

Abstract
Embryonic stem cells (ESCs) are maintained in an undifferentiated state through expression of the core transcriptional factors Nanog, Oct4, and Sox2. However, the epigenetic regulation of pluripotency is poorly understood. Differentiation of ESCs is accompanied by a global reduction of panacetylation of histones H3 and H4 suggesting that histone acetylation plays an important role in maintenance of ESC pluripotency. Acetylated lysine residues on histones are read by members of the bromodomain family that includes BET (bromodomain and extraterminal domain) proteins for which highly potent and selective inhibitors have been developed. In this study we demonstrate that the pan-BET bromodomain inhibitor JQ1 induces rapid spontaneous differentiation of murine ESCs by inducing marked transcriptional downregulation of Nanog as well as the stemness markers Lefty1 and Lefty2, but not Myc, often used as a marker of BET inhibitor activity in cancer. We show that the effects of JQ1 are recapitulated by knockdown of the BET family member BRD4 implicating this protein in Nanog regulation. These data are also supported by chromatin immunoprecipitation experiments which confirm BRD4 binding at the Nanog promoter that is known to require acetylation by the histone acetyltransferase MOF for transcriptional activity. In further support of our findings, we show that JQ1 antagonizes the stem cell-promoting effects of the histone deacetylase inhibitors sodium butyrate and valproic acid. Our data suggest that BRD4 is critical for the maintenance of ESC pluripotency and that this occurs primarily through the maintenance of Nanog expression.
Introduction
Embryonic stem cells (ESCs) exhibit dual unique properties: limitless self-renewal and pluripotency in differentiation [1]. Murine ESCs cultured in the presence of the cytokine leukemia inhibitory factor (LIF), which activates Stat3, are maintained in an undifferentiated state through the expression of critical transcription factors Oct4 (also known as Pou5f1), Sox2, and Nanog [2]. These factors form the ESC transcriptional core [3]. Ectopic expression of Oct4 and Sox2 together with Myc and Klf4 in terminally differentiated somatic cells can result in reprogramming and generation of induced pluripotent stem cells [4]. Recently, the histone acetyltransferase (HAT) known as MOF (also called MYST1 or KAT8) has been shown to be a key regulator of the ESC transcriptional network and required for self-renewal [5]. Deletion of Mof results in loss of ESC self-renewal and induction of differentiation with downregulation of the transcriptional core factors Oct4, Sox2, and Nanog and aberrant expression of differentiation marker genes. Overexpression of Nanog was shown to rescue the Mof null phenotype suggesting that Nanog is the key downstream target for MOF and largely mediates its function in ESCs, a conclusion supported by the considerable overlap of MOF and Nanog transcriptomes and also the finding that ∼80% of Nanog target genes have MOF binding sites [6]. Chromatin immunoprecipitation (ChIP) analysis has confirmed MOF binding and H4K16 acetylation at the Nanog promoter, but not in Mof null cells suggesting that MOF, unlike other HATs such as p300/CBP, TIP60, and GCN5, is able to regulate Nanog expression [6].
Acetylated lysine residues in histones are specifically recognized by proteins that contain a small helical interaction module known as a bromodomain [7]. Members of the BET (bromodomain and extraterminal domain) family of proteins read the differentially acetylated histones causing changes in gene transcription [8,9] and have particularly high affinity for the tail, including H4K16ac [10]. The BET family comprises four distinct genes, namely, BRD2, BRD3, and BRD4, which are ubiquitously expressed, and BRDT, which is restricted to the testis. Each BET protein contains two bromodomains, both of which can be prevented from binding acetyl-lysine by the prototypic bromodomain inhibitor JQ1. Enantiomerically pure (+)-JQ1, hereafter referred to as JQ1, binds with a Kd of approximately 50 nM and 90 nM to the first and second bromodomains of BRD4 and BRD3, respectively, whereas BRDT and BRD2 show about threefold weaker binding [11]. Inhibition of BRD4 by JQ1 has been shown to induce differentiation and death of human acute myeloid leukemia and multiple myeloma cells, possibly through the transcriptional downregulation of MYC [12–15] or by influencing MYC turnover [16]. In wild-type mice, JQ1 causes reversible sterility by inhibition of BRDT [17]. However, the effect of JQ1 on ESCs has not been previously reported.
In this study, we show that pharmacological inhibition of BRD4 and BRD4 knockdown causes morphological differentiation of ES cells. Microarray analysis of ES cells treated with JQ1 causes a robust downregulation of Nanog with little effect on the pluripotency genes Sox2, Oct4, and klf4. Furthermore, we show that this effect is mediated by BRD4 and that BRD4 binds to the Nanog promoter suggesting that BRD4 induces differentiation of murine ESCs through downregulation of Nanog.
Materials and Methods
Materials
DMEM, penicillin/streptomycin, and L-glutamine were from Fisher. Anti-Nanog and anti-BRD4 antibodies were from Bethyl laboratories, anti-c-MYC was from New England Biolabs. Taqman probes were from Applied Biosystems. Taq polymerase and wst-1 were from Roche. The Alkaline phosphatase kit, Leukemia inhibitory factor (LIF), trichostatin A (TSA), valproic acid, sodium butyrate, nonessential amino acids, and two mercaptoethanol were from Sigma. The HiPerFect and the Quantitect cDNA synthesis kit were from Qiagen. The pCMV Nanog expression plasmid was a kind gift from Professor Austin Smith (University of Cambridge). The LTx transfection reagent was from Life Technologies. Anti-acetylated histone H3 and acetylated histone H4 were from Millipore. Anti-SSEA1 (clone MS480) was from the Developmental Hybridoma Bank.
Embryonic stem cells
E14 ESCs were kindly provided by Dr Lesley Forrester (University of Edinburgh) and derived from the inbred mouse strain 129/Ola in 1985 by Dr Martin Hooper [18]. Early passage cells were used (<20 passages). The cell line was maintained in vitro and propagated in DMEM supplemented with 10% FBS, 100 nM L-glutamine, 100 IU/mL penicillin, 100 IU/mL streptomycin, 1% nonessential amino acids, 50 mM 2-mercaptoethanol, and 100 IU/mL of LIF. This medium is referred to as LIF-positive medium to distinguish it from LIF-negative medium that was used to induce ESC differentiation. Cells were incubated at 37°C in a humidified atmosphere containing 95% air and 5% CO2.
Compounds and experimental conditions
(+)-JQ1 (referred hereafter as JQ1) and the inactive enantiomer (−)-JQ1 was synthesized according to the published synthetic route [11]. Stock dilutions of 10 mM in dimethylsulfoxide (DMSO) were made and stored at −20°C. Dilutions to working concentrations were made with phosphate buffered saline (PBS) and also stored at −20°C. The appropriate DMSO dilution and the inactive enantiomer (−)-JQ1 were both used as negative controls. HDAC inhibitor concentrations used were 0.5 mM valproic acid and 0.2 mM sodium butyrate.
Alkaline phosphatase assay
The Alkaline phosphatase staining was performed using a Sigma-Aldrich kit (procedure no 85). Cells were plated on coverslips at 2.5×104 and 5×104 per well. Cells were treated with varying concentrations of JQ1 and analyzed after 72 h culture in LIF-positive medium. Cells were fixed by immersing in citrate buffered acetone for 30 s and incubated in an alkaline-dye mix (naphthol AS-MX phosphate alkaline solution and diazonium salt solution) for 30 min. ESC differentiation was evaluated morphologically and photomicrographs were produced.
Immunocytochemical analysis
Cells were plated on gelatin-coated coverslips at 2.5×104 cells per well in 24 well plates in 400 μL of LIF-positive medium in the presence or absence of 0.5 and 1 μM for 72 h. Preparations were fixed in 4% paraformaldehyde for 15 min then permeabilized with 0.2% TritonX100 for 3 min. Primary antibodies were applied for 1 h at room temperature. After washing thoroughly with PBS, cells were incubated in Alexa fluor-linked secondary antibodies for 30 min. After the final wash through PBS the slides were incubated in 0.1 μg/mL DAPI (4′,6-diamidino-2-phenylindole) for 5 min then mounted in antifade mounting medium and viewed by fluorescence microscopy using a Leica DM 5000B microscope fitted with a Leica DPC300FX digital camera. To illustrate the intensity of immunolabeling, digital images were captured at identical exposure times. In the case of the global histone acetylation immunolabeling experiment, semiquantitative analysis was performed by antibody titration.
Western blot analysis
E14 ESC were treated with 0.5 or 1 μM JQ1 for 24 h then harvested. Proteins were extracted in sample buffer containing protease inhibitor tablets. Samples were then diluted in loading buffer (with β-mercaptoethanol), boiled for 5 min, and subjected to gel electrophoresis on 10% SDS-polyacrylamide gels. A total of 36 μg of cell extract was loaded per lane. Proteins were electrophoretically transferred onto a PVDF membrane for 1 h at 150 mA constant current. Membranes were blocked with 5% milk for 1 h at room temperature and then rotated overnight at 4°C with primary antibody diluted 1:2,000 in TBS 0.1% Tween. HRP-linked secondary antibodies were used at a dilution of 1:20,000 for 1 h. Blots were developed using a Vectastain ABC kit or a chemiluminescent detection kit.
Quantitative RT-PCR
Total RNA was extracted from ESCs using the TRIzol reagent according to the manufacturer's instructions. RNA (1 μg) was reverse transcribed using a QuantiTect cDNA kit and the PCR reactions were performed using the TaqMan Gene Expression Assays (Applied Biosystems) c-MYC (Mn00487804_m1), Nanog (Mm02384862_g1), GAPDH (Mn99999915_g1), MYST-1 (Mn00458911_m1), Pou5f1 (Mn03053917_g1), RPS18 (Mn02601778_g1), Lefty1 (Mn03053915_s1) using the standard TaqMan reagents, and protocols on a MX3500 (Stratagene) qRT-PCR machine.
The ΔΔCt method was used for relative expression quantification using the average cycle threshold for GAPDH RNA and 18s RNA to normalize gene expression levels between samples. All experiments were performed in triplicate repeats and expression levels were compared between JQ1 and (−)-JQ1-treated cells using a two-tailed t test with P values ≤0.05.
Gene transcriptional profiling
Mouse ESCs were treated with 1 μM JQ1 or 1 μM (−)-JQ1 for 16 h in quadruplicate experimental replicates. RNA was extracted with the TRIzol reagent and analyzed using an Agilent 2100 bioanalyzer. All RNA used had a RIN value of greater than 8.0. Transcriptional profiling for control and JQ1-treated ESCs was performed using the Illumina expression microarrays. Normalization of the raw data included a variance stabilizing transformation followed by robust spline normalization using the R package Lumi. The expression changes of genes were calculated by comparing the mean expression levels of treatment versus control untreated.
Of note, the Illumina array used in the transcriptional profiling experiments was the MouseWG-6 v2.0 Expression BeadChips system, which allows 12 samples per array, thereby allowing all three quadruplicate experimental samples to be analyzed on a single array. The complete expression dataset is available through the MIAME-compliant GEO (Gene Expression Omnibus) NCBI public repository.
Transfection of cell lines with siRNAs
For siRNA studies, ESCs were seeded in 24-well plates at a density of 250,000 cells per well. Twenty-four hours after plating, cells were transfected with 37.5 ng ON-TARGETplusSMARTpool siRNA (Thermo Scientific) targeting BRD4 or a nontargeting scrambled control using the HiPerFect Transfection Reagent according to the manufacturer's instructions. RNA was prepared from cells 24 h after transfection.
CHiP analysis
E14 ESC were grown to 80% confluence before being treated with ethanol, 1 μM JQ1 or (−)-JQ1 for 24 h. Cells were crosslinked with 1% formaldehyde at 37°C for 10 min, and sonicated for 12×20 s with 1 min intervals at 2Aμ (Soniprep 150; MSE, Sanyo Gallenkamp PLC). Chromatin was precleared with 1 μg anti-rabbit IgG, 2 μg sheared salmon sperm DNA, and 50 μL of 50% protein-A-Agarose slurry for 4 h at 4°C. Immunoprecipitation was performed overnight at 4°C with 2 μg sheared salmon sperm DNA, 50 μL protein-A-Agarose, and either rabbit anti-Brd4 (Santa Cruz) or rabbit IgG. DNA fragments were amplified using the FastStart Universal SYBR-Green Master Mix (Roche Diagnostics) and previously published primers targeting the Nanog and GAPDH promoters [19]. Results are shown as a percentage of the input DNA.
Generation of Nanog overexpressing ESCs
ESCs plated in LIF+ve medium were transfected with 6 μg of pCAG-Nanog expression vector or control vector using the LTx reagent. Transient transfectants were used in experiments 24 h after transfection. Stable transfectants were made by selecting cell cultures with 300 μg/mL hygromycin for 10 days.
Statistical analysis
Results of each experiment represent mean±standard deviation (SD) of replicate experiments. Differences were determined using the Student's t-test. When there was a prior hypothesis results were considered significant when P<0.05. When there was no prior hypothesis and multiple tests were performed (microarray analysis), we also derived q values of significance using the Benjamini–Hochberg method.
Results
JQ1 induces differentiation of ESCs accompanied by downregulation of Nanog
Initial experiments were conducted to study the effect of JQ1 on ESCs cultured in LIF-containing medium. Using concentrations of JQ1 up to 1 μM we observed clear dose-dependent morphological changes over 72 h. JQ1-treated cells became flattened and elongated with reduced cell-to-cell contact and a fibroblastic morphology similar to cells grown in LIF-negative medium whereas cells grown in LIF-positive medium remained undifferentiated in small, spherical, well-delimited three dimensional phase-bright clusters (Fig. 1A). In further experiments, we demonstrated that exposure of ESCs to 1 μM JQ1 for as little as 6 h is sufficient to induce morphological differentiation indicating that JQ1 causes rapid differentiation of ESCs despite continued presence of LIF. Importantly, removal of JQ1 from the medium for 72 h did not reverse ESC differentiation. The (−)-JQ1 inactive enantiomer had no discernible morphological effect on the cells.
FIG. 1.

Effect of JQ1 on embryonic stem cells (ESCs). (A) Phase contrast micrograph of ESCs grown in LIF-containing medium exposure to 1 μM JQ1 for 72 h showing morphology (upper panel) and alkaline phosphatase staining (middle panel) and immunolabeling of SSEA1 (lower panel); (B) BRD4 immunolabeling of control and JQ1-treated ESCs with DAPI counterstain (upper panel) and Nanog immunolabeling showing JQ1-induced downregulation (lower panel). Color images available online at www.liebertpub.com/scd
We confirmed these observations by staining for alkaline phosphatase, a well-established marker of the undifferentiated state. This revealed that JQ1 treatment had caused a dose-dependent reduction in alkaline phosphatase expression when compared with untreated ESCs. The (−)-JQ1 enantiomer had no effect. These data show that JQ1 treatment promotes morphological differentiation of ESCs resulting in an apparent loss of pluripotency (Fig. 1A). Interestingly, the JQ1-treated ESCs retained expression of the embryonic surface marker SSEA1 at the 72 h time point used in this experiment (Fig. 1A). Importantly, induction of ESC differentiation by JQ1 occurred without any significant effect on cell viability up to a concentration of 1 μM as assessed by WST-1 proliferation assay (data not shown).
To further explore the role of BET proteins in stem cell maintenance we conducted immunocytochemistry of ESCs. This confirmed that BRD4 exhibits ubiquitous nuclear expression in both control and JQ1-treated ESCs (Fig. 1B). We also performed immunocytochemistry for Nanog given its role in maintenance of the core transcriptional network. This clearly demonstrated marked reduction in expression of Nanog in JQ1-treated ESCs suggesting that this gene had been significantly downregulated (Fig. 1B). Taken together, the addition of JQ1 to ESC cultures interferes with several characteristics of stem cells, including morphology and alkaline phosphatase staining, and that this is accompanied by loss of expression of Nanog suggesting that BET proteins are required for maintenance of the undifferentiated ESC phenotype.
BET bromodomain inhibition leads to Nanog downregulation in ESCs
To explore possible transcriptional deregulation of ESCs by JQ1, we performed expression profiling microarray experiments. We treated ESCs cultured in LIF-containing medium for 16 h with either JQ1, the inactive enantiomer (−)-JQ1 (both at 0.5 μM), or vehicle only. We found that JQ1 had significant effects on the ESC transcriptome, 5,697 of the 30,854 genes (18.4%) on the array being significantly deregulated (two-tailed t-test, P<0.05). Compared with vehicle only, 92 genes were differentially downregulated and 42 genes were differentially upregulated by greater than two-fold change for JQ1 with no significant transcriptional changes for (−)-JQ1. This data is illustrated by way of heatmaps for both compounds showing the relative expression level changes for the most significantly deregulated genes (n=1,113 genes at significance level of P<0.001) in Fig. 2A. Further analysis of these genes showed that the 50 that were most significantly deregulated (two-tailed t-test, P<2.24E-06) were significantly downregulated compared with the rest (mean log2=−0.6084 versus −0.0026, respectively, P=9.6E-11, two-tailed t-test). This, together with the fact that twice as many genes were downregulated by greater than two-fold than upregulated, suggests that the predominant effect of JQ1 is to induce transcriptional downregulation of genes. While upregulated genes were observed in the analysis, the extent of the changes was less and the statistical significance of those changes was reduced when compared with the downregulated genes (Fig. 2B).
FIG. 2.


JQ1 induces transcriptional deregulation of genes. Microarray profiling was performed using the Illumina platform to assess the transcriptional effects of 1 μM JQ1 and (−)-JQ1 at 16 h on ESCs. (A) Heatmaps (derived from the analysis of 4 replicates) showing expression level changes for the most deregulated genes (n=1,113 genes at t-test significance level of P<0.001) ordered by degree of deregulation (green=up; red=down); (B) heatmaps of the most significantly deregulated genes ordered by P values (JQ1 versus untreated control, two-tailed t-test) with analysis of the 50 most deregulated genes (*) revealing a predominant downregulatory effect compared with the rest (mean log2−0.6084 versus −0.0026, respectively, P=9.6×10−11, two-tailed t-test); (C) effect of JQ1 on transcription of germ layer-specific genes; (D) effect of JQ1 on selected pluripotency-related genes; (E) confirmatory qRT-PCR analysis of cDNA samples prepared from ESCs treated with 0.5 μM for up to 24 h was performed for Nanog, Lefty1, Mof, and Myc; (F) quantification of mRNA transcript levels for Fbox15, klf4, Oct4, Sox2, and Zfp42 was performed by qRT-PCR (data are representative of at least three independent experiments performed in triplicate±SD). Color images available online at www.liebertpub.com/scd
Nanog was markedly downregulated by 70.3% as assessed by microarray profiling consistent with our immunolabeling data. Interestingly, the most downregulated gene was Lefty1 (87.7%, q=7.1×10−4) with Lefty2 and Nodal also significantly downregulated (66.7%, q=3.4×10−3 and 45.4%, q=0.008, respectively). Other notable genes downregulated by JQ1 were the pluripotency marker genes Fbx15 and Rex1 (78.9%, q=1.6×10−3 and 56.7%, q=0.02, respectively). By contrast, expression of Oct4 was modestly downregulated by 36.3% (q=0.05) whereas the other core pluripotency genes Sox2 and Klf4 were not significantly affected. Importantly, and in contrast to published observations in leukemia and myeloma cell lines, we find that c-Myc was not significantly affected by JQ1 in ESCs where it is weakly transcribed suggesting this gene is not invariably downregulated by BET bromodomain inhibition. Of note, JQ1 upregulated Mof/Myst1 by 37.9% (q=0.01) and also the BET gene Brd2 by 118% (q=0.006). Brd3 and Brd4 were upregulated by 14.5% and 36.7%, respectively, but these latter differences were not statistically significant (q>0.05). These results are summarized in Table 1.
Table 1.
Microarray Data Showing the Effect of JQ1 on Expression of Selected Genes
| Gene function | Gene (selected genes shown) | Rank total genes on array=30,854) | Control Av. gene expression | JQ1 Av. gene expression | (−)-JQ1 Av. gene expression | Log2 change JQ1 vs control (+up; −down) | % change gene expression JQ1 vs control | Benjamini–Hochberg derived q-values for multiple testing |
|---|---|---|---|---|---|---|---|---|
| Pluripotency genes | Nanog | 24 | 699 | 207 | 740 | −1.753 | 70.3% down | N/A |
| Fbx15 | 7 | 1,321 | 279 | 1,256 | −2.245 | 78.9% down | q=1.6×10−3 | |
| Rex1 | 55 | 1,113 | 482 | 1,275 | −1.206 | 56.7% down | q=0.02 | |
| Embryonic patterning genes | Lefty1 | 1 | 706 | 87 | 676 | −3.028 | 87.7% down | q=7.1×10−4 |
| Lefty2 | 29 | 234 | 78 | 187 | −1.585 | 66.7% down | q=3.4×10−3 | |
| Nodal | 139 | 568 | 310 | 610 | −0.873 | 45.4% down | q=0.008 | |
| Bromodomain BET family genes | Brd4 | 30,345 | 184 | 252 | 181 | +0.451 | 36.7% up | q>0.05 |
| Brd2 | 30,832 | 497 | 1,084 | 482 | +1.125 | 118.0% up | q=0.006 | |
| Brd3 | 29,011 | 554 | 635 | 545 | +0.195 | 14.5% up | q>0.05 | |
| Brdt | 20,820 | 79 | 80 | 79 | +0.020 | 1.4% up | N/S | |
| Pluripotency core transcriptional genes | Oct4 | 318 | 3,284 | 2,092 | 3,544 | −0.651 | 36.3% down | q=0.05 |
| Sox2 | 27,978 | 129 | 139 | 126 | +0.114 | 8.2% up | N/S | |
| Klf4 | 1,312 | 122 | 101 | 122 | −0.007 | 17.2% down | N/S | |
| Mof | 30,380 | 293 | 404 | 301 | +0.466 | 37.9% up | q=0.01 | |
| Myc | 27,965 | 83 | 90 | 84 | +0.113 | 8.1% up | N/S |
Transcriptional profiling was performed using the Illumina microarray platform. ESCs were exposed to JQ1 or (−)-JQ1 or vehicle only for 16 h in independent quadruplicate experiments. Average gene expression data is shown with log change for JQ1 versus control and t-test. Percentage up or downregulation of the gene has been calculated and the P values have been subjected to a Benjamini–Hochberg correction for multiple testing of genes other than Nanog (N/A=not applicable; N/S=not significant). Lefty1 was the most downregulated gene. Selected other genes are shown, including ESC core transcriptional factors, BET protein family members, and various genes involved with embryonic patterning and pluripotency.
ESC, embryonic stem cell; BET, bromodomain and extraterminal domain.
To explore possible lineage-specificity of JQ1-induced ESC differentiation, we analyzed the microarray profiling data to explore transcriptional markers specific for the embryonic germ layers. This revealed a tendency to upregulation of ectodermal markers and a concomitant downregulation of endodermal markers (Fig. 2C). Further analysis of pluripotency-related genes, and their fold change with JQ1, is shown in Fig. 2D.
We confirmed the key microarray findings outlined above by qRT-PCR (Fig. 2E, F). A time course of JQ1 treatment of ESCs demonstrated a dramatic decrease in expression of both Nanog and Lefty1 genes with maximal downregulation occurring at 8–24 h. By contrast, levels of the HAT Mof/Myst1 were modestly increased by JQ1 over the same time course, whereas levels of c-Myc were largely unaffected by JQ1. Fbox15 and Zfp42 were also seen to be downregulated after 24 h JQ1 treatment, whereas the pluripotency genes Klf4, Oct4, and Sox2 were unaffected. The observation that morphological changes were first apparent several hours after Nanog downregulation suggests that inhibition of BET bromodomain proteins by JQ1 suppresses Nanog expression resulting in ESC differentiation. This is supported by the finding that downregulation of both Nanog and Lefty1 in ESCs was more pronounced after 24 h of JQ1 exposure than seen in ESCs following LIF withdrawal, where onset of morphological differentiation occurs over a longer time course (data not shown).
JQ1-induced differentiation of ESCs is independent of MYC
Based on the observations that JQ1 reduces levels of MYC in drug-sensitive cell lines and primary samples derived from hematological malignancies (AML and MM), we investigated whether JQ1 suppresses c-Myc expresssion in ESCs. Microarray results from JQ1-treated cells showed no significant effect on c-Myc transcripts, a result that was largely corroborated by qRT-PCR (Fig. 3A). Furthermore, western blotting showed that MYC protein levels were not significantly regulated by 24 h of 1 μM JQ1 exposure (Fig. 3B). Thus we show, that in murine ESCs, the mechanism of JQ1-induced differentiation is independent of MYC.
FIG. 3.
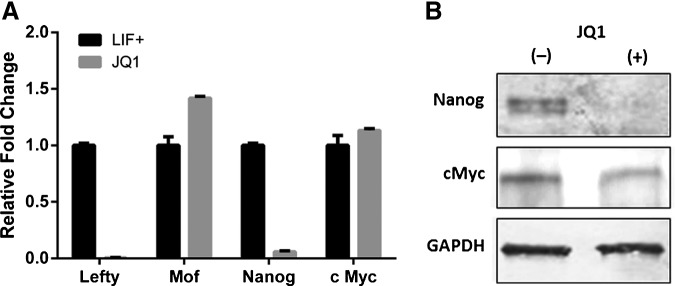
JQ1 downregulates Nanog and Lefty1, but not c-Myc in ESCs. ESCs were treated with 1 μM JQ1 for 24 h then analyzed: (A) Quantification of mRNA transcript levels for Lefty1, Mof, Nanog, and c-Myc was performed by qRT-PCR (data are representative of at least three independent experiments performed in triplicate±SD); (B) Quantification of protein levels of Nanog and MYC was performed by western immunoblot analysis (blots were probed with GAPDH antibodies as a loading control).
Nanog overexpression prevents JQ1-induced ESC differentiation
Nanog is a highly divergent homeodomain-containing protein that occupies a central position in the core transcriptional network to maintain pluripotency of ESCs. We, therefore, sought to test whether ectopic overexpression of Nanog was able to overcome JQ1-induced ESC differentiation. We used an expression vector in which the Nanog gene has been cloned downstream of a constitutively active chicken actin/globin promoter (pCAG-Nanog, or pNanog) designed for robust expression in ESCs (kind gift from Professor A. Smith). Both transient and stable transfection experiments were performed using a pCMVeGFP vector as the control. We observed that transient transfection of the Nanog expression vector significantly prevented ESC differentiation by 1 μM JQ1 at 24 h as assessed by morphology and alkaline phosphatase staining suggesting that JQ1-induced ESC differentiation results primarily from downregulation of the endogenous Nanog promoter (Fig. 4A). In further experiments, we generated stable pCAG-Nanog transfectants by pooling hygromycin-resistant ESC clones at 10 days posttransfection. As for the transient transfection experiment, the stable transfectants were clearly refractory to JQ1-induced differentiation. After exposure to 1 μM JQ1 for 24 h, the effects on Nanog gene expression were analyzed by qRT-PCR (Fig. 4B). While we found near-silencing of the endogenous Nanog in the control-transfected ESCs, Nanog expression persisted in the stable transfectants where the gene was under transcriptional control of the constitutive CAG promoter. Taken together, our results suggest that the Nanog locus is a major functional target of BET bromodomain proteins in ESCs and required for maintenance of pluripotency.
FIG. 4.

Overexpression of Nanog attenuates JQ1-induced downregulation of Nanog. (A) Photomicrographs showing morphology (above) and alkaline phosphatase staining (below) of ESCs transfected with pNanog (CAG vector) or pControl (empty vector) and treated with JQ1. (B) Nanog expression levels of stably transfected ESCs showing effect of JQ1 at 1 μM for 24 h as determined by qRT-PCR. (LIF- vs. LIF+ vs. LIF+/JQ1 for pNanog and pControl ESCs). Data are representative of at least three independent experiments performed in triplicate±SD. Color images available online at www.liebertpub.com/scd
Nanog is a transcriptional target of BRD4 in ESCs
Based on the fact that JQ1 has greater selectivity and bromodomain inhibitory activity for BRD4 than other members of the BET protein family [11], we hypothesized that this was the major effector of JQ1-induced ESC differentiation. We carried out RNAi experiments using Brd4-specific siRNAs. Twenty-four hours after transfection, a profound effect on ESC morphology was observed (Fig. 5A) with Brd4 siRNA-transfected cells having a differentiated morphology compared with the scrambled siRNA control. The qRT-PCR analysis revealed approximately 80% knockdown of Brd4 with a concomitant 70% knockdown of Nanog gene expression (Fig. 5B).
FIG. 5.

Brd4 knockdown induces ESC differentiation and downregulation of Nanog. (A) Phase contrast micrographs of ESCs transfected with scramble siRNA control versus Brd4-specific siRNA. (B) Graph showing level of Brd4 knockdown and effect on Nanog expression compared with siRNA control as determined by qRT-PCR. (C) Chromatin immunoprecipitation of BRD4 at the promoter regions of Nanog and GAPDH in ESC following 24 h growth in the presence of 1 μM JQ1 or (−)-JQ1, or following mock manipulation with ethanol. Enrichment is shown as a percentage of input, and in comparison to IgG enrichment. Data are representative of two independent experiments performed in triplicate±SD.
Next, we performed chromatin immunoprecipitation (ChIP) analysis to measure BRD4 binding to acetylated histones at the Nanog promoter to understand JQ1-induced transcriptional changes. We found that JQ1 caused approximately 40–50% reduction in BRD4 binding compared with the (−)-JQ1 negative enantiomer and also the mock control (both experiments performed twice) suggesting that JQ1 does indeed disrupt BRD4 binding at the Nanog promoter. This finding suggests that histone acetylation at the Nanog promoter is critically involved in gene transcription through recruitment of BRD4 (Fig. 5C).
Histone deacetylase inhibitors can overcome JQ1-induced differentiation
As JQ1 blocks binding of BET bromodomain proteins to acetylated histones, we investigated the activity of JQ1 with a number of histone deacetylase (HDAC) inhibitors. We employed two HDAC inhibitors, namely sodium butyrate and valproic acid, both with activity against class I and II HDACs [20]. We hypothesized that exposure to HDAC inhibitors at pluripotency maintaining concentrations might rescue JQ1-induced differentiation through histone hyperacetylation. In initial experiments we determined that 0.5 μM JQ1 had no significant effect on global histone H3 and H4 acetylation as assessed by semiquantitative immunocytochemistry using antibody titration (Fig. 6A).
FIG. 6.

JQ1 does not affect histone acetylation, but does antagonize HDAC inhibition in ESCs. (A) JQ1 has no significant effect on global histone H3 and H4 acetylation in ESCs. ESCs were cultured in the presence or absence of LIF (to induce differentiation) or in medium containing 1 μM JQ1 plus LIF for 24 h, then immunolabeled with antibodies against acetylated histone H3 or H4. (B) JQ1 overcomes the effects of low-dose HDAC inhibitors on ESCs. Morphological appearance by phase microscopy of ESCs cultured for 24 h in LIF-containing medium in the presence of: (a) no drug; (b) 0.5 μM JQ1; (c) 0.2 mM sodium butyrate; (d) 0.2 mM sodium butyrate plus 0.5 μM JQ1; (e) valproic acid 0.5 μM; (f) valproic acid 0.5 μM plus 0.5 μM JQ1. Color images available online at www.liebertpub.com/scd
We next studied the effects of combined JQ1 exposure and HDAC inhibition on ESC morphology. We observed that JQ1-induced morphological changes were less pronounced in the presence of 0.2 mM sodium butyrate or 0.5 mM valproic acid suggesting functional antagonism between JQ1 and low-dose HDAC inhibition. Of note, neither of the HDAC inhibitors alone had any discernible effects on ESC morphology although higher concentrations of sodium butyrate (2 mM) did induce differentiation (Fig. 6B).
Discussion
Recent studies have highlighted the importance of histone acetylation in the regulation of ESC pluripotency [6,21–23]. This epigenetic modification is read by a family of proteins containing bromodomains (BRDs) that recognize acetylated lysine residues such as those on certain proteins [24–26] and the N-terminal tails of histones [8]. Inhibition of the bromodomain–acetyl histone interaction, therefore, represents a novel mechanism of disrupting the epigenetic state of ESCs that underpins maintenance of self-renewal. In this article, we studied the effects of the bromodomain inhibitory molecule JQ1, which primarily inhibits members of the BET family of proteins [11], on ESCs to investigate the role of histone acetylation in the maintenance of stem cell pluripotency. We found that JQ1 induces differentiation of ESCs and overcomes the pluripotency-promoting effects of LIF as assessed by morphological, biochemical, and gene expression criteria. Central to this finding, we show that inhibition of BET bromodomain activity by JQ1 leads to downregulation of the core transcription factor Nanog at both the transcriptional and protein level. Given the greater affinity of JQ1 for BRD4 compared with BRD2 and BRD3 (Kd values of 49 nM versus 128 and 82 nM, respectively), we hypothesized that inhibition of BRD4 in particular mediates the effect of JQ1 on ESCs.
Microarray analysis demonstrated that the most significant effect of JQ1 on transcription was to induce downregulation of active genes with twice as many genes being significantly downregulated than upregulated. As JQ1 inhibits the binding of BET bromodomains these data suggest that the primary purpose of this binding to acetylated histones is to induce gene transcription. In addition to Nanog, microarray analysis of JQ1-treated cells also revealed significant downregulation of other notable genes of interest, including Lefty1, Lefty2, and Nodal, involved in embryonic patterning and left–right determination, and Fbx15 and Rex1, well-recognized markers of ESC pluripotency.
Analysis of germ lineage-specific genes showed that JQ1 induces a shift toward ectodermal differentiation and away from endodermal differentiation. Our experiment used ESCs exposed to JQ1 for 16 h. In view of the fact that transcriptional changes seen in ESCs following LIF withdrawal mostly occur at 48–72 h [6,21–23], we predict that the changes we observe with JQ1 would likely increase over time to establish a more definitive skew toward ectodermal differentiation.
In contrast to the downregulation of Nanog, we found that the expression of the other core pluripotency transcription factors Sox2 and Klf4 was not significantly altered whereas Oct4 was modestly suppressed by JQ1. Likewise, no significant downregulation of c-Myc expression was observed at either the transcriptional or protein level. These results are in contrast to several other studies of JQ1 on hematological cancer cell lines in which downregulation of MYC has been identified as the principal mechanism of action of BET bromodomain inhibitors. In these cells, ChIP experiments reveal that BRD4 actively binds to the MYC promoter and regulates signaling events leading to transcript elongation [12,14,15]. In response to BET inhibitors, BRD4 is released from acetylated histones at the MYC locus leading to transcriptional downregulation [15]. However MYC downregulation is neither necessary nor sufficient for the antiproliferative effects of BET bromodomain inhibitors: JQ1 induces MYC downregulation in the leukemia cell lines K562 and Jurkat and the breast cancer cell line MDA MB-231 without causing apoptosis [12,27]. Furthermore MYC is not downregulated in JQ1-sensitive lung cancer cell lines and in one cell line it is significantly upregulated [28].
In many previous studies, the effect of BET bromodomain inhibitors has been examined in the context of its antiproliferative effects. In our study, however, we show that JQ1 has minimal effect on ESC proliferation, but rather induces differentiation even when the cells are cultured in the presence of the pluripotency factor LIF. JQ1, through BRD4 inhibition, has been shown to induce markers of differentiation in some leukemia cell lines and primary bone marrow samples. Furthermore in NUT midline carcinoma, where a translocation results in fusion between BRD4 and NUT (nuclear protein in testis) genes, application of JQ1 decreased cell proliferation and the expression of BRD4 target genes and increased cellular differentiation suggesting that JQ1 may have therapeutic effects for this rare disease [29]. Our RNAi experiments using Brd4-specific siRNAs show that Brd4 knockdown induces morphological differentiation of ESCs and downregulation of Nanog. Therefore, it would appear that the effects of JQ1 on ESCs are mediated primarily through inhibition of BRD4 binding to acetylated histones within the Nanog promoter. While our experiments were conducted using transient BRD4 knockdown, this is not the same as producing a stable knockdown/knockout of BRD4, although we anticipate that such an experiment would yield similar findings. In addition, while BRD4 knockdown alone appears to be sufficient in inducing the morphological effects of JQ1 on ESCs, it is likely that inhibition of BRD2 and BRD3 may contribute to the transcriptional deregulation of genes observed on microarray profiling.
Our data clearly reveal an important role for BET bromodomain proteins in the maintenance of the pluripotent state of ESCs through binding to acetylated histones at discrete gene loci, notably Nanog. The importance of the acetylation status of histones has been demonstrated in several studies [6,30,31]. Although the majority of these studies suggest that histone acetylation is important in the maintenance of the pluripotent state, others suggest that histone acetylation increases during differentiation [21]. Over the limited time period of our experiment (48 h) no apparent change in global H3 and H4 acetylation levels were found in ESCs induced to differentiate with JQ1 showing that JQ1-induced ESC differentiation is not mediated through a global reduction of histone acetylation.
Profound defects in ESC differentiation have been observed after knockdown or knockout studies involving several HAT genes, including Tip60 [32,33], p300 [32,33], and Mof [5], and also the HAT cofactor Trrap [22,34]. Furthermore one study shows that differentiation of ESCs is accompanied by a global reduction in pan-acetylation of histones H3 and H4 [6]. The effect of HDAC inhibitors on ESC differentiation is less clear as it has been reported that they can promote either self-renewal or differentiation depending on the concentrations used [27,35]. It has been suggested that HDAC inhibitors exert an antidifferentiation effect when low doses are applied on cells that have already exited from self-renewal either as embryoid bodies or epiblast-like cells, whereas higher doses applied on undifferentiated cells provoke differentiation [35]. In our experiments, we found that JQ1-induced differentiation of ESCs was reduced in the presence of low-dose sodium butyrate or valproic acid suggesting functional biological antagonism. We speculate that low-dose HDAC inhibition induces histone hyperacetylation at relevant genomic loci to which BET proteins bind, including potentially Nanog, leading to some weakening of the bromodomain inhibitory action of JQ1.
The other notable finding from our work was the demonstration that JQ1 is a potent downregulator of Lefty1 (most downregulated gene by microarray) and its homologue Lefty2 in ESCs. Lefty1 and Lefty2 are both divergent members of the transforming growth factor-β (TGF-β) superfamily of proteins that function as diffusible morphogens involved in left–right determination and give rise to embryonic asymmetry [36]. Both proteins have been previously recognized as stemness markers in undifferentiated ESCs and in blastocysts. Moreover, Lefty1 and Lefty2 have been implicated in the maintenance of self-renewal and pluripotency of mouse ESCs and optimal expression of both genes appears to be critical for the balanced differentiation of ESCs into three germ layers [37]. In particular, Lefty1 knockdown in ESCs has been shown to result in enhanced phosphorylation of Smad2 and increased differentiation, which supports our own findings and suggests that JQ1-induced differentiation of ESCs may be mediated by Lefty1 downregulation as well as by Nanog. Taken together, we hypothesize that BET family proteins are fundamentally involved with embryonic patterning and left–right determination, an assertion supported by the observation that Nodal is also significantly downregulated by JQ1 in ESCs.
In summary, our results implicate members of the BET bromodomain family of proteins in the maintenance of ESC self-renewal and pluripotency primarily through the expression of Nanog and Lefty1. In the case of Nanog, our data suggest that expression of this gene requires BRD4 binding to the promoter and that this is itself dependent on histone acetylation. Based on the fact that the aberrant phenotype of Mof-deficient ESCs, namely defective self-renewal and induction of differentiation, is rescued by Nanog expression, we speculate that Mof normally acts to maintain transcriptional activity of the Nanog promoter through acetylation at H4K16 and this facilitates recruitment of BRD4. The rapid induction of differentiation induced by JQ1 suggests that ESC pluripotency is critically dependent on BRD4 binding at the Nanog locus to maintain its expression. Given the considerable overlap in signaling networks that exist between ESCs and cancer stem cells [38–40], our results are potentially pertinent not only to understanding ESC self-renewal and pluripotency, but also for novel cancer-directed therapies based on BET bromodomain inhibition.
Acknowledgments
The authors would like to thank Kathrin Bisling, Sophie Robinson, and Drs Tom Rider, John Brewin, and Rohit Ghurye for technical assistance. The authors would also like to thank Professor Austin Smith for gift of the pCAG-Nanog expression vector. This work was partly funded by the Elimination of Leukemia Fund and the Brighton and Sussex University Hospital Cancer Trust Fund. SK is supported by the SGC, a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, Takeda, AbbVie, the Novartis Research Foundation, the Ontario Ministry of Research and Innovation, and the Wellcome Trust [092809/Z/10/Z].
Statement of Regulatory Approval
This research was approved by the Brighton and Sussex Medical School at the University of Sussex.
Author Disclosure Statement
None of the authors has any commercial associations that might create a conflict of interest in connection with the submitted article and no competing financial interests exist.
References
- 1.Evans MJ. and Kaufman MH. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292:154–156 [DOI] [PubMed] [Google Scholar]
- 2.Chambers I. and Smith A. (2004). Self-renewal of teratocarcinoma and embryonic stem cells. Oncogene 23:7150–7160 [DOI] [PubMed] [Google Scholar]
- 3.Yamanaka SaKO. (2006). Intracellular signaling pathways regulating pluripotency of embryonic stem cells. Curr Stem Cell Res Ther 1:103–111 [DOI] [PubMed] [Google Scholar]
- 4.Takahashi K. and Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676 [DOI] [PubMed] [Google Scholar]
- 5.Li X, Corsa CAS, Pan PW, Wu L, Ferguson D, Yu X, Min J. and Dou Y. (2010). MOF and H4 K16 Acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol 30:5335–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Li L, Pandey R, Byun JS, Gardner K, Qin Z. and Dou Y. (2012). The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell 11:163–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng L. and Zhou MM. (2002). Bromodomain: an acetyl-lysine binding domain. FEBS Lett 513:124–128 [DOI] [PubMed] [Google Scholar]
- 8.Filippakopoulos PKS. (2012). The bromodomain interaction module. FEBS Lett 586:2692–2704 [DOI] [PubMed] [Google Scholar]
- 9.Dawson MA, Kouzarides T. and Huntly BJP. (2012). Targeting epigenetic readers in cancer. N Engl J Med 367:647–657 [DOI] [PubMed] [Google Scholar]
- 10.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, et al. (2012). Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149:214–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, et al. (2010). Selective inhibition of BET bromodomains. Nature 468:1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478:524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delmore Jake E, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146:904–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L. and Sims RJ. (2011). Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA 108:16669–16674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, Robson SC, Chung C-w, Hopf C, et al. (2011). Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478:529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picaud S, Da Costa D, Thanasopoulou A, Filippakopoulos P, Fish PV, Philpott M, Fedorov O, Brennan P, Bunnage ME, et al. (2013). PFI-1, a highly selective protein interaction inhibitor, targeting BET bromodomains. Cancer Res 73:3336–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matzuk Martin M, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno Julio E, Lemieux Madeleine E, Picaud S, Yu Richard N, et al. (2012). Small-molecule inhibition of BRDT for male contraception. Cell 150:673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hooper M, Hardy K, Handyside A, Hunter S. and Monk M. (1987). HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature 326:292–295 [DOI] [PubMed] [Google Scholar]
- 19.O'Neill LP, VerMilyea MD. and Turner BM. (2006). Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nat Genet 38:835–841 [DOI] [PubMed] [Google Scholar]
- 20.Ververis K, Rodd AL, Tang MM, El-Osta A. and Karagiannis TC. (2011). Histone deacetylase inhibitors augment doxorubicin-induced DNA damage in cardiomyocytes. Cell Mol Life Sci 68:4101–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCool KW, Xu X, Singer DB, Murdoch FE. and Fritsch MK. (2006). The role of histone acetylation in regulating early gene expression patterns during early embryonic stem cell differentiation. J Biol Chem 282:6696–6706 [DOI] [PubMed] [Google Scholar]
- 22.Sawan C, Hernandez-Vargas H, Murr R, Lopez F, Vaissière T, Ghantous AY, Cuenin C, Imbert J, Wang Z-Q, Ren B. and Herceg Z. (2013). HAT cofactor trrap maintains self-renewal and restricts differentiation of embryonic stem cells. Stem Cells 31:979–991 [DOI] [PubMed] [Google Scholar]
- 23.Saraiva NZ. (2010). Histone acetylation and its role in embryonic stem cell differentiation. World J Stem Cells 2:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamonica JM, Deng W, Kadauke S, Campbell AE, Gamsjaeger R, Wang H, Cheng Y, Billin AN, Hardison RC, Mackay JP. and Blobel GA. (2011). Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc Natl Acad Sci USA 108:159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu SY, Lee AY, Lai HT, Zhang H. and Chiang CM. (2013). Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 49:843–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drost J, Mantovani F, Tocco F, Elkon R, Comel A, Holstege H, Kerkhoven R, Jonkers J, Voorhoeve PM, Agami R. and Del Sal G. (2010). BRD7 is a candidate tumour suppressor gene required for p53 function. Nat Cell Biol 12:380–389 [DOI] [PubMed] [Google Scholar]
- 27.Herrmann H, Blatt K, Shi J, Rappaport AR, Gleixner KV, Cerny-Reiterer S, Peter B, Wang E, Sperr WR, et al. (2011). Small-molecule inhibition of BRD4 is a novel promising approach to therapeutically target leukemia stem cells in AML. ASH Annual Meeting Abstracts 118:3484 [Google Scholar]
- 28.Lockwood WW, Zejnullahu K, Bradner JE. and Varmus H. (2012). Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci USA 109:19408–19413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz BE, Hofer MD, Lemieux ME, Bauer DE, Cameron MJ, West NH, Agoston ES, Reynoird N, Khochbin S, et al. (2011). Differentiation of NUT midline carcinoma by epigenomic reprogramming. Cancer Res 71:2686–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ware CB, Wang L, Mecham BH, Shen L, Nelson AM, Bar M, Lamba DA, Dauphin DS, Buckingham B, et al. (2009). Histone deacetylase inhibition elicits an evolutionarily conserved self-renewal program in embryonic stem cells. Cell Stem Cell 4:359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.http://p53.free.fr/Database/Cancer_cell_lines/p53_cell_lines.html
- 32.Fazzio TG, Huff JT. and Panning B. (2008). An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 134:162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong X. and Jin Y. (2009). Critical roles of coactivator p300 in mouse embryonic stem cell differentiation and Nanog expression. J Biol Chem 284:9168–9175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loizou JI, Oser G, Shukla V, Sawan C, Murr R, Wang ZQ, Trumpp A. and Herceg Z. (2009). Histone acetyltransferase cofactor Trrap is essential for maintaining the hematopoietic stem/progenitor cell pool. J Immunol 183:6422–6431 [DOI] [PubMed] [Google Scholar]
- 35.Kretsovali A, Hadjimichael C. and Charmpilas N. (2012). Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int 2012:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meno C, Saijoh Y, Fujii H, Ikeda M, Yokoyama T, Yokoyama M, Toyoda Y. and Hamada H. (1996). Left-right asymmetric expression of the TGF beta-family member lefty in mouse embryos. Nature 381:151–155 [DOI] [PubMed] [Google Scholar]
- 37.Kim DK, Cha Y, Ahn HJ, Kim GI. and Park KS. (2013). Lefty1 and Lefty2 control the balance between self-renewal and pluripotent differentiation of mouse embryonic stem cells. Stem Cells Dev 23:457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dreesen O. and Brivanlou AH. (2007). Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev 3:7–17 [DOI] [PubMed] [Google Scholar]
- 39.Wong RCB, Pera MF. and Pébay A. (2008). Role of gap junctions in embryonic and somatic stem cells. Stem Cell Rev 4:283–292 [DOI] [PubMed] [Google Scholar]
- 40.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A. and Weinberg RA. (2008). An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40:499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]