

Abstract
The last few decades have witnessed a silent revolution in the war against NSCLC, thanks to the discovery of “oncogenic drivers” and the subsequent development of targeted therapies. The discovery of the EML4-ALK fusion gene in a subgroup of patients with NSCLC and the subsequent clinical development of crizotinib has been an amazing success story in lung cancer translational-research, and its accelerated approval [only 4 years from the discovery of ALK rearrangement in NSCLC to the approval by the Food and Drug Administration (FDA)] marked the beginning of the new decade of targeted therapy. However, common to all targeted therapies, despite an initial benefit, patients inevitably experience tumor progression, due to the development of resistance. Several molecular mechanisms are responsible for acquired resistance, such as secondary mutations of ALK kinase domain or amplification of ALK fusion gene, or the activation of other oncogenic drivers, which may cause resistance independently of ALK genetic alterations. Pre-clinical data and early clinical trials showed the promising efficacy of a new class of ALK-inhibitors in overcoming acquired resistance. The inhibition of the molecular chaperone, HSP90, represents another promising strategy to overcome crizotinib resistance in ALK-rearranged NSCLC. Several molecules are currently under investigation in order to establish their specific role in the treatment of ALK-rearranged NSCLC.
Keywords: ALK rearrangements, NSCLC, ALK inhibitors, resistance
Introduction
In the last few decades several advances have been made in translational research, thanks to the discovery of key-oncogene alterations, responsible for cancer cell proliferation and survival, and the subsequent advent of targeted drugs capable to inactivate them. This knowledge led to a gradual shift of lung cancer subtyping from a histological to a molecular basis. In fact, even if the histological subtype is considered in the choice between the different chemotherapies, the tumor molecular profile is crucial in order to personalize new targeted treatments. This was first demonstrated in 2004, with the discovery of sensitive epidermal growth factor receptor (EGFR) mutations and subsequent correlation with clinical responses to EGFR tyrosine-kinase-inhibitors (TKIs) (1,2), leading to the subsequent approval of gefitinib, erlotinib, and recently afatinib, as first line treatment in this subset of NSCLC patients (3). The discovery of the EML4-ALK fusion gene in a subgroup of patients with NSCLC (4), was followed by the development of a new class of agents, the ALK inhibitors, which dramatically improved the clinical outcome of those patients. This review will briefly discuss the biology of EML4-ALK gene and the clinical development of crizotinib in NSCLC, focusing on emerging mechanisms of acquired resistance and new treatment strategies to overcome it.
ALK-translocation in NSCLC
EML4-ALK chromosome rearrangement
Transforming rearrangement of the ALK gene was first reported in anaplastic large-cell lymphoma in 1994 (5), while in 2007 Soda et al. identified the ALK gene-rearrangement with EML4 in NSCLC (4). This rearrangement led to a fusion between the N-terminal portion of the EML4 protein with the intracellular region of the ALK receptor, resulting in a constitutive, ligand-independent activation of the tyrosine-kinase-domain of the rearranged ALK-receptor and downstream signaling pathways, such as Ras/MAPK, PI3K/Akt, and JACK/STAT3 pathways, responsible for both, tumor cell proliferation and survival (6). EML4-ALK fusion gene is a potent oncogenic driver, reported in about 3-7% of all NSCLC patients (7). Other fusion partners for ALK have been discovered in NSCLC, (such as TFG and KIF5B) (8-10), and multiple EML4-ALK variants have been identified, (such as E13;A20, and E6a/b;A20) (11), but their clinical significance still remains unknown. In vitro studies show some differences in sensitivity among the different variants to ALK-inhibition (12), but no correlation has been yet observed in this clinical setting. ALK rearrangement is a relatively rare event in the unselected NSCLC population. Data obtained by different studies show that it occurs more frequently in adenocarcinomas, in light or never smokers, and younger patients, but was also found, at a much lower rate, in squamous or adeno-squamous carcinomas and in smokers (7,13). ALK rearrangements are not fully mutually exclusive with EGFR or KRAS mutations; several simultaneously occurring EGFR and KRAS mutations in ALK-positive patients have been recently reported (14,15). ALK-positive tumors seem to be associated with a worse survival and an increased risk of brain and liver metastases (16), but an improved prognosis is observed in the context of treatment with ALK inhibitors (17). Several studies showed a lack of response to EGFR-TKIs (7,13,18). Finally, ALK rearrangements define a new molecular subtype of NSCLC that is exquisitely sensitive to a new class of tailored agents, the ALK inhibitors.
ALK-rearrangement detection
Several techniques are currently under investigation in order to detect ALK-rearrangements in NSCLC, such as fluorescence in situ hybridization (FISH), immunohistochemistry (IHC), and reverse transcriptase PCR (RT-PCR), each showing advantages and limitations. Currently, FISH using break-apart probes is considered the gold standard, approved by the Food and Drug Administration (FDA) to identify ALK-rearranged NSCLC. It was tested and validated in clinical trials that have led to the approval of crizotinib, an ALK TKI, and it is used to screen patients to enter the ongoing trials with second-generation ALK inhibitors. According to the definition of this test, ALK-FISH is considered positive if a split in more than 15% tumor cells occurs and counting at least 60 cells (19). Despite many positive features, the ALK FISH test has also several disadvantages. It requires laboratories with an experienced operator, and it results expensive. IHC is an easier and cheaper method, generally available in local laboratories, based on the use of ALK-specific monoclonal antibody. Several studies showed a strong correlation between ALK-rearrangement positivity, as detected by FISH, and ALK protein overexpression, as detected by IHC (20-26). These findings suggest that IHC could be used for screening of ALK rearrangements prior to FISH, leading to the development of new diagnostic algorithms, which need to be validated in large scale concordance studies. Finally RT-PCR is the most sensitive method of detecting not only ALK rearrangements, but also of characterizing their variant types, and the abundance of EML4-ALK positive cells in NSCLC tumor tissues (27). It also has the advantage of requiring limited amount of material for analysis and is relatively easy to perform, but the development of this method as a diagnostic tool has several limitations (28). The optimal diagnostic strategy for ALK rearrangement remains to date a matter of debate, but FISH may be still considered the gold standard to consider the patient to crizotinib or other ALK inhibitors. A properly validated immunohistochemical method could be considered as a screening test (Figure 1).
Figure 1.
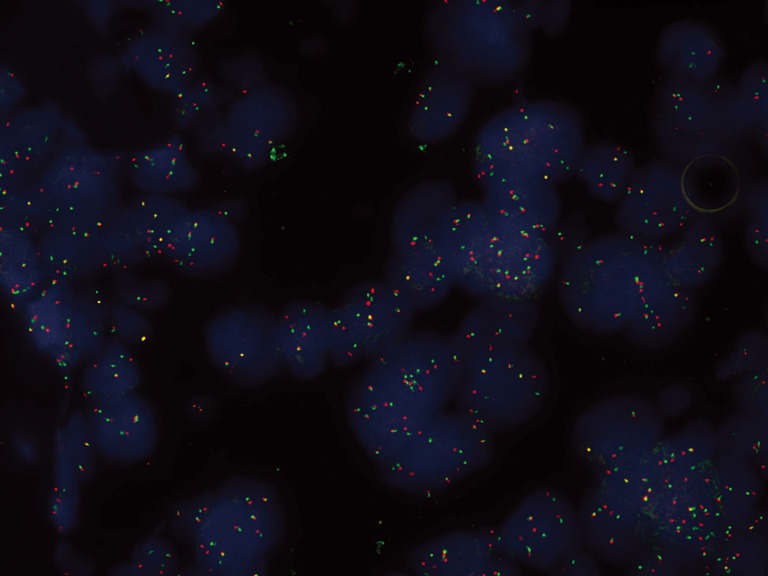
ALK translocation FISH break-apart positive in a NSCLC patient.
Clinical development of crizotinib
Clinical efficacy
Crizotinib is an oral, selective, ATP-competitive, small molecule, originally developed as c-MET inhibitor, approved for the treatment of ALK+, pre-treated NSCLC patients (3). In pre-clinical studies, it showed a great activity against c-MET-positive and ALK-positive cell lines (29), on the basis of which, a first in human, phase I, dose-escalation trial, evaluated crizotinib as an oral single agent in 37 patients with advanced solid tumors, identifying 250 mg twice daily as the recommended dose (30). There were two patients with NSCLC harboring EML4-ALK rearrangement treated with crizotinib who showed dramatic improvement in their symptoms during the dose escalation phase. That observation led to a large prospective screening of NSCLC patients and recruitment of those with ALK-positive NSCLC into an expanded molecular cohort at the maximum tolerated dose (MTD) of 250 mg twice daily (31). The updated results, reported by Camidge et al., including 149 previously treated and untreated, ALK+ NSCLC patients, showed three complete response (CR) and 84 partial response (PR), for an overall response rate (ORR) of 61% with a median duration of response of 49.1 weeks, and a median PFS of 9.7 months (95% CI, 7.7-12.8) (32). At the time of presentation, overall survival (OS) data were not mature. Noteworthy, 39 patients continued to receive crizotinib for more than 2 weeks after progression due to a perceived clinical benefit from the physician. The phase II trial (PROFILE1005) confirmed these striking results on 261 ALK+ pre-treated NSCLC patients. The ORR was 59.8%, with a median duration of response of 45.6 weeks, and a median PFS of 8.1 months (95% CI, 6.8-9.7) (33). On the basis of these impressive results, the FDA approved the use of crizotinib in October 2011, for the treatment of ALK+ advanced NSCLC. Two phase III trials have compared crizotinib to chemotherapy in a selected population. First, the PROFILE1007 trial compared crizotinib to chemotherapy (docetaxel or pemetrexed), in ALK+ NSCLC patients after one previous platinum-based chemotherapy regimen (34). The study met its primary endpoint, with a median PFS of 7.7 months for patients treated with crizotinib vs. 3.0 months for those treated with chemotherapy (HR: 049; 95% CI, 0.37-0.64; P<0.0001). The ORR was also significantly higher for the crizotinib arm compared to the chemotherapy group (65% vs. 20%; P<0.0001). Another interesting feature emerging from the PROFILE1007 trial is a strong correlation between ALK-rearrangement and pemetrexed efficacy. In fact, better results were reported in patients treated with pemetrexed, compared to those treated with docetaxel, both in terms of RR (29.3% vs. 6.9%) and PFS (4.2 vs. 2.6 months), respectively. This could be related to the lower concentration of thymidylate synthase (TS), the main target of pemetrexed, in ALK+ tumors (35), but this assumption needs further validation in prospective, randomized studies. Finally, the ongoing open label phase III clinical trial PROFILE1014 trial is comparing crizotinib against a platinum-based doublet (cisplatin or carboplatin plus pemetrexed) in previously untreated, ALK-positive, stage IIIB/IV NSCLC patients. Waiting for the results in first line-setting, the European Society of Medical Oncology (ESMO) guidelines recommend crizotinib as the treatment of choice for ALK+ NSCLC patients only after progression to a first-line chemotherapy regimen (3). The analysis of data from all phases I-III studies regarding the toxic effects of crizotinib shows a good tolerability profile. The most frequent treatment-related adverse effects (AEs) were visual disorders (grades 1,2). Elevated amino transferase, lymphopenia, neutropenia, and pneumonitis were the most common treatment-related grade 3 or 4 AEs, observed in 7-15%, 11.4%, 5.2%, and 1-5% of patients, respectively. Finally, about 69% of patients experienced at least one episode of sinus bradycardia (HR ≤60 bpm). Patients treated with crizotinib reported grater improvement in lung cancer symptoms and greater improvement in global quality of life when compared with chemotherapy (34) (Figure 2 and Table 1).
Figure 2.

A computerized tomography (CT) scan of the thorax showing an objective response in an ALK-rearranged lung tumor of a patient treated with crizotinib. (A) 49-year-old women, PS 4, with advanced, ALK-traslocated, NSCLC: baseline CT-scan; (B) 49-year-old women, PS 4, with advanced, ALK-traslocated, NSCLC: CT-scan two weeks after treatment initiation with crizotinib.
Table 1. Clinical trials with crizotinib in ALK+ patients.
| Study | Phase | Patients included | ORR (%) | PFS (months) | Author |
|---|---|---|---|---|---|
| PROFILE 1001 | Phase I | N: 149 pt (NSCLC; ALK+) | 60.8 (95% CI, 52.3-68.9) | 9.7 (95% CI, 7.7-12.8) | Camidge 2012 (32) |
| PROFILE 1005 | Phase II | N: 261 pt (NSCLC; ALK+) | 60 (95% CI, 53.6-65.9) | 8.1 (95% CI, 6.8-9.7) | Kim 2012 (33) |
| PROFILE 1007 | Phase III (CZT vs. P/D) | N: 346 pt (NSCLC; ALK+) | 65 vs. 19.5 P<0.0001 | 7.7 vs. 3.0 P<0.0001 | Shaw 2013 (34) |
| PROFILE 1014 | Phase III (CZT vs. C + P) | (NSCLC; ALK+) | Ongoing | Ongoing | (NCT01639001) |
ORR, objective response rate; PFS, progression free survival; CZT, crizotinib; P, pemetrexed; D, docetaxel; C, cisplatin or carboplatin; N.A, not available.
Crizotinib: also a ROS1 and c-MET inhibitor
Even though crizotinib has been approved for the treatment of ALK positive NSCLC, it has shown activity also in other subgroup of NSCLC patients whose tumors presented ROS1 gene rearrangements. ROS1 is a receptor tyrosine kinase of the insulin receptor superfamily, and it is most closely related to ALK. Multiple ROS1 fusion partners have been identified in NSCLC (TPM3, SDC4, SLC34A2, CD74, EZR, LRIG3, and FIG), resulting in a ligand-independent, constitutive activation of ROS1 fusion kinase responsible for both, cancer cell proliferation and survival (36,37). ROS1 rearrangements are reported in about 1-2% of the whole NSCLC population (38), mostly in young people, never smokers, with an adenocarcinoma histology subtype (39), and seems to be mutually exclusive with other common mutations, like EGFR, K-RAS and ALK. Although the amino-acid sequence of ROS1 does not completely correspond to ALK in the kinase domain, ROS1 positive tumors showed in vitro sensitivity to several ALK-Inhibitors (40). On the basis of these encouraging results in preclinical studies (39), an expansion cohort of the initial crizotinib phase I trial, included 23 patients with advanced NSCLC harboring ROS1 rearrangements, reporting an impressive ORR of 50% (10/20), with 9 PRs and 1 CR (41). Recruitment is still ongoing, but early results suggest that crizotinib could be also considered the standard of care for another subgroup of molecularly selected NSCLC patients. Finally, crizotinib could also represent an efficacy treatment for NSCLC with c-MET (mesenchymal-epithelial transition factor) amplifications, which have been reported to be oncogenic drivers in about 4-11% of patients (42,43), and are implicated in about 22% of the cases of acquired resistance to EGFR TKI (44). Although it was recently approved by the FDA as an ALK-inhibitor, it was synthesized primarily as a Met inhibitor. A recent case report described a rapid and durable response to crizotinib in a NSCLC patient carrying a de novo MET amplification and lacking an ALK rearrangement (43). Furthermore, several ongoing trials are investigating the combination of crizotinib with therapies targeting EGFR (erlotinib, NCT00965731; dacomitinib, PF-00299804, NCT01121575) to overcome EGFR-TKI acquired resistance, due to MET amplification.
Mechanisms of crizotinib resistance
Despite of a dramatic initial activity of crizotinib in molecularly defined, ALK-rearranged, NSCLC population, common to other targeted therapies used in NSCLC, such as EGFR-TKIs, acquired resistance to TKIs inevitably develops during the first year of treatment. In the clinic, relapses during crizotinib treatment commonly involve the central nervous system (CNS). The reasons for crizotinib failure at CNS are actually unknown, and data available from clinical studies are controversial. In fact, in the phase II PROFILE1005 study (33), only 2 out of 18 enrolled patients with brain metastasis developed CNS progression under crizotinib treatment, while in another study (45), 13 out of 28 ALK-positive patients (46%) treated developed brain metastasis. Whether and how acquired resistance alterations may contribute to resistance in the CNS is actually unknown. However, extremely lower crizotinib concentration in the cerebrospinal fluid (CSF) compared to the plasma, was recently reported in one patient with focal CNS progression during crizotinib treatment (46), suggesting a possible pharmacokinetic mechanism, like inadequate drug exposure in a sanctuary site, as possible cause of the high rate of CNS relapse in ALK-positive patients. We may distinguish between “intrinsic TKI resistance”, which usually occurs earlier, especially in “fast progressive” patients, and, “acquired TKI resistance”, which more frequently occurs, within 8-10 months from crizotinib initiation (34). However, some cases showing more than 24 months of clinical benefit have been reported (32).
Recently, in vitro studies have shown a different sensitivity to ALK-inhibition among the different variants of the ALK-fusion gene (12), suggesting a possible explanation for intrinsic resistance to crizotinib, but this correlation was not confirmed in a subgroup analysis from a phase I trial of crizotinib (31). Mechanisms of acquired resistance to crizotinib may be divided into two categories. The first one includes additional genetic alterations in the target, such as secondary mutations of the ALK Kinase domain or amplification of the ALK fusion gene, which preserve and facilitate the ALK signaling pathway activity (47,48). Regarding the secondary mutations, the most common and well characterized, is the L1196M mutation (49), consisting of a substitution of methionine for leucine in the “gatekeeper” residue, promoting formation of the active conformation of the protein and leading to an increased protein kinase activity (50). Common to the others gatekeeper mutations, like T790M in EGFR (51), the L1196M mutation interferes with the inhibition of the kinase activity by the targeted agent, promoting enzyme activation and not preventing the access of the drug into the hydrophobic binding pocket. Additional resistance mutations include: G1269A, C1156Y, L1152R, G1202R, S1206Y, 1151Tins, and recently F1174C and D1203N. In in vitro experiments, all these secondary mutations reduced crizotinib binding and/or increased affinity for ATP, conferring different sensitivity to ALK-Inhibitors. In particular, G1269A and 1151Tins mutations increase the ALK kinase affinity for the ATP (52). G1202R and S1206Y mutations induce conformational changes, causing hindrance of crizotinib binding. Finally, C1156Y mutations lead to specific conformational changes in the binding site of the drug, decreasing the affinity of crizotinib, while L1152R mutations reduce crizotinib activity interfering with the downstream signaling pathway phosphorylation (53,54). ALK fusion gene amplification, alone or in combination with secondary mutations, has also been identified as a sufficient cause for developing crizotinib resistance in cell lines experiments, and subsequent studies have confirmed ALK fusion gene amplification in resistant clinical specimens (48,55). It was supposed that normal doses of crizotinib could not inhibit a fraction of ALK fusion proteins, and that increased expression may allow sufficient downstream signaling for tumor cell survival. These so-called “ALK dominant mechanisms” are involved in about 30% of acquired resistance to crizotinib. The second category of mechanisms includes activation of other oncogenic drivers, which may cause resistance through reactivation of downstream signaling pathways via bypass tracts, independently of ALK genetic alterations.
Recently, in 3 out of 11 ALK+ specimens, EGFR phosphorylation and K-RAS mutation were found in the post crizotinib biopsy, and c-KIT amplification was identified in 2 of 13 resistant specimens (47,48), suggesting that under crizotinib treatment pressure, pre-existent oncogenic drivers present in the same cells or in other clones, may emerge leading to the development of the resistance. Furthermore, with regards to c-Kit activation, in vitro studies showed that it is completely dependent on the concentration of stroma derived stem cell factor (SCF) in the tumor specimen. In fact, the crizotinib plus imatinib combination may lead to a decrease in c-Kit+ cells proliferation only in presence of SCF, suggesting also the involvement of the microenvironment in the development of crizotinib resistance (47). Therefore, K-RAS mutations, KIT amplification and increased EGFR autophosphorylation and mutations, represent the so-called “ALK non-dominant mechanisms” involved in the development of the acquired resistance to crizotinib. Occasionally, multiple different resistance mechanisms may be found in the same tumor of the same patient, suggesting the need of combination therapies, such as ALK inhibitors, with therapies targeting EGFR, erlotinib or dacomitinib, to optimize the patients’ outcomes (56). Finally, the specific mechanism of acquired resistance development during crizotinib treatment remain still unknown in about 20% of patients, and loss of the ALK fusion oncogene has been supposed as an additional potential mechanism of resistance (47,48). The dynamic nature of the tumor and the higher heterogeneity of resistance mechanisms, underscore the need of a re-biopsy at each new phase of treatment, to better understand the specific molecular profile of each patient and personalize the treatment. The development of not invasive tools for monitoring resistance, such as mutational analysis of circulating free DNA or circulating tumor cells, will be crucial in the near future.
Emerging strategies to overcome resistance
Next-generation ALK inhibitors (e.g., AP26113, LDK378, CH5424802) and HSP90 inhibitors [e.g., STA-9090 (ganetespib), IPI-504, and AUY922] represent two different, promising, strategies to overcome the “ALK-dominant” resistance to crizotinib, both under investigation in early ongoing phase I/II clinical trials (Table 2). Next generation ALK inhibitors potently inhibit the ALK kinase and have activity against many of the resistance mutations in vitro and in vivo, also in the CNS. Although not ALK-specific, HSP90 inhibitors may overcome crizotinib resistance by decreasing proper folding of the oncogenic proteins, including ALK fusion protein. In the case of the activation of other oncogenic drivers, such as upregulation of EGFR and c-KIT, dual inhibition of ALK and these altered enzymes or the downstream effector pathways, such as PI3K/AKT/mTOR and MEK/ERK, represent potential therapeutic strategies. Finally, about 33% of patients enrolled in the PROFILE1007 trial who developed focal brain progression continuing on crizotinib treatment after progression in combination with local therapy. Median duration of crizotinib treatment beyond progression was 15.9 weeks, suggesting a new possible treatment strategy in this specific subset of patients (34). Recently, a case report by Matsuoka et al. (64), described a pronounced antitumor effect of crizotinib rechallenge in a Japanese woman with ALK rearranged NSCLC who initially responded to this drug but subsequently showed tumor progression. Analogously to the T790M mutation in the EGFR gene, the temporary drug withdrawal could restore cell sensitivity to the TKI, through a reduction in the proportion of cells harboring the acquired resistant mutation (65), allowing the subsequent retreatment with the same TKI.
Table 2. New drugs targeting EML4-ALK in NSCLC patients.
| Drug | MTD | DLTs | Patients [N] | ORR (%) | Author |
|---|---|---|---|---|---|
| AP26113 | 120 mg daily | ↑ALT, dyspnea, and hypoxia | CZT-resistant [16] | 76 | Camidge 2013 (57) |
| LDK378 | 750 mg daily | Diarrhea, nausea-vomiting dehydration | CZT-resistant [26] | 81 | Shaw 2012 (58) |
| CH5424802 | 300 mg BID | Not determined | CZT-naïve [49] | 85.7 | Nishio 2012 (59) |
| 900 mg BID | CZT-resistant [37] | 48 | Gadgeel 2013 (60) | ||
| IPI504 | N.A | Not determined | CZT-naïve [3] | 66.6 | Sequist 2010 (61) |
| Ganetespib | N.A | Not determined | CZT-naïve | 50 | El-Hariry 2012 (62) |
| AUY922 | 70 mg/m2 weekly | Anorexia, diarrhea, dark vision, asthenia, fatigue | CZT naïve | 50 | Felip 2012 (63) |
CZT, crizotinib; N.A, not available.
New ALK-inhibitors
LDK378
This compound is a novel, potent, and selective ALK-inhibitor also active against the C1156Y acquired mutation in xenograft models. It was synthesized based on the structure of TAE684, a previous ALK-inhibitor not suitable for clinical development, because of its potential to generate toxic reactive adducts. Extensive medicinal chemistry modifications were successfully undertaken, leading to the synthesis of novel derivatives that did not form reactive adducts and that potently inhibited ALK, like LDK378. Both in biochemical and cellular assays, LDK378 showed potent antiproliferative activity, with an IC50 value of 2.2 nmol/L, lower than the IC50 of crizotinib. It showed an excellent pharmacokinetics profile in animals, with an oral bioavailability of >50%, and also a good tolerability profile.
On the basis of encouraging pre-clinical data a phase I, first in human study was conducted in patients with ALK-positive advanced malignancies that were both treatment-naïve and also progressed on standard targeted therapy. The MTD was defined at 750 mg/daily and dose-limiting toxicities (DLT) included diarrhea, nausea-vomiting and dehydration. A total of 47 patients with NSCLC were evaluable for response; 51% of them achieved a PR. Interestingly, of the 26 patients with NSCLC who had progressed following crizotinib and were treated at dose of ≥400 mg/day, there were 21 (81%) responses (58). Another phase I trial exploring the MTD and/or recommended dose in Japanese patients with tumors harboring ALK alterations, mostly NSCLC (18/19) showed that the safety profile was tolerable and comparable to that of Western patients, with MTD: 750 mg. LDK378 exhibited antitumor activity not only in crizotinib naïve patients but also in resistant ALK+ NSCLC patients, with an ORR of 50%. PRs were observed in 7/9 (77.7%) crizotinib pretreated patients (66).
Considering the phase I promising results LDK378 received the breakthrough therapy designation by the FDA for the treatment of patients with ALK+ metastatic NSCLC who had progressed to crizotinib. Currently, two confirmatory phase II studies of LDK378 in crizotinib-naïve (NCT01685060) and crizotinib-resistant ALK+ (NCT01685138) NSCLC patients are ongoing. In addition, two phase III trials comparing this novel agent with standard chemotherapy in ALK+ NSCLC untreated patients (NCT01828099) or after platinum and crizotinib failure (NCT01828112), are also recruiting patients.
AP26113
This drug is a novel, synthetic, orally-active TKI, which potently inhibits mutant activated forms of ALK, ROS, and EGFR in cell lines, including the ALK-L1196M and EGFR-T790M mutations (67,68), but not native EGFR. In vitro studies showed a potent antiproliferative activity, with an IC50 of 15-45 nmol/L, lower than crizotinib for the wild type and the L1196M mutant ALK. Preliminary data available from a phase I/II ongoing trial showed that AP26113 was well tolerated with preliminary antitumor activity in ALK-positive patients who had failed prior crizotinib. As of April 17th 2013, among 24 evaluable patients, 14 responded: 2/4 (50%) ALK+ crizotinib naïve patients, and 12/16 (76%) ALK+, crizotinib resistant patients. Furthermore, 4/5 patients with untreated or progressing CNS lesions, experienced objective responses, that are currently maintained. The most common AEs included fatigue (40%), nausea (36%) and diarrhea (33%), while the most common grade 3/4 AE was pneumonia (5%). DLT were observed in two patients (grade 3 ALT increase, grade 4 dyspnea and grade 3 hypoxia) (57). Therefore, the dose of 180 mg was determined for the phase II expansion study, which will include five cohorts: ALK-positive NSCLC patients who are naïve or resistant to prior ALK-targeted therapy; EGFR mutated NSCLC patients who are resistant to EGFR targeted therapy; other cancers with abnormalities in the ALK gene or other AP26113 targets (such as ROS1), and finally an ALK+ brain metastasis dedicated cohort. This study is currently recruiting patients (69).
CH5424802
This compound is a highly potent (IC50 =1.9 nmol/L), selective, oral ALK inhibitor. It binds to the ATP site of ALK, preventing the ALK autophosphorylation, showing antitumor activity against NSCLC cells expressing EML4/ALK fusion in preclinical studies, but also against mostly of the second site mutations of the ALK domain, notably the L1196M gatekeeper mutation and C1156Y mutation (70). Preclinical studies in CNS implantation models also suggest a promising antitumor activity of CH5424802 against CNS lesions. A phase I/II trial has been recently presented using CH5424802 in ALK-positive, Japanese, NSCLC patients. In the phase I portion, dose was escalated using an accelerated titration scheme. During that phase, 15 patients were treated with CH5424802. When doses of 300 mg twice daily, the highest dose level defined in the protocol, were administered, MTD was not reached since a DLT was not determined. All patients (at all dose levels) achieved tumor regression and at dose levels of 240 mg twice daily or more under fasting conditions. All seven patients with measurable lesions achieved a PR (71). In the phase II portion of the trial, a total of 34 patients with no prior exposure to ALK inhibitor therapy were treated with CH5424802 at a 300 mg dose twice daily. The reported response rate in the first 15 evaluable patients was 73.3% with 1 CR and 10 PR. No treatment-related AEs led to dose reductions. Main treatment-related AEs among 34 patients were liver function test abnormalities, dysgeusia, rash, nausea and myalgia, most of them grade 1 except for neutropenia. Responses were seen in 3 patients with brain metastases (59). A phase II trial (NCT 01588028) is ongoing. Another phase I study of CH5424802 (900 mg), including 37 ALK+ NSCLC patients who have failed to crizotinib, showed preliminary efficacy and good tolerability profile also in this subset of patients, with PR in 48% and stable disease (SD) in 34%. The most common AEs were fatigue, CPK increase, myalgia, cough, ALT increase, peripheral edema and rash. Grade 3/4 AEs include GGT increase (n=3), neutrophil decrease (n=2), but no grade 3 nausea, vomiting, diarrhea or edema were reported (60). Furthermore, within 3-6 weeks after treatment initiation, CH5424802 dramatically shrank brain lesions previously progressed on crizotinib, suggesting a possible role in replacing or delaying the need of brain radiation in ALK-positive NSCLC patients (72).
HSP90-inhibitors
The inhibition of the molecular chaperone, HSP90, represents a new promising strategy to overcome the “ALK-dominant” crizotinib resistance (due to secondary mutations or amplification) in ALK-rearranged NSCLC. The inhibition of HSP90 prevents from regulating the activation and stability of its client proteins, including oncogenic proteins such as ALK, facilitating their proteasome mediated degradation (73). This degradation leads to a potent inhibition of downstream signaling pathways, the arrest of cell growth and the induction of apoptosis in cells carrying the EML4-ALK fusion (74). Some in vitro studies have shown that Hsp90 inhibitors are active against all types of acquired ALK mutations identified, particularly in the case of L1196M mutation, and also against ALK amplification (55). Furthermore, HSP90 inhibition led to a reduced expression of EML4-ALK in animal models (56). Several HSP-inhibitors have been developed and tested in the clinical setting. In fact, in those clinical trials usually including NSCLC patients with a heterogeneous molecular profile (EGFR-mutations, KRAS-mutations, ALK-rearrangements), promising results have been observed in the ALK-rearranged subgroup of patients. In a phase II study testing the HSP90 Inhibitor IPI-504, 2 out of 3 ALK+ NSCLC patients enrolled responded, with a median PFS of 7 months (61). This is probably related to the crucial role of HSP90 as a chaperone for ALK protein. In fact, preclinical studies showed high sensitivity of EML4-ALK+ NSCLC cells to the HSP90-inhibitor IPI-504 which induces tumor regression in a xenograft model, showing activity also against cells that have been selected for resistance to ALK kinase inhibitors (74). In a phase II clinical trial with another HSP90-Inhibitor, ganetespib, in monotherapy, a total of 99 NSCLC patients were included and divided according to their molecular profile into three cohorts: EGFR+, KRAS+, and EGFR/KRAS wild type. Among the wild type subgroup, only four responders have been reported, and all four patients have been subsequently found to be ALK+ (75). In another study, ganetespib showed an ORR of 50% in ALK+ crizotinib-naïve population (62). On the basis of these encouraging evidences, several clinical trials are currently exploring the activity of the HSP-inhibitor ganetesbip, alone (NCT01562015), or in combination with crizotinib (NCT01579994) in ALK+ NSCLC. A phase II study with AUY922 was conducted in patients with previously treated advanced NSCLC, stratified by molecular status (ALK-positive, EGFR-mutant, KRAS mutant or wild-type, EGFR/K-ras/ALK “triple negative”). Interestingly, clinical activity of AUY922 was mainly observed among patients with ALK-positive tumors and EGFR mutant NSCLC, with PR confirmed in 25% of ALK+ patients (50% in crizotinib-naive ALK+ patients), 18% in EGFR mutated patients, 13% in wild-type patients, and none of the KRAS-mutated patients. Estimated median PFS rates were 42% and 34% at 18 weeks in ALK-positive and EGFR-mutant patients, respectively. The most common AEs were diarrhea (73%), visual disorders (71%), and nausea (43%) (63). Based on the above mentioned data, there are multiple studies underway involving patients with NSCLC receiving AUY922 either alone or in combination with other targeted agents, especially for those with either ALK+ or EGFR-mutated tumors. Finally, AT13387 is another Hsp90 inhibitor which showed activity both alone and in combination with crizotinib against ALK+ NSCLC cells lines and xenograft models (76). In the phase I, dose-escalation study including all solid tumors, the DLT observed were infusion-related symptoms, gastrointestinal effects, and fatigue. A phase I/II trial of AT13387 in NSCLC with or without crizotinib is currently ongoing.
Conclusions
Crizotinib represents the last targeted agent approved for the treatment of NSCLC. In phase III randomized clinical trials it has shown to be superior to conventional cytotoxic agents in second line treatment of selected groups of patients with ALK-rearranged NSCLC. We are eagerly waiting for the results of PROFILE 1014 in the frontline setting. The clinical development of crizotinib has been an amazing success story in lung cancer translational-research. Its accelerated approval has benefited many patients whose tumors harbor this novel EML4/ALK translocation, marking the beginning of the new decade of targeted therapy, characterized by new ethical and scientific considerations.
Common to others targeted therapies that have revolutionized the therapy of NSCLC, such as EGFR-TKIs, acquired resistance to crizotinib inevitably develops during the treatment. The novel generation of ALK inhibitors and the Hsp90-inhibitors represent promising alternatives for those patients whose tumors develop the so called “ALK-dominant” mechanisms of resistance. Combination of different targeted agents may be considered to overcome the activation of alternative signaling pathways, such as EGFR, KRAS. To date, the causes of progression remain unknown in about one-third of patients receiving crizotinib. Therefore, elucidating acquired resistance mechanisms, preventing CNS progressions, and developing adequate therapeutic strategies such as the optimal sequence of treatment and the best combinations, are the crucial questions to be answered by dedicated translational research studies. Only a close collaboration between oncologists, pathologists and molecular biologists may help to find the right answers in an efficient and timely fashion.
Acknowledgements
Disclosure: The authors declare no conflict of interest.
References
- 1.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-39. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-500. [DOI] [PubMed] [Google Scholar]
- 3.Peters S, Adjei AA, Gridelli C, et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23Suppl 7:vii56-64. [DOI] [PubMed] [Google Scholar]
- 4.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561-6. [DOI] [PubMed] [Google Scholar]
- 5.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994;263:1281-4. [DOI] [PubMed] [Google Scholar]
- 6.Chiarle R, Voena C, Ambrogio C, et al. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 2008;8:11-23. [DOI] [PubMed] [Google Scholar]
- 7.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ouyang T, Bai RY, Bassermann F, et al. Identification and characterization of a nuclear interacting partner of anaplastic lymphoma kinase (NIPA). J Biol Chem 2003;278:30028-36. [DOI] [PubMed] [Google Scholar]
- 9.Riera L, Lasorsa E, Ambrogio C, et al. Involvement of Grb2 adaptor protein in nucleophosmin-anaplastic lymphoma kinase (NPM-ALK)-mediated signaling and anaplastic large cell lymphoma growth. J Biol Chem 2010;285:26441-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res 2009;15:3143-9. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki T, Rodig SJ, Chirieac LR, et al. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010;46:1773-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heuckmann JM, Balke-Want H, Malchers F, et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res 2012;18:4682-90. [DOI] [PubMed] [Google Scholar]
- 13.Klempner SJ, Cohen DW, Costa DB. ALK translocation in non-small cell lung cancer with adenocarcinoma and squamous cell carcinoma markers. J Thorac Oncol 2011;6:1439-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kris MG, Johnson BE, Kwiatkowski DJ, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI’s Lung Cancer Mutation Consortium (LCMC). J Clin Oncol 2011;29:abstr CRA7506.
- 15.Rosell R, Sureda BM, Costa C, et al. Concomitant actionable mutations and overall survival (OS) in EGFR-mutant non-small-cell lung cancer (NSCLC) patients (p) included in the EURTAC trial: EGFR L858R, EGFR T790M, TP53 R273H and EML4-ALK (v3). ESMO 2012: Abstract 929. [Google Scholar]
- 16.Yang P, Kulig K, Boland JM, et al. Worse disease-free survival in never-smokers with ALK+ lung adenocarcinoma. J Thorac Oncol 2012;7:90-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 2011;12:1004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koh Y, Kim DW, Kim TM, et al. Clinicopathologic characteristics and outcomes of patients with anaplastic lymphoma kinase-positive advanced pulmonary adenocarcinoma: suggestion for an effective screening strategy for these tumors. J Thorac Oncol 2011;6:905-12. [DOI] [PubMed] [Google Scholar]
- 19.Camidge DR, Kono SA, Flacco A, et al. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res 2010;16:5581-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wynes MW, Sholl LM, Dietel M, et al. An International Interpretation Study Using the ALK IHC Antibody D5F3 and a Sensitive Detection Kit Demonstrates High Concordance between ALK IHC and ALK FISH and between Evaluators. J Thorac Oncol 2014;9:631-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi ES, Boland JM, Maleszewski JJ, et al. Correlation of IHC and FISH for ALK gene rearrangement in non-small cell lung carcinoma: IHC score algorithm for FISH. J Thorac Oncol 2011;6:459-65. [DOI] [PubMed] [Google Scholar]
- 22.Selinger CI, Rogers TM, Russell PA, et al. Testing for ALK rearrangement in lung adenocarcinoma: a multicenter comparison of immunohistochemistry and fluorescent in situ hybridization. Mod Pathol 2013;26:1545-53. [DOI] [PubMed] [Google Scholar]
- 23.Sakai Y, Nakai T, Ohbayashi C, et al. Immunohistochemical profiling of ALK fusion gene-positive adenocarcinomas of the lung. Int J Surg Pathol 2013;21:476-82. [DOI] [PubMed] [Google Scholar]
- 24.Park HS, Lee JK, Kim DW, et al. Immunohistochemical screening for anaplastic lymphoma kinase (ALK) rearrangement in advanced non-small cell lung cancer patients. Lung Cancer 2012;77:288-92. [DOI] [PubMed] [Google Scholar]
- 25.Martinez P, Hernández-Losa J, Montero M, et al. Fluorescence in situ hybridization and immunohistochemistry as diagnostic methods for ALK positive non-small cell lung cancer patients. PLoS One 2013;8:e52261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conklin CM, Craddock KJ, Have C, et al. Immunohistochemistry is a reliable screening tool for identification of ALK rearrangement in non-small-cell lung carcinoma and is antibody dependent. J Thorac Oncol 2013;8:45-51. [DOI] [PubMed] [Google Scholar]
- 27.Wu YC, Chang IC, Wang CL, et al. Comparison of IHC, FISH and RT-PCR methods for detection of ALK rearrangements in 312 non-small cell lung cancer patients in Taiwan. PLoS One 2013;8:e70839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wallander ML, Geiersbach KB, Tripp SR, et al. Comparison of reverse transcription-polymerase chain reaction, immunohistochemistry, and fluorescence in situ hybridization methodologies for detection of echinoderm microtubule-associated proteinlike 4-anaplastic lymphoma kinase fusion-positive non-small cell lung carcinoma: implications for optimal clinical testing. Arch Pathol Lab Med 2012;136:796-803. [DOI] [PubMed] [Google Scholar]
- 29.Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther 2007;6:3314-22. [DOI] [PubMed] [Google Scholar]
- 30.Kwak EL, Camidge DR, Clark J, et al. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066. J Clin Oncol 2009;7:abstr 3509.
- 31.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol 2012;13:1011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim DK, Ahn MJ, Shi Y, et al. Updated results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol 2012;30:abstr 7533.
- 34.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385-94. [DOI] [PubMed] [Google Scholar]
- 35.Takeda M, Okamoto I, Sakai K, et al. Successful long-term treatment with pemetrexed of NSCLC associated with EML4-ALK and low thymidylate synthase expression. Clin Lung Cancer 2012;13:157-9. [DOI] [PubMed] [Google Scholar]
- 36.Rimkunas VM, Crosby KE, Li D, et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res 2012;18:4449-57. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med 2012;18:378-81. [DOI] [PubMed] [Google Scholar]
- 38.Pan Y, Zhang Y, Li Y, et al. ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: A comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer 2014;84:121-6. [DOI] [PubMed] [Google Scholar]
- 39.Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012;30:863-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ou SH, Tan J, Yen Y, et al. ROS1 as a ‘druggable’ receptor tyrosine kinase:lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther 2012;12:447-56. [DOI] [PubMed] [Google Scholar]
- 41.Ou S, Camidge R, Engelman J, et al. Clinical activity of crizotinib in patients with advanced non-small cell lung cancer harboring ROS1 gene rearrangement. ESMO 2012:abstract 2091. [Google Scholar]
- 42.Chen YT, Chang JW, Liu HP, et al. Clinical implications of high MET gene dosage in non-small cell lung cancer patients without previous tyrosine kinase inhibitor treatment. J Thorac Oncol 2011;6:2027-35. [DOI] [PubMed] [Google Scholar]
- 43.Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol 2011;6:942-6. [DOI] [PubMed] [Google Scholar]
- 44.Cappuzzo F, Jänne PA, Skokan M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol 2009;20:298-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weickhardt AJ, Scheier B, Burke JM, et al. Continuation of EGFR/ALK inhibition after local therapy of oligoprogressive disease in EGFR mutant (Mt) and ALK+ non-small cell lung cancer (NSCLC). J Clin Oncol 2012;30:abstr 7526.
- 46.Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443-5. [DOI] [PubMed] [Google Scholar]
- 47.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med 2012;4:120ra17. [DOI] [PMC free article] [PubMed]
- 48.Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 2012;18:1472-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010;363:1734-9. [DOI] [PubMed] [Google Scholar]
- 50.Lovly CM, Heuckmann JM, de Stanchina E, et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res 2011;71:4920-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azam M, Seeliger MA, Gray NS, et al. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol 2008;15:1109-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi YL, Soda M, Yamashita Y, et al. EML4–ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010;363:1734-9. [DOI] [PubMed] [Google Scholar]
- 53.Sun HY, FQ J. A molecular dynamics investigation on the crizotinib resistance mechanism of C1156Y mutation in ALK. Biochem Biophys Res Commun 2012;423:319-24. [DOI] [PubMed] [Google Scholar]
- 54.Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 2011;71:6051-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A 2011;108:7535-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ou S, Govindan R, Eaton KD, et al. Phase I/II dose-finding study of crizotinib (CRIZ) in combination with erlotinib (E) in patients (pts) with advanced non-small cell lung cancer (NSCLC). J Clin Oncol 2012;30:abstr 2610.
- 57.Camidge DR, Bazhenova L, Salgia R, et al. Updated results of a first-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies. J Thorac Oncol 2013:8:S296 Available online: http://journals.lww.com/jto/toc/2013/11001 [Google Scholar]
- 58.Shaw AT, Camidge DR, Felip E. Results of a first-in-human phase I study of the ALK inhibitor LDK378 in advanced solid tumors. Ann Oncol 2012;23:abstr 4400.
- 59.Nishio M, Kiura K, Nakagawa K. A phase I/II study of ALK inhibitor CH5424802 in patients with ALK-positive NSCLC; safety and efficacy interim results of the phase II portion. ESMO 2012: Abstract 4410. [Google Scholar]
- 60.Gadgeel S, Ou SH, Chiappori AA, et al. A Phase 1 Dose Escalation Study Of A New ALK Inhibitor, CH5424802/RO5424802, In ALK+ Non-Small Cell Lung Cancer (Nsclc) Patients Who Have Failed Crizotinib (Nct01588028). J Thorac Oncol 2013:8:S199 Available online: http://journals.lww.com/jto/toc/2013/11001 [Google Scholar]
- 61.Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol 2010;28:4953-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.El-Hariry I, Acquaviva J, Jiang Q, et al. Ganetespib: an effective strategy to overcome crizotinib resistence in ALK driven-cancers. 3rd ELCC: Abstract 251P.
- 63.Felip E, Carcereny E, Barlesi F, et al. Phase II activity of the HSP90 inhibitor AUY922 in patients with ALK-Rearrenged (ALK+) or EGFR mutated advanced non-small cell lung cancer. Ann Oncol 2012;23:ix152-74. [Google Scholar]
- 64.Matsuoka H, Kurata T, Okamoto I, et al. Clinical response to crizotinib retreatment after acquisition of drug resistance. J Clin Oncol 2013;31:e322-3. [DOI] [PubMed] [Google Scholar]
- 65.Chmielecki J, Foo J, Oxnard GR, et al. Optimization of dosing for EGFRmutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med 2011;3:90ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seto T, Murakami H, Hirai F, et al. Phase i study of the ALK inhibitor LDK378 in Japanese patients with advanced, ALK rearranged NSCLC and other tumors harboring genetic ALK alterations. J Thorac Oncol 2013:8:S595 Available online: http://journals.lww.com/jto/toc/2013/11001 [Google Scholar]
- 67.Zhang S, Wang F, Keats J, et al. AP26113, a potent ALK inhibitor, overcomes mutations in EML4-ALK that confer resistance to PF-02341066. Proceedings of the 101st Annual Meeting of the American Association for Cancer Research: 2010 Apr 17-21; Washington, DC 2010. [Google Scholar]
- 68.Rivera VM, Wang F, Anjum R, et al. AP26113 is a dual ALK/EGFR inhibitor: characterization against EGFR T790M in cell and mouse models of NSCLC. AACR 103rd Annual Meeting: AACR 2012. [Google Scholar]
- 69.Gettinger S, Weiss G, Salgia R, et al. A first-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies. Ann Oncol 2012;23Suppl 9:ix152-74. [Google Scholar]
- 70.Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679-90. [DOI] [PubMed] [Google Scholar]
- 71.Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, openlabel, phase 1-2 study. Lancet Oncol 2013;14:590-8. [DOI] [PubMed] [Google Scholar]
- 72.Ou SH, Gadgeel S, Chiappori AA, et al. Consistent therapeutic efficacy of CH5424802/RO5424802 in brain metastases among crizotinib-refractory alk-positive non-small cell lung cancer (NSCLC) patients in an ongoing phase I/II study (NCT01588028). J Thorac Oncol 2013:8:S200 Available online: http://journals.lww.com/jto/toc/2013/11001 [Google Scholar]
- 73.Neckers L, Workman P.Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 2012;18:64-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Normant E, Paez G, West KA, et al. The HSP90 Inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene 2011;30:2581-6. [DOI] [PubMed] [Google Scholar]
- 75.Socinski MA, Goldman J, El-Hariry I, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced nonsmall cell lung cancer. Clin Cancer Res 2013;19:3068-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smyth T, Munck J, Rodriguez-Lopez A, et al. Combining the Hsp90 inhibitor, AT13387, with crizotinib improves response in an ALK positive model of NSCLC. J Thorac Oncol 2013;8:S163. [Google Scholar]