

Abstract
It is widely acknowledged that there is a need for molecular profiling in non-small-cell lung cancer. For example, treatment based on EGFR mutation status has attained successful results. However, in spite of excellent initial response to oral EGFR tyrosine kinase inhibitors (TKIs), progression-free survival is still limited. Current research has focused mostly on acquired resistance mechanisms, such as overexpression of AXL and loss of the Mediator MED12. In this review, in contrast, we discuss adaptive, rather than acquired, resistance.
Adaptive resistance can occur almost immediately after starting targeted therapy through a rapid rewiring of cancer cell signaling. By losing ERK negative feedback on receptor tyrosine kinase (RTK) expression, cancer cells are exposed to the stimuli of several ligands, and the ensuing activation of several RTKs reprograms all the canonical signaling pathways. The overexpression of several RTKs was observed in breast cancer cell lines treated with a MEK inhibitor and in BRAFV600E melanoma cell lines treated with BRAF inhibitors. This rebound effect of overexpression of several RTKs, including ERBB3, also occurs in lung cancers driven by Kras or EGFR mutations when treated with MEK, PI3K or dual PI3K/mTOR inhibitors. Synthetic lethality can be effectively induced by co-targeting these overexpressed RTKs.
We speculate that in patients with EGFR mutations, adaptive resistance occurs in a significant proportion of patients. Rebiopsies performed hours after starting treatment with EGFR TKIs can identify which RTKs are overexpressed after treatment. Efficient co-targeting of these RTKs can induce synthetic lethality and help overcome the limited effect of EGFR TKI monotherapy.
Key Words: Adaptive resistance, RTK reprogramming, signalability, synthetic lethality
Kinase inhibitors have limited benefit in cancer treatment because of a rebound effect, as exemplified by the inhibition of mitogen-activated protein-extracellular signal-regulated kinase (MEK) in triple-negative breast cancer cells. MEK inhibition causes acute loss of extracellular signal-regulated kinase (ERK), resulting in rapid c-Myc degradation, which induces the expression and activation of several receptor tyrosine kinases (RTKs) (1) (Figure 1). The transduction of signals from activated RTKs has been termed “signalability”. This signalability is markedly suppressed in BRAFV600E melanomas (2), which have high levels of ERK-dependent feedback and markedly decreased sensitivity to extracellular ligands. BRAF-driven tumors are relatively insensitive to secreted growth factors because of the inability of ligands to induce signaling. However, ERK inhibition reduces the ERK-dependent feedback, growth factors can signal, and the antitumor effects of the inhibitor are attenuated. Thus, the signaling network is radically changed and reactivated as an adaptation to inhibition of ERK signaling (2).
Figure 1.
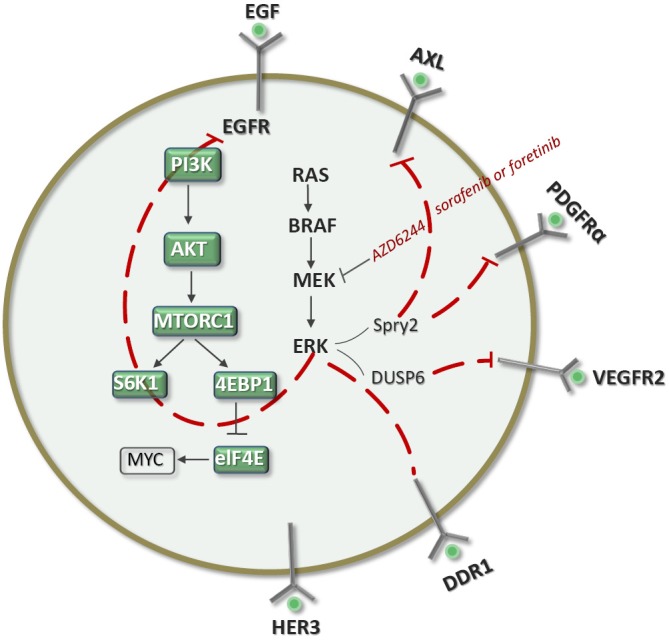
In breast cancer cell lines, MEK inhibition (2), Akt inhibition (3) or mTOR inhibition (4) causes kinome reprogramming in different breast cancer subtypes.
This dynamic reprogramming of signalability was also described in triple-negative breast cancers. MEK inhibition with AZD6244 increased RTK expression and downstream survival signaling, coinciding with the reactivation of RAF-MEK-ERK signaling. AZD6244 induces expression of platelet-derived growth factor receptor beta (PDGFRb), VEGFR2, HER3, AXL and DDR1 (Figure 1). Sorafenib inhibits PDGFRa/b, VEGFR2, DDR1/2, BRAF and RAF (1), and the combination of AZD6244 with sorafenib and foratenib caused a synthetic lethal effect. This kinase rewiring has also been observed in EGFR-mutant non-small-cell lung cancer (NSCLC) resistant to erlotinib, where AXL was overexpressed in 20% of cases. The tumor growth was reverted with the addition of an AXL inhibitor to erlotinib (5).
Induction of signalability when ERK-dependent feedback is relieved requires the presence of RTKs. Multiple ligands contribute to ERK rebound in melanomas exposed to BRAF inhibitors, including EGF, hepatocyte growth factor (HGF), NRG and FGF, which antagonized vemurafenib sensitivity in several BRAFV600E melanomas tested (Figure 2). In fact, several reports have shown that ligands, particularly HGF, can cause resistance to RAF inhibitors (6,7). In contrast, other growth factors, such as PDGF and insulin growth factor (IGF), have a minimal effect and some, like transforming growth factor beta, accentuated vemurafenib-induced growth inhibition (2). BRAFV600E melanoma cells have high levels of pMEK and pERK and high levels of dual specificity phosphatase (DUSP) 6 and Sprouty (Spry) 2. After 24 hours of exposure to vemurafenib, DUSP6 and Spry2 levels were markedly diminished, leading to relief of ERK-dependent feedback and potentiating a permissive signaling environment. Tumor growth of melanoma-derived xenografts was abrogated with the combination of vemurafenib and either lapatinib or neratinib (2).
Figure 2.
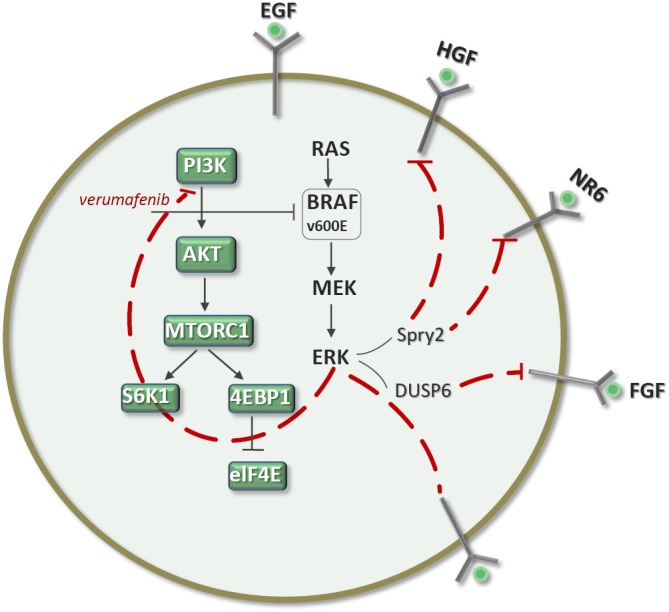
Ligand stimulation of ERK and PI3K signaling in BRAFV600E melanomas. In untreated melanomas, there is low signalability. However, hours after the ERK pathway is inhibited, the transduction of the signal is markedly potentiated (restored signalability). The induction of signalability could be blocked with the addition of an HER kinase inhibitor, such as neratinib (2).
In order to understand how EGFR-driven tumors adapt to pathway inhibition and design more effective therapies, it is necessary to identify the pathways that can be reactivated in the patients. This will require comparison of pretreatment biopsies with biopsies obtained hours after treatment and the development of new technologies to determine which ligands are present and which pathways become reactivated. This can permit the rational combination of therapies aimed at inhibiting adaptive resistance to the targeted therapy.
ERK signaling
ERK signaling plays an important role in regulating pleiotypic cellular functions. Activation of RTKs causes Ras to adopt an active, guanosine triphosphate (GTP)-bound conformation in which it induces the dimerization and activation of members of the RAF kinase family. Activated RAF phosphorylates and activates MEK1/2, which phosphorylate and activate ERK1/2, which in turn regulate cellular function by phosphorylating multiple substrates. A complex network of negative-feedback interactions limits the amplitude and duration of ERK signaling. Negative feedback is mediated directly by ERK-dependent inhibitory phosphorylation of components of the pathway, including EGFR, SOS and RAF. Importantly, ERK activation induces the expression of proteins that negatively regulate the pathway, including members of the Spry and DUSP families. BRAF-mutant colorectal cancers, which express greater levels of pEGFR than BRAF-mutant melanomas, are less sensitive to vemurafenib in spite of transient inhibition of pERK, since rapid ERK reactivation occurs through EGFR-mediated activation of Ras and CRAF. Interestingly, combined RAF and EGFR inhibition blocked reactivation of ERK signaling. Spry4 levels decreased after treatment with vemurafenib, coinciding with induction of pCRAF and pERK. Tumor growth in BRAF colorectal cancer cell lines was abrogated with the combination of vemurafenib and erlotinib. High pEGFR levels were observed in 60% of patients with colorectal cancer, who may benefit from combined RAF/EGFR or RAF/MEK inhibition (8). Similarly, in multiple BRAFV600E colon cancers, the inhibition of EGFR by cetuximab or gefitinib or erlotinib was strongly synergistic with BRAF inhibition both in vitro and in vivo. BRAF inhibition causes a rapid feedback activation of EGFR, supporting continued proliferation in the presence of BRAF inhibition (9).
In Kras-driven NSCLCs, ERK activity is negatively regulated by DUSP1, which can be abrogated by crosstalk with the Notch pathway. A significant proportion of patients with NSCLC have a hyperactivated Notch pathway, which is evidenced by higher levels of active g-secretase complex, increased levels of NICD (the activated form of Notch1), decreased Numb mRNA (a negative regulator of the Notch pathway), and increased HES1 protein (a downstream target of the Notch pathway). NSCLC patients with high HES1 expression had shorter overall survival. The Notch pathway regulates ERK phosphorylation by abrogating DUSP1 (10). These observations support the therapeutic potential of targeting g-secretase with MEK inhibitors in Kras-driven NSCLCs.
ERK activation is a common feature of tumors with RTK dysregulation stemming from EGFR, Kras or BRAF mutations. MEK inhibitors induced activation of phosphoinositide 3-kinase (PI3K)-Akt in EGFR-mutant cancer models via loss of an inhibitory threonine phosphorylation in the conserved juxtamembrane domains of EGFR and HER2 (11). Phosphorylation of the threonine residue impairs EGFR activation through disruption of receptor dimerization. Recent findings suggest that direct ERK-mediated phosphorylation of EGFR T669 and HER2 T677 suppresses activation of ERBB3. Suppressing ERK with AZD6244 prevented the effects of EGFR T669A on ERBB3/PI3K/Akt signaling in an EGFR-mutant cancer cell line. These findings suggest that combining MEK inhibition with ERBB3 or PI3K inhibitors may be a valuable strategy. The combination of MEK inhibitors with ERBB3 antibodies would block feedback activation of Akt and induce synthetic lethality (Figure 3). Interestingly, in Kras-mutant cancers that initially respond to single-agent RAF and MEK inhibitors, chronic inhibition of this pathway may lead to persistent activation of EGFR or HER2 (12).
Figure 3.
The combination of MEK inhibitors with ERBB3 antibodies blocks feedback activity of Akt in multiple cancer models (12).
Other feedback systems involving insulin growth factor receptor (IGFR) and mammalian target of rapamycin (mTOR)
The PI3K, RAF/MEK/ERK and mTORC1 pathways transmit signals from RTKs to downstream effector networks regulating cell growth, metabolism, survival and proliferation. One of the first feedback mechanisms indicating a rewiring phenomenon was the inhibition of mTORC1, which relieves protosomal degradation of IRS1, leading to feedback upregulation of IRS1/PI3K/Akt, reducing the efficacy of mTORC1 inhibitors as single agents and prompting the use of combination therapies (13,14). Briefly, mTORC1 activation leads to PI3K and MAPK inhibition through a negative feedback loop stemming from S6K1, while treatment with mTORC1 inhibitors results in a hyperactive RTK/IRS1/PI3K pathway, increasing the signal toward the Ras-RAF1-MEK1/2-ERK pathway. Hence, the combination of MEK and mTORC1 inhibitors could induce synthetic lethality by attenuating the rebound effect (13).
mTOR is an intracellular serine threonine kinase involved in the control of translational initiation. PI3K/Akt-dependent phosphorylation signaling occurs through tuberin, the protein product of TSC1/TSC2 complex, which leads to activation of mTOR. mTOR phosphorylates S6 kinase, which phosphorylates the ribosomal protein S6. mTOR also phosphorylates eukaryotic translation initiation factor 4E-binding protein (4E-BP1). This causes 4E-BP1 to disassociate from the eukaryotic translation initiation factor 4E (eIF4E), allowing activation of protein translation. Hyperactivation of mTOR signaling frequently occurs in cancer in more than 70% of patients, making the targeting of mTOR an attractive anticancer therapy. Rapamycin, a naturally occurring allosteric inhibitor of mTORC1, and its analogs (rapalogs) are clinically approved for treatment of renal cell cancer, mantle cell lymphomas and pancreatic neuroendocrine tumors. However, the overall success of rapalog monotherapy is limited. In a feedback mechanism, rapamycin inhibits the function of mTOR, leading to the downregulation of S6 kinase and maintenance of the 4E-BP1 eukaryotic translation initiation factor 4E complex. The inhibition of mTOR leads to downregulation of pS6K and p4E-BP1, leading to a rebound of Akt, since the negative feedback loop is lost. This allows activation of IRS1 (15,16). IGF1 receptor inhibition could prevent rapamycin-induced Akt activation and sensitize tumor cells to inhibition of mTOR (16). Erlotinib also blocked the rapamycin-induced increase in Akt phosphorylation in lung cancer cells lines (15).
About 30% of cancers exhibit elevated eIF4E levels, which correlate with poor prognosis. eIF4E overexpression induces cell transformation by selectively augmenting translation of mRNAs referred to as eIF4E-sensitive mRNAs, which encode proliferation- and survival-promoting proteins (cyclins, c-MYC and Bcl-xL). Multiple factors can induce the expression of eIF4E in cancer cells, including gene amplification and transcriptional upregulation by c-MYC, and lead to an increase in eIF4E mRNA stability by HuR. HuR increases Musashi1 mRNA stability, and high Musashi1 expression is associated with Notch1 expression. Musashi1 represses the regulation of NUMB mRNA, a negative regulator of Notch (17). Resistance to BEZ235, a dual PI3K/mTOR inhibitor) can be acquired through amplification of eIF4E. A model has been proposed whereby an elevated eIF4E/4E-BP ratio renders tumors resistant not only to a TOR inhibitor but also to dual PI3K/mTOR inhibitors. These findings suggest that the eIF4E/4E-BP ratio could serve as a predictive marker for tailoring personalized treatment with TOR inhibitors (18).
PI3K and Akt inhibitors relieve negative feedback on ERBB receptors and other RTKs, leading to partial reactivation of PI3K/Akt signaling, MEK/ERK signaling, and other downstream pathways, potentially limiting the use of PI3K inhibitors as single agents. For example, receptor activation of PI3K-Akt causes Akt-dependent phosphorylation of FOXO proteins, which downregulate the expression of some of the receptors that are tightly coupled to PI3K, including HER3, IGF1R and IR. In addition, Akt activation leads to activation of TORC1 and S6K, which inhibits IRS1 expression, as mentioned above. Akt inhibitors will result in activation of FOXO-dependent transcription of receptors and, importantly, inhibition of S6K-dependent inhibition of signalability with resultant activation of multiple receptors. In fact, all drugs that inhibit components of dysregulated mitogenic signaling pathways would be expected to relieve feedback inhibition of other components of the signaling network. Combined inhibition of the oncoprotein and key pathways reactivated by inhibition of negative feedback would thus enhance antitumor activity. Akt inhibitors cause tumor regression when combined with low doses of HER kinase of HSP90 inhibitors, which prevent or attenuate induction of receptor phosphorylation (3). The combination of ganetespib, a HSP90 inhibitor, with BEZ235, a dual pan-PI3K/mTOR inhibitor, displayed significant efficacy in a Kras-mutant NSCLC xenograft model (19).
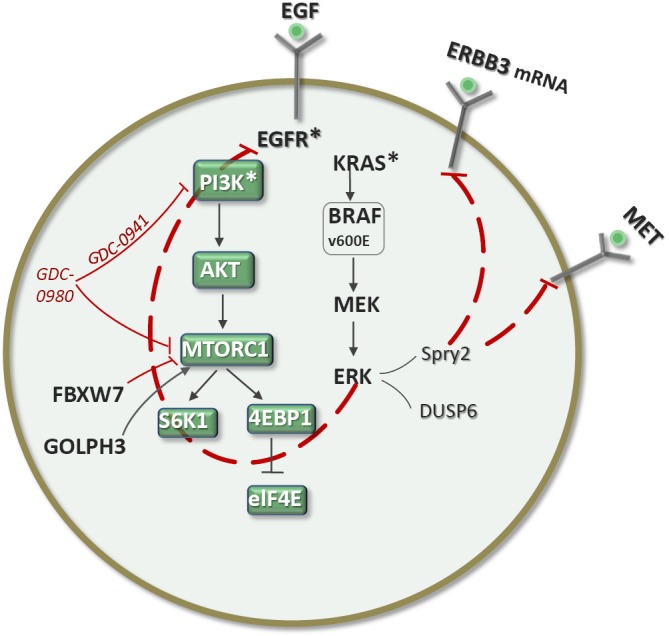
NSCLC cell lines harboring PI3K pathway alterations (RTK activation, PI3K mutation or amplification, or PTEN loss) are sensitive to the PI3K inhibitor GDC0941. Also, a dual PI3K/mTOR inhibitor, GDC0980, has shown even broader activity across cell lines and tumor xenografts. The combination of GDC0941 with paclitaxel, erlotinib, or a MEK inhibitor had greater effects on cell viability than PI3K inhibition alone (20). PIK3CA mutations and PTEN loss are predictors of sensitivity to PI3K inhibitors, as are FBXW7, which encodes a ubiquitin ligase that degrades mTOR, and GOLPH3, which is amplified in NSCLC and associated with mTOR inhibitor sensitivity. Tumor cells harboring deletions or mutations in FBXW7 are sensitive to rapamycin treatment (21). Rapamycin was significantly more effective against xenograft tumors expressing high levels of GOLPH3. GOLPH3, a Golgi oncoprotein, is a bona fide oncoprotein that is amplified in several types of cancers (22). In two EGFR-mutant NSCLC cell lines (HCC827 and H1975), GDC0941 showed greater than 100% tumor growth inhibition. The dual PI3K/mTOR inhibitor GDC0980 had greater antitumor activity than GDC0941 in models harboring Kras mutations or dual PIK3CA/Kras mutations (22). Intriguingly, treatment of inhibitor-sensitive cell lines with GDC0941 and GDC0980 resulted in increased expression of RTKs such as ERBB3 and MET, akin to the abovementioned examples (Figure 4). Co-targeting EGFR or downstream at the level of MEK could result in synergistic inhibition of cell growth, consistent with the hypothesis that such co-targeting may block PI3K inhibitor-induced pathway reactivation (Figure 4). It is particularly intriguing that the NSCLC cell line H1975 (16 copies of MYC) was very sensitive to GDC0941, which suggests that the biology of PI3K resistance may differ between breast cancer and NSCLC (20). In fact, a new rewiring mechanism of resistance to dual PI3K/mTOR inhibitors has been reported in breast cancer. Following PI3K-mTOR inhibition with BEZ235, increasing IRS1-dependent activation of JAK2/STAT5 and secretion of IL-8 was observed in several cell lines and primary breast tumors (Figure 5). Pharmacological inhibition of JAK2 abrogated this feedback loop and combined inhibition of PI3K/mTOR (with BEZ235) and JAK2 (with NVP-BSK805) synergistically reduced cancer cell number and tumor growth. The findings pave the way for co-targeting of the PI3K/mTOR and JAK2/STAT5 pathways in triple-negative breast cancer (23).
Figure 4.
Co-targeting EGFR or downstream at the level of MEK results in synergistic inhibition of cell growth in NSCLC cell lines treated with PI3K inhibitors or dual PI3K/mTOR inhibitors (20).
Figure 5.
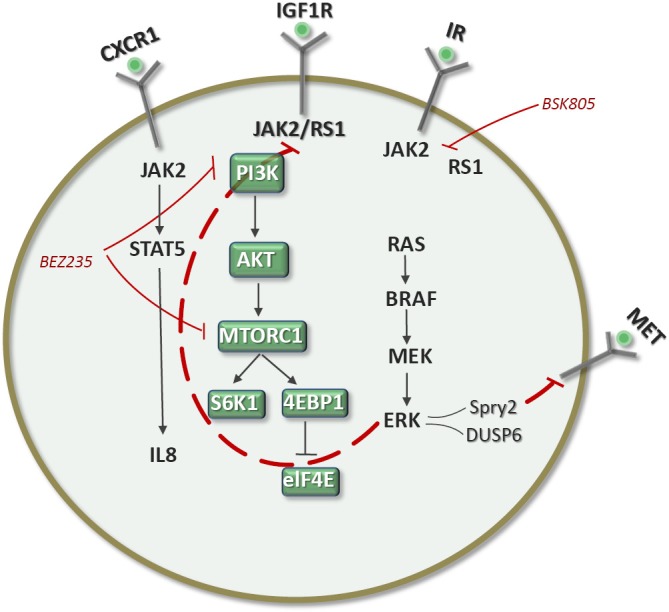
In triple-negative breast cancer, PI3K/mTOR inhibition elicits a positive feedback loop by activating JAK2/STAT5. Inhibition of JAK2 abrogates this feedback and provides a rationale for combining inhibition of PI3K/mTOR and JAK2/STAT5 pathways (23).
Treatment with erlotinib increased the levels of EGFR/IGF1R heterodimer localized on cell membrane, activated IGF1R and its downstream signaling mediators, and stimulated mTOR-mediated de novo protein synthesis of EGFR and survivin in NSCLC cells. These data suggest that enhanced synthesis of survivin protein mediated by the IGFR/EGFR heterodimer counteracts the antitumor action of erlotinib, suggesting the need for integrating IGF1R targeted agents with EGFR tyrosine kinase inhibitors (TKIs) for patients with NSCLC (24). Although this evidence is compelling, no trials have investigated this possibility. What is intriguing is that IGF1R targeted therapy has so far failed to give the expected results. Interestingly, the anti-IGF1R antibody figitumumab induced IGF1R/b-arrestin association, allowing b-arrestin1-dependent activation of ERK signaling. In consequence, the addition of an ERK1/2 inhibitor increased sensitivity to figitumumab (25). b-arrestin acts as an E3 ligase adaptor in response to IGF stimulation. After IGF binds to the tetrameric IGF1R, b-arrestin recruits Mdm2 to the receptor. Mdm2 ubiquitinates IGF1R, causing its internalization. Once internalized, IGF1R is degraded by the protoesome and b-arrestin mediates the activation of ERK from internalized signalosomes. ERK then translocates to the nucleus and activates transcription (26). In short, b-arrestin1 recruitment to IGF1R leads to ERK signaling activation and receptor downregulation. The combination of IGF1R targeting antibodies and MAPK inhibitors could be a new treatment strategy (25). Furthermore, Klotho inhibits the IGF1 pathway. Low Klotho expression has been found in breast cancer. Studies in breast cancer cells revealed increased activation of the FGF pathway following Klotho overexpression. Therefore, Klotho is an inhibitor of the IGF1 pathway and an activator of the FGF pathway in human breast cancer (27).
Adaptive resistance in NSCLC driven by EGFR mutations
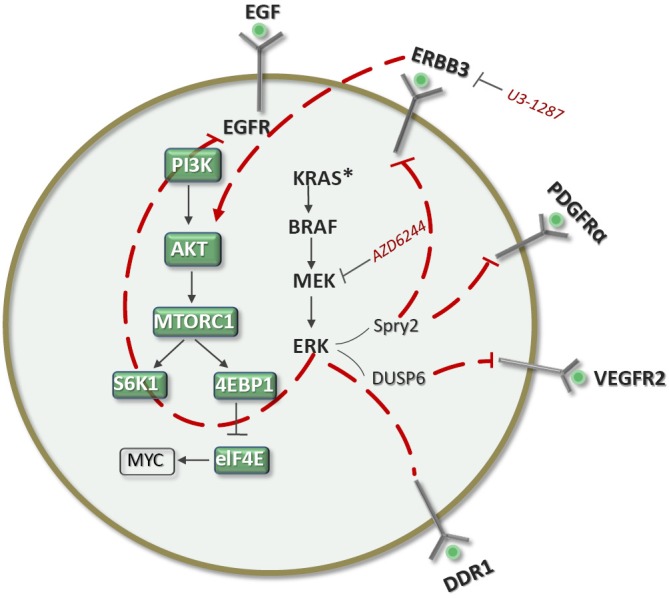
EGFR mutant-driven NSCLC responds very well to EGFR TKIs such as erlotinib. However, the response rate is around 60% and progression-free survival around one year or less, and all patients will eventually relapse (28,29). There are thus intrinsic resistance mechanisims, at least for the 30-40% of patients who do not respond initially, which can be attributed to crosstalk with other signaling pathways. For responders, the limited progression-free survival indicates that adaptive mechanisms of resistance can develop. Our hypothesis is that responders have high expression levels of BIM and that attenuation of the ERK pathway caused by erlotinib can lead to an effect similar to that observed with BRAF inhibitors in melanomas driven by BRAFV600E mutations (Figure 2) or with MEK inhibitors in triple-negative breast cancers (Figure 1). The loss of ERK function can lead to the stimulation of several RTKs and ligands (Figure 6). It is thus absolutely vital to perform a rebiopsy several hours following erlotinib treatment in order to understand the dynamic changes that cancer cells can undergo. The experience acquired in breast cancers with MEK inhibitors and in melanoma with BRAF inhibitors can be reproducible in EGFR-mutant NSCLC responders. Understanding the phenomenon of adaptive resistance from the beginning can lead to combining EGFR TKIs with other inhibitors blocking other RTKs, which presumably will include ERBB3. The correct identification of adaptive resistance mechanisms will allow us to implement the correct combination of targeted therapies for a true synthetic lethal effect that will increase the currently limited progression-free survival times attained with EGFR TKIs alone.
Figure 6.
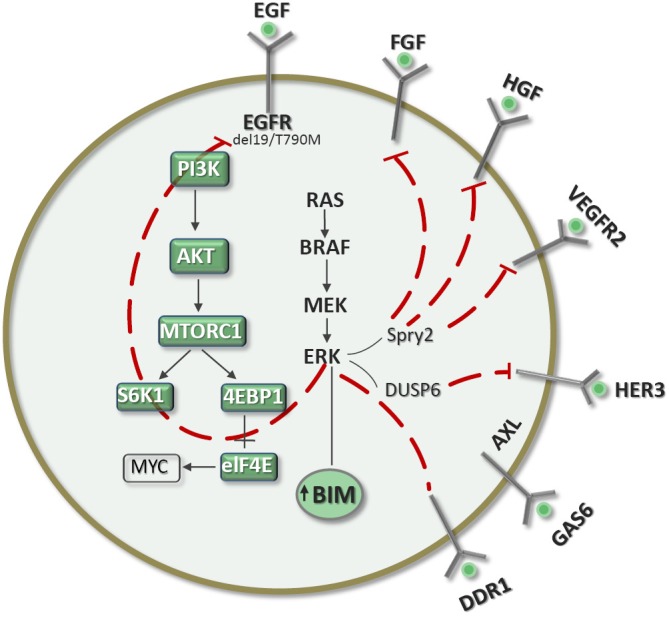
Hypothetical model for tumors with EGFR mutations, where erlotinib can abrogate ERK and adaptive resistance can be expected in tumors expressing BIM. Rebiopsies hours after initiating treatment will help us to understand the correct RTKs for co-targeting.
Acknowledgements
Work in Dr Rosell’s laboratory is partly supported by a grant from La Caixa Foundation, which had no role in writing the manuscript or in the decision to publish.
Disclosure: This manuscript has not been published or submitted for publication elsewhere. The authors declare no conflicts of interest.
References
- 1.Duncan JS, Whittle MC, Nakamura K, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012;149:307-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lito P, Pratilas CA, Joseph EW, et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell 2012;22:668-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandarlapaty S, Sawai A, Scaltriti M, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011;19:58-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov 2011;1:248-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012;44:852-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487:500-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012;487:505-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012;2:227-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100-3. [DOI] [PubMed] [Google Scholar]
- 10.Maraver A, Fernandez-Marcos PJ, Herranz D, et al. Therapeutic effect of γ-secretase inhibition in KrasG12V-driven non-small cell lung carcinoma by derepression of DUSP1 and inhibition of ERK. Cancer Cell 2012;22:222-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Red Brewer M, Choi SH, Alvarado D, et al. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell 2009;34:641-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turke AB, Song Y, Costa C, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res 2012;72:3228-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008;118:3065-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 2008;27:5527-41. [DOI] [PubMed] [Google Scholar]
- 15.Johnson BE, Jackman D, Jänne PA. Rationale for a phase I trial of erlotinib and the mammalian target of rapamycin inhibitor everolimus (RAD001) for patients with relapsed non small cell lung cancer. Clin Cancer Res 2007;13:s4628-31. [DOI] [PubMed] [Google Scholar]
- 16.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006;66:1500-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vo DT, Abdelmohsen K, Martindale JL, et al. The oncogenic RNA-binding protein Musashi1 is regulated by HuR via mRNA translation and stability in glioblastoma cells. Mol Cancer Res 2012;10:143-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alain T, Morita M, Fonseca BD, et al. eIF4E/4E-BP Ratio Predicts the Efficacy of mTOR Targeted Therapies. Cancer Res 2012;72:6468-76. [DOI] [PubMed] [Google Scholar]
- 19.Acquaviva J, Smith DL, Sang J, et al. Targeting KRAS-Mutant Non-Small Cell Lung Cancer with the Hsp90 Inhibitor Ganetespib. Mol Cancer Ther 2012;11:2633-43. [DOI] [PubMed] [Google Scholar]
- 20.Spoerke JM, O’Brien C, Huw L, et al. Phosphoinositide 3-Kinase (PI3K) Pathway Alterations Are Associated with Histologic Subtypes and Are Predictive of Sensitivity to PI3K Inhibitors in Lung Cancer Preclinical Models. Clin Cancer Res 2012;18:6771-83. [DOI] [PubMed] [Google Scholar]
- 21.Mao JH, Kim IJ, Wu D, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008;321:1499-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott KL, Kabbarah O, Liang MC, et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 2009;459:1085-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Britschgi A, Andraos R, Brinkhaus H, et al. JAK2/STAT5 Inhibition Circumvents Resistance to PI3K/mTOR Blockade: A Rationale for Cotargeting These Pathways in Metastatic Breast Cancer. Cancer Cell 2012;22:796-811. [DOI] [PubMed] [Google Scholar]
- 24.Morgillo F, Woo JK, Kim ES, et al. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res 2006;66:10100-11. [DOI] [PubMed] [Google Scholar]
- 25.Zheng H, Shen H, Oprea I, et al. β-Arrestin-biased agonism as the central mechanism of action for insulin-like growth factor 1 receptor-targeting antibodies in Ewing’s sarcoma. Proc Natl Acad Sci U S A 2012;109:20620-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kovacs JJ, Hara MR, Davenport CL, et al. Arrestin development: emerging roles for beta-arrestins in developmental signaling pathways. Dev Cell 2009;17:443-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf I, Levanon-Cohen S, Bose S, et al. Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 2008;27:7094-105. [DOI] [PubMed] [Google Scholar]
- 28.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735-42. [DOI] [PubMed] [Google Scholar]
- 29.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-46. [DOI] [PubMed] [Google Scholar]