

Abstract
Human pluripotent stem cells (hPSCs) display a very short G1 phase and rapid proliferation kinetics. Regulation of the cell cycle, which is linked to pluripotency and differentiation, is dependent on the stem cell environment, particularly on culture density. This link has been so far empirical and central to disparities in the growth rates and fractions of self-renewing hPSCs residing in different cycle phases. In this study, hPSC cycle progression in conjunction with proliferation and differentiation were comprehensively investigated for different culture densities. Cell proliferation decelerated significantly at densities beyond 50×104 cells/cm2. Correspondingly, the G1 fraction increased from 25% up to 60% at densities greater than 40×104 cells/cm2 while still hPSC pluripotency marker expression was maintained. In parallel, expression of the cycle inhibitor CDKN1A (p21) was increased, while that of p27 and p53 did not change significantly. After 4 days of culture in an unconditioned medium, greater heterogeneity was noted in the differentiation outcomes and was limited by reducing the density variation. A quantitative model was constructed for self-renewing and differentiating hPSC ensembles to gain a better understanding of the link between culture density, cycle progression, and stem cell state. Results for multiple hPSC lines and medium types corroborated experimental findings. Media commonly used for maintenance of self-renewing hPSCs exhibited the slowest kinetics of induction of differentiation (kdiff), while BMP4 supplementation led to 14-fold higher kdiff values. Spontaneous differentiation in a growth factor-free medium exhibited the largest variation in outcomes at different densities. In conjunction with the quantitative framework, our findings will facilitate rationalizing the selection of cultivation conditions for the generation of stem cell therapeutics.
Introduction
Self-renewing human pluripotent stem cells (hPSCs) are characterized by rapid proliferation and a significantly short G1 phase [1]. Stem cell commitment may be triggered by and coincides with lengthening of the G1 phase as hPSCs are more prone to differentiate during this segment of the cell cycle [2–5]. The interplay between specification and proliferation has also been documented in various organs, especially during development [6]. However, the cycle variability of hPSCs in conditions supporting their self-renewal has received less attention. Interestingly, the fractions of hPSCs in the G1 phase reported in different studies are not consistent [1,3,7–9]. Considering that cell cycle duration is intimately linked to proliferation kinetics, the wide range of doubling times (Td; 18 to over 60 h) of hPSCs in many studies [10–12] also exemplifies the variability of proliferation and cycle of self-renewing hPSCs.
One of the potential factors contributing to such discrepancies is the culture density of hPSCs. Density-induced contact inhibition causes cell cycle arrest in epithelial cells [13]. Recently, it was reported that cell cycle changes with local culture density and an increased G1 phase fraction, after exposure to dimethyl sulfoxide (DMSO), lead to higher differentiation efficiency [14]. Yet, the effects of culture density on hPSC cycle and proliferation have been described empirically at best. Typically, a high confluency is suggested for mesoderm differentiation, whereas lower density monolayers favor endoderm commitment more efficiently [15]. As a vehicle for differentiation, embryoid body (EB) cultures also exhibit high local densities [16]. The length of the G1 phase of human embryonic stem cells (hESCs) in differentiating media has been reported to be inversely proportional to the cell density at plating, possibly due to the role of auto/paracrine factors and cell–cell contact [3]. With the assumption of relatively fixed S and G2/M phase durations [17], hPSCs should grow at an accelerated rate as their density increases, but this contradicts experimental observations, that is, that hPSCs at high densities do not grow faster than sparsely plated cells.
Therefore, the link between hPSC culture density, cycle, and proliferation is still unclear. The need for elucidation becomes more compelling due to the routine differentiation of hPSCs as EBs with aggregate size-dependent commitment proclivities [18]. Systems for large-scale cultivation of stem cells are also characterized by high cell densities [19,20]. In this study, we have started to address the association of cell culture density with changes in cycle and proliferation dynamics. For this purpose, the experimental design was coupled to a quantitative model of the evolution of the hPSC population dependent on density-linked changes in hPSC proliferation and the state of pluripotency. We addressed the following questions: (a) How does the proliferation rate of self-renewing hPSCs change with culture density? (b) Are there concomitant changes in the expression of pluripotency markers and cycle regulators? (c) Can the propensity of hPSCs for commitment under different culture conditions (mainly media types) be quantified?
Our findings show a clear correlation between hPSC culture density and cycle dynamics. Cells at higher densities lengthen the time they spend in the G1 phase concomitant with reduction in NANOG expression and become more susceptible to (even aberrant) differentiation, while a fraction of cells at high density also enter quiescence (G0). The increase in G1 duration is accompanied by an increase in the cyclin-dependent kinase inhibitor 1A (p21). The differentiation strength of a particular medium on cultured hPSCs is predicted through quantitative analysis and validated with experiments. This type of framework will be essential both for better understanding of the hPSC cycle regulation and for the rational design of hPSC commitment strategies and pertinent processes for producing therapeutic progeny.
Materials and Methods
hPSC culture
The hESC lines, H9 and H1 (passages 30–60), and the human induced pluripotent stem cell (hiPSC) line IMR90 [iPSC(IMR90)-4; passage 40–70] were obtained from the WiCell Research Institute (Madison, WI). Cells were cultured in dishes coated with Matrigel (BD Biosciences, San Jose, CA) and in the mTeSR1 medium (StemCell Technologies, Vancouver, BC). StemPro (hESC serum-free medium) (Life Technologies, Grand Island, NY) and mouse embryonic fibroblast-conditioned medium (CM) [21] were also used as stated. The cultures were maintained in 5% CO2/95% air at 37°C. The medium was replaced every day, and the cells were passaged every 5–6 days by enzymatic dissociation with dispase (Life Technologies). After dispase treatment, 50 μL of the cell suspension was set aside and incubated with 1 mL of TrypLE to generate a suspension of single dispersed cells for enumeration (see also section “Determination of hPSC culture density”).
Flow cytometry
Cells on Matrigel-coated dishes were dissociated with Accutase (Innovative Cell Technologies, San Diego, CA) and collected by centrifugation at 200 g for 5 min. Cells were fixed for 10 min with 3.7% paraformaldehyde solution (Sigma-Aldrich, St. Louis, MO), washed with phosphate-buffered saline (PBS; Mediatech, Manassas, VA), and permeabilized with Cytonin (Trevigen, Gaithersburg, MD) for 1 h before blocking with 5% normal donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h. Then, cells were incubated for 1 h at room temperature with monoclonal-conjugated antibodies against Nanog, Ki-67 (both from BD Biosciences), p21, p27, and phospho-p53 (Ser37) (all three from Cell Signaling Technology, Danvers, MA). Samples with appropriate isotype control antibodies (BD Biosciences) were included. Data were acquired on a FACS Calibur flow cytometer with the CellQuest software (BD Biosciences) and analyzed with the Flow Express V4.0 suite (De Novo Software, Los Angeles, CA). Quantum PE MESF beads (Bangs Laboratories, Fishers, IN) were used for calibration.
Immunocytochemistry
Cells plated on glass slides were fixed with 4% paraformaldehyde (Sigma-Aldrich) in PBS and blocked/permeabilized in PBS with 0.1% Triton X-100 (Mallinckrodt Baker, Phillipsburg, NJ) and 1% bovine serum albumin for 30 min. Samples were incubated overnight at 4°C with primary antibodies. After three washes with PBS, cells were incubated with appropriate secondary antibodies for 1 h at room temperature. Donkey anti-rabbit and -mouse secondary antibodies conjugated with Dylight 488 or 549 (Jackson ImmunoResearch Laboratories) were used. Nuclear DNA was stained with Hoechst 33342 (Life Technologies). The mounting medium was added (Life Technologies), and the slides were sealed with coverslips. Fluorescence images were acquired with a Zeiss Observer Z1 fluorescence microscope with an AxioCam MRm camera (Carl Zeiss, Thornwood, NY) and processed with the AxioVision software.
Cell cycle analysis
Cells were stained with propidium iodide (PI), examined by flow cytometry, and the acquired histogram data were analyzed for cell cycle statistics. Specifically, hPSCs were dissociated with Accutase, immediately fixed in ice-cold 70% ethanol for 1 h, and then washed twice with PBS before staining with PI/RNase (Trevigen) for 30 min and analysis by flow cytometry. The distribution of cells in the cell cycle was determined with the Multicycle module of the Flow Express 4.0 suite.
Alternatively, hPSCs were incubated with EdU solution (Life Technologies) for 30 min and then with Click-iT fixative for 15 min. The Click-iT reaction cocktail was added to label cells that underwent DNA synthesis during incubation. Then, PI was added for total DNA staining. Cells not treated with EdU served as controls for gating the EdU+ population and the cell cycle data analysis was carried out with the FCS Express 4.0 suite.
Reverse transcription–polymerase chain reaction and quantitative polymerase chain reaction
Total RNA was isolated using Trizol (Life Technologies) according to the manufacturer's instructions, and reverse transcription was performed using the ImPromII reverse transcriptase (Promega, Madison, WI).
Quantitative polymerase chain reaction (qPCR) was performed on a Bio-Rad CFX96 (Bio-Rad, Hercules, CA) machine using the Dynamo qPCR Mix (Thermo Scientific, Waltham, MA) for 40 cycles with primers (Integrated DNA Technologies, Coralville, IA) listed in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/scd). All reactions were run in triplicates with samples from five independent experiments. Amplification specificity was verified by the melting curve method and gel electrophoresis. Relative gene expression was calculated by normalizing to the expression of endogenous β-actin using the ΔΔCt method [22]. The Ct for the housekeeping gene did not vary under different experimental conditions when equal amounts of RNA were used.
Determination of hPSC culture density
The hPSC culture densities were measured by both cell counting and microscopic image analysis. For endpoint cell density measurement, the density was measured with a TC20 cell counter (Bio-Rad). During differentiation, light microscopy images were taken every day and analyzed by a MATLAB module developed in our laboratory for confluency analysis. At least 15 images per 10 cm2 of culture surface were acquired from randomly selected regions. A linear calibration curve was also constructed to convert confluency (%) to density (×104 cells/cm2). Of note, we determined the upper limit for confluency (100% of the surface covered by a cell monolayer) to be around 60×104 cells/cm2.
hPSC differentiation
Four types of media were used to induce hPSC differentiation: unconditioned medium (UM: consisting of 80% Dulbecco's modified Eagle's medium/F12, 20% Knockout serum replacer [Life Technologies], 1 mM l-glutamine, 1% nonessential aminoacids, 0.1 mM β-mercaptoethanol [12]), UM with 100 ng/mL activin A (R&D Systems, Minneapolis, MN), CM with 5 ng/mL BMP4 (R&D Systems), and growth factor-free mTeSR medium (mTeSRGF−) (StemCell Technologies) with 5 ng/mL BMP4. Specifically, hPSCs were seeded on Matrigel-coated dishes at split ratios of 1:1 to 1:10. The culture densities were monitored every day by image analysis. Induction of differentiation started when the culture density reached a certain level designated as low (<10×104 cells/cm2), medium (10–20×104 cells/cm2), and high (>20×104 cells/cm2). Differentiation medium was replaced daily.
Quantitative model for hPSC cycle, proliferation, and differentiation
A model of two populations balance equations (PBEs) was developed to simulate hPSC proliferation and differentiation. Cells were categorized into two types: PSCs (ie, hPSCs), which were NANOG+ (Eq. 1), and NANOG− cells (Eq. 2), which either lost their pluripotency or were primed for commitment. The latter type was termed primed cells.
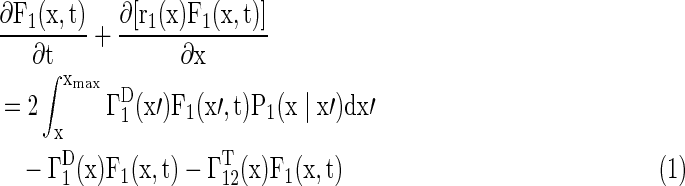 |
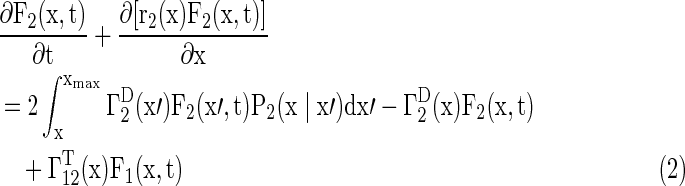 |
where x=[v, τ]T with v and τ being the cell size and age, respectively.
F1(x,t) represents the hPSC cell number with cell property x, and F2(x,t) represents the primed cell number with property x. Determination of the initial F1(x,t) and F2(x,t) was carried out by cell counting or image analysis (total cultured cell density in cells/cm2) and by flow cytometry (for the NANOG+ and NANOG− cell fractions). Flow cytometry was also employed to obtain the cell size (v) distributions. The rates of growth [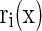 ], division [
], division [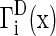 ], and partitioning [
], and partitioning [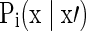 ] are described in the Supplementary Materials (see also Supplementary Table S4).
] are described in the Supplementary Materials (see also Supplementary Table S4).
The doubling time Td and the length of the G1 phase (TG1) in low and high culture densities were measured from experiments (growth curves; Supplementary Fig. S7). We have previously determined the parameter values of the partitioning function for hESCs [23]. To account for the varying division rates with culture density in the model, the Td value was switched from the low to the high-density Td (Table 1) when the culture density reached a critical threshold.
Table 1.
Summary of Td and TG1 Values
| Parameter name | Low-density condition (h) | High-density condition (h) |
|---|---|---|
| Td (hPSC) | 22.7±4.0 | 244.2±49.5 |
| Td (primed) | 26.4±2.9a | 244.2±49.5b |
| TG1 (hPSC) | 8.0a | 48.0 |
| Threshold density | 50×104/cm2 | |
The values were measured in this study, except as indicated: afrom Seguin et al. [10]; bassumed the same value as for hPSCs at high density. Details about the G1 phase length determination can be found in the Supplementary Methods.
hPSC, human pluripotent stem cell.
In addition, the differentiation of hPSCs into primed cells was described by a differentiation rate 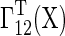 . Stem cell fate decisions transpire during the G1 phase [2,24,25], and thus we considered that no differentiation occurs in the S and G2/M phases by setting the rate to zero. In contrast, the specification rate while in G1 was assumed to follow zeroth order kinetics (kdiff):
. Stem cell fate decisions transpire during the G1 phase [2,24,25], and thus we considered that no differentiation occurs in the S and G2/M phases by setting the rate to zero. In contrast, the specification rate while in G1 was assumed to follow zeroth order kinetics (kdiff):
 |
where TG1 is the length of the G1 phase.
The PBE system was computed by a Monte Carlo method [23,26] based on the calculation of the interval of quiescence interrupted by cell division or differentiation events simulated as Markov processes. The culture density was updated during each quiescence interval and, once it exceeded a certain threshold, the Td was increased to account for slower proliferation and division. The same framework was implemented to screen ranges of initial densities and kdiff values for the calculation of the fractions of NANOG+ and NANOG− (loss of pluripotency) cells after 4 days of culture (see Supplementary Materials). Alternatively, simulation was run with fixed initial density at 6×104 cells/cm2, which is a typical passage density for hPSCs, and kdiff ranging between 10−3 and 10−1 h−1 for the calculation of NANOG+ cell fractions. The results were used to lookup the kdiff value corresponding to each experiment from the initial density and day 4 NANOG+ cell fraction for cultures of different hPSC lines and media. Final kdiff values were calculated using leave-one-out cross-validation (LOOCV) [27]. Training and testing errors were calculated as the mean squared error between model-predicted (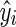 ) and experiment-derived NANOG+ cell fractions (yi) from n sets (independent experiments):
) and experiment-derived NANOG+ cell fractions (yi) from n sets (independent experiments):
 |
Standard errors were used to define the 95% confidence interval (CI) for kdiff and the corresponding 95% CI of the predicted differentiation outcome. FORTRAN 90 code with embedded Intel MPI directives was written for all calculations. More details are provided in the Supplementary Materials.
Statistical analysis
Data are expressed as mean±standard deviation unless stated otherwise. Analysis of variance and the post-hoc Tukey test were performed using Minitab (Minitab, Inc., State College, PA). P values <0.05 were considered as significant.
Results
hPSC proliferation rate correlates inversely with culture density
We first investigated the proliferation kinetics of self-renewing hPSCs cultured in Matrigel-coated dishes. In CM, StemPro, and mTeSR media (Fig. 1A), which are typically used in hPSC maintenance, cells proliferated with a Td of 22.6±4 h over multiple 5-day passages (Supplementary Table S2). However, when the cells were cultivated longer between splittings, their growth decelerated significantly, especially after reaching∼50×104 cells/cm2 (Fig. 1B). Even when cells were cultivated in twice the volume of the medium typically used (Supplementary Fig. S1), the proliferation curve did not change, excluding the possibility of slower growth caused by medium depletion. Note that the initial dip in density (24 h) was due to the <100% seeding efficiency (see also Supplementary Table S3). Initial seeding at a higher density (∼30×104 cells/cm2) confirmed that the slower proliferation was density dependent rather than time dependent (Supplementary Fig. S1). After 4 days at high density (days 5–9) without passaging, the fraction of NANOG+ cells dropped from over 80% to∼50% (P<0.05) (Fig. 1C).
FIG. 1.

Proliferation of cultured human pluripotent stem cells (hPSCs) slows down with increasing density. (A) Five-day growth of hPSCs in different media. (B) Proliferation and viability of hPSCs in the mTeSR medium over 8 days of culture. The Td in the exponential phase was calculated based on four independent experiments. (C) Determination of the NANOG+ hPSC fraction by flow cytometry at different times from at least three independent experiments. (D) Expression of NANOG and Ki67 by cultured hPSCs at different densities and times. *P<0.05 compared to all other conditions. Color images available online at www.liebertpub.com/scd
The expression of NANOG was monitored in conjunction with that of Ki-67, which marks proliferative cells (Fig. 1D). Almost 98% of hPSCs from cultures with <50×104 cells/cm2 were Ki67+. In contrast, denser cultures exhibited about 14.2±2.8% of Ki-67− cells after 4 days, pointing to the emergence of a quiescent (G0) subpopulation. This was corroborated by EdU pulse experiments revealing that ∼10% of the day 9 cells were not entering the S phase even after 48 h (data not shown). Moreover, both Ki-67−/NANOG− and Ki-67+/NANOG− cell populations were detectable by flow cytometry, while there were no Ki-67−/NANOG+ cells, suggesting that pluripotent hPSCs did not enter a quiescent state. It should be noted that other markers (eg, TRA-1-60 and TRA-1-81) also exhibited a similar profile as NANOG (Supplementary Fig. S2), supporting the selection of the latter as a representative marker of the pluripotent state of hPSCs.
Taken together, these findings show that hPSCs proliferated rapidly until their density reached a certain threshold (50×104/cm2 for H9 cells). Beyond this, proliferation slowed down along with loss of NANOG expression and entry into a quiescent state even in the self-renewal medium. hPSCs also became more heterogeneous with the appearance of three subpopulations based on the expression of Ki-67 and NANOG.
Increased culture density results in the extended G1 phase and higher p21 expression
Given the significant changes in proliferation kinetics with culture density, we next investigated the hPSC distribution in the different cycle phases. The NANOG+ fraction was over 75% in the cultures we examined, excluding confounding results from differentiating cells. hPSCs cultured under various densities were analyzed after PI staining (Fig. 2A, B) and/or EdU treatment (Fig. 2C, D) to ensure data consistency. At 10–20×104 cells/cm2, <30% of the hPSCs were in the G1 phase compared with 50–60% at densities greater than 40×104 cells/cm2 (Fig. 2E, F, P<0.05). Since the S and G2 phases are considered to have relatively fixed durations [17], the increase of G1 cell fractions suggested that the slower growth under high density was due to a lengthened G1 phase.
FIG. 2.

Cell cycle analysis of hPSCs at different culture densities. Analysis was performed by (A, B) propidium iodide (PI) or (C, D) EdU staining at low (<20×104 cells/cm2) and high (>40×104 cells/cm2) densities. (E) Scatter plot of the fractions of cells corresponding to the G1 phase versus culture density. (F) The fraction of the G1 phase cells increases with the culture density (n≥3). *P<0.05 compared to all other conditions. Color images available online at www.liebertpub.com/scd
Therefore, the reduced proliferation rate was attributed to both changes in the duration of the cell cycle and the emergence of differentiated and quiescent cells as shown earlier (Fig. 1).
It has been reported that both CDK2 and CDK4 are required for hPSCs to enter the S phase [1,7] and overexpression of cyclin-dependent kinase inhibitors 1A (CDKN1A; p21) and 1B (CDKN1B; p27) leads to a protracted G1 phase [28,29]. Besides, the cell cycle regulator p53 (TP53) induces differentiation of hPSCs [30,31]. When measured by qPCR on day 5 or 6, CDKN1A expression increased fivefold at densities above 50×104 cells/cm2 (P<0.05) compared with that in cells from more sparse cultures (<20×104 cells/cm2) (Fig. 3A), while CDKN1B and TP53 did not vary significantly. Moreover, POU5F1 and NANOG levels were similar, suggesting that the extension of the G1 phase happened before the loss of pluripotency observed on day 9 (Fig. 1C). The upregulation of p21 was further confirmed by western blotting, immunocytochemistry, and flow cytometry analysis (Fig. 3B–D). The fractions of CDKN1A+ in low and high densities showed a difference of 14%, but no differences were noted in CDKN1B and phosphorylated (Ser37) TP53 (Fig. 3B and Supplementary Fig. S3), indicating that p21 was activated in a p53-independent manner [32].
FIG. 3.

Expression of hPSC pluripotency markers and cycle regulators at different densities. Human embryonic stem cells (hESCs) were cultured in the mTeSR medium at low (<30×104 cells/cm2) or high density (>40×104 cells/cm2). (A) Expression profile of POU5F1, NANOG, CDKN1A (p21), CDKN1B (p27), and (TP53) p53 from five independent experiments. *P<0.01 compared to low-density (LD) expression of p21. (B) Western blotting analysis of p21, p27, and phospho-p53 expression in confluent HEK293T cells (HEK), LD and high-density (HD) cultured H9 hESCs. (C) Immunostaining for p21 and NANOG. Nuclear DNA staining is also shown. Bar: 100 μm. (D) NANOG and p21 expression probed by flow cytometry. Fractions of p21+ are shown in the plots with the gate set by using isotype controls. Color images available online at www.liebertpub.com/scd
The differentiation propensity of hPSCs increases at higher culture densities
The early G1 phase is considered to be the period during which hPSCs are responsive to differentiation signals [2,24]. We therefore hypothesized that culturing hPSCs at high density would prolong their G1 phase, enhancing their propensity for commitment in the presence of appropriate stimuli. To better monitor and quantify the density of hPSCs cultures over time, we developed a software module for automated acquisition of culture confluency data (%) and determination of density (cells/cm2) as shown in Fig. 4A and Supplementary Fig. S4. This tool enabled on-line measurements of culture density without the need for cell dissociation. Use of the module eliminated errors associated with density estimation from split ratios with seeding efficiencies varying between 20% and 50% (Supplementary Table S3).
FIG. 4.

Effect of initial culture density on the commitment of hPSCs. (A) Estimation of the culture confluency (%) through analysis of images from phase-contrast light microscopy. Bar: 200 μm. (B) Tracking of confluency changes over time in unconditioned medium (UM) and mTeSR media for H9 hESCs. (C) Fractions of NANOG+ hPSCs after culture in UM for 4 days at different initial densities for H9 and H1 hESCs and IMR90 human induced pluripotent stem cells (hiPSCs). For each cell line, NANOG+ fractions were measured for low (<10×104/cm2), middle (10–20×104/cm2), and high initial densities (>20×104/cm2). The percentages of NANOG+ hPSCs in the mTeSR medium are also shown. Results are from at least three independent experiments. *P<0.05; **P<0.05 compared to all other conditions. (D) Box plot illustrating the variability in NANOG+ cell fractions after 4 days in UM. Results are shown for different hPSC lines and initial densities. (E) Four-day culture increase of the total cell number cultured in UM. Data from all three lines are plotted together along with a curve representing a power-law fit. Color images available online at www.liebertpub.com/scd
Spontaneous differentiation of hPSCs was induced by incubation with UM for 4 days. Human H9 cells exhibited similar proliferation and viability characteristics as in the mTeSR medium (Fig. 4B). Compared with hPSCs under self-renewal conditions, the NANOG+ cell fraction at all initial densities showed a significant drop after 4 days in the UM (P<0.05) (Fig. 4C). More importantly, the percent of NANOG- cells after 4 days was greater in hPSCs at initial densities over 20×104/cm2 compared with sparser cultures (P<0.05). The distribution of NANOG expression was bimodal in all measurements attesting to the consistency and accuracy of the data (Supplementary Fig. S5).
Similar findings were obtained for other hESC (H1) and hiPSC (IMR90) lines confirming the density effect on differentiation propensity (Fig. 4C). Statistical analysis revealed lower variations in differentiation efficiencies when the density variation was more limited (Fig. 4D). In addition, the 4-day increase in cell number was inversely related to the initial culture density (Fig. 4E). Taken together, these findings support that higher culture densities lead to reduced hPSC proliferation and enhanced propensity for differentiation.
Quantitative model of hPSC proliferation and commitment
We constructed a quantitative model in combination with our experiments to gain further insight into the differentiation proclivity of cells in heterogeneous hPSC populations under various contexts [23] (Fig. 5A). Two cell types were considered: Pluripotent hPSCs and cells primed for commitment based on NANOG expression. The proliferation kinetics of each subpopulation were determined by the density-dependent rate parameters k1 and k2, respectively. Transition of hPSCs into primed cells was quantified by a kdiff and occurred only in the G1 phase (cell age <G1 phase duration). The kdiff represents the differentiation-inducing strength of a particular condition.
FIG. 5.

Quantitative analysis of hPSC cycle progression, proliferation, and commitment. (A) Schematic of cell cycle regulation in conjunction with culture density, proliferation, and differentiation. Cells were categorized as undifferentiated (hPSCs) and primed for commitment proliferates with specific doubling times. Cells in G1 may transition into primed cells with a rate kdiff (B, C). Model simulations run for kdiff ranging from 1×10−3 h−1 to 1×10−1 h−1 and different (B) initial densities or (C) time points. In (B) the NANOG+ cell fractions were captured at day 4, whereas in (C) the initial (seeding) density was fixed at 6×104/cm2. Color images available online at www.liebertpub.com/scd
The model was first employed to screen differentiation outcomes at a wide range of kdiff (10−3–10−1 h−1) at different initial densities and time points. The model output was the NANOG+ cell fraction for 4-day cultures (Fig. 5B, C). Cultures with low initial densities (<10×104/cm2) exhibited higher NANOG+ cell fractions in agreement with experimental data (Fig. 5B). Higher kdiff values corresponded to more prominent differentiation (lower NANOG+ cell percentages), whereas maintenance of hPSC pluripotency was linked to low kdiff (Fig. 5C).
With the data mapping the NANOG+ cell fraction to the kdiff and initial density domains, we proceeded to predict the kdiff values of media commonly used in hPSC culture. Estimation of kdiff was performed for particular initial culture densities and NANOG+ cell fractions. Moreover, the data were divided into training and testing sets. As shown in Fig. 6, values of kdiff calculated from the training set can be used in the prediction of the differentiation outcomes based on the testing data. It should be noted that H1 hESCs showed higher initial NANOG+ fractions (Fig. 4C), suggesting potentially different kinetics from the H9 and IMR90 cells. In fact, the latter two hPSC lines exhibited similar parameter values in UM (Fig. 6A), and their differentiation propensity was the highest (of all conditions tested) in 5 ng/mL BMP4. Although studies showed that BMP4 in CM and mTeSRGF− induced commitment into disparate lineages [12,33], the kdiff values were similar. Taken together, these findings demonstrate that the framework developed in this study coupled with information from experiments allows the prediction of the differentiation strength (kdiff) of various culture conditions typically used for stem cells.
FIG. 6.

Comparison of model and experimental results for differentiation outcomes at different initial densities. (A) Results are shown for 4-day cultures for H9 hESCs and IMR90 hiPSCs, and H1 hESCs in UM, as well as for H9 hESCs in conditioned medium (CM)+5 ng/mL BMP4 or mTeSRGF−+5 ng/mL BMP4. Both the model predictions and 95% confidence interval (CI) of prediction are shown. Detailed statistics from model training and testing are provided in Table 2. (B) Differentiation outcome variation caused by culture densities in different types of media. The variation was quantified as the difference of NANOG+ hPSC fractions between low and high densities on day 4. The types of media tested are shown. Horizontal error bars represent the standard error of kdiff for each condition.
For the media employed in our study, the kdiff values are summarized in Table 2 and training and testing errors are shown from LOOCV pertaining to the accuracy of the model used in our analysis. As expected, the UM, which is typically utilized for spontaneous differentiation of hPSCs had a kdiff between those of media for self-renewing hPSCs (mTeSR) and directed differentiation (CM+BMP4 and mTeSRGF−+BMP4). The kdiff of the UM with 100 ng/mL Activin A fell between those for mTeSR and UM alone. This may be because Activin A not only causes differentiation to the endoderm [15,21] but is also shown to promote self-renewal [34]. The lowest kdiff was calculated for the mTeSR medium, which is typically used for hPSC maintenance. In contrast, the highest kdiff was associated with media supplemented with BMP4, in which commitment was readily seen. Calculations of kdiff were also performed in scalable microcarrier bioreactor cultures of hPSCs [35], demonstrating the wider applicability of our approach. Based on data obtained from IMR90 hiPSCs cultured on peptide-conjugated microcarriers in stirred-suspension bioreactors [35], the kdiff value was low (mTeSR medium) and similar to that for two-dimensional (2D) static cultures.
Table 2.
Summary of Calculated kdiff Values (h−1)
| Medium type | Cell line | kdiff ×102 (h−1) | Training/testing error (×102) |
|---|---|---|---|
| UM | H9 and IMR90 | 1.86±0.20 | 1.09/1.19 |
| UM | H1 | 1.35±0.17 | 0.99/1.22 |
| mTeSR | H9 | 0.22±0.029 | 1.05/1.21 |
| CM+5 ng/mL BMP4 | H9 | 2.87±0.33 | 0.98/1.09 |
| mTeSRGF−+5 ng/mL BMP4 | H9 | 2.86±0.31 | 0.91/0.98 |
| UM+100 ng/mL AA | H9 | 0.69±0.34 | N/A |
| mTeSR (bioreactor)a | IMR90 | 0.20±0.070 | N/A |
Listed values are shown as the mean±standard error from multiple simulations (n≥3). LOOCV was used to calculate the training and testing errors, which were mean squared errors between model predictions and the experimental data.
IMR90 hiPSCs were cultured on peptide-conjugated microcarriers in stirred-suspension bioreactors [36].
UM, unconditioned medium; CM, conditioned medium; N/A, data not available; LOOCV, leave-one-out cross-validation; hiPSCs, human induced pluripotent stem cells.
Given the effects of the culture density on hPSC commitment, the PBE model was further used to predict the change in the differentiation outcome by varying the culture density along the full range of kdiff (10−3–10−1 h−1). A plot of the variation in density-induced differentiation versus kdiff (for relevant media) is shown in Fig. 6B. The fluctuation was minimal (<10%) at low and high kdiff values, but peaked (>20%) for values around 10−2 h−1. Cells in the UM (spontaneous commitment) or UM+AA were more sensitive to density effects than in media supporting self-renewal (mTeSR) or directed specification (CM+BMP4 and mTeSRGF−+BMP4). This finding may be explained by the competing relationship between proliferation and differentiation. At the extremes of the kdiff range proliferation or differentiation became dominant. When the kinetics of proliferation and commitment were comparable, the differentiation outcomes showed higher fluctuations with changes in culture conditions (culture density, time, etc.).
Because the simulation results above were based on measured Td and G1 phase duration, scenario analysis was performed to show the effect of these variables on specification. Changes in either the Td or G1 length under high-density conditions did not alter the outcome to a significant extent, especially for the first 3 days (Supplementary Fig. S6). In our simulations, the marginal effect from the increase in G1 duration was diminished beyond a G1/Td of 30% (with Td for high-density cultures). In fact, we and others [14,25,29] have not observed less than 20% of self-renewing hPSC populations in the G1 phase. Hence, the model is robust to the variations in the Td and G1 phase duration.
Discussion
The distinguishing features of stem cell cycle have attracted much attention recently, especially in connection with hPSC fate specification. Through experimental evidence coupled with quantitative analysis, we showed that hPSC cycle progression is linked to culture density. At higher densities, a larger fraction of hPSCs resides in the G1 phase and is more prone to differentiation. Recent findings [25] corroborate ours on the lengthening of the G1 phase suppressing hPSC self-renewal and leading to more extensive differentiation. Hence, controlling, in part, the propensity for commitment through modulation of the cell cycle may be an appealing alternative to growth-arresting agents, such as nocodazole and DMSO, which often induce aberrant differentiation and apoptosis [8,36]. Cells at a low or high density display distinct profiles of the CDK inhibitor p21 (CDKN1A), warranting further investigation into its participation in molecular mechanisms regulating self-renewal or commitment. The associated model framework allowed the quantification of the effect of the culture conditions (mainly medium and culture density) on the hPSC differentiation proclivity.
Our findings support the notion that fast hPSC self-renewal is facilitated by low culture density. However, sparsely seeded cells (typically <2×104/cm2) may not form colonies of appropriate size leading to loss of pluripotency (at least in 2D cultures) [5]. Moreover, the presence of fast-proliferating hESC variants in cultures of normal hPSCs has been reported [37,38]. Both hPSC types share similar properties, but the fast-growing subpopulation is less or not responsive to differentiation cues and its emergence is concomitant with changes in cell cycle regulators (eg, CDK2, CKD4).
Stem cell subpopulations were characterized as self-renewing and primed based on the expression of NANOG. As a safeguard of hPSC pluripotency [39–41], NANOG is more sensitive to differentiation stimuli, in part, due to its rapid degradation (half-life ∼2 h [42,43]) potentially facilitated by a PEST motif identified toward the N-terminus of the protein [42]. Analysis by flow cytometry revealed that NANOG+ cells were POU5F1+ (OCT4+), but not all OCT4+ cells were NANOG+ (data not shown). However, whether NANOG- cells are differentiated is still unclear. For instance, subpopulations of Nanog− mouse embryonic stem cells (mESCs) revert to a Nanog+ state [44]. Allelic regulation of Nanog [26,45], which is debatable [46,47], may also contribute to Nanog fluctuations. Whether these findings can be directly translated to human stem cells is still an open question. Nevertheless, Nanog- cells exhibit significantly dissimilar phenotypes from Nanog+ cells [48] and respond differently to specification factors [44,49]. Consequently, in this study, we considered Nanog- cells as primed for differentiation.
A fraction of NANOG− cells was also found to be Ki67−, indicating a state of quiescence (Fig. 1). NANOG+ cells are copositive for Ki-67, and only when the NANOG expression is substantially reduced (NANOG−), cells may become Ki-67−. Adult stem cells can reside in the quiescent state for prolonged periods of time [50]. Our findings show that hPSCs maintain an active cell cycle and quiescence (G0) only occurs in NANOG- cells. If these hPSCs can return to a pluripotent state then a parallel can be drawn with their adult counterparts. However, if loss of NANOG denotes irreversible withdrawal from pluripotency, then entering G0 before hPSC commitment seems implausible. These issues in conjunction with the exact nature of the NANOG- hPSC population require detailed analysis.
In our experiments, the fraction of NANOG+ cells dropped from ∼80% to ∼50% when hPSCs were maintained at a high density (>50×104 cells/cm2) for 4 days. Becker et al. [3] suggested that hESCs are primed to go through two successive rounds of cell division and enter a second S phase in the absence of external factors. This short-term autonomous cell cycle progression for hESCs may explain why a significant reduction in the NANOG+ cell fraction is not seen before 4 days of culture. Of note, significant changes in the transcriptional regulatory network associated with pluripotency and a fate switch are not observed for 3–5 days after the doxycycline-regulated reduction of Nanog expression in mESCs [51].
Among the CDK inhibitors, p15 and p16 induce lengthening of the average G1+G2+M duration when overexpressed in H9 hESCs [29]. Interestingly, overexpression of p21 leads to an increase in the G1 fraction, permanent cell cycle arrest, and differentiation with concomitant loss of OCT4 expression. These findings are aligned with our observations of p21 upregulation accompanying elongation of the G1 phase at high culture densities. In addition, p21 expression increased with no changes in p53, indicating that elevated p21 may be more relevant to stem cell differentiation [52]. Similar to the effect of high culture density, 1% DMSO enhances the portion of human stem cells in G1 favoring differentiation [14]. The underlying mechanism is still unclear, but DMSO treatment of hybridoma B cells augments the expression of p21 causing early G1 growth arrest [53]. Taken together, p21 is a candidate factor for the control of cycle progression in hPSCs, although further investigation is necessary about its role in fate specification.
The development of our quantitative model aimed at rationalizing the effects of cell proliferation and cycle variability on hPSC expansion and differentiation. For this analysis, the cell doubling time (Td) and the duration of the G1 phase (TG1) at low (rapid growth phase) and high (decelerated growth) densities can be determined from experiments. The PBE framework is especially suitable for capturing single-cell events and traits (eg, division, gene expression, position in the cell cycle) and their effects on the asynchronous hPSC population. Ensemble-wide (eg, ordinary differential equation-based) models are simpler than PBEs [54,55], but do not afford high enough resolution to reflect stem cell heterogeneity. Gene regulatory network modules, which focus on single-cell phenotypic changes and not on population-level changes [56], can be embedded in the PBEs [57]. Such modules may be used to describe p21 interactions pertaining to the hPSC cycle regulation as suggested by our findings.
Other model extensions can also be considered, including the addition of terms, to account for cell death or apoptosis. Indeed, the basal media used in this study support cell survival and maintenance, but factors employed in directed differentiation of hPSCs have been linked to significant cell death, especially at the onset of commitment. Activin A (100 ng/mL) not only supports hPSC pluripotency [34] but also induces endoderm differentiation [15,21]. This divergent role of Activin A has been attributed to tuning of signals from pathways such as PI3K [58,59] and Smad2/3 [34,60]. To that end, lineage specification can be incorporated in the model. Although we categorized cells into PSCs and cells primed for commitment, the latter can be further divided into particular phenotypes. Of course, this will require information on lineage markers, but such models will facilitate the standardization and optimization of hPSC culture methods depending on the intended application. For instance, a PBE model accounting for the hPSC growth and commitment in heterogeneous populations may provide insights on strategies for optimizing the purity and biomass (cell number) of stem cell therapeutics in bioprocesses.
In fact, the kdiff was calculated for IMR90 hiPSCs in a microcarrier stirred-suspension culture, illustrating the wider applicability of the presented methodology. The density-dependent proliferation kinetics can be easily translated to a microcarrier system, based on the number of cells per surface area, allowing for the simulation of bioreactor cultures with respect to different seeding densities and time intervals. However, the use of the same framework for cultures of hPSCs as aggregates [19] or after encapsulation [61] requires additional considerations. Aggregate cultures exhibit high local densities with extensive cell–cell rather than cell–substratum contacts within a complex three-dimensional culture environment with variations in fluid shearing stresses and nutrient, O2, and factor concentrations. Many of the same considerations apply as well to in vivo niches hosting stem and progenitor cells. Application of PBE models to such environments [62] is an interesting topic of significant practical value.
Supplementary Material
Acknowledgment
Funding support has been provided by the National Institutes of Health (NHLBI, R01HL103709) to E.S.T.
Author Disclosure Statement
All authors declare that no competing financial interests exist.
References
- 1.Becker KA, Ghule PN, Therrien JA, Lian JB, Stein JL, van Wijnen AJ. and Stein GS. (2006). Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J Cell Physiol 209:883–893 [DOI] [PubMed] [Google Scholar]
- 2.Salomoni P. and Calegari F. (2010). Cell cycle control of mammalian neural stem cells: putting a speed limit on G1. Trends Cell Biol 20:233–243 [DOI] [PubMed] [Google Scholar]
- 3.Becker KA, Stein JL, Lian JB, van Wijnen AJ. and Stein GS. (2010). Human embryonic stem cells are pre-mitotically committed to self-renewal and acquire a lengthened G1 phase upon lineage programming. J Cell Physiol 222:103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filipczyk AA, Laslett AL, Mummery C. and Pera MF. (2007). Differentiation is coupled to changes in the cell cycle regulatory apparatus of human embryonic stem cells. Stem Cell Res 1:45–60 [DOI] [PubMed] [Google Scholar]
- 5.Sela Y, Molotski N, Golan S, Itskovitz-Eldor J. and Soen Y. (2012). Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of retinoblastoma protein. Stem Cells 30:1097–1108 [DOI] [PubMed] [Google Scholar]
- 6.Fuchs E. (2009). The tortoise and the hair: slow-cycling cells in the stem cell race. Cell 137:811–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neganova I, Vilella F, Atkinson SP, Lloret M, Passos JF, von Zglinicki T, O'Connor JE, Burks D, Jones R, Armstrong L. and Lako M. (2011). An Important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells 29:651–659 [DOI] [PubMed] [Google Scholar]
- 8.Kallas A, Pook M, Maimets M, Zimmermann K. and Maimets T. (2011). Nocodazole treatment decreases expression of pluripotency markers Nanog and Oct4 in human embryonic stem cells. PloS One 6:e19114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Neganova I, Przyborski S, Yang CB, Cooke M, Atkinson SP, Anyfantis G, Fenyk S, Keith WN, et al. (2009). A role for NANOG in G1 to S transition in human embryonic stem cells through direct binding of CDK6 and CDC25A. J Cell Biol 184:67–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seguin CA, Draper JS, Nagy A. and Rossant J. (2008). Establishment of endoderm progenitors by SOX transcription factor expression in human embryonic stem cells. Cell Stem Cell 3:182–195 [DOI] [PubMed] [Google Scholar]
- 11.Melkoumian Z, Weber JL, Weber DM, Fadeev AG, Zhou YE, Dolley-Sonneville P, Yang JW, Qiu LQ, Priest CA, et al. (2010). Synthetic peptide-acrylate surfaces for long-term self-renewal and cardiomyocyte differentiation of human embryonic stem cells. Nat Biotechnol 28:606–610 [DOI] [PubMed] [Google Scholar]
- 12.Xu RH, Peck RM, Li DS, Feng XZ, Ludwig T. and Thomson JA. (2005). Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat Methods 2:185–190 [DOI] [PubMed] [Google Scholar]
- 13.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM. and Koff A. (1994). P27(Kip1), a cyclin-Cdk inhibitor, links transforming growth-factor-beta and contact inhibition to cell-cycle arrest. Gene Dev 8:9–22 [DOI] [PubMed] [Google Scholar]
- 14.Chetty S, Pagliuca FW, Honore C, Kweudjeu A, Rezania A. and Melton DA. (2013). A simple tool to improve pluripotent stem cell differentiation. Nat Methods 10:553–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E. and Baetge EE. (2005). Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol 23:1534–1541 [DOI] [PubMed] [Google Scholar]
- 16.Murry CE. and Keller G. (2008). Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell 132:661–680 [DOI] [PubMed] [Google Scholar]
- 17.Roccio M, Schmitter D, Knobloch M, Okawa Y, Sage D. and Lutolf MP. (2013). Predicting stem cell fate changes by differential cell cycle progression patterns. Development 140:459–470 [DOI] [PubMed] [Google Scholar]
- 18.Bauwens CL, Peerani R, Niebruegge S, Woodhouse KA, Kumacheva E, Husain M. and Zandstra PW. (2008). Control of human embryonic stem cell colony and aggregate size heterogeneity influences differentiation trajectories. Stem Cells 26:2300–2310 [DOI] [PubMed] [Google Scholar]
- 19.Kehoe DE, Jing D, Lock LT. and Tzanakakis EM. (2010). Scalable stirred-suspension bioreactor culture of human pluripotent stem cells. Tissue Eng Part A 16:405–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodrigues CA, Fernandes TG, Diogo MM, da Silva CL. and Cabral JM. (2011). Stem cell cultivation in bioreactors. Biotechnol Adv 29:815–829 [DOI] [PubMed] [Google Scholar]
- 21.Lock LT. and Tzanakakis ES. (2009). Expansion and differentiation of human embryonic stem cells to endoderm progeny in a microcarrier stirred-suspension culture. Tissue Eng Part A 15:2051–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan JS, Reed A, Chen F. and Stewart CN. (2006). Statistical analysis of real-time PCR data. BMC Bioinformatics 7:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J. and Tzanakakis ES. (2012). Contribution of stochastic partitioning at human embryonic stem cell division to NANOG heterogeneity. PloS One 7:e50715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orford KW. and Scadden DT. (2008). Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet 9:115–128 [DOI] [PubMed] [Google Scholar]
- 25.Pauklin S. and Vallier L. (2013). The cell-cycle state of stem cells determines cell fate propensity. Cell 155:135–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu J. and Tzanakakis ES. (2013). Distinct allelic patterns of nanog expression impart embryonic stem cell population heterogeneity. PLoS Comput Biol 9:e1003140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geisser S. (1975). The predictive sample reuse method with applications. J Am Stat Assoc 70:320–328 [Google Scholar]
- 28.Menchon C, Edel MJ. and Belmonte JCI. (2011). The cell cycle inhibitor p27(Kip1) controls self-renewal and pluripotency of human embryonic stem cells by regulating the cell cycle, Brachyury and Twist. Cell Cycle 10:1435–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruiz S, Panopoulos AD, Herrerias A, Bissig KD, Lutz M, Berggren WT, Verma IM. and Belmonte JCI. (2011). A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr Biol 21:45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin TX, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E. and Xu Y. (2005). P53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 7:165–171 [DOI] [PubMed] [Google Scholar]
- 31.Qin H, Yu TX, Qing TT, Liu YX, Zhao Y, Cai J, Li J, Song ZH, et al. (2007). Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem 282:5842–5852 [DOI] [PubMed] [Google Scholar]
- 32.Yanes O, Clark J, Wong DM, Patti GJ, Sanchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S. and Siuzdak G. (2010). Metabolic oxidation regulates embryonic stem cell differentiation. Nat Chem Biol 6:411–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu PZ, Pan GJ, Yu JY. and Thomson JA. (2011). FGF2 sustains NANOG and switches the outcome of BMP4-induced human embryonic stem cell differentiation. Cell Stem Cell 8:326–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vallier L, Mendjan S, Brown S, Chng Z, Teo A, Smithers LE, Trotter MW, Cho CH, Martinez A, et al. (2009). Activin/nodal signalling maintains pluripotency by controlling Nanog expression. Development 136:1339–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan Y, Hsiung M, Cheng C. and Tzanakakis ES. (2014). Facile engineering of xeno-free microcarriers for the scalable cultivation of human pluripotent stem cells in stirred suspension. Tissue Eng Part A 20:588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J. and Gepstein L. (2001). Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest 108:407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Werbowetski-Ogilvie TE, Bosse M, Stewart M, Schnerch A, Ramos-Mejia V, Rouleau A, Wynder T, Smith MJ, Dingwall S, et al. (2009). Characterization of human embryonic stem cells with features of neoplastic progression. Nat Biotechnol 27:91–97 [DOI] [PubMed] [Google Scholar]
- 38.Barta T, Dolezalova D, Holubcova Z. and Hampl A. (2013). Cell cycle regulation in human embryonic stem cells: links to adaptation to cell culture. Exp Biol Med 238:271–275 [DOI] [PubMed] [Google Scholar]
- 39.Hatano S, Tada M, Kimura H, Yamaguchi S, Kono T, Nakano T, Suemori H, Nakatsuji N. and Tada T. (2005). Pluripotential competence of cells associated with Nanog activity. Mech Dev 122:67–79 [DOI] [PubMed] [Google Scholar]
- 40.Loh YH, Wu Q, Chew JL, Vega VB, Zhang WW, Chen X, Bourque G, George J, Leong B, et al. (2006). The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38:431–440 [DOI] [PubMed] [Google Scholar]
- 41.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M. and Yamanaka S. (2003). The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113:631–642 [DOI] [PubMed] [Google Scholar]
- 42.Ramakrishna S, Suresh B, Lim KH, Cha BH, Lee SH, Kim KS. and Baek KH. (2011). PEST motif sequence regulating human NANOG for proteasomal degradation. Stem Cells Dev 20:1512–1520 [DOI] [PubMed] [Google Scholar]
- 43.Chae HD, Lee MR. and Broxmeyer HE. (2012). 5-Aminoimidazole-4-carboxyamide ribonucleoside induces G(1)/S arrest and Nanog downregulation via p53 and enhances erythroid differentiation. Stem Cells 30:140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalmar T, Lim C, Hayward P, Munoz-Descalzo S, Nichols J, Garcia-Ojalvo J. and Martinez Arias A. (2009). Regulated fluctuations in nanog expression mediate cell fate decisions in embryonic stem cells. Plos Biol 7:e1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyanari Y. and Torres-Padilla ME. (2012). Control of ground-state pluripotency by allelic regulation of Nanog. Nature 483:470–473 [DOI] [PubMed] [Google Scholar]
- 46.Faddah DA, Wang H, Cheng AW, Katz Y, Buganim Y. and Jaenisch R. (2013). Single-cell analysis reveals that expression of nanog is biallelic and equally variable as that of other pluripotency factors in mouse ESCs. Cell Stem Cell 13:23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Filipczyk A, Gkatzis K, Fu J, Hoppe PS, Lickert H, Anastassiadis K. and Schroeder T. (2013). Biallelic expression of nanog protein in mouse embryonic stem cells. Cell Stem Cell 13:12–13 [DOI] [PubMed] [Google Scholar]
- 48.Lu R, Markowetz F, Unwin RD, Leek JT, Airoldi EM, MacArthur BD, Lachmann A, Rozov R, Ma'ayan A, et al. (2009). Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature 462:358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang YS, Sevilla A, Wan LQ, Lemischka IR. and Vunjak-Novakovic G. (2013). Patterning pluripotency in embryonic stem cells. Stem Cells 31:1806–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheung TH. and Rando TA. (2013). Molecular regulation of stem cell quiescence. Nature reviews. Mol Cell Biol 14:329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacArthur BD, Sevilla A, Lenz M, Muller FJ, Schuldt BM, Schuppert AA, Ridden SJ, Stumpf PS, Fidalgo M, et al. (2012). Nanog-dependent feedback loops regulate murine embryonic stem cell heterogeneity. Nat Cell Biol 14:1139–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B. and Jacks T. (1995). p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev 9:935–944 [DOI] [PubMed] [Google Scholar]
- 53.Ponzio G, Loubat A, Rochet N, Turchi L, Rezzonico R, Farahi Far D, Dulic V. and Rossi B. (1998). Early G1 growth arrest of hybridoma B cells by DMSO involves cyclin D2 inhibition and p21[CIP1] induction. Oncogene 17:1159–1166 [DOI] [PubMed] [Google Scholar]
- 54.Chang HH, Hemberg M, Barahona M, Ingber DE. and Huang S. (2008). Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 453:544–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tyson DR, Garbett SP, Frick PL. and Quaranta V. (2012). Fractional proliferation: a method to deconvolve cell population dynamics from single-cell data. Nat Methods 9:923–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu J, Rostami MR. and Tzanakakis ES. (2013). Stem cell modeling: from gene networks to cell populations. Curr Opin Chem Eng 2:17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu J. and Tzanakakis ES. (2013). Deconstructing stem cell population heterogeneity: single-cell analysis and modeling approaches. Biotechnol Adv 31:1047–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McLean AB, D'Amour KA, Jones KL, Krishnamoorthy M, Kulik MJ, Reynolds DM, Sheppard AM, Liu HQ, Xu Y, Baetge EE. and Dalton S. (2007). Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells 25:29–38 [DOI] [PubMed] [Google Scholar]
- 59.Singh AM, Reynolds D, Cliff T, Ohtsuka S, Mattheyses AL, Sun Y, Menendez L, Kulik M. and Dalton S. (2012). Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 10:312–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown S, Teo A, Pauklin S, Hannan N, Cho CH, Lim B, Vardy L, Dunn NR, Trotter M, Pedersen R. and Vallier L. (2011). Activin/nodal signaling controls divergent transcriptional networks in human embryonic stem cells and in endoderm progenitors. Stem Cells 29:1176–1185 [DOI] [PubMed] [Google Scholar]
- 61.Jing DH, Parikh A. and Tzanakakis ES. (2010). Cardiac cell generation from encapsulated embryonic stem cells in static and scalable culture systems. Cell Transplant 19:1397–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morchain J, Gabelle JC. and Cockx A. (2013). Coupling of biokinetic and population balance models to account for biological heterogeneity in bioreactors. AIChE J 59:369–379 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.