

The challenges in making biological products, starting from tissue sourcing to the processes involved in manufacture, storage, and distribution, are discussed. A model is described that has been proposed by the U.S. NIH for reducing the costs and permitting flexibility and innovation by individual investigators that has the potential to significantly accelerate implementation of pluripotent stem cell therapy.
Keywords: Stem cells, Pluripotent stem cell, Therapy, Current Good Manufacturing Practice
Abstract
The field of pluripotent stem cells (PSCs) is in a state of dynamic flux driven by significant advances in the derivation of specific phenotypes from embryonic stem cells, breakthroughs in somatic cell nuclear transfer, and dramatic improvements in generating induced PSCs using zero footprint methods. Spurred by these technological advances, companies have begun to plan clinical studies using human PSC derivatives manufactured in current Good Manufacturing Practice-compliant conditions. In the present review, we discuss the challenges in making these biological products, starting from tissue sourcing to the processes involved in manufacture, storage, and distribution. Additional challenges exist to meeting the regulatory requirements and keeping costs affordable. A model is described that has been proposed by the U.S. National Institutes of Health for reducing the costs and permitting flexibility and innovation by individual investigators. This model, combined with small adjustments in the regulatory processes tailored to address the unique properties of PSCs, has the potential of significantly accelerating the implementation of PSC-based cell therapy.
Developing Therapies Based on Pluripotent Stem Cells
Pluripotent stem cell (PSC)-based therapy is a very young field. Although we have known how to derive and culture mouse embryonic stem cells (ESCs) successfully for many years, we were unable to adapt those techniques to human cells until recently. The pioneering studies performed in Dr. Thomson’s laboratory were built on earlier work by Drs. Bongso and Eldor and others that have enabled investigators to develop techniques for the derivation of ESCs from nonhuman primates, followed by the generation of ESC lines from human blastocysts [1]. The relatively inefficient, early techniques were soon improved and refined. As a result, the production of human ESC (hESC) lines is now straightforward, and more than 1,000 lines have been derived worldwide. At least 200 of these are on the NIH registry that records the lines that have been determined to be eligible for federal funding in the United States [2].
Since the initial derivation of ESCs in 1998, several key technical advances have enabled the widespread use of ESC-based technology [3, 4]. These include techniques for deriving lines without destroying the embryo, generating lines from different stages of embryonic development (inner cell mass, morula, and late blastocyst stage), and deriving parthenogenetic lines (Table 1). These advances, coupled with improvements in the techniques used in culturing cells and identifying the growth factors required for maintaining undifferentiated cells, suggested that it would be possible to derive human PSC (hPSC) lines that comply with the regulations set by the Food and Drug Administration (FDA) for the use of these cells in a clinical setting. Such lines could then be used as the starting material for producing differentiated cell products that can pass FDA scrutiny. Several companies and university-based investigators have generated such PSC banks (Table 2).
Table 1.
A brief list of possible pluripotent cells
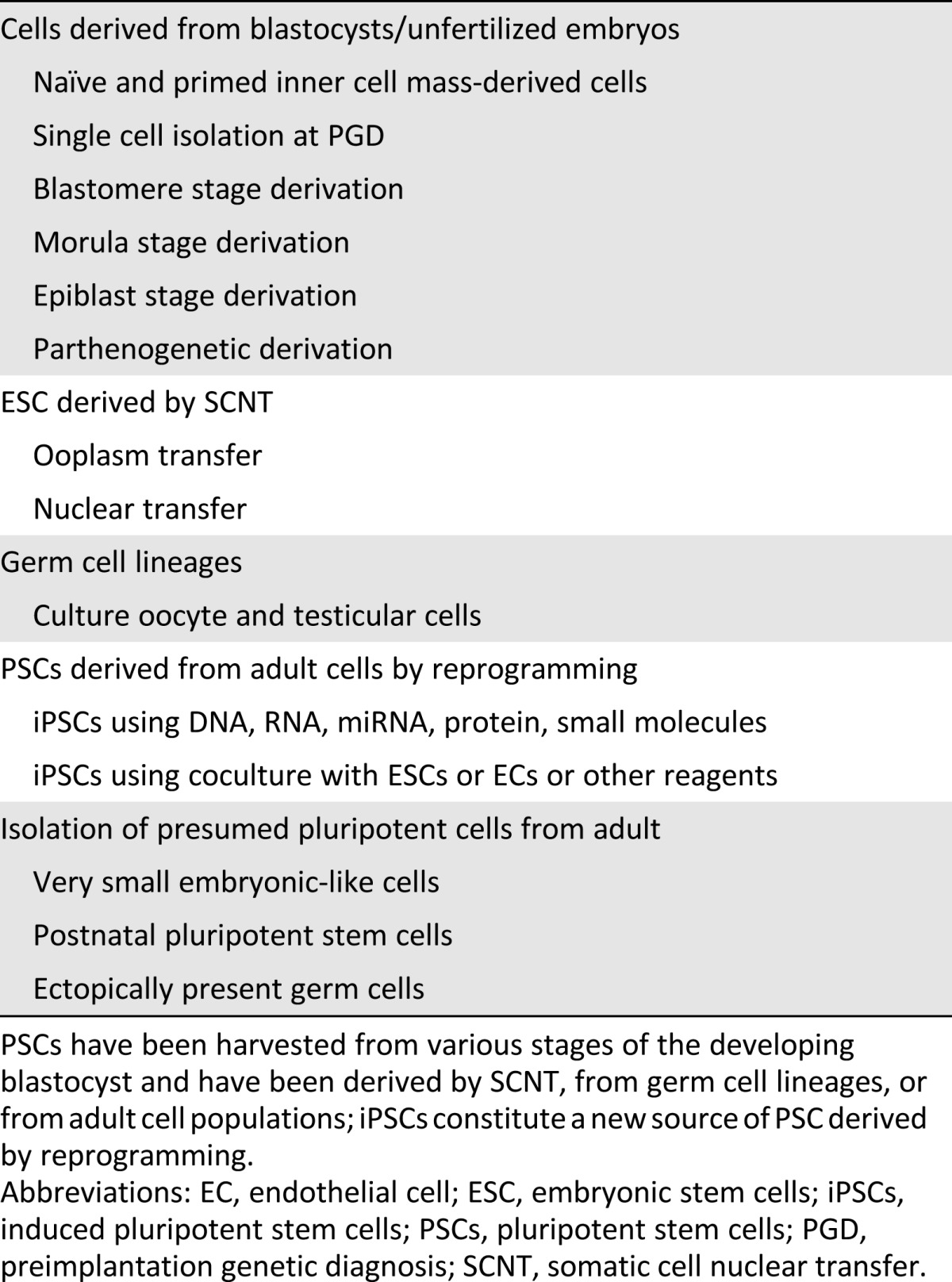
Table 2.
A brief list of organizations that have generated ESC or iPSC cell banks under cGMPs

In planning cell therapies that involve the transplantation of PSC-derived products, a major concern has been the presence of residual undifferentiated PSCs that can form teratomas in the recipient. This issue has been addressed by companies such as Geron Corporation, Advanced Cell Technology, and ViaCyte, Inc. and has been discussed in detail in other publications [5]. The reader is referred to the original references in the review for a detailed discussion of this important topic. In brief, the results of the preclinical studies and the published rodent data reported by these groups suggest that once PSCs are differentiated, few residual pluripotent cells will persist, and these have not appeared to generate teratomas in animal studies. These data have been considered sufficient to demonstrate the safety, and these groups have obtained FDA clearance for phase I clinical trials.
The use of ESC derivatives for allogeneic transplants raises another issue—that of immune matching and tolerance. Years of clinical studies involving nonautologous cell and organ transplantation have shown that no truly immune-privileged sites exist. In the absence of immune suppression, most grafted, allogeneic cells will provoke rejection. The emerging consensus is that, at the very least, short-term suppression would be required. However, even short-term immune suppression introduces its own risks and difficulties that need to be considered in designing trials and performing adequate follow-up studies. Investigators have attempted to resolve the issue of rejection using several different methods. One strategy involves generating cell banks of human leukocyte antigen (HLA)-matched cells that will serve as an extensive library for selecting close-to-optimal matches for patients. Debates concerning the size of these banks indicate that such an effort is possible, although expensive. Other investigators have offered engineering techniques to reorganize the entire HLA locus in an attempt to produce universal donor lines [6–8]. Still other groups have suggested nuclear transfer techniques pioneered in frog embryos by Dr. Gurdon and his colleagues [9, 10]. This approach has been successful in a number of species and has resulted in the cloning of “Dolly” the sheep and “SNUPI” the dog and the cloning of pigs and cows. However, extending nuclear transfer techniques to derive human embryos with the desired genotype met with difficulties [9]. These early studies faced technical issues, because the investigators could not maintain the integrity of the mitotic spindle that would allow successful cell division and growth. The recent work of Noggle et al. [11] and Tachibana et al. [12] has shown that these hurdles can be overcome, and hESCs have been derived from embryos created using nuclear transfer.
Despite the tremendous progress within a short span of 15 years, these hard-earned advances have highlighted the practical difficulties involved in using ESC-derived products in cell therapy. Such challenges, coupled with the lingering ethical concerns expressed by some groups, have steered the field to focus on using existing lines for nontherapeutic applications, on understanding the basic biology of stem cells and the pluripotent state, and on exploring other sources of pluripotent cells.
The recent landmark discovery that pluripotency can be induced via a handful of transcription factors or by modulation of key pathways using RNA, microRNA, protein, or small molecules has dramatically changed the field [13–15]. Although induced PSCs (iPSCs) can be similar to ESCs in their ability to generate multiple cell types, they offer the ability to generate ESC-like, pluripotent cells from any individual with fidelity and high efficiency [16]. This possibility is a game-changer and offers the tantalizing promise of personalized therapy and a novel solution to the immune rejection issue. As discussed, one of the significant roadblocks to cell therapy has been immune rejection; alternative solutions to bypass rejection have thus far proved unsuccessful. Despite some initial concerns [17], autologous iPSCs do not appear to cause immune rejection and thus might not require either short- or long-term immune suppression [18–20]. This strategy has recently been used for a clinical trial in Japan in which autologous iPSC-derived retinal pigment epithelial (RPE) cells are being implanted into patients with age-related macular degeneration (AMD). This new approach to cell-based therapy has fueled an exponential growth in the PSC field, and the importance of this discovery was confirmed by the award of the Nobel Prize for this discovery to two pioneers Drs. Gurdon and Yamanaka.
The potential of the pluripotent stem cell field has been further enhanced by recent breakthroughs in cell engineering. Building on the Nobel Prize-winning work of Oliver Smithies and Mario Capecchi, several investigators examined whether it was possible to engineer human ESC to correct gene defects or to introduce genes to provide a missing enzyme or trophic support [21, 22]. What was exciting was that it appeared that the efficiency was about the same as that for mice, suggesting that, technically, no fundamental unresolved issues to performing ex vivo gene therapy in humans remained. However, one major hurdle remains because, unlike mice in which heterozygous-engineered cells can be passaged although a chimera stage to obtain homozygous lines, alternative techniques would need to be developed to generate homozygous lines efficiently in humans. Recently, several methods to enhance the efficiency and fidelity of homologous recombination have been described. These include the use of zinc finger nuclease, transcription activator-like effector nuclease, and clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9/CRISPR systems [23, 24]. Efficiencies as great as 40% for a specific site for knockouts have been reported, which are an order of magnitude greater than what was achievable previously [25], allowing for single allele and double allele knockouts. Thus, for the first time, the technologies required for autologous cell therapy with gene engineered cells are available.
Given these advances, several models of cell based therapy can now be proposed. One model is to use autologous PSC-derived cell products or engineered PSC-derived cells for cell replacement or as a vehicle for the delivery of an enzyme or drug. Similar to other autologous cell therapies, the use of patient-specific PSCs will bypass the issue of immune rejection. Alternatively, if HLA-matched banks of iPSC cells are available, this “hybrid” model will allow the selection and use of optimally matched cells to produce graft material that will only require limited immune suppression. Differences between these autologous and allogeneic models are highlighted in Figure 1. Each model can be further subdivided based on associated genetic engineering, such as knockouts to treat gain-of-function diseases or knock-ins to restore a missing or nonfunctioning gene, or to create cells that have a novel method of ameliorating disease.
Figure 1.

Differences between autologous and allogeneic cell therapy models are highlighted. A “universal cell” is a concept of a line that can be used for all patients because it is immunologically modified to not generate an immune response. A new licensing model will likely need to be developed for autologous therapy as current regulations make it difficult to get approval for a variety of reasons (details given in text). Abbreviations: GMP, Good Manufacturing Practice; HLA, human leukocyte antigen; iPSC, induced pluripotent stem cell; pt., patient.
The PSC field is not just being shaped by the rapid pace of technological change, it is also strongly influenced by the requirements set by regulatory authorities and by the ethical and social constraints in different countries and states. Likewise, it has been influenced by patents and regulations governing the delivery of cell-based therapy and by the enormous expectations that people have for this novel type of therapy [26–31]. Countries with stricter regulations on use of ESCs have focused on iPSCs, and others are continuing a more balanced approach and awaiting further results from a longer term analysis of iPSCs. Nevertheless, a few pioneers have already initiated efforts to use iPSCs for therapy, and a clinical trial has been initiated in Japan [32].
Regulatory and Manufacturing Issues in Pluripotent Stem Cell-Based Therapy
In general, the same set of regulatory requirements apply for all cell-based therapies, irrespective of the source of cells used. The FDA guidance reports include requirements concerning cell sourcing, manufacturing, product characterization, and safety and efficacy testing. For allogeneic therapies, to comply with FDA requirements, the cell and/or tissue must be obtained from a donor that meets donor eligibility requirements. This includes obtaining the medical history from the donor, donor screening, and donor testing. Next, the PSC starting material and the final cell products that will be used for commercialization must be generated according to the current Good Manufacturing Practices (cGMPs). In these respects, no difference exists between a protocol that uses PSCs and one that uses adult cells such as mesenchymal stem cells or fibroblasts.
The different types of PSC-derived cell products have different challenges in complying with these regulations. When developing hESC-derived therapies, donor eligibility information can be challenging or impossible to obtain. In addition, generating hESCs under cGMPs is limited, because the in vitro fertilization process used to create the blastocyst is not generally a cGMP-compliant procedure. However, even without completely complying with these regulations, the FDA has allowed some groups to initiate clinical trials without this information. It remains unclear whether these groups will be able to successfully commercialize these therapies.
Although all cell therapies have the same regulatory requirements, PSC-based therapies have some unique properties that influence the manner in which they are regulated. A critical issue unique to therapies using PSC derivatives is that of residual, undifferentiated PSCs in the final product. By definition, PSCs are naïve cells capable of developing an entire individual, provided all the correct signals are present in time and space. When transplanted into an adult environment, they will not find all the instructive cues that direct their differentiation. Therefore, if sufficient numbers of pluripotent cells are present, they will grow stochastically to form a benign tumor termed a teratoma [5]. These benign tumors pose a problem, because they will have mass effects, and a small, but definite, possibility of malignant transformation. Paradoxically, in the case of autologous or matched cells, this problem could be exacerbated because the immune system might remain passive. However, we do not have any easy models to study this issue in vivo. Transplanting human cells in xeno models requires immune suppression or the use of immunologically compromised mice (although these might still retain some residual immunological responses), which reduces the fidelity of the model. Matched teratoma experiments, in which the iPSCs are of the same species as the animal model, were difficult to perform and ultimately might not reflect human responses. These issues make the process of manufacturing cells from PSCs much more difficult and significantly more expensive than that for adult cells.
Another issue for PSC-based therapy and, in particular, for iPSC-based therapy is the combination of ex vivo gene correction with cell delivery [33]. Currently, we do not have a defined approval process for the novel technologies being considered for gene correction. If we impose the same regulations that we have for viral-based gene therapy, or add regulations in addition to those for cell-based therapies, we might be creating an impossible barrier for pioneers developing such innovative, combination therapies. In addition, our manufacturing and testing processes for manufacturing these products must be carefully designed so as to not impose an unacceptable cost for the novel, autologous therapies.
In terms of manufacturing, PSC-derived therapies have some unique issues. Humans have a long lifespan; thus, human cells mature more slowly in culture, taking months to reach an immature but transplantable stage. This results in added cost and time, because the cells must be maintained for longer periods in a cGMP environment. Residual pluripotent cells in the final product need to be depleted; this too will increase the costs in time and development and testing. Additional caveats to manufacturing cell products from PSCs include the large number of steps involved in differentiation and the length of time required for the process. This raises the risk of introducing karyotypic changes owing to the stresses of the cell culture. Furthermore, each differentiated product requires a specific set of reagents, a different sequence of steps, and, often, different techniques [34–36]. Although several groups have successfully generated cell products using these lengthy processes, none of our current cGMP facilities are designed to efficiently perform long-term processes or to be rapidly modified to accommodate the multiple different processes required for differentiation of different cells. A second, perhaps less obvious, contributor to the cost is that each cell type for therapy, in addition to requiring a different manufacturing process, often requires a special final formulation and a different method of delivery that has been tailored to the cell type and disease state. Thus, one cannot standardize the equipment, standard operating procedures (SOPs), or training when manufacturing different PSC-derived products. The burden of this specificity is illustrated by comparing the making of influenza vaccines with producing RPE and dopaminergic neurons from PSCs. It is possible to create a different vaccine every year using the same facility, same personnel, and same processes; this conservation of resources keeps the manufacturing and training costs manageable. However, it is impossible to have a common process for creating RPE sheets and also a dopaminergic neuron, even when using the same cell line as the starting material. The protocols, reagents, techniques, and timeframes for producing RPE are completely different from those for differentiating dopaminergic neurons. This issue will be further compounded by the differences in the total number of cells needed for each disease condition. Treating macular degeneration might require 200K RPE per patient but treating heart disease might require in the neighborhood of 10 billion cells per procedure.
Autologous, Allogeneic, or Hybrid Models Have Different Tissue Sourcing and Manufacturing Constraints
In designing cell-based treatments starting with PSCs, a key decision is whether to use an autologous-, an allogeneic-, or a hybrid-matched approach (Fig. 1). This choice has important implications for the manufacturing process and the associated infrastructure and could affect the design of the preclinical studies. Nevertheless, these issues are similar to those faced by any other cell product and have been well covered elsewhere. We emphasize two important issues. First, the expense related to manufacturing PSC-derived cells will be further exacerbated when an autologous therapy is considered, as discussed above. One of the key concerns with this type of therapy is the variability among patients; iPSCs derived from different patients could have different characteristics and different safety profiles. With this type of therapy, the costs cannot be amortized over a large lot size, and the current regulations might necessitate performing tests, such as long-term stability and detailed preclinical studies for every single patient’s sample. The regulators themselves are constrained by rules and cannot provide blanket exemptions; it will be very important to determine whether specific regulations can inhibit this technology from moving forward [37].
Second, if plans exist to use HLA-matched and banked samples that have already been generated, we will need to scrutinize the original consent forms for tissue donation. The consent requirements for stored samples often did not anticipate their use for the generation of iPSCs or clinical and commercial applications. Without the appropriate consent, we could lose the ability to use such valuable samples. In theory, it might be possible to contact the donors for their consent; however, this might not be possible given the conflicting regulations on anonymization. A requirement for contact might, therefore, prove insurmountable. If the desired material is in another country, transportation could pose another obstacle. Shipping human cells is closely regulated, and the regulations governing transportation could prohibit harmonization or use in different countries [38–40].
Although these issues apply to existing samples, we should also be concerned about samples collected for future use and decide which tests are necessary, or represent acceptance criteria, and when they need to be performed. For example, in an autologous setting, does transplanting a prion protein-bearing sample represent an unacceptable hazard? Is cytomegalovirus contamination an issue or even required in autologous transplants? Is an existing karyotypic abnormality an issue in an autologous setting?
Another practical decision involves the point at which a sample enters a cGMP environment. In general, multiple iPSC colonies are harvested during the iPSC derivation process. After expansion, one clone is selected for all future work, such as differentiation, with or without genetic engineering. This selected clonal cell line can be entered into the cGMP manufacturing facility after suitable qualification. In contrast, the costs will rapidly escalate and become prohibitive if the entire derivation process must be conducted in a cGMP environment. Furthermore, the cell lines initially generated in a research laboratory and then transferred to a cGMP facility might be suitable for clinical entry in phase I trials; however, it is not clear whether these cells will be acceptable for licensure. More importantly, no consensus worldwide on this issue has been reached.
Preclinical Studies for Autologous Therapy Pose Challenges in Cost and Time
Conducting preclinical studies to evaluate cells for therapy faces numerous challenges, including the lack of good models, the lack of homologous cells in appropriate species, and the immune issue in xenotransplants. These have been discussed previously [32, 34, 35]. In the present report, we emphasize one point that will affect the timing, cost, and development of autologous therapy using iPSCs. The FDA usually requires that critical preclinical studies be performed on the final product, manufactured using the appropriate cGMP-qualified process. The cGMP process is costly and time consuming and accompanied by a large, upfront expense. The manufacturing process is likely to require adjustments as improvements occur and manufacturing technologies change. Although this is similar to the manufacture of other biologics, the manufacturing process for PSC-based cell products is generally longer and more complicated and requires many more novel technologies; thus, it carries a lot more risk.
Reasons exist why it might not be prudent to commit to a single, cGMP-qualified PSC line too early in the process. A change might be necessary, because the line was altered in some fashion or new clinical or genetic findings have revealed a concern that might limit the use of the line. Replacing one PSC line with another is relatively straightforward; however, if regulations require all tests be repeated with each change, this will impose a burden on developers. The burden involves the cost and time required. It is important to note that Takahashi et al [14] have just initiated a clinical trial in Japan using autologous iPSC-derived RPEs for the treatment of AMD. This group has performed extensive preclinical testing and has implanted the first patient [19, 41, 42].
NIH Model Offers a Partial Solution to the Burden of Cost
In an attempt to help reduce the cost of manufacture without restricting individuals to the use of a particular cell line, the NIH has proposed the following model of cell development (Fig. 2). The NIH, by developing clinically compliant protocols for generating, storing, and distributing human PSC lines, has ensured that the SOPs and protocols will be accessible to all individuals and that each individual or laboratory will not have to reinvent these protocols de novo. Equally important, the actual cells made by these processes will also be available for testing and evaluation in individual protocols, which will allow individual investigators to test an off-the-shelf product at a relatively modest cost. Importantly, this process will reduce the risk of unexpected problems surfacing later when the final cell line using the same processes has been generated. This is particularly important for autologous products for which one needs some data on the process and procedure for therapy before one moves forward to the clinic. One can imagine having a small bank of 10–12 lines, manufactured under a process that can be transferred to a cGMP facility, that is widely available for preclinical testing and for developing modified differentiation protocols. Once a reproducible differentiation has been demonstrated in 3–4 such lines, one can be reasonably comfortable that a new autologous PSC line or one with a new unique therapeutic profile can be used to generate a differentiated final product. These studies will also provide the regulatory authorities with data to use to compare different PSC lines, data that could be useful in determining regulatory compliance. In the long term, this could reduce the time required for development and, by extension, the costs of development. Thus, by providing well-characterized lines to a large community of researchers, the NIH, at a relatively modest initial investment, has enabled a large community of researchers to test the efficacy of a potential therapeutic agent. The NIH has also reduced the risk of failure that is inherent in translating processes from a research laboratory environment to a cGMP environment.
Figure 2.

The NIH model funds the process of making pluripotent stem cell lines and bears the cost. The process and the lines generated using this process can then be used by nonacademic entities without any restrictions from the NIH. This reduces the cost of development, and because a common process may be used by some of the entities, standardization and comparison of results are feasible. Abbreviations: CCRM, Colorado Center for Reproductive Medicine; cGMP, current Good Manufacturing Practice; ESC, embryonic stem cell; iPSC, induced pluripotent stem cell; PACT, Production Assistance for Cellular Therapies; PSC, pluripotent stem cell.
The benefits of the NIH model, however, will depend on how the regulatory authorities view and use such a model. Will they accept the comparability data provided, and will they provide clarity on this acceptance so that developers can mitigate risk? Equally important, will sufficient scientific data be available to suggest that comparability and predictability data can be generated? Tests are currently available to inform these questions. The PluriTest and GeneCard (Weizmann Institute of Science, Rehovat, Israel, http://www.weismann.ac.il) data provide information about the quality of the cell lines [43, 44]. Recently generated data from a direct comparison of multiple lines have suggested that a number of important variables, such as the tissue source, errors introduced during the iPSC-derivation process, or donor age, do not significantly alter the quality of the iPSCs generated, provided the same process and integration-free methods are used [45].
The NIH model offers additional potential benefits. Using commercial cell manufacturing organizations (CMOs) will allow the same experienced facility to create new lines in the future or to manufacture the same end product from a unique line, using the same SOPs, the same quality control tests, and preclinical models. This can lead to significant cost savings for any potential cell therapy provider, because they will not have to be committed, early on, to a specific cell line or manufacturing process. Also, they will not be required to disclose their specific modifications or improvements to a particular process.
Other Innovative Ways to Accelerate Translation
The Japanese government has taken a different approach to reducing the cost of development and accelerating the process of bringing PSC therapies to patients. In 2013, the Pharmaceutical Affairs Act was revised and the Act for Ensuring Regenerative Medicines was enacted. These Acts promote the practical application of advanced science such as the iPSC technology by modifying the existing Pharmaceutical and Medical Devices Agency regulations [46], such that hospitals and other qualified institutions will be able to introduce an experimental therapy, provided it was safe, and could expect to be reimbursed during the initial experimental studies in humans. Institutions have up to 7 years to provide data to the regulatory authorities for a full commercial license. To further increase the number of eligible institutions, the authorities made an additional and crucial change. Recognizing that individual institutions might not have the capability to manufacture a clinically qualified, experimental product, they have allowed institutions to contract out such services to eligible vendors.
These key policies will substantially reduce the enormous cost of conducting clinical trials, mitigate the risk that small companies take, and dramatically reduce the time to market.
Although it is unclear whether any of these modified regulatory paths would be approved in the United States, it is important to realize that the issue of access to these therapies is being debated. An important component of the debate is cost and development time while keeping the safety considerations in mind.
A Slightly Different Interpretation of the Rules Might Resolve Some of the Issues Facing the Field
Attempting to retrofit the existing rules to a new model of therapy could be a part of the challenge in implementing stem cell-based therapy today. An alternative approach that might solve many of these issues is to consider PSCs as just another reagent used to manufacture the end product, a reagent no different from that of qualified serum or a growth factor required to make a therapeutic product. The use of starting materials such as these follows regulations that are straightforward and well defined. If similar regulations could be applied to cellular input products, this could greatly simplify and expedite the conduct of cell-based therapies. For example, Certificates of Analysis and validation and Quality Control can be performed as a part of a supplier verification process. In the use of reagents for a “typical” manufacturing process, one supplier could be replaced with another as long as the material met all the qualifying specifications. Similarly, with appropriate change documentation, one could change the input cell, provided it met the same specifications as the original cell. This would obviate the time and cost of repeated consent issues and, importantly, would allow the use of a single cell line to generate multiple clinical products. It would also enable the straightforward transfer of the approved, starting cell line from one facility to another. Residual starting material in the end product is considered a contaminant, and the standard rules for contaminants would apply. The advantages of such a change would be that it avoids the automatic, required testing and the repeated preclinical studies that might otherwise be necessary. Making the same product at two sites is a manageable issue, and replacing the starting material or seed stocks would become practical when such a clear change path exists. In essence, approval would not be based on the source of a cell product (i.e., a particular cell line). Rather, approval would be for a particular end product, defined by its potency assay and composition, not its source. To be clear, no one is suggesting the use of undifferentiated cells for therapy nor that donor eligibility criteria be waived. The process described is, conceptually, not dissimilar to that used for cord blood or marrow banks, nor with some more recently approved cell therapies.
This logic can be applied to the case of RPE cell therapy for the treatment of AMD. The input cell comes from different individuals; however, the end product is a formulation of RPE with a defined composition and a defined function. This distinction would mean that preclinical tests could be conducted with a number of established lines or a subset of these lines. One would then accept the comparability of the product manufactured using the approved process. The logic of this approach would allow autologous therapies to be developed in a simple, rational method, with testing and characterization focused on the final cell product, rather than the starting material. The regulatory authorities might not be adverse to this concept; several autologous therapies have been approved. In these cases, the starting sample was different, but the manufacturing process was the same, and the end products were functionally similar, although by no means genetically identical. Two such approved products are LAVIV (Fibrocell Science, Exton, PA, http://www.fibrocellsicence.com) and Carticel (Sanofi-Aventis, Paris, France, http://www.carticel.com). If such a process is extended, it could allow many of the existing ESC lines to be used as input cells for therapy.
Some potential risks could be associated with such a model. Perhaps the greatest is analogous to what has been termed off-label use of small molecule drugs. This happens when a cell product has been approved and made available as a therapeutic agent for a specific condition, and clinicians wish to use it for some other condition. One could imagine a cell therapy company obtaining approval for one indication and then making the cells available for multiple indications, in particular, in the case of autologous cell-based therapy. This is a reasonable concern, and although it requires vigilance and adequate checks and balances in the system, it is not of sufficient magnitude to consider requiring the development of entirely new regulations.
What Might the Future Look Like
If regulatory changes were implemented along the lines we have discussed and some of the manufacturing issues were resolved, it might well be feasible to have a bank of HLA-matched PSC lines that would be readily available to all users. A clinical investigator who has decided on a specific cell-based therapy for a patient and has determined the required differentiated cell type and dosage could request shipment of HLA-matched cells from the bank to an appropriate CMO or university-based cGMP facility, where the shipped cells would be differentiated to generate the appropriate cell phenotype using an SOP that has been approved for the production of cells for therapeutic use. Alternatively, if time permitted, or if a clinician believed that autologous cells were required, the patient could go to an approved collection site to donate a tissue sample to be used to generate autologous iPSCs for that patient’s use and for contribution to an HLA-typed bank.
Acquiring regulatory approval for PSC-based cell therapy would be more akin to current practice in using an approved product for a new indication. Investigator-initiated trials would be approved by institutional review boards and posted on registries, such as clinicaltrials.gov, with full transparency; the regulatory authorities could thus be kept informed. Universities, biotech companies, and even pharmaceutical companies could initiate clinical trials using the standard regulatory path.
New studies and new uses would be developed by investigators initiating trials under the current regulations, and widespread use of a particular therapy would depend on rigorous trials conducted in an investigational new drug format. In one sense, the future would not be very different for the regulatory authorities; however, for the clinicians and patients and manufacturers, it would mean clarity, a reduction of risk, and widespread adoption of this exciting new technology.
Conclusion
We have discussed three current models of PSC-based cell therapy, each faced with different regulatory requirements. The first is a personalized medicine approach in which autologous cells are harvested from the patient, processed as required under appropriate guidelines, and then returned to the patient as a treatment. A second, allogeneic model uses a single, well-characterized, high-quality cell population that can be manufactured on a large scale. In this model, the end product would be subjected to rigorous tests akin to those used for a small molecule or biologic agent. Once tested and approved for clinical application, the product could then treat hundreds, even thousands, of individuals. The third is a hybrid model in which a bank consisting of different HLA-matched cell lines is established. Differentiated cells could then be derived from selected lines on an “as per need” basis for use in a limited number of patients at any one time.
Each model faces unique challenges that are exacerbated because the technology is also evolving rapidly. Even iPSCs, which were developed less than a decade ago, might be superseded by modifications in the basic iPSC approach, such as direct differentiation or differentiation in situ. These new methods might offer faster and less expensive alternatives, bypassing the stable intermediate pluripotent stage in iPSC protocols.
Nevertheless, for these efforts to be viable, it will likely require changes in the regulations. These changes should not compromise patient safety, which is the paramount concern of the regulators. By mitigating the twin burdens of cost and time, these strategies will incentivize developers to establish novel, cell-based therapies that are so critically needed for the many intractable diseases that continue to afflict people worldwide.
Acknowledgment
This work was supported by Q Therapeutics Inc., Carpenter Group Consulting Inc., and NxCell Inc.
Author Contributions
M.K.C. and M.S.R.: manuscript writing, final approval of the manuscript.
Disclosure of Potential Conflicts of Interest
M.S.R. worked at the NIH and played an important role in developing the NIH model, and at Thermo Fisher, which sells the GeneCard system developed at Harvard. M.S.R. is currently a compensated employee of Q Therapeutics and NxCell. M.K.C. is a compensated consultant for companies that work in the field of stem cell-based therapies.
References
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.NIH. Grants & funding: NIH Human Embryonic Stem Cell Registry. Available at: http://grants.nih.gov/stem_cells/registry/current.htm. Accessed September 15, 2014.
- 3.Rao M. Scalable human ES culture for therapeutic use: Propagation, differentiation, genetic modification and regulatory issues. Gene Ther. 2008;15:82–88. doi: 10.1038/sj.gt.3303061. [DOI] [PubMed] [Google Scholar]
- 4.Rao M, Condic ML. Alternative sources of pluripotent stem cells: Scientific solutions to an ethical dilemma. Stem Cells Dev. 2008;17:1–10. doi: 10.1089/scd.2008.0013. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter MK, Frey-Vasconcells J, Rao MS. Developing safe therapies from human pluripotent stem cells. Nat Biotechnol. 2009;27:606–613. doi: 10.1038/nbt0709-606. [DOI] [PubMed] [Google Scholar]
- 6.Bradley JA, Bolton EM, Pedersen RA. Stem cell medicine encounters the immune system. Nat Rev Immunol. 2002;2:859–871. doi: 10.1038/nri934. [DOI] [PubMed] [Google Scholar]
- 7.Turner M, Leslie S, Martin NG, et al. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell. 2013;13:382–384. doi: 10.1016/j.stem.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Zimmermann A, Preynat-Seauve O, Tiercy JM, et al. Haplotype-based banking of human pluripotent stem cells for transplantation: Potential and limitations. Stem Cells Dev. 2012;21:2364–2373. doi: 10.1089/scd.2012.0088. [DOI] [PubMed] [Google Scholar]
- 9.Gurdon JB, Wilmut I. Nuclear transfer to eggs and oocytes. Cold Spring Harb Perspect Biol. 2011;3:3. doi: 10.1101/cshperspect.a002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian XC, Kubota C, Enright B, et al. Cloning animals by somatic cell nuclear transfer—Biological factors. Reprod Biol Endocrinol. 2003;1:98. doi: 10.1186/1477-7827-1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noggle S, Fung HL, Gore A, et al. Human oocytes reprogram somatic cells to a pluripotent state. Nature. 2011;478:70–75. doi: 10.1038/nature10397. [DOI] [PubMed] [Google Scholar]
- 12.Tachibana M, Amato P, Sparman M, et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell. 2013;153:1228–1238. doi: 10.1016/j.cell.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malik N, Rao MS. A review of the methods for human iPSC derivation. Methods Mol Biol. 2013;997:23–33. doi: 10.1007/978-1-62703-348-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi K, Okita K, Nakagawa M, et al. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc. 2007;2:3081–3089. doi: 10.1038/nprot.2007.418. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 16.Yamanaka S. Induced pluripotent stem cells: Past, present, and future. Cell Stem Cell. 2012;10:678–684. doi: 10.1016/j.stem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Zhao T, Zhang ZN, Rong Z, et al. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–215. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- 18.Araki R, Uda M, Hoki Y, et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 2013;494:100–104. doi: 10.1038/nature11807. [DOI] [PubMed] [Google Scholar]
- 19.Kamao H, Mandai M, Okamoto S, et al. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Reports. 2014;2:205–218. doi: 10.1016/j.stemcr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morizane A, Doi D, Kikuchi T, et al. Direct comparison of autologous and allogeneic transplantation of iPSC-derived neural cells in the brain of a non-human primate. Stem Cell Reports. 2013;1:283–292. doi: 10.1016/j.stemcr.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 22.Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 23.Li M, Suzuki K, Kim NY, et al. A cut above the rest: Targeted genome editing technologies in human pluripotent stem cells. J Biol Chem. 2014;289:4594–4599. doi: 10.1074/jbc.R113.488247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue H, Wu J, Li S, et al. Genetic modification in human pluripotent stem cells by homologous recombination and CRISPR/Cas9 system. Methods Mol Biol. 2014 doi: 10.1007/7651_2014_73. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 25.Peters DT, Cowan CA, Musunuru K. Genome Editing in Human Pluripotent Stem Cells. StemBook. Cambridge, MA: Harvard Stem Cell Institute; 2013. [PubMed] [Google Scholar]
- 26.Caulfield T, McGuire A. Athletes’ use of unproven stem cell therapies: Adding to inappropriate media hype? Mol Ther. 2012;20:1656–1658. doi: 10.1038/mt.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Georgieva BP, Love JM. Human induced pluripotent stem cells: A review of the US patent landscape. Regen Med. 2010;5:581–591. doi: 10.2217/rme.10.43. [DOI] [PubMed] [Google Scholar]
- 28.Ikegaya H. Bioethics: Tighten up Japan’s stem-cell practices. Nature. 2012;489:33. doi: 10.1038/489033c. [DOI] [PubMed] [Google Scholar]
- 29.Magnus D. Translating stem cell research: Challenges at the research frontier. J Law Med Ethics. 2010;38:267–276. doi: 10.1111/j.1748-720X.2010.00487.x. [DOI] [PubMed] [Google Scholar]
- 30.Sipp D, Turner L. Stem cells. U.S. regulation of stem cells as medical products. Science. 2012;338:1296–1297. doi: 10.1126/science.1229918. [DOI] [PubMed] [Google Scholar]
- 31.Vrtovec KT, Scott CT. Patenting pluripotence: The next battle for stem cell intellectual property. Nat Biotechnol. 2008;26:393–395. doi: 10.1038/nbt0408-393. [DOI] [PubMed] [Google Scholar]
- 32.Andrews PW, Cavanagro J, Deans R, et al. Harmonizing standards for producing clinical-grade therapies from pluripotent stem cells. Nat Biotechnol. 2014;32:724–726. doi: 10.1038/nbt.2973. [DOI] [PubMed] [Google Scholar]
- 33.Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12:301–315. doi: 10.1038/nrg2985. [DOI] [PubMed] [Google Scholar]
- 34.Frey-Vasconcells J, Whittlesey KJ, Baum E, et al. Translation of stem cell research: Points to consider in designing preclinical animal studies. Stem Cells Translational Medicine. 2012;1:353–358. doi: 10.5966/sctm.2012-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleitman N, Rao MS, Owens DF. Pluripotent stem cells in translation: A Food and Drug Administration-National Institutes of Health collaboration. Stem Cells Translational Medicine. 2013;2:483–487. doi: 10.5966/sctm.2013-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panchision DM. Meeting report: Using stem cells for biological and therapeutics discovery in mental illness, April 2012. Stem Cells Translational Medicine. 2013;2:217–222. doi: 10.5966/sctm.2012-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jonlin EC. Differing standards for the NIH Stem Cell Registry and FDA approval render most federally funded hESC lines unsuitable for clinical use. Cell Stem Cell. 2014;14:139–140. doi: 10.1016/j.stem.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 38.U.S. Food and Drug Administration. Available at: http://www.fda.gov/regulatoryinformation/guidances/ucm122049.htm. Accessed September 15, 2014.
- 39.Lomax GP, Hull SC, Lowenthal J, et al. The DISCUSS Project: Induced pluripotent stem cell lines from previously collected research biospecimens and informed consent: Points to consider. Stem Cells Translational Medicine. 2013;2:727–730. doi: 10.5966/sctm.2013-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowenthal J, Lipnick S, Rao M, et al. Specimen collection for induced pluripotent stem cell research: Harmonizing the approach to informed consent. Stem Cells Translational Medicine. 2012;1:409–421. doi: 10.5966/sctm.2012-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Assawachananont J, Mandai M, Okamoto S, et al. Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice. Stem Cell Reports. 2014;2:662–674. doi: 10.1016/j.stemcr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanemura H, Go MJ, Shikamura M, et al. Tumorigenicity studies of induced pluripotent stem cell (iPSC)-derived retinal pigment epithelium (RPE) for the treatment of age-related macular degeneration. PLoS One. 2014;9:e85336. doi: 10.1371/journal.pone.0085336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bock C, Kiskinis E, Verstappen G, et al. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Müller FJ, Schuldt BM, Williams R, et al. A bioinformatic assay for pluripotency in human cells. Nat Methods. 2011;8:315–317. doi: 10.1038/nmeth.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rouhani F, Kumasaka N, de Brito MC, et al. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014;10:e1004432. doi: 10.1371/journal.pgen.1004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pharmaceuticals and Medical Devices Agency (PMDA). Japan's gatekeeper of medical products. Available at: http://www.pmda.go.jp/english/about/pdf/nature_20140501_e.pdf. Accessed September 15, 2014.