol-Grade, Anatrace, >98%; Affymetrix, cat. no. D310S)
Glycerol (Sigma-Aldrich, cat. no. G5516)
Trifluoroacetic acid (TFA) (Fisher scientific, cat. no. A116-10X1AMP) Caution: TFA is harmful by inhalation. It causes severe burns.
Dithiothreitol (DTT, Roche, cat. no. 03117014001) Caution: DTT solid is harmful by inhalation, in contact with skin and if swallowed.
Iodoacetimide (IAA) (Sigma-Aldrich, cat. no. I1149) Caution: IAA is toxic if swallowed. It may cause sensitivity by inhalation and skin contact.
Chymotrypsin (Sigma-Aldrich, cat. no. C6423)
Trypsin (modified, sequencing grade) (Promega, cat. no. v5111)
α-cyano-4-hydroxycinnamic acid (CHCA) (Sigma-Aldrich, cat. no. C8982)
All other reagents are of analytical grade obtained from various suppliers and used without further purification unless indicated otherwise.
EQUIPMENT
ABI 4700, 4800 or 5800 MALDI-TOF/TOF mass spectrometer (Applied Biosystems, Framingham, MA)
Benchtop microcentrifuge
Vacuum manifold
Vortex
Lyophilizer
Speed-vac
RAININ GPS-L250 tip (Anachem, cat. no. GPS-L250)
5 mL syringe
1.5 mL Protein LoBind microcentrifuge tube (Eppendorf, cat. no. 22431081)
0.6 mL Protein LoBind microcentrifuge tube (Eppendorf, cat. no. 22431064 )
REAGENT SETUP
Protein expression and purification
Depending on the protein of interest, expression system and purification strategy may vary. For purification of the human β2AR we typically express it in baculovirus infected Sf9 insect cells as N-terminus FLAG- and C-terminus 6xHis-tagged protein and subsequently purify it after solubilizing it in n-Dodecyl-β-D-maltoside (DDM) detergent using three step affinity-chromatographic procedures as described previously65. Purity and activity of the purified protein is assessed by Coomassie-Blue staining resolved on SDS–PAGE and radio-ligand binding assays, respectively.
Test ligand stock solutions
Weigh out sufficient dry compound to make a 100 mM ligand stock in DMSO. Dispense into small aliquots and store at −20°C. Use one aliquot each time, do not freeze and thaw. Prepare working solutions of each ligand, immediately before use (within 5 minutes) from the stored stock solutions.
Buffer for differential labeling reactions for cysteines and lysines
Differential labeling reactions with protiated and deuterated reagents should be done under identical conditions. Purified receptor protein can be diluted to 2.5-5.0 μM in 50 mM potassium phosphate buffer (pH 7.5), containing 100 mM NaCl and 0.02% (w/v) n-Dodecyl-β-D-maltoside. The diluted receptor protein can be incubated with carrier solvent or test ligands (50 μM) for 30 min at 25 °C in a 110 μL total reaction volume.
Detergent solution: 10% (w/v) stock solution of n-Dodecyl-β-D-maltoside (DDM). Store at −20°C in 0.5 mL aliquots for 6 months.
Protein solubilization solution: 20 mM HEPES, pH 7.5, 100 mM NaCl, 1% DDM. Freshly prepare whenever it is required and filter-sterilize. Chill it on ice before needed.
Chymotrypsin digestion buffer: 50 mM NH4HCO3, pH 8.0, 1 mM CaCl2, 0.001% DDM. Freshly prepare the buffer by supplementing with DDM to the final concentration using 10% (w/v) stock solution.
Trypsin digestion buffer: 50 mM Tris-HCl pH 8.0, 0.001% DDM. Freshly prepare the buffer by supplementing with DDM to the final concentration using 10% (w/v) stock solution.
Saturated solution of α-cyano-4-hydroxycinnamic acid (approximately 10 mg in 0.25 ml volume) in 20 mM ammonium phosphate, 50% ACN and 0.2% TFA. Freshly prepare immediately before use.
1% TFA stock solution prepared in HPLC grade H2O: 1 ml in HPLC grade H2O, 10 μL TFA. Prepare and keep at room temperature (RT, 20–25 °C) for no longer than 4 weeks.
Digested sample reconstitution buffer (0.1% to 1% TFA in HPLC grade H2O to attain pH of peptide mixture to < 2). For example to prepare 0.5% TFA use 500 μL of 1% TFA in 500 μL HPLC grade H2O. Prepare freshly on the day of the experiment.
Activation (50% ACN in 0.1% TFA): 100 μL 1% TFA/ in 400 μL HPLC grade H2O and 500 μL acetonitrile. Prepare freshly on the day of the experiment.
Equilibration (5% ACN in 0.1% TFA): 100 μL 1% TFA/ 50 μL 100% ACN in 850 μL HPLC grade H2O. Prepare freshly on the day of the experiment.
Washing (0.1% TFA in HPLC grade H2O): 100 μL 1% TFA/ in 900 μL HPLC grade H2O. Prepare freshly on the day of the experiment.
Elution (50% ACN in 0.1% TFA): 100 μL 1% TFA/ in 400 μL HPLC grade H2O and 500 μL acetonitrile. Prepare freshly on the day of the experiment.
EQUIPMENT SETUP
MALDI-TOF-MS
Mass spectrometry analyses of the peptide digest can be performed on an ABI 4700 MALDI-TOF/TOF mass spectrometer (Applied Biosystems, Framingham, MA) equipped with a nitrogen laser operating at 337 nm, a video system, and a 4000 Series Data Explorer software for spectra acquisition and instrument control. Peptide samples (0.4 μL) are spotted onto MALDI target plate followed by adding an equal volume of the matrix solution; the mixture is allowed to co-crystallize at room temperature. All mass spectra can be obtained in the reflection positive-ion mode, at an acceleration voltage of 20 kV and a detector voltage of 2 kV. MS data can be automatically acquired over the mass-to-charge (m/z) range of 700 - 4000 m/z and laser intensity adjustment to obtain optimized resolution.
PROCEDURE
Protein sample preparation and receptor-ligand binding
-
1| Dialyze the protein solution overnight at 4°C against 50 mM potassium phosphate buffer (pH 7.5), containing 100 mM NaCl, 0.02% (w/v) DDM. Change the dialysis buffer twice.
- Δ CRITICAL STEP It is important to avoid buffers that contain primary amines, such as Tris, because these will compete for conjugation with the amine-reactive reagent such as succinic anhydride and affect labeling efficiency.
-
2
| Add glycerol to a concentration of 15% (v/v) and dispense protein sample into single-use aliquots in 1.5 ml centrifuge tubes. Each tube should contain of accurately measured protein amount of 65 μg, sufficient to perform a single experimental condition stable isotope labeling i.e., one sample with light isotope and the other with heavy isotope. These protein samples are then stored at –80°C until needed for a year.
■ PAUSE POINT Aliquots of protein samples can be stored at –80°C for a year.
? TROUBLESHOOTING
-
3
| Adjust the protein concentration to 2.5-5.0 μM with 50 mM potassium phosphate buffer (pH 7.5), containing 100 mM NaCl and 0.02% (w/v) DDM. Approximately 65 μg of protein in a 250 μL final volume is required per condition (i.e., receptor/ligand) to perform a single differential stable isotope labeling experiment with both ‘light’ and ‘heavy’ chemical reagents.
▲CRITICAL STEP. Labeling experiments are performed at least in triplicate to ensure reliability in measurements. Each labeling condition requires about 65 μg of protein, in triplicate would be ~ 200 μg. Avoid combining different batches of proteins, use a single batch, validated, and dialyzed receptor protein for a given set of experimental conditions (example in this study for three receptor:ligand pairs and receptor alone; plus validation experiments would require a total of ~ 1mg protein).
-
4
| To the 250 μL diluted protein solution in a 1.5 mL microcentrifuge tube, incubate with a test ligand or control (50-100 μM; 10–100-fold molar excess or equivalent amount of carrier solvent for control) for 30 minutes at room temperature.
-
5
| After incubation, the protein/ligand solution is then distributed into two 1.5 ml microcentrifuge tubes in a volume of 120 μL. Label one of the tubes as (A) “Ref Reaction”, and the other one as (B) “Exp Reaction”. It is important to note here that pipetting has to be as precise as possible to prevent any loss of samples.
? TROUBLESHOOTING
Differential NEM and SA labeling reactions
-
6
| Perform differential labeling reactions by using protiated reagents (NEM-H5 or SA-H4) for (A) “Ref Reaction” and deuterated reagents (NEM-D5 or SA-D4) for (B) “Exp Reaction” or vice versa (swap labeling) (Fig. 2). Use 20 mM DTT in NH4HCO3 to quench the NEM labeling reactions and 100 mM free L-lysine in 200 mM Tris-Cl (pH 8.0) to quench the SA labeling reactions.
▲ CRITICAL STEP “Ref Reaction” (A) and “Exp Reaction” (B) can be performed side by side. A pilot experiment should be performed to determine what kind of time points should be used for the protein of interest.
(A) Reference labeling reaction (Ref Reaction)
-
(i)
Add 2 mM of protiated NEM (NEM-H5) or SA (SA-H4) to the protein mixture and allow the labeling reaction to take place at 25°C.
-
(ii)
At the selected last time point for the “Exp Reaction” (A) (for example 60 minutes), remove precisely measured 10 μL aliquots and add each to a series of pre-chilled 1.5 mL Protein LoBind microcentrifuge tubes containing 1 μL of dithiothreitol (DTT, 20 mM final concentration from 50× stock in NH4HCO3) or 100 mM free L-lysine in 200 mM Tris-Cl (pH 8.0) (for SA labeling) and flash freeze samples in liquid N2 until further use.
▲ CRITICAL STEP The quenching solution should be pre-aliquoted in individual 1.5 mL Protein LoBind microcentrifuge tubes.
(B) Experimental time-point labeling reaction (Exp Reaction)
-
(i)
Add 2 mM of deuterated NEM (NEM-D5) or SA (SA-D4) to the protein mixture and let the reaction take place at 25°C.
-
(ii)
Remove 10 μL fractions at each of the selected time points (e.g., total time points of ten: 0, 0.5, 1, 2, 3, 5, 10, 20, 40 and 60 minutes) from the labeling reaction. Add these fractions to a series of pre-chilled 1.5 mL Protein LoBind microcentrifuge tubes containing 1 μL of dithiothreitol (DTT, 20 mM final concentration from 50× stock in NH4HCO3) (for NEM labeling) or 100 mM free L-lysine in 200 mM Tris-Cl (pH 8.0) (for SA labeling) to quench the reaction and flash freeze the fractions in liquid nitrogen.
▲ CRITICAL STEP The total number of aliquots from “Ref Reaction” (A) should be the same as that for the “Exp Reaction” (B). It is important that pipetting is as precise as possible to ensure equal mixing of protiated- and deuterated-labeled samples in the next step.
-
7
| Remove labeled reaction samples, spin down quickly for 10 seconds and mix equal aliquots from the “Ref Reaction” (A) and “Exp Reaction” (B) in fresh 1.5 mL Protein LoBind microcentrifuge tubes to obtain ~ 20 μL final mixed volume of the ‘light’ and ‘heavy’ labeled samples. Flash-freeze samples in liquid nitrogen and store at –80°C indefinitely without freeze and thaws.
▲ CRITICAL STEP 1.5 mL Protein LoBind microcentrifuge tubes should be used for mixing.
■ PAUSE POINT At this point one can continue with Step 5 immediately or pause at this point leaving samples stored at –80°C.
? TROUBLESHOOTING
CHCl3/MeOH precipitation
-
8
| Get labeled samples from −80°C freezer and keep them on dry ice until ready to move to the next step.
-
9
| Add 4 volumes of MeOH, mix by tumbling and vortex for 30 seconds.
-
10
| Add 1 volume of CHCl3, mix layers and vortex for 30 seconds.
-
11
| Add 3 volumes of double distilled H2O, mix layers rigorously by vortexing for 30 seconds.
-
12
| Centrifuge at 12,000 × g at room temperature in a bench-top microcentrifuge for 2 minutes.
-
13
| Carefully remove the aqueous top layer.
▲ CRITICAL STEP Be sure to mix and vortex the sample before centrifugation to reach equilibrium of extraction. Using gel-loading tips, carefully remove the top phase without touching the finer emulsion that is created between the aqueous and chloroform layers, which contains the protein of interest.
? TROUBLESHOOTING
-
14
| Add 4 volumes of MeOH and vortex for 30 seconds.
-
15
| Centrifuge at 12,000 × g at room temperature in a bench-top microcentrifuge for 5 minutes.
-
16
| Carefully remove the MeOH without disturbing the solution as the pellet may be difficult to see and speed-vac the precipitate for 5 minutes.
Protein solubilization, reduction and alkylation
-
17
| Add 20 μL of protein solubilization solution (8 M urea, 50 mM Tris-HCl, pH 8.0, 5 mM EDTA, 0.001% DDM) to the dried precipitate. Vortex for 30 seconds.
▲ CRITICAL STEP: Be sure to pipette up and down including around the inner wall of the microcentrifuge tubes as many times as needed to solubilize the precipitate.
-
18
| Add 0.5 μL of 100 mM DTT to the above solubilization solution. The final concentration of DTT is 2.5 mM. Let the reduction take place at 37°C for 30 minutes.
-
19
| Allow the reduction reaction to cool to room temperature. Add 1.5 μL of freshly prepared 200 mM iodoacetimide (IAA) to a final concentration of 15 mM. Incubate the alkylation reaction in dark at room temperature for 30 minutes.
In-solution protein digestion
-
20
| Dilute chymotrypsin in chymotrypsin digestion buffer (50 mM NH4HCO3, pH 8.0, 1 mM CaCl2, 0.001% DDM) to ~ 100 ng/ μL to be used for cleavage of the differentially labeled samples involving NEM or SA. Note that protease such as trypsin can also be used, not to the SA-labeled samples (as trypsin cleavage would be inhibited because of modification at lysines by SA) but to cleave for the NEM labeled samples, and identify additional labeled residues that might be missed out from chymotrypsin cleavage analysis. For use in such cleavage reactions, dilute trypsin in trypsin digestion buffer (50 mM Tris-HCl pH 8.0, 0.001% DDM) to ~ 100 ng/ μL.
-
21
| For digestion reaction with chymotryptic digestion add 5 μL (final working concentration at ~10 ng/μL). For digestion with trypsin add 2.5 μL diluted trypsin (final working concentration at ~5 ng/μL). Dilute the final volumes of digestion mixture to 30 μL using respective buffers and incubate for 16 h at 37°C.
-
22
| When digestion reactions are completed, add 50 μL of 100% acetonitrile (ACN), and dry samples under speed-vac.
■ PAUSE POINT Product from 19 can be stored at −80°C for 1 year.
Peptide desalting
-
23
| Reconstitute peptide mixtures in 40 μL of 0.3% TFA, adjust the pH below 2 prior to performing peptide desalting. Check pH of the reconstituted peptide mixture by pipetting 1 μL onto a pH strip.
-
24
| Perform peptide desalting using one of the following options, which the research feels comfortable with: A, B or C. All are made from C18 reversed-phase resin. A full description of the cost effective-stage-tip peptide desalting procedures (A and B) are found in 69. Zip-Tip C18 is a commercially available desalting column.
Option A: Stage-Tip using syringes
Column packing: pack two C18 Empore disk membranes into GPS-L250 tip (1 disk for ~4 μg peptides)
Column cleaning: Add 40 μL of MeOH to the StageTip column.
Push out 50% of the solution using a 5 mL syringe.
Leave the column at room temperature for 5 minutes.
Push out the solution slowly (20-40 μL/min), leave ~ 5% solution in the stage-tip column.
Column activation: Activate stage-tip column with 40 μL of 50% ACN, 0.1% TFA.
Push out the solution slowly (20-40 μL/min), leave ~ 5% solution in the stage-tip column.
Column equilibration: Add 40 μL of 0.1% TFA in 5% ACN to the stage-tip column.
Push out the solution slowly (20-40 μL/min), leave ~ 5% solution in the stage-tip column.
Peptide binding: Load the peptides from step 20 to the equilibrated stage-tip column.
▲ CRITICAL STEP Peptides from Step 23 should be reconstituted in 40 μL of 0.3% TFA. Check pH of the reconstituted peptide solution with a pH indicator as described in Step 23. If pH is higher than 2, add more diluted TFA.
? TROUBLESHOOTING
-
xi
Push out the peptide solution slowly (10-20 μL/min), leave ~ 5% solution in the stage-tip column.
-
xii
Repeat Steps x and xi three times.
-
xiii
Column wash: Add 40 μL 0.1% TFA to the stage-tip column.
-
xiv
Push out the peptide solution slowly (10-20 μL/min), leave ~ 5% solution in the stage-tip column.
-
xv
Repeat Steps xiii and xiv.
-
xvi
Peptide elution: Add 40 μL 50% ACN, 0.1% TFA to the stage-tip column.
-
xvii
Push out the entire liquid slowly (10-20 μL/min) to a 1.5 mL Protein LoBind microcentrifuge tube.
-
xviii
Sample reconstitution: Speed-vac the samples to dryness.
-
xix
Reconstitute the peptides in 4 μL of 50% ACN, 0.2% TFA.
▲ CRITICAL STEP: Do not dry the stage-tip columns during column cleaning, activation, equilibrating, peptide binding, and column washing steps.
Option B: Stage-tip using a benchtop microcentrifuge
Column packing: pack two to three C18 Empore disk membranes into GPS-L250 tip (1 disk for ~ 4 μg peptides)
Column cleaning: Add 80 μL of MeOH to the stage-tip column.
Transfer the column to a clean 1.5 mL microcentrifuge tube.
Spin the microcentrifuge tube at 2,500 × g for 30 to 60 seconds (leave half of the MeOH on the column)
Leave the column at room temperature for 5 minutes.
Centrifuge the column (in 1.5 ml microcentrifuge tube) at 2,500 × g for 2 minutes to remove 95% of the MeOH.
Aspirate the flow-through in the microcentrifuge tube.
Column activation: Add 40 μL of 50% ACN, 0.1% TFA to the stage-tip column.
Place the column back to the microcentrifuge tube and centrifuge at 2,500 × g for 2 minute to remove 95% of the solution.
Column equilibration: Add 40 μL of 5% ACN, 0.1% TFA to the stage-tip column. Place the column back to the microcentrifuge tube.
Centrifuge the microcentrifuge tube at 2,500 × g for 2 minutes to remove 95% of the solution.
Aspirate the flow-through in the microcentrifuge tube.
Repeat Steps x and xii twice.
Peptide binding: Load the peptides from Step 23 to the equilibrated stage-tip column and place the column back to the micro-centrifuge tube.
▲ CRITICAL STEP: Peptides from Step 23 should be reconstituted in 40 μL of 0.3% TFA. Check pH of the reconstituted peptide solution with a pH indicator as described in Step 23. If pH is higher than 2, add more diluted TFA.
? TROUBLESHOOTING
-
xv
Centrifuge the micro-centrifuge tube at 2,500 × g for 3 to 4 minutes to remove 95% of the solution.
-
xvi
Aspirate the flow-through in the micro-centrifuge tube.
-
xvii
Repeat Steps xiv through xv twice.
-
xviii
Column wash: Add 20 μL 0.1% TFA to the stage-tip column.
-
xix
Centrifuge the microcentrifuge tube at 2,500 × g for 3 to 4 minutes to remove 95% of the solution.
-
xx
Aspirate the flow-through in the microcentrifuge tube.
-
xxi
Peptide elution: Add 40 μL 50% ACN, 0.1% TFA to the stage-tip column.
-
xxii
Transfer the column to a new 1.5 mL Protein LoBind microcentrifuge tubes.
-
xxiii
Centrifuge the tube at 4,000 × g for 3-4 minutes to elute the peptide sample.
-
xxiv
Sample reconstitution: Speed-vac the samples to dryness.
-
xxv
Reconstitute the peptides in 4 μL of 50% ACN, 0.2% TFA.
▲ CRITICAL STEP: Do not dry the stage-tip column during the column cleaning, wetting, equilibrating, peptide binding and column washing steps.
Option C: Zip-Tip C18 desalting
The Zip-Tip desalting is done essentially according to manufacturer's protocol with slight modifications.
▲ CRITICAL STEP: Use a P10 or P20 pipetter set to 10μl for Zip-Tips. The resin bed of Zip-Tip C18 provides back pressure so set pipetter to 10 μL, depress plunger to dead stop and slowly release or dispense plunger throughout operation.
Zip-Tip C18 column cleaning: Clean Zip-Tip with 10 μL of MeOH.
Zip-Tip C18 column activation: Wash Zip-Tip with 10 μL 0.1% TFA in 50% ACN.
Zip-Tip C18 column equilibration: Equilibrate the Zip-Tip three times with 10 μL 0.1% TFA.
Peptide Binding: Pass the sample through the Zip-Tips repeatedly by pipeting in and out to bind the sample to the column.
Repeat Step iv 10 times.
▲ CRITICAL STEP: Peptides from Step 23 should be reconstituted in 40 μL of 0.3% TFA. Check pH of the reconstituted peptide solution with a pH indicator as described in Step 23. If pH is higher than 2, add more diluted TFA.
? TROUBLESHOOTING
-
vi
Zip-Tip C18 column washing: Wash the Zip-Tip three times with 0.1% TFA.
-
vii
Peptide elution: Elute samples from the Zip-Tip in 3 μL of 50% ACN, 0.2% TFA.
-
viii
Dispense to a new 0.6 mL Protein LoBind microcentrifuge tubes.
▲ CRITICAL STEP: Don not dry the Zip-tip column during the column cleaning, activation, equilibrating, peptide binding and column washing steps.
■ PAUSE POINT For MALDI-TOF MS analysis, it is highly recommended proceeding to spot samples on plates immediately following elution otherwise samples can be stored at −20 °C for several weeks.
MALDI-TOF/TOF-MS and data analysis
-
25
| Prepare matrix solution: prepare saturated solution of α-cyano-4-hydroxycinnamic acid (CHCA) in 20 mM ammonium phosphate, 50% ACN and 0.2% TFA and use it as ionization matrix.
-
26
| Spot 0.4 μL of peptide sample onto a MALDI target plate in triplicate followed by addition of equal volume of the supernatant matrix solution.
-
27
| Allow the mixture to co-crystallize70 at room temperature.
? TROUBLESHOOTING
-
28
| Load the MALDI-TOF/TOF target plate into a MALDI-TOF/TOF mass spectrometer
-
29
| Acquire spectra, using a MALDI-TOF/TOF mass spectrometer, over the range 700 - 4000 m/z. See EQUIPMENT SETUP for details about configuration and settings.
▲ CRITICAL STEP: It is important to set spectral acquisition over a mass range of 700 – 4000 m/z to analyze suitably sized proteolytic peptides. Analyses of very short peptides that fall below 700 m/z are unreliable due to matrix interferences.
? TROUBLESHOOTING
-
30
| Mass spectra data analyses are performed with Data Explorer software (version 4.3.0.0, Applied Biosystems, Framingham, MA). To confirm for mass accuracy and identify uniquely labeled peptides, we preliminary depend on the appearance of protiated (“light”) and deuterated (“heavy”) pairs of peptides that give a mass shift of 5 Daltons (for NEM labeling) or 4 Daltons (for SA labeling).
-
31
| Perform calibration of mass spectra with known mass of proteolytic autolysis fragments (typically using peaks like at 705.48 m/z for chymotrysin or at 842.51 m/z for trypsin for example) and extract mass list of mono-isotopic peptide peaks.
-
32
| Perform database search using GPS software or peptide mass fingerprinting analysis using program of the Protein Prospector database search algorithm Mass list of peptides (http://prospector.ucsf.edu, UCSF, San Francisco, CA) in mass range of 700 to 4,000 Da.
When using this, or similar, software: Allow search parameters for a maximum of two missed cleavages and a mass tolerance of 50 ppm. For identification of NEM modified peptides, either oxidation of methionines, N-ethylmaleimide alkylation, or carbamidomethylation of cysteines can be used as variable modifications. For identification of SA modified peptides, oxidation of methionines as well as succinylation, and carbamylation of lysines can be used as variable modifications; while carbamidomethylation of cysteines as constant modification.
-
33
| In addition, to confirm sites of modification by the specific modifying reagents, MS/MS amino acid perform sequence analyses of all the differentially labeled peptides. The resulting spectra can be interpreted manually with the help of MS-PRODUCT program in Protein Prospector or other available software packages.
-
34
| After calibration, extract data lists of mono-isotopic peptide peak mass and intensity from baseline corrected spectra.
■ PAUSE POINT At this point, data can be stored and analyzed later.
Quantitation of site-specific labeling kinetics
-
35
| Perform quantification at the MS level and measure the relative signal intensity ratios of the monoisotopic peaks of the deuterated (“heavy”) to protiated (“light”) reagent-modified peptides to get percent intensity ratios (% R), for each time point and amino acid residue in the protein. Such data analysis can be performed manually using Data Explorer or other compatible automated software packages that are usedfor very large numbers of data sets. Mono-isotopic peaks should be used for the measurement. Since the mass differences between labeled peptides are 5 and 4 Daltons, overlap between isotope envelopes of the differentially labeled peptides is usually negligible for most of the peptides. However, for peptides which are bigger than 3,000 m/z, significant overlap between isotope envelops may occur; in this case, depending on the significance and quality of the data, the peak signal intensity ratios can be corrected for by performing further kinetic analysis or can be totally excluded. The percentage of overlap between isotope envelops can be calculated using program available in: http://prospector.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msisotope.
-
36
| Plot percent intensity ratios, denoted as %R (for the ratio between peak intensities of the heavy and light peptides) as a function of time for every labeled residue in a protein to generate time-course labeling curves. In our experience, we find it convenient to plot this data as a semi-logarithmic graph where time is on the log scale.
-
37
| Fit the labeling curves of each site to exponential equations (2-4), starting with a single exponential (eq 2) to determine the appropriate fitting model. The model used to fit the data should be the simplest possible that provides a similar goodness of fit as assessed statistically (e.g., by R2 values).
-
38
| Tabulate all the kinetic parameters calculated from the fits (amplitudes of the phases and relaxation times).
-
39
| We find that using relaxation times, best interprets the labeling kinetics at different sites. Transform the fast phase relaxation time to negative logarithmic value (-log τ1) to obtain what we refer to as labeling reactivity factor (L-factor). Note that, although it may vary from protein to protein, most of our labeling curves were sufficiently described by a double exponential function.
-
40
| For a given site, compare the L-factor values obtained when the receptor/protein is in its unliganded state and ligand-bound state. Compute the difference between each of these L-factors. Note that reactivity measurements are sensitive to the conformational state of the receptor/protein, thus change in L-factor computed would reflect ligand-dependent conformational rearrangements in the vicinity of the site (i.e., a large L-factor reflects higher reactivity relative to unliganded receptor/protein-only, and vice versa).
-
41
| Alternatively, the percent intensity ratios (%R (t)) for each site can also be normalized according to equation 1 using the term reactivity factor (Rr), a normalization factor determined experimentally (Step 57).
▲ CRITICAL STEPS: Note that normalization of percent intensity ratios (as in Steps 41-57) is important to compare reactivities between different residues within a given protein. An alternative approach to normalization is to use a synthetic stable isotope-labeled internal standard of identical sequence to replace “Ref Reaction” in the experimental scheme shown in Figure 2.
(Optional) Estimating the fraction of individual site labelled at reaction completion
▲ CRITICAL: The purpose of limited proteolysis of the receptor in the detergent environment is to partially denature the protein and make cysteine or lysine residues fully available to labeling reagents (NEM or SA). However, care must be taken in designing such experiments with SA labeling at lysines, as full proteolytic cleavage followed by labeling can cause further mass shift due to labeling of the neo-N-terminal amine group.
<CRITICAL> Estimate the fraction of individual site labeled at the longest time point chosen for the labeling experiment. While this experiment is optional; it allows a normalization of percent intensity ratios (%R (t)) to compare reactivity between different residues.
-
42
| Native protein preparation labeling Prepare the protein of interest in native conditions as described in Steps 1-2.
-
43
| React with 2 mM NEM-D5 or SA-D4 for one hour at 25°C (Step 3).
-
44
| Quench the NEM-D5 labeling reactions by adding 1M DTT to a final concentration of 20 mM DTT in NH4HCO3 (pH 8.0) and the SA-D4 labeling reactions by adding 1M L-lysine to a final concentration of 100 mM in a 200 mM Tris-Cl (pH 8.0).
-
45
| Flash freeze samples in liquid nitrogen until further use.
-
46
| Partially proteolyzed/unfolded sample labeling Prepare the protein of interest as described in Steps 1-2.
-
47
| Incubate protein sample with a relatively low concentration of appropriate proteases (final concentrations of 1 ng/μL for chymotrypsin or 0.5 ng/μL for trypsin) at 37°C for 10-30 min.
-
48
| React the partially proteolyzed/unfolded sample with 5 mM NEM-D5 or SA-D4 at 25°C for 14 h.
-
49
| Quench the NEM-D5 labeling reactions by adding 1M DTT to a final concentration of 25 mM DTT in NH4HCO3 (pH 8.0) and the SA-D4 labeling reactions by adding 1M L-lysine to a final concentration of 100 mM in a 200 mM Tris-Cl (pH 8.0).
-
50
| Flash freeze samples in liquid nitrogen until further use.
-
51
| Mixing “light” and “heavy” chemical reagent labeled protein samples, alkylation and reduction Combine equal amounts of labeled protein mixtures from Steps 45 and 50 (“light” and “heavy”and perform chloroform/methanol precipitation of the labeled receptor sample mixture as described in Steps 8-16.
-
52
| Reconstitute, and reduce any remaining cysteines by adding 100 mM DTT to a final concentration of 2.5 mM DTT. Let the reduction take place at 37°C for 30 minutes.
-
53
| Alkylate cysteines by adding 200 mM IAA to final concentration of 15 mM. Incubate sample at room temperature in the dark for 30 minutes.
-
54
| In-solution protein digestion, desalting and MS analysis Perform in-solution protein digestion as described in Steps 17-19 using appropriate proteases. When digestion reactions are completed, add 50 μL of 100% acetonitrile, and dry samples under speed-vac.
-
55
| Reconstitute peptide mixtures in 40 μL of 0.3% TFA, adjust the pH below 2 and perform peptide desalting using pipette tips containing C18 media.
-
56
| Perform MALDI-TOF MS analysis of spotted samples.
-
57
| Measure the peak intensities and quantify the ratios of the light-to-heavy reagent modified peptides for each site to obtain reactivity ratio (Rr).
-
58
| Use reactivity ratio (Rr) obtained from Step 57 to normalize the percent intensity ratios (%R (t)) described in Step 36, applying equation 1 to obtain fraction sites labeled [(% F(t)].
-
59
| Plot the percent reactivity ratios (%F) versus time to establish new labeling curves and fit each time-dependent labeling profile to an appropriate exponential kinetic equation (eq. 2 to 4).
-
60
| Repeat Steps 37-39 to compute L-factor values for individual time-dependent labeling profiles.
-
61
| Statistical significance of the differences between pairs of treatments can be assessed using one-way analysis of variance (ANOVA) followed by Bonferroni's post-test, or a student's two-tailed paired t-test, as appropriate using Prism 5.01 (GraphPad Software, San Diego, CA) or an equivalent software package.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
• TIMING
| Protein sample preparation and protein-radio-ligand binding assay | Step 1-2 | 1 day |
| Differential NEM and SA labeling reactions | Step 6-7 | 5-6 hours |
| CHCb/MeOH precipitation | Step 8-16 | 3-5 hours |
| Protein solubilization, reduction and alkylation | Step 17-19 | 3-4 hours |
| In-solution protein digestion | Step 20-23 | 16 hours |
| Peptide desalting | Step 24 | 4-5 hours |
| MALDI-TOF/TOF-MS and data analysis | Step 25-34 | 4-6 hours |
| Quantitation of site-specific labeling kinetics | Step 35-61 | 1 day |
TABLE 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 1 | Labeling reaction is not successful or efficiency too low | Protein samples may contain primary amine components that compete with labeling reagents | Perform complete dialysis of protein sample to make sure protein samples are free of primary amines bearing reagents. Prepare fresh regents |
| 2-4 | The “heavy” to “light” ratio of the end time point mixture is not 1:1 | Problem during labeling rection and mixing of “heavy” and “light” samples | Ensure precision during pipetting, resuspension, mixing and transferring of samples |
| Problem of not performing “Experimental time-point” and “Reference” labeling reactions under identical condition | Perform “Experimental time-point” and “Reference” labeling reactions side by side, and essentially, under identical conditions | ||
| Problem during mixing of “heavy” and “light” samples | Be sure to equally mix experimental time-point and reference labeling samples | ||
| 10 | No signal, or poor signal intensity | Possible protein sample loss during extraction and precipitation | Be sure to mix and vortex as directed before centrifugation to reach equilibrium of extraction. Using gel-loading tips, carefully remove the aqueous layer without touching the finer emulsion that is created between the layers, containing protein of interest |
| 21: A (x), B (xiv) and C (iv) | No signal, or poor signal intensity | Problem during desalting process | Ensure that all buffers needed for desalting are appropriately prepared, as directed |
| Ensure that the pH of the reconstituted peptide is below 2 by pipetting 1 μL onto a pH strip; if not add more TFA | |||
| Ensure that air is not drawn into the column-tip and that it does not dry during the process | |||
| 24 | No signal, or poor signal intensity | Sample-matrix crystallization process is not optimized or irregularities of spotting on MALDI plates | Optimize sample-matrix crystallization conditions (See ref. Cohen et. al.) |
| Always use high-quality, ultra pure reagents and freshly prepared matrix | |||
| Take extra care when spotting and spot as uniform as possible | |||
| 24 | No signal, or poor signal intensity | Possible problem of having low signal-to-noise of peptide ions on MS | Increase laser to obtain optimized signal |
ANTICIPATED RESULTS
The CDSiL-MS strategy presented here is a versatile technique that can be applied to different soluble and membrane proteins for elucidating conformational rearrangments at a site-specific resolution. We have recently applied the CDSiLMS methodology to study the dynamics of various ligand-occupied structural conformations of the human β2-adrenergic receptor (β2AR), a well-studied G protein-coupled receptor (GPCR)55. Initial requirement for the method is to characterize the functional and ligand-binding properties of the protein of interest to ensure its structural integrity is preserved during labeling experiments. In addition, the method also relies on obtaining optimal signal ion intensity (i.e., high abundance and signal-to-noise ratio) of the labeled peptide ion pairs under study for accurate quantification. As determined by linear response experiment, a typical labeled-peptide ion pair shows a strong linear relationship between measured peptide ion pair peak intensity ratios and applied dilution of labeled protein concentrations (slope = 0.9785 ± 0.13; R2 = 0.9996; p < 0.0001) (Fig. 4).
To illustrate the procedures of the CDSiL-MS strategy, we show the results on two residues located in conformational sensitive regions of β2AR (Fig. 5a). Cys 327, located in the intracellular portion of transmembrane helix 7 (TM7) near NPxxY motif; and Lys 263, located in the third intracellular loop (ICL3) at the cytoplasmic end of TM6, where larger conformational change occurs during receptor activation and G-protein binding61,63. We use three functionally distinct ligands consisting of an agonist isoproterenol (iso), an inverse agonist ICI-118,551 (ICI), and a biased ligand carvedilol (carv; a β antagonist for G protein activation with selective stimulation of β-arrestin–mediated signaling64). The labeling of Cys 327 residue by NEM is identified by a singly charged ion ([M+H]+) peaks (327CRSPDFRIAF336) at m/z 1336.6 and 1341.7(Fig. 5b). While, the labeling of Lys 263 residue by SA is identified by a singly charged ion ([M+H]+) peaks (259RRSSKF264) at m/z 880.5 and 884.5 corresponding to peptide modification by SA-H4 and SA-D4, respectively (Fig. 5c). Figures 6a and 6d show plots of percent intensity ratios [%R(t)], i.e., the ratios of intensities of monoisotopic peaks of the deuterated (“heavy”) to protiated (“light”) NEM or SA-labeled peptides as a function of time, respectively. Such time-course labeling curves in the form of percent intensity ratios (%R(t)) can be used directly to fit with a suitable exponential kinetic model (equations 2-4) or normalized. It is important to normalize %R (t) to % F (t) using reactivity factor (Rr) via equation 1 when comparison in the reactivities between different residues within a given protein is desired. For example, the Rr values (which is fraction of individual residues reacted by the last time point of the experiment) in the unliganded, iso, ICI, and carv-bound-β2AR, are obtained to be > 80% at Cys 327 (0.84 ± 0.01, 0.81 ± 0.01, 0.88 ± 0.02 and 0.86 ± 0.01, respectively); while > 90% at Lys 263 (0.94 ± 0.04, 0.94 ± 0.04, 0.95 ± 0.03 and 0.99 ± 0.03, respectively); indicating a higher proportion of these sites being reacted under different ligand bound states by the longest time point. Such Rr values, although insignificant, are used as normalization factors to obtain labeling curves in the form of %F (t) using equation 1 (Figure 6b, e). These labeling data (%F (t)) are well described by double exponentials (equation 3) with fast and slow relaxation times; and produce excellent fits to the data with R2 values greater than 0.994. The fast phase relaxation times (τ1) in min at Cys 327 were obtained to be 3.87 ± 0.1 (none), 1.89 ± 0.2 (iso), 5.32 ± 0.6 (ICI), and 5.32 ± 0.5 (carv); while at Lys 263, 0.68 ± 0.1 (none), 0.50 ± 0.1 (iso), 0.59 ± 0.1 (ICI), and 0.22 ± 0.1 (carv).
Figure 5. CDSiL-MS based monitoring of conformational rearrangements at critical structural elements of the β2AR.

(a) Cytoplasmic view of β2AR (PDB: 2RH1), showing relative positions of C327 at TM7 (yellow sphere), and K263 (blue sphere) at TM6. (b) Singly charged ion ([M+H]+) isotope-peak pair corresponding to a chymotryptic peptide (327CRSPDFRIAF336) modified at C327 by a light and heavy NEM (m/z 1336.6 and 1341.7, respectively; Δm/z = 5). (c) Singly charged ion ([M+H]+) isotope-peak pair corresponding to a chymotryptic peptide (259RRSSKF264) modified at K263 by light and heavy succinic anhydride (m/z 880.5 and 884.5, respectively; Δm/z = 4).
Figure 6. CDSiL-MS based strategy in measuring functional residue-specific conformational rearrangements in β2AR.

Time-course curves for the extent of NEM reactivity at C327 (a) and SA reactivity at K263 (d) expressed as percent intensity ratio plotted vs. labeling time (%R (t)) on a logarithmic scale, for unliganded (black circles), iso (red squares), ICI (green triangles) or carv (blue diamond). (b, e) Same labeling curves expressed in the form of percent of sites labeled, %F (t) following normalization of %R (t). Bar graphs summarizing the effects of each ligand on the changes in the L-factors of the C327 (c) and K263 (f) on the β2AR, expressed relative to the unliganded receptor. Inset indicates position of labeled residue in the β2AR snake like diagram. Data correspond to the means ± standard errors from at least three independent experiments. Asterisk indicates statistical significance (*p < 0.05) compared to control receptor alone by one-way ANOVA.
To quantitatively compare the labeling kinetics of the residues and associated conformational re-arrangement upon ligand binding, the fast relaxation times are transformed to negative logarithmic value (-log τ1) to obtain labeling reactivity factor (L-factor) (Fig. 6 c, f). Reactivity measurements are sensitive to the conformational state of a protein, thus change in L-factor computed would reflect ligand-dependent conformational rearrangements in the vicinity of the site (a large L-factor reflects higher reactivity relative to unliganded protein, and vice versa). As illustrated in Figure 6c, the full agonist (isoproterenol)-bound β2AR induce the highest reactivity (L-factor) at Cys 327 comparing to others (p < 0.01, iso vs. none or ICI, one way ANOVA), suggesting structural rearrangements upon agonist binding and receptor activation55,61,63. These results are in agreement with the observations of large conformational rearrangements at the cytosolic end of TM7 (where Cys 327 is located) in the activated form of the β2AR in the X-ray crystal structures61,63 and from a conformational dynamics study by HDX/MS23. Similarly, at Lys 263, a markedly higher L-factor in carvedilol-bound β2AR complex is observed relative to that of pharmacologically similar ligand, ICI, for G protein inactivation or unliganded-β2AR (p < 0.05, carv vs. none) (Fig. 6f), consistent with ligand-specific structural rearrangments at these regions of the receptor.
We have demonstrated here the method using a pair of residues; similar reactivity measurements can be done to demonstrate in a time-dependent manner upon binding to different ligands, for several other residues located in different regions of the receptor55 (Fig. 7a); including the ones that are absent in the X-ray crystal structures, obtained from engineered protein/GPCR constructs (Fig. 7b). Taken together, the data that the ability of functionally different ligands to alter local conformational rearrangements (intracellular or extracellular regions), suggest the presence of multiple ligand specific functional conformations, which may play a role in biased agonism or functional selectivity. The ability of functionally selective ligands to stabilize unique functional conformations was also recently demonstrated by NMR and HDX/MS experiments9,10,23,71. This highlights the complementary nature of different structural biology methods to provide a comprehensive understanding of conformational changes associated with protein's function.
Figure 7. Multiple β2AR residues from different structural elements that can be monitored using CDSiL-MS strategy.
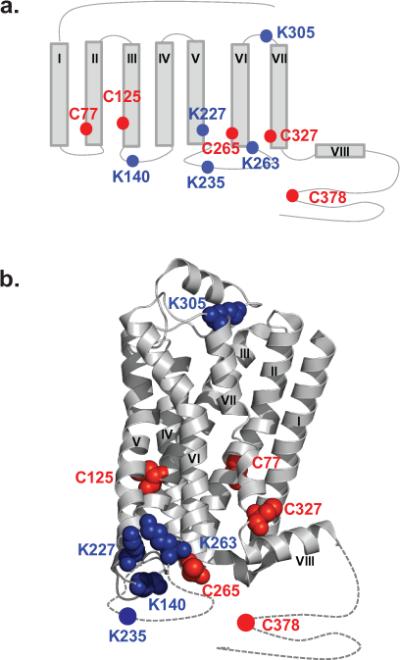
Cartoon (a) and ribbon (PDB: 3SN6) diagram (b) of the β2AR, showing locations of Cys (red) and Lys (blue) residues for which reactivities can be monitored. In (b), the Cys and Lys residues shown in sphere format are the ones that are available in the X-ray crystal structure. Dotted lines indicating highly flexible regions of the receptor which are absent in the X-ray crystal structure.
In conclusion, the CDSiL-MS strategy described here provides a reliable and cost effective way for monitoring-and precisely measuring site-specific conformational dynamics of proteins via stable isotope labeling coupled with MS-based quantitative analysis. As MS instrumentation and methods of quantitative stable isotope covalent labeling strategies continue to evolve it is anticipated the protocol presented here will also improve quickly for broad range proteome applications. In the near future, we believe that the approach can be applicable to moderate-or high-throughput systems where protein targets that undergo function-dependent conformational changes can be interrogated structurally to allow drug screening.
BOX 1 Pilot experiments for time point selection.
The following steps describe the pilot experiment for the determination of numbers of and distribution between time points for the protein in study.
Choose a series of time points, for example, 0, 0.5, 1,2, 3, 5, 10, 20, 40, and 60 minutes for the experiments.
Perform the labeling experiments as described in “Differential stable isotope labeling reactions” and Figure 2.
Plot “heavy”-to-“light” percent intensity ratios (%R) of each examined peptide vs. time to make it easier to determine which time points are most appropriate..
Acknowledgements
We thank Drs. Robert J. Lefkowitz and Brian K. Kobilka for invaluable guidance; Dr. Terrence G. Oas for enthusiastic support; Dr. Arun K. Shukla and Seungkirl Ahn for stimulating ideas; Dr. Ryan T. Strachan and Ms. Adi Blanc for critically reading the manuscript. We gratefully acknowledge Dr. Timothy Haystead (Duke University) and David Loiselle for valuable assistance with the mass spectrometry experiments; we are also grateful to Christoph H. Borchers (University of Victoria, Victoria, BC, Canada) and Igor A. Kaltashov (University of Massachusetts) for helpful discussions; we also thank Xinrong Jiang for excellent technical assistance. This work was supported, in whole or in part, by NIH grant HL-075443 Proteomics Core support to K. Xiao.
Footnotes
Author contributions
A.W.K. and K.X. designed and conducted the experiments; A.W.K., K.X., S.R., and J.S. analyzed data; A.W.K., S.R. and K.X. wrote the paper; all authors read, edited and discussed the paper.
Competing financial interests
The authors declare no competing financial interests.
REFERENCES
- 1.Uphoff S, et al. Monitoring multiple distances within a single molecule using switchable FRET. Nat Methods. 2010;7:831–836. doi: 10.1038/nmeth.1502. [DOI] [PubMed] [Google Scholar]
- 2.Kajihara D, et al. FRET analysis of protein conformational change through position-specific incorporation of fluorescent amino acids. Nat Methods. 2006;3:923–929. doi: 10.1038/nmeth945. [DOI] [PubMed] [Google Scholar]
- 3.Taraska JW, Puljung MC, Olivier NB, Flynn GE, Zagotta WN. Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat Methods. 2009;6:532–537. doi: 10.1038/nmeth.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Islas LD, Zagotta WN. Short-range molecular rearrangements in ion channels detected by tryptophan quenching of bimane fluorescence. J Gen Physiol. 2006;128:337–346. doi: 10.1085/jgp.200609556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK. Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci U S A. 2001;98:5997–6002. doi: 10.1073/pnas.101126198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao X, et al. Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat Chem Biol. 2006;2:417–422. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 7.Sprangers R, Velyvis A, Kay LE. Solution NMR of supramolecular complexes: providing new insights into function. Nat Methods. 2007;4:697–703. doi: 10.1038/nmeth1080. [DOI] [PubMed] [Google Scholar]
- 8.Chill JH, Naider F. A solution NMR view of protein dynamics in the biological membrane. Curr Opin Struct Biol. 2011;21:627–633. doi: 10.1016/j.sbi.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Bokoch MP, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nygaard R, et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 12.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 13.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 14.Cravatt BF, Simon GM, Yates JR., 3rd The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 15.Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 16.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 17.Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 18.Heck AJ, Krijgsveld J. Mass spectrometry-based quantitative proteomics. Expert Rev Proteomics. 2004;1:317–326. doi: 10.1586/14789450.1.3.317. [DOI] [PubMed] [Google Scholar]
- 19.Mendoza VL, Vachet RW. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Analytical chemistry. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics. 2011;8:43–59. doi: 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morgan CR, Engen JR. Investigating solution-phase protein structure and dynamics by hydrogen exchange mass spectrometry. Curr Protoc Protein Sci. 2009 doi: 10.1002/0471140864.ps1706s58. Chapter 17, Unit 17 16 11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West GM, et al. Ligand-dependent perturbation of the conformational ensemble for the GPCR beta2 adrenergic receptor revealed by HDX. Structure. 2011;19:1424–1432. doi: 10.1016/j.str.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 25.Kaltashov IA, Bobst CE, Abzalimov RR. H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: is there a need for a top-down approach? Analytical chemistry. 2009;81:7892–7899. doi: 10.1021/ac901366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.England J, Britovsek GJ, Rabadia N, White AJ. Ligand topology variations and the importance of ligand field strength in non-heme iron catalyzed oxidations of alkanes. Inorg Chem. 2007;46:3752–3767. doi: 10.1021/ic070062r. [DOI] [PubMed] [Google Scholar]
- 27.Katta V, Chait BT. Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun Mass Spectrom. 1991;5:214–217. doi: 10.1002/rcm.1290050415. [DOI] [PubMed] [Google Scholar]
- 28.Yan X, Zhang H, Watson J, Schimerlik MI, Deinzer ML. Hydrogen/deuterium exchange and mass spectrometric analysis of a protein containing multiple disulfide bonds: Solution structure of recombinant macrophage colony stimulating factor-beta (rhM-CSFbeta). Protein Sci. 2002;11:2113–2124. doi: 10.1110/ps.0204402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, et al. Dynamics of the beta2-adrenergic G-protein coupled receptor revealed by hydrogen-deuterium exchange. Analytical chemistry. 82:1100–1108. doi: 10.1021/ac902484p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niwayama S, Kurono S, Matsumoto H. Synthesis of d-labeled N-alkylmaleimides and application to quantitative peptide analysis by isotope differential mass spectrometry. Bioorg Med Chem Lett. 2001;11:2257–2261. doi: 10.1016/s0960-894x(01)00452-8. [DOI] [PubMed] [Google Scholar]
- 31.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A. 1999;96:6591–6596. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 33.Wu CC, MacCoss MJ, Howell KE, Matthews DE, Yates JR., 3rd Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Analytical chemistry. 2004;76:4951–4959. doi: 10.1021/ac049208j. [DOI] [PubMed] [Google Scholar]
- 34.Ong SE, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 35.Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- 36.Shiio Y, Aebersold R. Quantitative proteome analysis using isotope-coded affinity tags and mass spectrometry. Nat Protoc. 2006;1:139–145. doi: 10.1038/nprot.2006.22. [DOI] [PubMed] [Google Scholar]
- 37.Thompson A, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical chemistry. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 38.Dayon L, et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Analytical chemistry. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 39.McAlister GC, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Analytical chemistry. 2012;84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye X, Luke B, Andresson T, Blonder J. 18O stable isotope labeling in MS-based proteomics. Brief Funct Genomic Proteomic. 2009;8:136–144. doi: 10.1093/bfgp/eln055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc. 2009;4:484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 42.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 43.Pratt JM, et al. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat Protoc. 2006;1:1029–1043. doi: 10.1038/nprot.2006.129. [DOI] [PubMed] [Google Scholar]
- 44.Kleifeld O, et al. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat Biotechnol. 2010;28:281–288. doi: 10.1038/nbt.1611. [DOI] [PubMed] [Google Scholar]
- 45.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 46.Lefkowitz RJ. A brief history of G-protein coupled receptors (Nobel Lecture). Angew Chem Int Ed Engl. 2013;52:6366–6378. doi: 10.1002/anie.201301924. [DOI] [PubMed] [Google Scholar]
- 47.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 48.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 49.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 50.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 51.Shukla AK, et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 53.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 55.Kahsai AW, et al. Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat Chem Biol. 2011;7:692–700. doi: 10.1038/nchembio.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galandrin S, Oligny-Longpre G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–430. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 57.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 58.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 59.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 61.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warne T, et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wisler JW, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci U S A. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kobilka BK. Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor. Anal Biochem. 1995;231:269–271. doi: 10.1006/abio.1995.1533. [DOI] [PubMed] [Google Scholar]
- 66.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 67.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kettenbach AN, Rush J, Gerber SA. Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat Protoc. 2011;6:175–186. doi: 10.1038/nprot.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Analytical chemistry. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 70.Cohen SL, Chait BT. Influence of matrix solution conditions on the MALDI-MS analysis of peptides and proteins. Analytical chemistry. 1996;68:31–37. doi: 10.1021/ac9507956. [DOI] [PubMed] [Google Scholar]
- 71.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F NMR. Science. 2012;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]