

Abstract
Background
Epigenetic modifications play important roles in the regulation of plant development. DNA methylation is an important epigenetic modification that dynamically regulates gene expression during developmental processes. However, little studies have been reported about the methylation profiles of photoperiod- and thermo-sensitive genic male sterile (PTGMS) rice during the fertility transition.
Results
In this study, using methylated DNA immunoprecipitation sequencing (MeDIP-seq), the global DNA methylation patterns were compared in the rice PTGMS line PA64S under two different environments (different temperatures and day lengths). The profiling of the DNA methylation under two different phenotypes (sterility and fertility) revealed that hypermethylation was observed in PA64S (sterility), and 1258 differentially methylated regions (DMRs) were found between PA64S (sterility) and PA64S (fertility). Twenty differentially methylated genes of them were further validated through bisulfite sequencing, and four of these genes were analyzed by qRT-PCR. Especially, a differentially methylated gene (LOC_Os08g38210), which encoded transcription factor BIM2, is a component of brassinosteroid signaling in rice. The hypermethylated BIM2 gene may suppress some downstream genes in brassinosteroid signaling pathway, and thus affect the male fertility in PA64S.
Conclusions
The results presented here indicated that hypermethylation was observed in PA64S (sterility). Gene Ontology (GO) analysis and KEGG analysis revealed that flavone and flavonol biosynthrsis, circadian rhythm, photosynthesis and oxidative phosphorylation pathways were involved in sterility-fertility transition of PA64S.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1317-7) contains supplementary material, which is available to authorized users.
Keywords: Oryza sativa, DNA methylation, PTGMS, Fertility transition, Gene expression
Background
DNA methylation is an indispensable epigenetic modification of plant genomes. It occurs predominantly in the CG context in both plants and animals [1-3]. The state of CpG methylation regulates and stabilizes chromatin structure, and possibly regulates the accessibility of these DNA regions to the transcriptional machinery [4]. Studies have shown that DNA methylation plays an important role in many plant processes, including transposon silencing, virus defence, and gene imprinting [5-7].
In plant, DNA methylation is mainly distributed in heterochromatic regions that are composed of tandem or inverted repeats, i.e., transposons. Transposable elements (TEs) are usually highly methylated along their entire length, but DNA methylation within genes is distributed away from the 5′ and 3′ ends. It has been proposed that DNA methylation is primarily used to inhibit the transcription and movement of TEs to protect genomes against uncontrolled insertions. Some studies have reported that transposons or retrotransposons are activated in response to environmental stress and may generate true genetic or epigenetic changes for adapting to stresses [8]. In rice, epigenetic regulation has been reported to affect the transposition activity of Tos17 and modulates the activity of the neighboring genes [9]. Highly methylated transposons may affect transcription of neighboring genes. For example, the patterns of DNA methylation in long terminal repeat (LTR) transposable elements differed between rice leaves and roots and affected the transcription of the neighboring genes [10].
Recently, three DNA treatment techniques have been typically employed for DNA methylation detection: (i) methylation sensitive amplified polymorphism (MSAP), (ii) methylated DNA enrichment (MeDIP), (iii) bisulfite-sequencing. With the advent of new technologies, the elucidation of the genome-wide methylation profiles is possible. In this regard, the immunoprecipitation of methylated DNA by monoclonal antibodies specific to 5-methylcytidine (5mC) combined with high-throughput sequencing (MeDIP-seq) has been used as a valuable tool to map methylated DNA at the genomic scale [11,12]. The MeDIP-seq approach employs an antibody against 5-methylcytosine or methyl-binding domain proteins to capture methylated DNA, which are subsequently subjected to next generation sequencing [11]. After methylated DNA enrichment, the unmethylated DNA fragments are removed from the genome samples for reducing the redundancy. Thus, a relatively lower sequencing throughput is required, compared with bisulfite-sequencing. In addition, MeDIP is more unbiased and less limited than MSAP. Due to these advantages, MeDIP-seq is considered to have great potential for the development of cost-effective and unbiased strategies for whole genome DNA methylation profiling.
Rice (Oryza sativa L.) is one of the most important grain crops worldwide and provides a staple food for almost half of the world’s population [13]. Recently, hybrid rice has made a tremendous contribution to food security both in China and many other countries which are losing arable land. Hybrid rice technologies are mainly based on two well-known male sterility systems, namely CMS (Cytoplasmic Male Sterile) and EGMS (Environmentally sensitive Genic Male Sterile). Compared with the three-line system (CMS system), the two-line system, is based on the discovery and application of EGMS lines, which serve as both the male sterile lines and maintainer lines under different environmental conditions [14]. PTGMS (Photoperiod- Thermo-Sensitive Genic Male Sterile) is the major type of EGMS germplasm resources and has been widely used for breeding two-line hybrid rice. The PTGMS system is advantageous for broad restoration ability, easy maintenance, and multiplication [15-17]. Thus, the utilization of PTGMS lines is an important approach for the better exploitation of heterosis in rice.
Peiai 64S (PA64S) is one of the most important indica rice genic male sterile lines in two-line system. Its paternal line is Peiai64, and its maternal line is Nongken58, which is the first photoperiod- sensitive genic male sterile line discovered by Mingsong Shi in Hubei, China in 1973 [18]. Using PA64S as the maternal line, more than 10 two-line hybrid rice varieties have been developed since 1996 in China. Among these new varieties, LiangYouPei9 (LYP9) is an elite super hybrid rice and increases grain yield by approximately 20% per hectare compared with other hybrid rice lines in the past years [7,19]. The fertility transition of PA64S was controlled by the photoperiod and temperature: the sterility trait exhibited at temperatures higher than 23.5°C during the anther development and long-day (LD) (≥14 h) conditions can suppress the fertility transition. Therefore, under lower temperatures (~21-23°C) and short-day (SD) (<14 h) conditions during anther development, PA64S can transform from male sterility to fertility [20,21].
To date, some studies have reported that a substitution of C-to-G in the male-fertility-associated noncoding RNA p/tms12-1 in PA64S produced a mutant small RNA, namely osa-smR5864m. This mutated noncoding small RNA gene may lead to PTGMS in PA64S [21]. Furthermore, the transcriptome of the PGMS rice Nongken 58S was significantly suppressed under LD condition and the circadian rhythm pathway had been shown to be involved in the male sterility transition [22]. Another noncoding RNA called LDMAR was also found in Nongken 58S and was required for the fertility transition. A siRNA (Psi–LDMAR) was detected to associate with the DNA methylation pattern of the LDMAR promoter in Nongken 58S. And increasing methylation in the promoter of LDMAR of Nongken 58S also reduced the expression of LDMAR, leading to male sterility [23]. These results suggested that DNA methylation or RNA-dependent DNA methylation (RdDM) might be involved in the regulation of PTGMS.
Brassinosteroids (BRs), a class of steroid hormones, have been reported to control male fertility by regulating the expression of several key genes involved in anther and pollen development, such as SPL/NZZ, TDF1, AMS, AtMYB103 and MS1 [24,25]. The basic helix-loop-helix (bHLH) protein BIM1 (BES1-interacting Myc-like1) is a brassinosteroid signaling component involved in regulating BR-induced genes in Arabidopsis [26]. BRI1- EMSSUPRESSOR1 (BES1) is shown to interact with BIM1 and together bind to the E-box of a BR-induced gene [27].
In this study, we investigated the global DNA methylation alterations in young panicles of the PTGMS line PA64S under two different environmental conditions using the MedIP-seq method. The aim of this work was to explore the DNA methylation patterns and their influences on gene expression during the transition from sterile to fertile in PA64S. The results will provide some information for a better understanding of the role of DNA methylation in PTGMS rice.
Results
Cytological observation of pollen morphology
Under higher temperature and LD, anthers from PA64S were light yellow, and its pollen wizened as well as unstained by I2-KI (Figure 1a, b), displaying sterility (S). However, under lower temperatures (~21-23°C) and SD condition, PA64S showed male fertility. The anthers of PA64S (fertility, F) were dark yellow, and its spherical pollen could be darkly stained by I2-KI, indicating much starch accumulation (Figure 1c, d).
Figure 1.
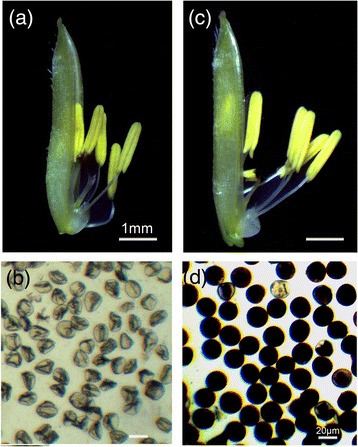
Cytological observation of pollen morphology. (a) and (c), the anther phenotypes of PA64S under two environmental conditions. (b) and (d) 1% I2-KI staining of the pollen grains of PA64S under two environmental conditions.
Genome-wide DNA methylation profiles of PA64S (S) and PA64S (F)
To decipher the genome-wide DNA methylome of PA64S under two different environmental conditions, we immunoprecipitated sheared genomic DNA with an antibody which specifically recognizes 5-methylcytosine and sequenced the enriched methylated DNA. Only the uniquely mapped reads were used in the scanning of the methylation peak. A total of 24,489,796 raw reads were generated for the two samples, and more than 92% of the reads were mapped, and about 58% of the reads in each sample were uniquely mapped to the rice genome in each sample (Table 1 and Additional file 1).
Table 1.
Data generated by MeDIP-seq in PA64S (S) and PA64S (F)
| Total MeDIP-Seq data | Percentage of mapped reads in total reads | Unique mapped reads | Unique mapped bases | Percentage of unique mapped reads | |
|---|---|---|---|---|---|
| PA64S(S) | 24,489,796 | 92.05% | 14,676,561 | 719,151,489 | 59.93% |
| PA64S(F) | 24,489,796 | 92.20% | 14,410,405 | 706,109,845 | 58.84% |
According the principle of MedIP method, analysis of the methylation enrichment in different components of the genome showed that 2 kb region upstream and 2 kb region downstream of the CpG Island and intragenic had enriched more methylation reads, especially, when departing from the CpG Island or intragenic regions (Figure 2). Furthermore, the upstream 2k and 5’UTR or downstream 2k and 3’UTR were enriched more reads (Figure 3a).
Figure 2.
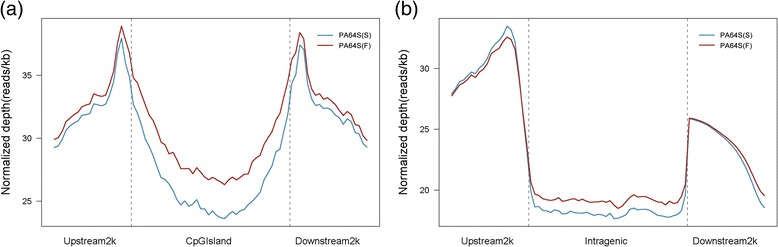
Distribution of reads around CpG Islands and gene body. (a) Distribution of reads around CpG Islands; (b) Distribution of reads around gene body. The upstream and downstream 2 kb regions were split into 20 equal regions. In the gene body, each gene was split into 40 equal regions. For each region, the normalized number of reads was calculated. The “Y” axis is the average of the normalized depth for each region.
Figure 3.
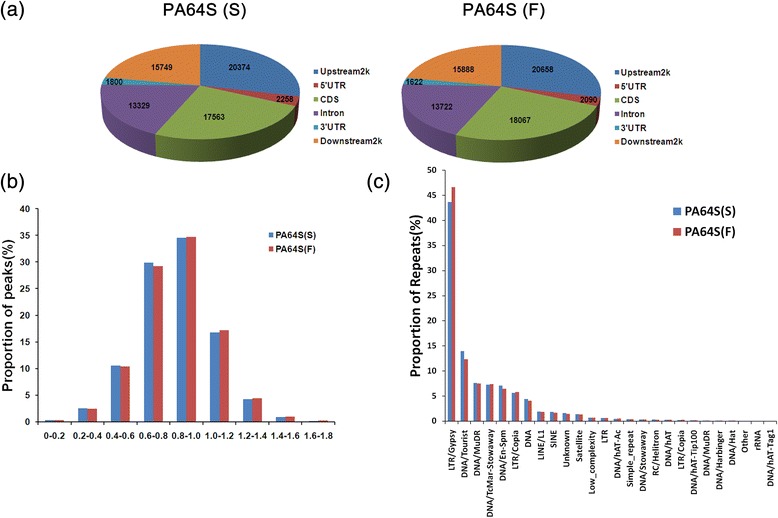
Different distributions of PA64S(S) and PA64S (F). (a) Peak distribution in different components of the PA64S(S) and PA64S (F) genome. (b) Distribution of methylated peaks of different repeat types in PA64S (S) and PA64S (F). (c) Distribution of CpG (O/E) in PA64S(S) and PA64S (F).
The CpG observed/expected (O/E) ratio of PA64S (S) and PA64S (F) was mainly in a range of 0.4 to 1.2. There was no significant difference in the CpG (O/E) ratio between PA64S(S) and PA64S (F). However, PA64S(S) had more peaks distributed at CpG (O/E) ratios of 0 ~ 0.8, while the peaks of PA64S (F) were inclined to CpG (O/E) ratios of 1.0 ~ 1.8 (Figure 3b). The CpG O/E ratio is a widely used parameter to predict the DNA methylation level based on C → T transition mechanisms resulting from deamination of mCs over the course of evolution [2,28]. In our study, more peaks of PA64S (S) were distributed in 0 ~ 0.8 (Figure 3c), and the methylation level of PA64S (S) was significant higher than that of PA64S (F) (Figure 4). These results were consistent with previous studies, showing that genes with higher methylation levels tend to have lower CpG O/E ratios [28,29].
Figure 4.
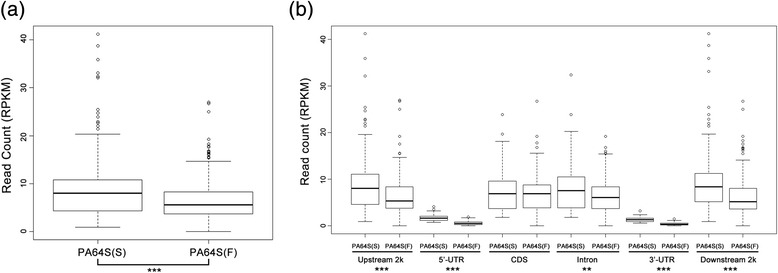
Boxplots showing the 5mC content (read count, y axis) in 1 kb tiled windows for PA64S(S) and PA64S (F). (a) Total reads of PA64S(S) and PA64S (F); (b) Reads for each elements of PA64S(S) and PA64S (F). The asterisks indicate significant differences between PA64S(S) and PA64S (F), as determined by Student’s t test (**P < 0.01, ***P < 0.001).
To get insight into the DNA methylation status of PA64S (S) and PA64S (F), RPKMs (Reads per 1 kb window per million mapped reads) were calculated for each sample and different elements (Figure 4). The results showed that PA64S(S) had higher methylation level than that of PA64S (F). And the DNA methylation levels between PA64S(S) and PA64S (F) were observed to be significantly different on the upstream 2k, 5’UTR, intron, 3’UTR, and downstream 2k regions of the gene body (p < 0.01, p < 0.001). However, there were not significant differences on CDS (coding DNA sequence) of the differential methylated regions (DMRs).
DNA methylation patterns in genes and TE regions
Comparison of the gene methylation status showed that there were 725 genes hypermethylated and 533 genes hypomethylated in PA64S (S). Most of these DMRs were observed on the upstream 2k and downstream 2k of genes and many of them were hypermethylated in PA64S (S) (Table 2 and Figure 4). The number of hypermethylated genes showed significant difference between PA64S (S) and PA64S (F) in the upstream2k, 5’UTR, 3’UTR, and downstream 2k regions comparing with the hypomethylated genes (Table 2 and Figure 4). Interestingly, the differentially methylated genes of the CDS regions between PA64S (S) and PA64S (F) exhibited almost no differences (Table 2 and Figure 4).
Table 2.
Numbers of differentially methylated genes between PA64S(S) and PA64S (F) in different gene regions
| Contrast | PA64S(S) VS. PA64S(F)- Hyper | PA64S(S) VS. PA64S(F)- Hypo |
|---|---|---|
| Upstream 2k | 245 | 122 |
| 5’UTR | 25 | 10 |
| CDS | 86 | 84 |
| Intron | 93 | 69 |
| 3’UTR | 23 | 10 |
| Downstream 2k | 253 | 106 |
| Total | 725 | 533 |
The significant enrichment of down-regulated genes in PA64S (S) was annotated (Table 3 and Additional file 2). These differential genes included: transposon, retrotransposon, phosphate carrier protein, NB-ARC domain containing protein, ent-kaurene synthase, PsbP and so on. They were involved in disease resistance, signal transduction, transport, chloroplast development and photosynthesis, energy metabolism, and pollen development. Further research on these genes may elucidate the functions of hypermethylation in the fertility transition of PA64S.
Table 3.
Significant GO enrichment of hypermethylated genes in PA64S (S)
| Locus ID | DMR location | Annotation | p-value |
|---|---|---|---|
| LOC_Os01g70080 | Upstream 2k/ Intron | NB-ARC domain containing protein | 1.43E-42 |
| LOC_Os07g04950 | Upstream 2k | Early nodulin 75-like protein | 2.33E-29 |
| LOC_Os01g70100 | Upstream 2k/5'-UTR | zinc finger DHHC domain-containing protein | 1.21E-22 |
| LOC_Os09g38100 | Upstream 2k | phosphate carrier protein, mitochondrial precursor | 1.84E-15 |
| LOC_Os09g39570 | Upstream 2k | beta-amylase | 2.29E-15 |
| LOC_Os03g25840 | Upstream 2k | Amino acid permease family protein | 2.8E-14 |
| LOC_Os12g30520 | Upstream 2k | pumilio-family RNA binding repeat containing protein | 1.06E-13 |
| LOC_Os12g35730 | Upstream 2k | transposon protein, putative, CACTA, En/Spm sub-class | 1.36E-12 |
| LOC_Os10g31780 | Upstream 2k | oxidoreductase | 2.25E-12 |
| LOC_Os01g04330 | Upstream 2k | Probable calcium-binding protein CML16 | 2.77E-11 |
| LOC_Os06g42420 | Upstream 2k | transposon protein, putative, unclassified | 1.08E-10 |
| LOC_Os01g70360 | Upstream 2k | Long chain base biosynthesis protein 2b | 1.21E-10 |
| LOC_Os12g40130 | Upstream 2k | phosphoribosyl transferase | 6.86E-10 |
| LOC_Os01g18620 | Upstream 2k | transferase family protein | 7.06E-10 |
| LOC_Os12g30520 | 5'-UTR | pumilio-family RNA binding repeat containing protein | 1.06E-13 |
| LOC_Os04g30490 | 5'-UTR/ CDS/ Intron | MATE efflux family protein | 3.05E-11 |
| LOC_Os01g62890 | 5'-UTR | prenylated rab acceptor | 3.75E-11 |
| LOC_Os01g70540 | CDS/ Intron | retrotransposon protein | 3.94E-31 |
| LOC_Os01g20880 | CDS | OsWAK3 | 1.06E-15 |
| LOC_Os01g70040 | CDS/Intron | transposon protein, CACTA, En/Spm sub-class | 1.08E-14 |
| LOC_Os01g70030 | CDS/Intron | transposon protein, CACTA, En/Spm sub-class | 1.5E-14 |
| LOC_Os04g02920 | CDS/3'-UTR | leucine-rich repeat family protein | 2.2E-14 |
| LOC_Os04g35210 | CDS | Leucine Rich Repeat family protein | 9.11E-12 |
| LOC_Os06g03850 | CDS | impaired sucrose induction 1 | 8.63E-11 |
| LOC_Os07g41610 | CDS/Intron | retrotransposon protein, Ty1-copia subclass | 1.86E-10 |
| LOC_Os06g36540 | CDS | retrotransposon protein, Ty3-gypsy subclass | 9.81E-10 |
| LOC_Os07g04940 | Intron | PE-PGRS family protein precursor | 2.33E-29 |
| LOC_Os08g21920 | Intron | retrotransposon protein | 5.37E-23 |
| LOC_Os06g21950 | Intron | inorganic phosphate transporter | 7.93E-15 |
| LOC_Os12g19549 | Intron | telomerase reverse transcriptase | 1.36E-12 |
| LOC_Os09g23650 | Intron | FAM10 family protein | 2.88E-11 |
| LOC_Os06g03850 | Intron | impaired sucrose induction 1 | 8.63E-11 |
| LOC_Os10g30090 | Intron | amino acid permease | 4.79E-10 |
| LOC_Os09g21770 | 3'-UTR | ES43 protein | 1.03E-12 |
| LOC_Os04g47410 | 3'-UTR | DHHC zinc finger domain containing protein | 5.04E-12 |
| LOC_Os01g70190 | Downstream 2k | exostosin family domain containing protein | 9.18E-65 |
| LOC_Os01g70550 | Downstream 2k | heparan-alpha-glucosaminide N-acetyltransferase | 3.94E-31 |
| LOC_Os12g36410 | Downstream 2k | transposon protein, Pong sub-class | 9.30E-24 |
| LOC_Os01g70810 | Downstream 2k | homeobox domain containing protein | 2.97E-23 |
| LOC_Os01g70270 | Downstream 2k | auxin response factor | 2.93E-21 |
| LOC_Os04g12060 | Downstream 2k | retrotransposon protein | 2.75E-19 |
| LOC_Os01g67980 | Downstream 2k | cysteine proteinase EP-B 1 precursor | 1.81E-16 |
| LOC_Os01g71070 | Downstream 2k | xylanase inhibitor | 1.73E-15 |
| LOC_Os09g39560 | Downstream 2k | genetic modifier | 2.29E-15 |
| LOC_Os01g70020 | Downstream 2k | DEK C terminal domain containing protein | 3.62E-15 |
| LOC_Os05g23610 | Downstream 2k | protein phosphatase inhibitor 2 containing protein | 2.20E-14 |
| LOC_Os01g70690 | Downstream 2k | Rapid ALkalinization Factor RALF family protein | 7.42E-14 |
| LOC_Os02g17780 | Downstream 2k | ent-kaurene synthase, chloroplast precursor | 8.65E-14 |
| LOC_Os05g12210 | Downstream 2k | chalcone synthase | 1.15E-12 |
| LOC_Os04g47420 | Downstream 2k | transmembrane amino acid transporter protein | 5.04E-12 |
| LOC_Os07g17390 | Downstream 2k | PsbP | 2.81E-11 |
| LOC_Os10g34910 | Downstream 2k | secretory protein | 3.37E-11 |
| LOC_Os02g15690 | Downstream 2k | polygalacturonase | 5.54E-11 |
| LOC_Os01g01840 | Downstream 2k | helix-loop-helix DNA-binding domain containing protein | 6.27E-11 |
| LOC_Os06g40200 | Downstream 2k | calcium-binding mitochondrial carrier | 1.04E-10 |
| LOC_Os01g70370 | Downstream 2k | serine palmitoyltransferase 2 | 1.21E-10 |
| LOC_Os01g45750 | Downstream 2k | bile acid sodium symporter family protein | 7.05E-10 |
| LOC_Os01g18630 | Downstream 2k | aspartic proteinase oryzasin-1 precursor | 7.06E-10 |
| LOC_Os06g36550 | Downstream 2k | retrotransposon protein, Ty3-gypsy subclass | 9.81E-10 |
| LOC_Os01g70900 | Downstream 2k | retrotransposon protein | 9.84E-10 |
Transposable elements (TEs) are frequently methylated in plant genomes. The analysis of 26 repeat types revealed that LTR /Gypsy had the highest 5mC content in PA64S and was increased in PA64S (F) (Figure 3c). Further analysis of four types of LTR and LINE, SINE showed that all of the LTR retroelements were highly methylated in PA64S (F) (Table 4). In contrast, the two types of retroelements (LINE, SINE) had higher methylation in PA64S (S) than in PA64S (F) (Table 4).
Table 4.
Transposable element methylation of PA64S(S) and PA64S (F) based on 10 kb tiled genome windows
| Type | PA64S(S) | PA64S(F) | ||
|---|---|---|---|---|
| reads | % | reads | % | |
| LTR/Copia | 521,175 | 5.62 | 533,088 | 5.79 |
| DNA/En-Spm | 658,162 | 7.1 | 676,617 | 7.35 |
| LTR/Gypsy | 4,045,323 | 43.63 | 4,293,192 | 46.61 |
| LTR | 55,692 | 0.6 | 55,798 | 0.61 |
| LINE/L1 | 174,518 | 1.88 | 171,516 | 1.86 |
| SINE | 167,785 | 1.81 | 154,945 | 1.68 |
Validation of MedIP data by bisulfite sequencing
In this study, the DNA methylation patterns of 20 DMR-associated genes were selected to carry out bisulfite sequencing for the validation of MedIP data, including LOC_Os10g10560 (invertase/pectin methylesterase inhibitor, IPMI), LOC_Os12g08770 (photosystem I reaction center subunit N), LOC_Os03g51200 (H2A), LOC_Os08g38210 (transcription factor BIM2) and so on (Figures 5 and 6). We found that many of the DMR-associated genes showed hyper-methylation in PA64S (S), and the results were almost in accordance with the MeDIP-seq data (Figures 5 and 6, and Additional file 3). For example, LOC_Os10g10560 (IPMI), LOC_Os03g01820 (vacuolar ATP synthase subunit E), LOC_Os06g23980 (MADS-AGL16) and LOC_Os04g37619 exhibited a higher methylation level in PA64S(S) compared with PA64S (F) (Additional file 3). However, the DMR-associated genes LOC_Os03g51200 (H2A) and LOC_Os03g27770 (heme oxygenase 2) were observed to be hypomethylation in PA64S(S) (Figure 6a, b).
Figure 5.
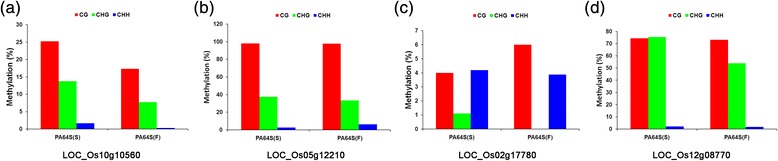
DNA methylation patterns of four selected DMR-associated genes validated by bisulfite sequencing. (a) LOC_Os10g10560 (invertase/pectin methylesterase inhibitor, IPMI); (b) LOC_Os05g12210 (chalcone synthase); (c) LOC_Os02g17780(ent-kaurene synthase, chloroplast precursor); and (d) LOC_Os12g08770 (photosystem I reaction center subunit N). The red, green and blue columns in the histograms refer to the collective methylation levels (in percentage) of CG, CHG, and CHH, respectively.
Figure 6.
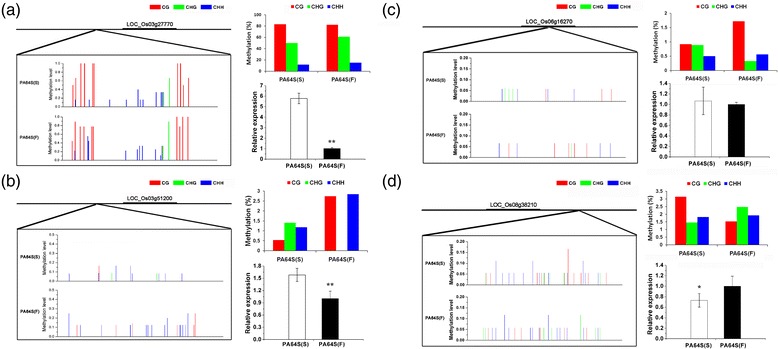
DNA methylation patterns and gene expression of the four selected DMR-associated genes. (a) Bisulfite sequencing and qRT-PCR analysis of LOC_Os03g27770 (heme oxygenase 2); (b) Bisulfite sequencing and qRT-PCR analysis of LOC_Os03g51200 (H2A); (c) Bisulfite sequencing and qRT-PCR analysis of LOC_Os06g16270 (heat shock factor binding protein 2); and (d) Bisulfite sequencing and qRT-PCR analysis of LOC_Os08g38210 (transcription factor BIM2). The red, green and blue columns in the histograms refer to the collective methylation levels (in percentage) of CG, CHG, and CHH, respectively.
Association of DNA methylation and gene expression
Analysis of the relationship between DNA methylation and gene expression revealed that hypermethylation in some regions were related to the expression of neighboring genes. DNA methylation level of PA64S(S) on the upstream 2k, 5’UTR, intron, 3’UTR, and downstream 2k regions were hypermethylated compared with PA64S (F) (p < 0.001) (Figure 4b and Table 2). Correspondingly, many DMR-associated genes may be down-regulated in PA64S (S).
The DNA methylation patterns and gene expression of four DMR-associated genes, namely LOC_Os03g51200 (H2A), LOC_Os08g38210 (transcription factor BIM2), LOC_Os06g16270 (heat shock factor binding protein 2) and LOC_Os03g27770 (heme oxygenase 2), were further examined in this study (Figure 6). Three of these had lower DNA methylation levels, but higher expression in PA64S (S). Furthermore, The DMR-associated gene LOC_Os08g38210 showed a higher DNA methylation level and a lower expression in PA64S (S) (Figure 6d).
Expression of the genes involved in brassinosteroid signaling pathway
One of the hypermethylated gene OsBIM2 (LOC_Os08g38210), which is homolog with BIM gene family in Arabidopsis, was thought to be involved in BR signaling pathway. Comparison with the structure of BIM1 suggested that OsBIM2 had higher similarity to BIM1 of Arabidopsis, with an extended N-terminal domain that was lacking in BIM2 and BIM3 [30]. Comparing the respective genomic loci also supported the idea that OsBIM2 and Arabidopsis BIM1 probably share a more ancestral intron-exon structure (Figure 7). And BIM1 was reported to be cooperated with BES1 to regulate BR-induced genes in Arabidopsis [26]. Therefore, we postulated that BR may participate in regulating male fertility in PA64S. To test this hypothesis, we examined the expression of some genes related to BR signaling pathway and the genes that regulated by BR. Expression analysis of these genes showed that some of them had lower expression in PA64S (S) (P < 0.01), including OsBAK1, OsBZR1, OsSPL8 (Figure 8). The expression of OsMS2, one of the regulating genes for BR signaling in the tapetum and microspore development, was also dramatically reduced in PA64S (S) (P < 0.01) (Figure 8).
Figure 7.
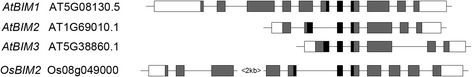
The genomic organization of the rice OsBIM2 (LOC_Os08g38210) loci is compared to those of the BIM1 , BIM2 and BIM3 genes in Arabidopsis. Note: This genomic organization is modified on the basis of Xing et al., 2013 [30].
Figure 8.
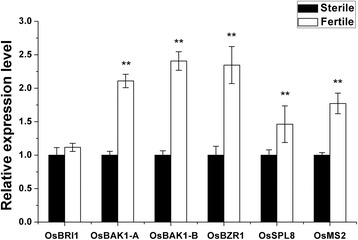
Expression analysis of genes related to BR signaling pathway.
Gene ontology (GO) and KEGG pathway enrichment analysis
In this study, genes that overlapped with the methylation peaks in the upstream 2k, downstream 2k or gene body regions were termed as methylated genes. A total of 36,321 methylated genes were found in the two samples: 32,678 in PA64S (S) and 32,990 in PA64S (F). Of them, 1258 methylated genes were identified to be differentially expressed genes between PA64S (S) and PA64S (F), and these included 725 genes that were hypermethylated and 533 genes that were hypomethylated in PA64S (S) (Table 2).
We further used WEGO (http://wego.genomics.org.cn) to functionally categorize the methylated genes and observed significant differences (Figure 9 and Additional file 4). Many genes enriched hypermethylation in PA64S(S), such as translation regulator, development process, multi-organism process and so on (Figure 9). The results showed that some genes were significantly down-regulated genes in PA64S (S), including 12 genes for “cellular component” (9), 16 genes for “molecular function” (21) and 64 genes for “biological process” (50) (Table 5 and Additional file 5). Methylated genes tend to be enriched in transferase activity, including transcription regulator as well as electron transporter. With respect to biological processes, the genes are enriched in functions associated with photorespiration, cellular metabolism, oxidative phosphorylation and mitochondrial ATP synthesis. Interestingly, the methylated genes of “response to stimulus” were increased in PA64S (S) (Figure 9). In contrast, for cellular component, the significantly differentially down-regulated genes in PA64S (S) were enriched in mitochondrial respiratory chain and mitochondrial inner membrane (Table 5).
Figure 9.
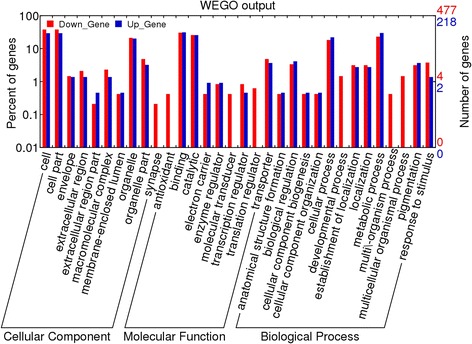
Gene categories and distribution of differential methylated genes between PA64S(S) and PA64S (F).
Table 5.
Overrepresented Gene Ontology (GO) categories in the hypermethylated genes in PA64S (S)
| GO term | Ontology a | Element | Ontology description | Number in input list | Number in BG/Ref | p-value |
|---|---|---|---|---|---|---|
| GO:0005750 | C | Down2k_ Down | mitochondrial respiratory chain complex III | 4 | 14 | 2.74e-05 |
| GO:0005746 | C | Down2k_ Down | mitochondrial respiratory chain | 3 | 22 | 0.00104 |
| GO:0005743 | C | Down2k_ Down | mitochondrial inner membrane | 5 | 78 | 0.00223 |
| GO:0016742 | F | 3’-UTR_ Down | hydroxymethyl-, formyl- and related transferase activity | 4 | 4 | 1.61e-14 |
| GO:0016741 | F | 3’-UTR_ Down | transferase activity, transferring one-carbon groups | 4 | 84 | 3.12e-08 |
| GO:0030528 | F | Intron_ Down | transcription regulator activity | 4 | 178 | 1.09e-05 |
| GO:0045153 | F | Down2k_ Down | electron transporter, transferring electrons within CoQH2-cytochrome c reductase complex activity | 4 | 13 | 4.58e-05 |
| GO:0009853 | P | 3’-UTR_ Down | photorespiration | 4 | 4 | 3.65e-13 |
| GO:0046653 | P | 3’-UTR_ Down | tetrahydrofolate metabolic process | 4 | 4 | 3.65e-13 |
| GO:0006760 | P | 3’-UTR_ Down | folic acid and derivative metabolic process | 4 | 8 | 2.56e-11 |
| GO:0042558 | P | 3’-UTR_ Down | pteridine and derivative metabolic process | 4 | 11 | 1.20e-10 |
| GO:0043094 | P | 3’-UTR_ Down | cellular metabolic compound salvage | 4 | 19 | 1.41e-09 |
| GO:0006732 | P | 3’-UTR_ Down | coenzyme metabolic process | 4 | 63 | 2.17e-07 |
| GO:0009259 | P | 3’-UTR_ Down | ribonucleotide metabolic process | 4 | 81 | 6.08e-07 |
| GO:0051186 | P | 3’-UTR_ Down | cofactor metabolic process | 4 | 102 | 1.55e-06 |
| GO:0009117 | P | 3’-UTR_ Down | nucleotide metabolic process | 4 | 111 | 2.19e-06 |
| GO:0006725 | P | 3’-UTR_ Down | cellular aromatic compound metabolic process | 4 | 129 | 4.02e-06 |
| GO:0032787 | P | 3’-UTR_ Down | monocarboxylic acid metabolic process | 4 | 142 | 5.94e-06 |
| GO:0046483 | P | 3’-UTR_ Down | heterocycle metabolic process | 4 | 189 | 1.88e-05 |
| GO:0006122 | P | Down2k_ Down | mitochondrial electron transport, ubiquinol to cytochrome c | 4 | 6 | 2.35e-06 |
| GO:0042775 | P | Down2k_ Down | mitochondrial ATP synthesis coupled electron transport | 4 | 18 | 0.00045 |
| GO:0006119 | P | Down2k_ Down | oxidative phosphorylation | 4 | 28 | 0.00289 |
| GO:0042773 | P | Down2k_ Down | ATP synthesis coupled electron transport | 4 | 28 | 0.00289 |
a, C: cellular component; F, molecular function; P, biological process.
To generate further insight view of pathway perturbation in PA64S (S) under long day length and high temperature conditions, we performed KEGG pathway analysis of the differentially methylated genes. As a result, 58 predicted pathways were enriched, including metabolism, circadian rhythm, photosynthesis, porphyrin and chlorophyll metabolism, peroxisome and oxidative phosphorylation (Additional files 6). Through the GO term enrichment and KEGG pathway analysis of the differentially regulated genes, we observed that certain genes were hypermethylated in PA64S (S) and that they were involved in photorespiration and mitochondrial ATP synthesis pathways. These pathways were closely connected to photosynthesis and energy synthesis. For instance, cytochrome c1-1 (LOC_Os05g23620) was involved in mitochondrial electron transport.
Discussion
Although global DNA methylation surveys have been performed in some plants [3,31,32], little studies have been reported about the methylation profiles of PTGMS rice (for example PA64S) during the fertility transition. In this study, the objective was to perform a genome-wide identification of the methylated genes that affect the sterility-fertility transition in PTGMS rice PA64S. To confirm the results from MeDIP-seq, the methylation patterns of twenty regions in each sample were analyzed by bisulfite sequencing. And the methylation levels obtained by the two methods were generally in accord with each other. The read distribution analysis performed in this study found that uniquely mapped reads were enriched in the upstream 2k, 5’UTR, 3’UTR and downstream 2k regions. In addition, the rice sterile line PA64S(S) had higher methylation level than that of PA64S (F), and many DMR-associated genes were down-regulated in PA64S (S).
Transposable elements (TEs) are abundant features in plant genomes that are frequently marked by cytosine methylation. In this study, we examined many categories of transposable elements from the RepBase transposable element annotation database. LTR retroelements are the predominant type of transposable element sequences in plant genomes [33]. Our results showed that the LTR categories had higher 5mC content in PA64S (F) than in PA64S (S). And the LINE and SINE retroelement categories had an increased in 5mC content in PA64S (S) compared with PAS64 (F), although this difference was not significant. Previous studies using methylation-deficient mutants have demonstrated that hypomethylation can result in transposable element mobilization [34]. The increased 5mC content of TEs found in the present study suggested that the operation of adaptive genomic mechanisms might help protect genomes against transposable element spread. It would be of interest to measure the relationship of DNA methylation and TE elements during the fertility transition of PA64S.
In this study, the DNA methylation patterns of twenty DMR-associated genes were validated through bisulfite sequencing and we found that hypermethylation in some regions affected the expression of neighboring genes (Figures 5 and 6, Additional file 3). Although hypermethylated DMR-associated genes did not show significant enrichment of down-regulated genes, there were still some down-regulated genes with higher methylation levels (Figures 5 and 6). Some flower/anther development related genes were hypermethylated in PA64S (S), for example, LOC_Os06g23980 (putative MADS-box protein AGL16) and LOC_Os10g10560 (IPMI) (Additional file 3). Previous studies of a male sterile mutant (Ms-cd1) in Brassica oleracea have demonstrated that IPMI was specifically expressed in anther and filament and was not expressed in CMS NILs [35]. The increased 5mC content in IPMI of PA64S(S) found in the present study might lead to a lower expression of IPMT and most likely disturbed the anther cell wall metabolism. Furthermore, photosystem and mitochondrial ATP synthesis associated genes, including LOC_Os12g08770 (photosystem I reaction center subunit N), LOC_Os07g17390 (PsbP) and LOC_Os03g01820 (vacuolar ATP synthase subunit E) were also hypermethyalted in PA64S (S) (Additional file 2). High temperature (>23.5°C) and LD (14 h) conditions increased the methylation levels of these genes and down-regulated their expression during anther development in PA64S, which might in turn lead to PA64S sterility.
In contrast, some heat shock proteins, such as LOC_Os08g43490 (heat shock protein DnaJ), LOC_Os06g16270 (heat shock factor binding protein 2) were found to be hypomethylated in PA64S (S) in our study, and the expression level of LOC_Os06g16270 was also higher in PA64S (S) (Figure 6c). Because heat shock proteins are always expressed in response to stress, their higher expression in PA64S (S) suggested that the adverse environment might affect anther development. These findings suggested that different environments might regulate the gene expression through DNA methylation in the gene body or flanking sequence of fertility associated genes.
In Arabidopsis and rice, the BR signaling pathway is similar. Generally, BRs bind the extracellular domain of BRI1, and then BRI1 heteromerizes with the BRI1-associated kinase 1 (BAK1). The receptor complex then activates the BR-signaling kinases (BSKs) and inactivates the glycogen synthase kinase-3-like kinase BIN2, resulting in nuclear accumulation of unphosphorylated forms of two transcription factors BZR1 and BES1, which activate the expression of BR-responsive genes [36-38]. In this study, some of the components in BR signaling pathway, such as OsBRI, OsBAK1, OsBZR and OsBIM2 were dramatically reduced in male sterile line PA64S (S) (Figure 8). Our results indicated that BR signaling involved in regulating the sterility-fertility transition of PA64S under different environments.
On the other hand, OsBIM2 (LOC_Os08g38210) showed a higher DNA methylation level in PA64S(S) and its expression dramatically reduced (Figure 6d). In Arabidopsis, BIM1 and SPL8 act cooperatively in a common complex or via synergistic pathways to promote male fertility [30]. In our study, both OsBIM2 and OsSPL8 were downregulated in PA64S(S) (Figure 8). Since OsBIM2 is homolog with BIM1, the lower expression level will affect BR signaling pathway and then regulated the downstream genes which involved in anther and pollen development. Therefore, DNA methylation of BR signaling related genes may affect the expression of key genes (for example, OsMS2) for anther development, leading to male sterility under long-day and high temperature conditions. However, whether the hypermethylated OsBIM2 directly suppress key genes need to be further investigated.
The GO term significant enrichment analysis showed that many genes, including the transposons and retrotransposons, were hypermethylated in PA64S (S) (Tables 3 and 5 and Additional files 3 and 4). For example, LOC_Os09g38100 (phosphate carrier protein mitochondrial precursor) and LOC_Os06g40200 (calcium-binding mitochondrial carrier) were observed higher methylation levels in PA64S (S) than PA64S (F) (Table 3 and Additional file 3). These two genes participate in mitochondrial respiratory chain and facilitate mitochondrial ATP synthesis [39]. Thus, their hypermethylated would affect the energy synthesis during pollen development. Another hypermethylated gene LOC_Os02g17780 (ent-kaurene synthase, chloroplast precursor) was an early intermediate in GA biosynthesis [40]. Other significantly differentially expressed DMR-associated genes included LOC_Os12g40130 (phosphoribosyl transferase), LOC_Os04g30490 (MATE efflux family protein) and LOC_Os05g12210 (chalcone synthase). Some studies have reported that the adenine phosphoribosyl transferase activity deficient mutants of Arabidopsis thaliana were male sterile due to abortion of pollen development after the meiotic divisions of pollen mother cells [41]. The MATE (Multidrug And Toxic Compound Extrusion) efflux protein family has also been reported to be differentially expressed in sterile and fertile NILs of Brassica oleracea [35]. Chalcone synthase catalyzes the initial step of the branch of phenylpropanoid pathway which leads to flavonoids. The absence of chalcone synthase activity was found to have a pleiotropic effect in maize and petunia mutants, and pollen fertility/pollen germination and flavonoid synthesis was disrupted [42]. Therefore, these differentially expressed genes might be involved in fertility transition.
To uncover the regulatory mechanisms of the fertility transition, the regulatory network of differentially methylated genes was examined in this study. Several important pathways were found, including flavone and flavonol biosynthesis, circadian rhythm, photosynthesis, porphyrin and chlorophyll metabolism, and oxidative phosphorylation (Additional files 6). Previous studies have reported that flavone and flavonol biosyntheses are associated with pollen development. Flavonoids are required for pollen viability in many species [42-44]. In addition, flavonols can promote pollen germination frequency and pollen tube growth in tobacco [45]. Many higher plants use the day length as an environmental cue to switch from vegetative to reproductive growth and plants with defects in their circadian regulation cannot properly regulate the timing of the floral transition [46]. In PGMS rice, the circadian rhythm genes OsPRR37 in inflorescences have been reported to directly affect the male sterility transition in Nongken 58S [22]. In this study, some DMR-associated genes, such as LOC_Os05g12210 (chalcone synthase) and LOC_Os08g38210 (transcription factor BIM2), were found to be involved in the circadian rhythm. The other three pathways, namely photosynthesis, porphyrin and chlorophyll metabolism, and oxidative phosphorylation were related to carbon or energy metabolism [47,48]. The down-regulated genes of these pathways in PA64S (S) may lead to insufficient energy and materials for pollen development. Therefore, these five pathways were regarded as potentially related to the fertility transition of PA64S in the present study.
Conclusions
We systematically investigated the complete DNA methylome of photoperiod- and thermo-sensitive male sterile rice Peiai 64S under two different environments (different temperatures and day lengths). From the whole-genome DNA metylation map, hypermethylation was observed in PA64S (S) and many pathways such as circadian rhythm, photosynthesis and oxidative phosphorylation were participated in the fertility transition of PA64S. Furthermore, the hypermethylated OsBIM2 may be involved in brassinosteroid signaling pathway and affected the male fertility in PA64S.
Methods
Plant materials
The rice photo-thermo-sensitive genic male sterile line PA64S, which was maintained at Wuhan University, was used in this study. The seeds of PA64S were sown under two different natural ecological conditions: in Hainan (18°48′ N, 110°02′ E) from December, 2010 to March, 2011 (fertility, F), and in Wuhan (30°30′ N, 114°18′ E) from May 2011 to August 2011 (sterility, S). Young panicles of PA64S of the two phenotypes at the meiosis stage were collected, and the pollen fertility was assayed by staining with potassium iodide (1% I2-KI).
DNA preparation and methylated DNA immunoprecipitation
Total genomic DNA was extracted using a Genomic DNA Miniprep Kit (Axygen) following the manufacturer’s instructions. The DNA quality was then evaluated by agarose gel electrophoresis and a BioPhotometer Plus spectrophotometer (Eppendorf, Germany).
Subsequently, the two samples were sonicated to produce DNA fragments ranging from 100 to 500 bp. After end repairing, phosphorylating and A-tailing with Paired-End DNA Sample Prep kit (Illumina, USA), the DNA was ligated to an Illumina sequencing primer adaptor. Then the fragments were used for methylated DNA immunoprecipitation (MeDIP) enrichment using a Magnetic Methylated DNA Immunoprecipitation kit (Diagenod, Belgium) following the manufacturer’s recommendations and the qualifying DNA was used for PCR amplification. Then bands between 220 and 320 bp were excised from the gel and purified with a QIAquick Gel Extraction Kit (Qiagen, Germany). The products were quantified with a Quant-iT™ dsDNA HS Assay Kit (Invitrogen, USA) using an Agilent 2100 Analyzer (Agilent Technologies, USA). Following qPCR qualification, DNA libraries were sequenced on the Illumina HiSeq 2000 (Illumina, CA, USA) to generate paired-end 50-bp reads by the Beijing Genomics Institute (BGI, China).
Bioinformatic analysis
The raw data obtained from Illumina sequencing were first processed to filter out reads containing adapters, unknown or low quality bases and were then were mapped to the rice reference genome (ftp://ftp.plantbiology.msu.edu/pub/data/Eukaryotic_Projects/o_sativa) by SOAPaligner v 2.21 (http://soap.genomics.org.cn/) with no more than 2 bp mismatches [49]. The uniquely mapped data were retained for read distribution analysis, including the distribution in rice chromosomes and the distribution in different components of the genome.
Subsequently, the regions of differential methylation (DMRs) with DNA methylation peaks were employed for differential DNA methylation analysis. Methylated regions were deemed significant differentially methylated between PA64S (S) and PA64S (F) with a p-value < 0.05, a false discovery rate (FDR) < 0.05 and at least a 2.0-fold change in sequence counts. The base data manipulations and statistical analysis were performed using the R package (http://www.r-project.org). Significant differences of DNA methylation level between PA64S (S) and PA64S (F) were evaluated using Student’s t test.
Gene Ontology and KEGG pathway analysis
Significantly DMRs in the whole gene body (DMGs) common to comparisons between PA64S (S) and PA64S (F) with opposing directions of methylation difference (for example, down-methylated or up-methylated in PA64S (S) relative to PA64S (F)) were screened and annotated with Gene Ontology (GO), and functionally annotated through KEGG Orthology (KO) enrichment analysis. The GO enrichment analysis of the functional significance applies a hypergeometric test to map all of the differentially expressed genes (DEGs) to terms in the GO database, looking for significantly enriched GO terms in the differentially expressed genes compared with the complete genome. The formula used in this analysis is the following:
where N is the number of all genes with a GO annotation in the rice genome, n is the number of those N genes that are differentially expressed, M is the number of all of the genes that are annotated to certain GO terms, and m is the number of differentially expressed genes in M. The hypergeometric test was also performed using a user written program in R (R Core Team, Vienna, Austria). The KEGG pathway enrichment analysis identifies significantly enriched metabolic pathways or signal transduction pathways in differentially expressed genes comparing with the whole genome background. The calculating formula and the applied program were the same as that used in the GO analysis. Then, FDR (Q-value) ≤0.05 was used as the threshold to determine the most significantly enriched pathways in DEGs.
Bisulfite sequencing
Genomic DNA was modified using an EpiTect Bisulfite kit (Qiagen, Germany) according to the manufacturer’s instructions. Briefly, 1 μg of DNA was first treated with bisulfite containing a C-T conversion reagent and then incubated at 95°C for 5 min, and 64°C for 5 h. The modified DNA was purified using a MinElute DNA spin columns (Qiagen, Germany) and stored at -20°C until use. Primers for selected genic sequences for bisulfite sequencing were designed by the Meth-Primer Program (http://www.urogene.org/methprimer/) and were given in Additional file 7. For each PCR reaction, 1.0 μl of bisulfite treated DNA was used in a 50 μl reaction system. These PCR products were gel-purified using a Gel Extraction Kit (Omega, USA), and then cloned into the pMD18-T vector (Takara, Dalian, China) and sequenced. At least fifteen clones were sequenced for each sample. The methylation levels which are expressed as percentage (%) per site for each of the three types of cytosines, CG, CHG and CHH, were calculated by dividing the number of non-converted (methylated) cytosines by the total number of cytosines within the assay. The analyses of the bisulfite sequencing results were conducted using the Kismeth website (http://katahdin.mssm.edu/kismeth).
RNA isolation and real time RT-PCR
Total RNA was extracted from young panicles using an RNA isolation kit (BioDev, Beijing, China) and treated with DNase I (Fermentas, Canada) to remove the genomic DNA contamination. Approximately 2 μg of RNA from each sample was used for reverse transcription using the RevertAid First Strand cDNA Synthesis Kit (Fermentas, Canada). The reaction was carried out in a total volume of 20 μl, including 2 μg of RNA, 1 μl of oligo (dN)18 primers (100 μmol/L), 4 μl 5× reaction buffer, 2 μl 10 mmol/L dNTP mix, 1 μl of RiboLock RNase Inhibitor (20 U/μl), and 1 μl of M-MuLV Reverse Transcriptase (Fermentas, Canada). The procedure was as follows: the mixture was first denatured at 70°C for 10 min and then incubated at 37°C for 5 min, followed by 42 °C for 60 min and at last incubated at 70°C for 15 min.
Quantitative real-time PCR (qPCR) was performed using a StepOne Plus real-time PCR system (Applied Biosystems) in a total volume of 10 μl reaction mixture, which included 1 μl cDNA, 0.4 μl each primer (5 μmol/L each), 0.2 μL ROX Reference Dye (50×) and SYBR premix Ex Taq (Tli RNaseH plus) (2×) (Takara, Dalian, China). The primers used for the qPCR experiments are given in Additional file 7. The qPCR reactions were repeated three times for each sample from three independent experiments. After the amplification steps, the melting curve was determined for each primer pair to verify that only one specific product had been amplified. Relative quantification was performed using the 2△△Ct method [50]. Rice actin1 was used as a reference for the mRNA expression analyses.
Availability of supporting data
All MeDIP-seq raw data supporting the results of this article were deposited in the sequence read archive repository (http://www.ncbi.nlm.nih.gov/sra/) under Sequence Read Archive (SRA) database (accession # SRP050401).
Acknowledgements
This work was supported by the “973” Program of China (2013CB126900) and the National Nature Science Foundation of China (31471464).
Additional files
Chromosome distribution of reads in PA64S (S) and PA64S (F). The distribution of reads in the chromosomes 1-12 of the rice genome was shown with a red color for each sample. The MeDIP-seq reads were plotted in 10 kb windows along chromosome.
Annotation of DMR-associated genes between PA64S (S) and PA64S (F).
Bisulfite sequencing of twenty selected DMR-associated genes. The red, green and blue columns in the histograms refer to the collective methylation levels (in percentage) of CG, CHG, and CHH, respectively.
Overrepresented Gene Ontology (GO) categories in genes that are hypermethylated in PA64S(S).
KEGG pathways of all of the differentially methylated genes between PA64S(S) and PA64S (F).
Pathways related to the sterility-fertility transition in PA64S.
Primers for bisulfite sequencing and qRT-PCR.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Designed the experiments: YD, JH, XC. Performed the experiments: JH, XC, HZ, Analyzed the data: JH, XC, HZ. Wrote the paper: YD, JH. All authors read and approved the final manuscript.
Contributor Information
Jihong Hu, Email: hujh2010@163.com.
Xiaojun Chen, Email: clingg@whu.edu.cn.
Hongyuan Zhang, Email: hongyuanzhang2011@163.com.
Yi Ding, Email: yiding@whu.edu.cn.
References
- 1.Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiang H, Zhu J, Chen Q, Dai FY, Li X, Li M, Zhang HY, Zhang GJ, Li D, Dong Y, Zhao L, Lin Y, Cheng DJ, Yu J, Sun JF, Zhou XY, Ma KL, He YH, Zhao YX, Guo SC, Ye MZ, Guo GW, Li YR, Li RQ, Zhang XQ, Ma LJ, Kristiansen K, Guo QH, Jiang JH, Beck S, Xia QY, Wang W, Wang J. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat Biotechnol. 2010;28(5):516–520. doi: 10.1038/nbt.1626. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SWL, Chen HM, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, Ecker JR. Genome wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell. 2006;126(6):1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Ruike Y, Imanaka Y, Sato F, Shimizu K, Tsujimoto G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immune- precipitation combined with high-throughput sequencing. BMC genomics. 2010;11:137. doi: 10.1186/1471-2164-11-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphitesequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang CJ, Chu HJ, Chen GX, Shi DW, Zuo M, Wang J, Lu CG, Wang P, Chen L. Photosynthetic and biochemical activities in flag leaves of a newly developed super high-yield hybrid rice (Oryza sativa) and its parents during the reproductive stage. J Plant Res. 2007;120:209–217. doi: 10.1007/s10265-006-0038-z. [DOI] [PubMed] [Google Scholar]
- 8.Reinders J, Wulff BB, Mirouze M, Mari-Ordonez A, Dapp M, Rozhon W, Bucher E, Theiler G, Paszkowski J. Compromised stability of DNA methylation and transposon immobilization inmosaic Arabidopsis epigenomes. Genes Dev. 2009;23:939–950. doi: 10.1101/gad.524609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng C, Daigen M, Hirochika H. Epigenetic regulation of the rice retrotransposon Tos17. Mol Genet Genomics. 2006;276:378–390. doi: 10.1007/s00438-006-0141-9. [DOI] [PubMed] [Google Scholar]
- 10.Kashkush K, Khasdan V. Large-scale survey of cytosine methylation of retrotransposons and the impact of readout transcription from long terminal repeats on expression of adjacent rice genes. Genetics. 2007;177:1975–1985. doi: 10.1534/genetics.107.080234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, Thorne NP, Bäckdahl L, Herberth M, Howe KL, Jackson DK, Miretti MM, Marioni JC, Birney E, Hubbard TJP, Durbin R, Tavaré S, Beck S. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomazou EM, Rakyan VK, Lefebvre G, Andrews R, Ellis P, Jackson DK, Langford L, Francis MD, Bäckdah L, Miretti M, Coggill P, Ottaviani D, Sheer D, Murrell A, Beck S. Generation of a genomic tiling array of the human major histocompatibility complex (MHC) and its application for DNA methylation analysis. BMC Med Genomics. 2008;1:19. doi: 10.1186/1755-8794-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delseny M, Salses J, Cooke R, Sallaud C, Regad F, Lagoda P, Guiderdoni E, Ventelon M, Brugidou C, Ghesquiere A. Rice genomics: Present and future. Plant Physiol Bioch. 2001;39:323–334. doi: 10.1016/S0981-9428(01)01245-1. [DOI] [Google Scholar]
- 14.Yuan LP. Purification and production of foundation seed of rice PGMS and TGMS lines. Hybrid Rice. 1994;6:1–3. [Google Scholar]
- 15.Xu JJ, Wang BH, Wu YH, Du PN, Wang J, Wang M, Yi CD, Gu MH, Liang GH. Fine mapping and candidate gene analysis of ptgms2-1, the photoperiod- thermo- sensitive genic male sterile gene in rice (Oryza sativa L.) Theor Appl Genet. 2011;122:365–372. doi: 10.1007/s00122-010-1452-0. [DOI] [PubMed] [Google Scholar]
- 16.Yuan LP. Progress of two-line system hybrid rice breeding. Sci Agric Sin. 1990;23(3):1–6. [Google Scholar]
- 17.Yang Q, Liang C, Zhuang W, Li J, Deng HB, Deng QY, Wang B. Characterization and identification of the candidate gene of rice thermo-sensitive genic male sterile gene tms5 by mapping. Planta. 2007;225:321–330. doi: 10.1007/s00425-006-0353-6. [DOI] [PubMed] [Google Scholar]
- 18.Shi MS. The discovery and study of the photosensitive recessive male sterile rice. Sci Agric Sin. 1985;2:44–48. [Google Scholar]
- 19.Wang WW, Meng B, Ge XM, Song SH, Yang Y, Yu XM, Wang LG, Hu SN, Liu SQ, Yu J. Proteomic profiling of rice embryos from a hybrid rice cultivar and its parental lines. Proteomics. 2008;8:4808–4821. doi: 10.1002/pmic.200701164. [DOI] [PubMed] [Google Scholar]
- 20.Xu ML, Zhou GQ, Chen LB. Response of fertility of Pei’ai 64S to temperature and photoperiod in rice. Sci Agric Sin. 1999;25:772–776. [Google Scholar]
- 21.Zhou H, Liu QX, Li J, Jiang DG, Zhou LY, Wu P, Lu S, Li F, Zhu LY, Liu ZL, Chen LT, Liu YG, Zhuang CX. Photoperiod and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res. 2012;22:649–660. doi: 10.1038/cr.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang W, Liu ZW, Guo ZB, Song GY, Cheng Q, Jiang DM, Zhu YG, Yang DC. Comparative transcriptomes profiling of photoperiod sensitive male sterile rice Nongken 58S during the male sterility transition between short-day and long-day. BMC Genomics. 2011;12(1):462. doi: 10.1186/1471-2164-12-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding JH, Shen JQ, Mao HL, Xie WB, Li XH, Zhang QF. RNA-directed DNA methylation is involved in regulating photoperiod sensitive male sterility in rice. Mol Plant. 2012;5:1210–1216. doi: 10.1093/mp/sss095. [DOI] [PubMed] [Google Scholar]
- 24.Kutschera U, Wang ZY. Brassinosteroid action in flowering plants: A darwinian perspective. J Exp Bot. 2012;63:3511–3522. doi: 10.1093/jxb/ers065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye QQ, Zhu WJ, Li L, Zhang SS, Yin YH, Ma H, Wang XL. Brassinosteroids control male fertility by regulating the expression of key genes involved in Arabidopsis anther and pollen development. Proc Natl Acad Sci USA. 2010;107:6100–6105. doi: 10.1073/pnas.0912333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin YH, Vafeados D, Tao Y, Yoshida SG, Asami T, Chory J. A new class of transcription factors mediates brassinosteroid-regulated gene expression in Arabidopsis. Cell. 2005;120:249–259. doi: 10.1016/j.cell.2004.11.044. [DOI] [PubMed] [Google Scholar]
- 27.Nemhauser JL, Mockler TC, Chory J. Interdependency of brassinosteroid and auxin signaling in Arabidopsis. PLoS Biol. 2004;2(9):e258. doi: 10.1371/journal.pbio.0020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elango N, Hunt BG, Goodisman MA, Yi SV. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc Natl Acad Sci USA. 2009;106:11206–11211. doi: 10.1073/pnas.0900301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki MM, Kerr AR, De Sousa D, Bird A. CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 2007;17:625–631. doi: 10.1101/gr.6163007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xing SP, Quodt V, Chandler J, Hohmann S, Berndtgen R, Huijser P. SPL8 acts together with the brassinosteroid-signaling component BIM1 in controlling Arabidopsis thaliana male fertility. Plants. 2013;2:416–428. doi: 10.3390/plants2030416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Zhu JD, Hu FY, Ge S, Ye MZ, Xiang H, Zhang GJ, Zheng XM, Zhang HY, Zhang SL, Li Q, Luo RB, Yu C, Yu J, Sun JF, Zou XY, Cao XF, Xie XF, Wang J, Wang W. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics. 2012;13:300. doi: 10.1186/1471-2164-13-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vining K, Pomraning KR, Wilhelm LJ, Priest HD, Pellegrini M, Mockler TC, Freitag M, Strauss SH. Dynamic DNA cytosine methylation in the Populus trichocarpa genome: tissue-level variation and relationship to gene expression. BMC Genomics. 2012;13:27. doi: 10.1186/1471-2164-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar A, Bennetzen JL. Plant retrotransposons. Annu Rev Genet. 1999;33:479–532. doi: 10.1146/annurev.genet.33.1.479. [DOI] [PubMed] [Google Scholar]
- 34.Vining K, Pomraning KR, Wilhelm LJ, Ma C, Pellegrini M, Di YM, Mockler TC, Freitag M, Strauss SH. Methylome reorganization during in vitro dedifferentiation an regeneration of Populus trichocarpa. BMC Plant Biol. 2013;13:92. doi: 10.1186/1471-2229-13-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lou P, Kang J, Zhang G, Bonnema G, Fang Z, Wang X. Transcript profiling of a dominant male sterile mutant (Ms-cd1) in cabbage during flower bud development. Plant Sci. 2007;172:111–119. doi: 10.1016/j.plantsci.2006.07.015. [DOI] [Google Scholar]
- 36.Clouse SD. Brassinosteroid signal transduction: from receptor kinase activation to transcriptional networks regulating plant development. Plant Cell. 2011;23:1219–1230. doi: 10.1105/tpc.111.084475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He JX, Gendron JM, Yang YL, Li JM, Wang ZY. The GSK3-like kinase BIN2 phosphorylates and destabilizes BZR1, a positive regulator of the brassinosteroid signaling pathway in Arabidopsis. Proc Natl Acad Sci USA. 2002;99:10185–10190. doi: 10.1073/pnas.152342599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang XL, Chory J. Brassinosteroids regulate dissociation of BKI1, a negative regulator of BRI1 signaling, from the plasma membrane. Science. 2006;313:1118–1122. doi: 10.1126/science.1127593. [DOI] [PubMed] [Google Scholar]
- 39.Stael S, Rocha AG, Robinson AJ, Kmiecik P, Vothknecht UC, Teige M. Arabidopsis calcium-binding mitochondrial carrier proteins as potential facilitators of mitochondrial ATP-import and plastid SAM-import. FEBS Lett. 2011;585:3935–3940. doi: 10.1016/j.febslet.2011.10.039. [DOI] [PubMed] [Google Scholar]
- 40.Yamaguchi S, Sun TP, Kawaide H, Kamiya Y. The GA2 Locus of Arabidopsis thaliana Encodes ent-Kaurene Synthase of Gibberellin Biosynthesis. Plant physiol. 1998;116:1271–1278. doi: 10.1104/pp.116.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moffatt B, Somerville C. Positive selection for male-sterile mutants of Arabidopsis lacking adenine phosphoribosyl transferase activity. Plant physiol. 1988;86:1150–1154. doi: 10.1104/pp.86.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mo YY, Nagel C, Taylor LP. Biochemical complementation of chalcone synthase mutants defines a role for flavonols in functional pollen. Proc Natl Acad Sci USA. 1992;89:7213–7217. doi: 10.1073/pnas.89.15.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor LP, Jorgensen R. Conditional male-fertility in chalconesynthase -deficient petunia. J Hered. 1992;83:11–17. [Google Scholar]
- 44.Buer CS, Imin N, Djordjevic MA. Flavonoids: new roles for old molecules. J Integr Plant Biol. 2010;52(1):98–111. doi: 10.1111/j.1744-7909.2010.00905.x. [DOI] [PubMed] [Google Scholar]
- 45.Ylstra B, Touraev A, Moreno RMB, Stöger E, van Tunen AJ, Vicente O, Joseph NMM, Heberle-Bors E. Flavonols stimulate development, germination, and tube growth of tobacco pollen. Plant physiol. 1992;100(2):902–907. doi: 10.1104/pp.100.2.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doyle MR, Davis SJ, Bastow RM, McWatters HG, Kozma-Bognár L, Nagy F, Millar AJ, Amasino RM. The ELF4 gene controls circadian rhythms and flowering time in Arabidopsis thaliana. Nature. 2002;419(6902):74–77. doi: 10.1038/nature00954. [DOI] [PubMed] [Google Scholar]
- 47.Bazzaz FA, Carlson RW, Harper JL. Contribution to reproductive effort by photosynthesis of flowers and fruits. Nature. 1979;279:554–555. doi: 10.1038/279554a0. [DOI] [Google Scholar]
- 48.Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem. 1985;54(1):1015–1069. doi: 10.1146/annurev.bi.54.070185.005055. [DOI] [PubMed] [Google Scholar]
- 49.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 50.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (-Delta Delta C) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]