

SUMMARY PARAGRAPH
MicroRNAs (miRNAs) are short non-coding RNAs expressed in different tissue and cell types that suppress the expression of target genes. As such, miRNAs are critical cogs in numerous biological processes1,2, and dysregulated miRNA expression is correlated with many human diseases. Certain miRNAs, called oncomiRs, play a causal role in the onset and maintenance of cancer when overexpressed. Tumors that depend on these miRNAs are said to display oncomiR addiction3–5. Some of the most effective anticancer therapies target oncogenes like EGFR and HER2; similarly, inhibition of oncomiRs using antisense oligomers (i.e. antimiRs) is an evolving therapeutic strategy6,7. However, the in vivo efficacy of current antimiR technologies is hindered by physiological and cellular barriers to delivery into targeted cells8. Here we introduce a novel antimiR delivery platform that targets the acidic tumor microenvironment, evades systemic clearance by the liver, and facilitates cell entry via a non-endocytic pathway. We found that the attachment of peptide nucleic acid (PNA) antimiRs to a peptide with a low pH-induced transmembrane structure (pHLIP) produced a novel construct that could target the tumor microenvironment, transport antimiRs across plasma membranes under acidic conditions such as those found in solid tumors (pH ~6), and effectively inhibit the miR-155 oncomiR in a mouse model of lymphoma. This study introduces a new paradigm in the use of antimiRs as anti-cancer drugs, which can have broad impacts on the field of targeted drug delivery.
Silencing aberrantly expressed miRNAs in vivo has been achieved using antisense with various nucleic acid analogs involving locked nucleic acids (LNA), 2′-O-methyl oligonucleotides (e.g. antagomiRs), and PNAs or nanoencapsulated PNAs5,9,10. As with most RNA-based therapies, each of these strategies is stymied by non-specific organ biodistribution, reticuloendothelial system (RES) clearance, and endolysosomal trafficking8,11. Acidosis is a hallmark of tumors12. The pHLIP peptide forms an inducible transmembrane alpha-helix under acidic conditions13, has the ability to translocate membrane-impermeable molecules into cells via a non-endocytic route13,14, and when administered systemically, can target a variety of epithelial tumors15. Exploiting acidity as a general property of the tumor microenvironment we find that the pHLIP peptide can localize to tumors of lymphoid origin in a subcutaneous flank model (Fig. 1a) and a model of disseminated lymphadenopathy (Fig. 1b), while avoiding the liver. Although pHLIP also shows kidney targeting, much of the peptide is cleared by renal excretion (Extended Data Fig. 1). To exploit these targeting and delivery properties we developed a tumor-targeted antimiR delivery vector (pHLIP-antimiR).
Figure 1. Targeting miR-155-addicted lymphoma using pHLIP.

a,b, Targeting distribution of pHLIP labeled with Alexa Fluor 750 (A750-pHLIP) 36 hours after systemic administration to (a) nude mouse with miR-155 flank tumors (n=3) and (b) mir-155LSLtTA mouse with lymphadenopathy (n=3), Alexa Fluor 750 conjugated to cysteine was the control. c, Schematic of pHLIP-mediated PNA antimiR delivery. (1) At pH less than 7, the C-terminus of pHLIP inserts across lipid bilayers, which facilitates delivery of attached antimiR-155. (2) The disulfide between pHLIP and antimiR-155 is reduced in the cytosol. (3) Intracellular antimiR-155 is free to inhibit miR-155.
PNAs are nucleic acid analogs comprising nucleobases joined by intramolecular amide bonds. This backbone imparts stability, nuclease resistance, and an increased binding affinity for complementary nucleic acids16. We hypothesized that pHLIP would facilitate the intracellular delivery of charge-neutral PNA antimiRs (Fig. 1c), which lack anionic phosphodiester groups, to cells within the tumor microenvironment. Tethering PNA antimiRs to pHLIP represents a unique approach because the multifunctional peptide component both targets tumors and mediates lipid membrane translocation13.
Fabrication of pHLIP-antimiR was verified by RP-HPLC, tricine SDS-PAGE, EMSA, and mass spectrometry (Extended Data Fig. 2a–c). In our constructs, the linkage between the PNA and peptide comprised a disulfide bond, which can be cleaved in the reducing environment of the cytosol (Fig. 1c)17; therefore, attachment to the inserting C-terminus of pHLIP promotes the intracellular delivery of the PNA antimiR. When administered to A549 cells (Fig. 2a and Extended Data Fig. 2d) and Toledo diffuse large-B cell lymphoma (DLBCL) cells (Extended Data Fig. 2e,f), which express elevated levels of miR-155 compared to other DLBCL cells18, a pHLIP-antimiR modified with a TAMRA label attached resulted in enhanced cellular delivery at acidic extracellular pH compared to neutral pH. PNA delivery to cells by pHLIP does not appear to be greatly affected by sequence since uptake has been demonstrated with numerous miRNAs including miR-182 (Fig. 2a and Extended Data Fig. 2d), miR-155 (Extended Data Fig. 2e,f), scrambled miR-155, miR-21, and miR-210. Delivery of antimiR-155 by pHLIP (pHLIP-anti155) derepressed luciferase in miR-155-overexpressing19 KB cells that stably expressed a miR-155-targeted dual luciferase sensor (Extended Data Fig. 2g). Additionally, inhibition of miR-155 by pHLIP-anti155 reduced KB cell viability at a dose comparable to LNA (15-mer, Exiqon) antimiR-155 delivered by lipofection (Fig. 2b). To demonstrate the adaptability of this antimiR delivery technology to silencing other miRNAs, pHLIP was attached to a PNA antimiR against miR-21, which derepressed a miR-21 luciferase sensor (Extended Data Fig. 2h). Together, these data suggest that pHLIP-antimiR is effective at delivering PNA antimiRs to multiple cancer cell types, in which endocytosis is hypothesized to be relegated to a supplemental mode of cell uptake due to the transport properties of pHLIP.
Figure 2. Intracellular translocation of PNA antimiRs mediated by pHLIP.
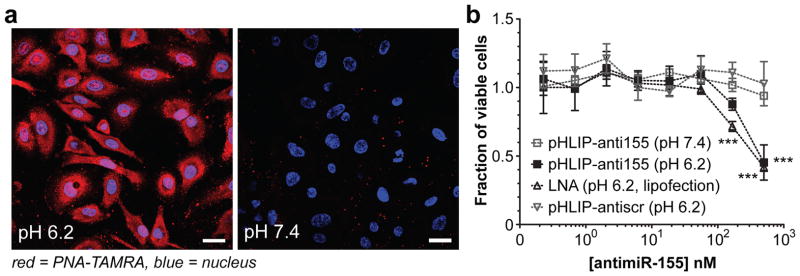
a, Confocal projections of A549 cells incubated with labeled pHLIP-antimiR (against control miR-182); scale bars represent 25 μm. b, Effects of miR-155 inhibition on KB cell viability; all data are normalized to cells treated with vehicle buffer. Data are shown as mean ± s.d., with n = 3; statistical analysis performed with two-way ANOVA; three asterisks, P < 0.001.
Certain oncomiRs have emerged as pharmacological targets. For example, ectopic expression of miR-155 in mice provided the first evidence that dysregulation of a single miRNA could cause cancer20. Although aberrant expression of miR-155 is characteristic of numerous cancers, miR-155 is notorious for its oncogenic involvement in lymphomas21. We previously developed a Tet-Off-based mouse model in which miR-155 expression is induced in hematological tissues and can be attenuated with the addition of doxycycline (DOX)5. Between 2–3 months of age, these mir-155LSLtTA mice develop disseminated lymphoma, in which lymphoid tissues progress from normal histology, to follicular hyperplasia, to follicular lymphoma, to DLBCL (Extended Data Fig. 3a,b). Although these are aggressive cancers comprising neoplastic B cells with a high Ki-67 proliferative index, the disease dramatically regresses upon DOX-induced miR-155 withdrawal (Extended Data Fig. 3b,c). Therefore, this is a model of oncomiR addiction in which tumorigenesis is dependent on expression of miR-155 and its removal leads to cancer regression22.
We assessed the therapeutic efficacy of pHLIP-anti155 in vivo using two tumor models based on mir-155LSLtTA mice: (1) nude mice subcutaneously implanted with neoplastic B cells derived from the enlarged spleens of mir-155LSLtTA mice (Extended Data Fig. 4a) and (2) mir-155LSLtTA mice after progression to conspicuous lymphadenopathy (Extended Data Fig. 4b). Continuous suppression of miR-155 via DOX-impregnated mouse chow or a cocktail of chemotherapeutics and anti-inflammatory steroids (CHOP) served as positive controls that each cause tumor regression (Extended Data Fig. 5a). Since CHOP is part of the current standard of care for human lymphomas23, the similar response to treatment with DOX and CHOP demonstrated the potential utility of antimiR-155 cancer therapy. Accordingly, intravenous administration of pHLIP-anti155 to the flank tumor model resulted in a significant reduction in tumor growth (Fig. 3a). In a subsequent study at a higher dose, pHLIP-anti155 showed a significant survival advantage compared to a commercially-available LNA (Exiqon) antimiR optimized for in vivo miR-155 silencing (Fig. 3b and Extended Data Fig. 5b). After administration of pHLIP-anti155, mice exhibited no clinical signs of distress, toxicity and renal damage (Extended Data Fig. 5c). Note that the dose of pHLIP-anti155 used in this study was much lower (ranging from 17- to 40-fold) than what has been used in other antimiR delivery reports10,24.
Figure 3. Targeted silencing of miR-155 has beneficial effects in mice with subcutaneous mir-155LSLtTA tumors.

a, Tumor growth response to treatment; arrows represent 1 mg/kg PNA dose per intravenous injection; all with n = 3, except for pHLIP-anti155 group with n=4. b, Survival in response to antimiR treatment; cutoff criteria include tumor volume greater than 1 cm3 or clinically mandated euthanasia. Symbols represent 2 (arrowhead) or 1 (arrow) mg/kg intravenous injections; LNA is a fully phosphorothioated LNA antimiR against miR-155; n = 4 for all groups; (*) for pHLIP-anti155 compared to LNA. c, Representative histologic analysis of livers (H&E, 200x magnification) harvested from early endpoint study (Fig. 3a and Extended Data Fig. 5a). d, Mass range of spleens from mice in early endpoint study; all with n = 3, except for pHLIP-anti155 group with n=4. e, Time to development of conspicuous lymphadenopathy in survival study; (**) for pHLIP-anti155 compared to mock. Data are shown as mean ± s.d., statistical analysis performed with (a) two-way ANOVA or (b,e) Mantel-Cox test or (d) two-tailed Student’s t-test; asterisk, P < 0.05; two asterisks, P < 0.01; three asterisks, P < 0.001.
In addition to delaying tumor growth, pHLIP-anti155 treatment suppressed the metastatic spread of neoplastic lymphocytes to other organs. The liver, lymph nodes, and spleen were common targets for metastatic lymphocytes. In a blinded pathological assessment, livers from mice treated with pHLIP-anti155 and DOX had rare scattered aggregates of 1–3 neoplastic lymphocytes, while livers in the negative control groups typically had dense tumoral aggregates of up to two dozen cells scattered throughout the entire organ (Fig. 3c)—note that these tissues were harvested at an early endpoint (i.e. when the negative controls reached a tumor size of 1 cm3, Fig. 3a) in relation to the survival study (Fig. 3b) in order to resolve pharmacological effects. Early endpoint treatment with pHLIP-anti155, DOX, and CHOP reduced the onset of splenomegaly (as judged by spleen mass), which occurred in all of the negative control groups (Fig. 3d). Additionally, pHLIP-anti155 significantly delayed the development of conspicuous lymphadenopathy (Fig. 3e), which was particularly evident in the inguinal and axillary lymph nodes throughout all of the groups (Extended Data Fig. 5d).
Based on a blinded complete blood count (CBC) analysis, the negative control groups comprised a large number of atypical mononuclear cells of lymphoid origin—consistent with the leukemic phase of lymphoma. Treatment with pHLIP-anti155 and DOX had levels of circulating lymphocytes similar to wild-type, while CHOP treatment resulted in lymphocyte levels much lower than wild type (Extended Data Fig. 5e). Although pHLIP can target to metastasized lymph node tumors (Extended Data Fig. 1c), the therapeutic effects on the levels of circulating lymphocytes suggest that the lower incidence of metastatic spread is likely due to antimiR activity at the primary tumor. These findings support the effective targeting of systemic antimiR-155 therapy to neoplastic cells (Extended Data Fig. 5f). The additional lymphopenia caused by CHOP treatment likely reflects the general toxicity of non-targeted conventional chemotherapy drugs (Extended Data Fig. 5e). The absence of systemic toxicity may represent an important advantage for pHLIP targeted antimiR therapy. Importantly, when healthy C57BL/6 mice were treated at the highest dose and frequency used in this study, pHLIP-anti155 showed no significant impairment of liver and kidney function (Extended Data Fig. 6a). Additionally, WBC levels (Extended Data Fig. 6b), body mass (Extended Data Fig. 6c), and organ mass (Extended Data Fig. 6d) were all within normal ranges.
In addition to the miR-155-addicted lymphoma subcutaneous tumor model, pHLIP-anti155 was also effective at treating KB cell xenograft tumors, which stably expressed luciferase for intravital monitoring of tumor bioluminescence (Extended Data Fig. 7), as well as disseminated tumors in mir-155LSLtTA mice. Although implanted subcutaneous tumor models are effective for evaluating tumor growth, spontaneous cancer models arising in endogenous tissues are a more clinically relevant means of assessing therapeutic efficacy. Remarkably, systemically administered pHLIP-anti155 accumulated in the enlarged lymph nodes of the transgenic mir-155LSLtTA mice (Fig. 4a). Furthermore, like most therapeutics, PNA oligomers are known to be cleared by the RES11, which results in accumulation in the liver; pHLIP-anti155 showed ~10-fold reduction in liver accumulation compared to anti155 alone (Fig. 4a and Extended Data Fig. 8a,b). The therapeutic impact of pHLIP-anti155 in mir-155LSLtTA mice was supported by a statistically significant decrease in spleen size and a non-statistically significant reduction in lymph node tumor burden (Fig. 4b and Extended Data Fig. 8c–e). A non-statistically significant increase in apoptosis was also observed in the lymph nodes of treated mice (Extended Data Fig. 8f,g). Interestingly, blinded histopathological analysis revealed that spleens in pHLIP-anti155-treated mir-155LSLtTA mice had differentiated red and white pulp (similar to wild type mice with no treatment), while the splenic architecture of mir-155LSLtTA mice treated with pHLIP-antiscr was almost completely effaced (Fig. 4c and Extended Data Fig. 8h). As with the subcutaneous tumor studies, pHLIP-anti155 treatment also showed a 12-fold reduction in liver metastasis (Fig. 4d and Extended Data Fig. 8i,j), while flow cytometric analysis revealed reductions in populations of B220-expressing spleen cells (Extended Data Fig. 8k). Consistent with a lack of systemic toxicity, treatment with pHLIP-anti155 produced no histopathological kidney damage (Extended Data Fig. 8l). Lastly, all mice that developed lymphoma-induced paresis showed improved motor skills after pHLIP-anti155 treatment (Supplementary Videos 1–4, n = 3).
Figure 4. Delivery of pHLIP-anti155 to mir-155LSLtTA mice with lymphadenopathy.

a, Confocal projections of systemic, tumor-targeted delivery of antimiR-155 to mir-155LSLtTA mice using pHLIP; scale bars represent 25 μm (top, enlarged cervical lymph node) and 250 μm (bottom, liver), n = 3. b, Representative mir-155LSLtTA mouse before and after treatment with pHLIP-anti155, n = 6. c,d, Representative H&E analysis of (c) spleens and (d) livers harvested from diseased littermate mir-155LSLtTA mice after treatment, n = 6, control spleen represents wild type mice with no treatment. e, Heatmap showing selected upregulated genes upon miR-155 withdrawal. f, qPCR determination of gene expression levels in lymphoid tissue from mir-155LSLtTA mice. Data are shown as mean ± s.d., n = 3; statistical analysis performed with two-tailed Student’s t-test, asterisk, P < 0.05; two asterisks, P < 0.01.
For a more direct assessment of miR-155 silencing in mir-155LSLtTA mice, we monitored the levels of miR-155 targets in response to antimiR treatment. As an oncogene in lymphoma, miR-155 suppresses genes involved in processes such as apoptosis, proliferation, immune response regulation, as well as cell differentiation and development21. However, the addiction mechanisms by which lymphoma regresses upon miR-155 withdrawal are unknown. Typically, miR-155 targets have been identified by differential gene expression analysis of an overexpression condition compared to wild-type25. To uncover the genes required for miR-155 addiction, we performed RNA-seq analysis on miR-155-addicted lymphoid tumors compared to regressing tumors (Extended Data Fig. 9a). This is the first study to identify miRNA cancer targets that directly result from oncomiR withdrawal. Out of 29,209 mouse genes, 2,101 showed significant upregulation or downregulation in response to miR-155 attenuation (Extended Data Fig. 9b, Supplementary Table 1). KEGG analysis of upregulated genes revealed that 41% have been associated with cancer pathways (Extended Data Fig. 9c). Additionally, 25% have been implicated in cell adhesion and migration pathways such as leukocyte transendothelial migration. We compared the upregulated genes to known and putative miR-155 targets (Supplementary Table 2) identified using the miRWalk target prediction algorithm26. At the intersection of these screens, several genes are known to have tumor suppressor characteristics (Fig. 4e, Extended Data Fig. 9d, Supplementary Table 3). One notable gene is Bach1, a transcription factor that has been validated as a miR-155 target in renal cancer and cultured B cells25,27. Gene expression analysis was used to validate Bach1 as a miR-155 target in Toledo cells treated with pHLIP-anti155 (Extended Data Fig. 9e) and in mir-155LSLtTA mice undergoing DOX-induced miR-155 withdrawal (Extended Data Fig. 10). Furthermore, diseased mir-155LSLtTA mice treated with pHLIP-anti155 showed an increase in Bach1 levels in cancerous axillary, cervical, and inguinal lymph nodes (Fig. 4f). A known miR-155 target in lymphoma, Mafb24, was also upregulated in response to pHLIP-anti155 treatment (Fig. 4f). Therefore, pHLIP-anti155 can target to lymph node neoplasms and cause effective blockage of miR-155 activity.
While oncomiRs are proving to be potent anticancer targets, in theory, using this approach, every miRNA is a “druggable” target. Through targeted antagonism of miRNAs, pHLIP-antimiR has vast therapeutic potential for cancer and many other pathological conditions that produce localized acidic environments such as ischemia, myocardial infarcts, stroke, tissue trauma, and sites of inflammation and infection. The main limitation of this transmembrane delivery approach involves the need for the drug cargo to have limited charge, such as PNA antimiRs. While other antimiR delivery and targeting strategies have been described28,29, utilization of pHLIP to target the acidic tumor microenvironment is a widely applicable technology that will present new therapeutic and mechanistic opportunities for effective targeting of miRNA silencing.
Methods
PNA synthesis
Regular Boc-protected PNA monomers were purchased from ASM Research Chemicals. All the given oligomers were synthesized on solid-support using standard Boc chemistry procedures30. The oligomers were cleaved from the resin using m-cresol:thioanisole:TFMSA:TFA (1:1:2:6) cocktail solution. The resulting mixtures were precipitated with ether (3X), purified and characterized by RP-HPLC and MALDI-TOF, respectively. All PNA stock solutions were prepared using nanopure water and the concentrations were determined at 90°C on a Cary 3 Bio spectrophotometer using the following extinction coefficients: 13,700 M−1 cm−1 (A), 6,600 M−1 cm−1 (C), 11,700 M−1 cm−1 (G), and 8,600 M−1 cm−1 (T). The 23-mer PNA oligomer complementary to miR-155 has an estimated Tm of 77.8°C. Single-isomer 5-Carboxytetramethylrhodamine (TAMRA) purchased from VWR was exclusively conjugated to the N-terminus of PNAs with a hydrophilic bifunctional linker, Boc-miniPEG-3Tm (11-Amino-3,6,9-Trioxaundecanoic Acid, DCHA, denoted in the sequences by -ooo-) purchased from Peptide International. Cysteine was also conjugated to C-terminus of PNAs using a Boc-miniPEG-3 linker.
The following PNA antimiR sequences were used:
anti155: TAMRA-ooo-ACCCCTATCACAATTAGCATTAA-ooo-Cys
antiscr: TAMRA-ooo-ACCCAATCGTCAAATTCCATATA-ooo-Cys
anti21: TAMRA-ooo-TCAACATCAGTCTGATAAGCTA-ooo-Cys
anti182: TAMRA-ooo-CGGTGTGAGTTCTACCATTGCCAAA-ooo-Cys.
Full length PNA antimiRs were used throughout this study. While current technologies such as “tiny” LNAs have seen efficacy with miRNA seed-targeted 8-mer antimiRs31, truncated PNA antimiRs should be similarly effective due to their high binding affinity, which can be further enhanced with chemical modifications32.
Synthesis and characterization of pHLIP-antimiR
For the generation of pHLIP-antimiR constructs, the following pHLIP sequence (New England Peptide) was synthesized: AAEQNPIYWARYADWLFTTPLLLLDLALLVDADEGT(CNPys)G; conjugation of the C-terminus to thiolated-PNA was facilitated by incorporating a cysteine group derivatized with 3-nitro-2-pyridinesulphenyl (NPys). To synthesize pHLIP-antimiR constructs, pHLIP-Cys(NPys) and antimiR PNA (peptide:PNA 1:1.3) were reacted overnight in the dark in a mixture of DMSO/DMF/0.1mM KH2PO4 pH 4.5 (v/v 3:1:1) under argon. Note that this protocol was adapted from a general method of conjugating peptides to PNAs. Aside from pHLIP, attaching molecules, such as cell-penetrating peptides, to PNAs can increase cellular uptake and in vivo delivery efficacy33,34. However, these conjugates typically require high doses and distribute to tissues throughout the body, which can result in off-target effects11,35. Similarly, pHLIP can be attached to other antimiR compositions (such as LNA), which would likely improve tumor targeting; however, physicochemical properties of PNA make them more amenable to pHLIP-mediated membrane translocation. A750-pHLIP was fabricated as previously described15.
Purification and verification of pHLIP-antimiR
After conjugation, pHLIP-antimiR was purified by RP-HPLC (Shimadzu) using a C18 column and a mobile phase gradient of water and acetonitrile with 0.1% trifluoroacetic acid. Purified pHLIP-antimiR was further characterized using MALDI-TOF. Concentrations of pHLIP-antimiR were determined on a Nanodrop Spectrophotometer (Thermo Scientific) at 260 nm corrected for peptide and TAMRA absorbance. Gelshift analysis was performed using a 20% TBE gel and Bolt electrophoresis system (Life Technologies); prior to loading, samples were incubated with an equamolar amount of miR-155, denatured at 95°C for 2 min, and allowed to anneal at 37°C for 30 min. SYBR Gold (Life Technologies) was used to visualize miR-155; pHLIP and free PNA were not detected by the stain. Tricine SDS-PAGE was performed using a 16% tricine gel (Life Technologies) and standard SDS-PAGE procedures. Samples were visualized first using TAMRA fluorescence on a Maestro 2 Multispectral Imaging System (PerkinElmer), and then using Simply Blue Coomassie stain (Life Technologies). For disulfide reduction studies, pHLIP-antimiR was reduced for 30 min in 200 mM DTT for HPLC and EMSA, and 5 mM TCEP for tricine SDS-PAGE. For all in vitro and in vivo studies, pHLIP-antimiR was heated at 65°C for 10 min to prevent aggregation.
Animals
All mice were maintained at Yale University in accordance with Yale Animal Resource Center (YARC) and the Institutional Animal Care and Use Committee (IACUC) guidelines. The mir-155LSLtTA mice were generated as previously described5. For transplant studies, 5–6 week-old female CrTac:NCr-Foxn1 nude mice (Taconic) were used. For toxicology studies, 8–9 week-old female C57BL/6J mice (Jackson) were used. For treatment of mir-155LSLtTA mice, a sample size of at least four was appropriate based on post hoc power analysis using quantitation of spleen size (Extended Data Fig. 8d) with a 95% confidence interval. For all animal studies, group allocations were randomized and all pathological analyses were blinded to treatment groups and expected experimental outcomes.
Cell culture
For all pH-controlled cell culture experiments, cells (previously tested for mycoplasma and supplied from ATCC) were incubated with 10% FBS in RPMI buffered at pH 7.4 with HEPES or pH 6.2 with MES, and treated with pHLIP-antimiR suspended in reaction buffer which constituted no more than 1% of the final volume.
Histology and other techniques
Harvested tissues were fixed in 10% formalin and processed by Yale Pathology Tissue Services for H&E and TUNEL staining. Retro-orbitally collected whole blood preserved in EDTA or serum separated using lithium heparin was sent to Antech Diagnostics for complete blood count (CBC) or clinical chemistry analysis, respectively. Image quantification performed using ImageJ 1.47v (NIH) and Color Deconvolution plugin (A.C. Ruifrok). Intravital and ex vivo fluorescence imaging was performed on either an IVIS Spectrum System (Caliper) or Maestro 2 Multispectral Imaging System using near-infrared or TAMRA filter sets. Live mice were anesthetized using isoflurane during image acquisition. For whole organ studies, organs were harvested and fixed in 10% formalin before imaging.
Flank tumor establishment
To establish mir-155LSLtTA lymphoma subcutaneous flank tumors, first enlarged spleens were extracted from 2–3 month-old mir-155LSLtTA mice with obvious lymphadenopathy (which generally correlated with incidence of splenomegaly). Using a 100 μm pore size cell strainer technique, spleen tissue was dispersed into a single cell suspension in 5% FBS in PBS on ice. Red blood cells were lysed using ammonium chloride lysis buffer (Stem Cell Technologies), and 5 × 106 cells were subcutaneously injected into nude mice. Tumors were generally palpable within 10 days; tumor volume was calculated as (Length x Width2)/2.
For bioluminescent xenograft tumors, KB cells were stably transfected with firefly luciferase and clonally selected via hygromycin B selection; 5 × 106 cells were subcutaneously injected into nude mice to establish tumors. RediJect D-Luciferin Ultra Bioluminescent Substrate (PerkinElmer) was administered via the manufacturer’s protocol for intravital monitoring of tumor bioluminescence using IVIS Spectrum (Caliper). It was pre-established that for all flank tumor studies, animals were excluded if their tumors had not reached a volume of 50–100 cm3 by the time of treatment. Animals were randomized into experimental arms by minimizing the differences in mean tumor size and standard deviation.
Confocal imaging and flow cytometry
For fixed cell confocal preparation, after treatment for 1 hour at with 5 μM of pHLIP-anti155 (Fig. 2a), cells were washed with 1% BSA in PBS, fixed in 4% paraformaldehyde, and permeabilized using 0.1% Triton X-100 in PBS. All washes were performed using PBS at pH 7.4 to wash away surface-bound pHLIP. Actin and nuclei were stained with Texas Red-X phalloidin (Life Technologies) and Hoechst 33342 (Life Technologies), respectively. Cells were mounted in Slow Fade Gold (Invitrogen). Alternatively, Toledo cells were treated with 500 nM pHLIP-anti155 (Extended Data Fig. 2e), washed with 1% BSA in PBS, and imaged live without fixation or permeabilization. For tumor and liver tissues, organs were harvested and fixed in 10% formalin, and then incubated overnight in 30% sucrose in PBS. Tumors were flash frozen in OCT before slicing into 10 μm-thick sections, permeabilization, staining, and mounting in Vectashield (Vector Labs). Cell and tissue confocal imaging was performed using TCS SP5 Spectral Confocal Microscope (Leica); confocal projections were constructed using LAS AF software (Leica) with 0.9 μm-stack height. For live cell flow cytometry, after 48 hours of treatment, cells were washed 5x with 1% BSA in PBS on ice and then analyzed on a FACScan (BD Biosciences) using FlowJo software (Tree Star); for B220 studies, freshly harvested spleen cells (see Flank Tumor Establishment section) were blocked with 10% FBS (20 min); stained with Alexa488-anti-CD45R/B220 (BD clone RA3-6B2, 20 min incubation at room temperature at 1 μg/ml concentration); washed 3x with PBS on ice, and transferred to 1% BSA 0.1% NaN3 in PBS on ice before analysis.
Luciferase reporter and cell viability
For dual luciferase reporter experiments, the miRNA sensor was generated by inserting the target sequence for miR-155 into the 3′UTR of Renilla luciferase on a psiCHECK™-2 vector (Promega). KB cells were stably transfected using Lipofectamine 2000 (Life Technologies) and co-transfection with a Linear Hygromycin Marker (Clontech) followed by clonal selection. Utilization of stable clones was more reliable than transiently transfected cells for antimiR studies. Cell lysates were measured for luciferase activity 48 hours after treatment using the Dual-Luciferase Reporter Assay System (Promega). Control LNA antimiR-155 (Exiqon) was delivered by lipofectamine RNAiMAX. Optimal sensor activity was seen at a 500 nM dose, though inhibition of miR-155 was also observed at lower doses. For analysis of miR-21 inhibition A549 cells were similarly treated with pHLIP-anti21 and relevant controls; however, the cells were instead transfected with a miR-21-specific LightSwitch miRNA Target GoClone Luciferase Reporter (Active Motif). Cell viability was measured 96 hours after treatment using CellTiter-Glo (Promega). For both luciferase and viability assays, all treatments were performed at the indicated pH for 24 hours, then media was replaced with 10% FBS in RPMI at physiological pH for extended incubation.
qPCR
For qPCR analysis of tissue after treatment with two 2 mg/kg injections of pHLIP-anti155 or pHLIP-antiscr spaced 48 hours apart; tissues were harvested 24 hours after the last injection and divided into at least five representative 1 mg slices. Tissue slices were pooled into Trizol (Life Technologies) and homogenized using a Precellys 24 Homogenizer. Per the manufacturer’s protocol, chloroform was added to facilitate phase separation, and the RNA-containing aqueous phase was collected. An equal volume of 200 proof ethanol was added, and RNA was purified from this mixture using RNeasy kit (Qiagen) and standard procedures with on-column DNase I digestion; standard RNeasy purification was followed for RNA extraction from cells. RT-PCR was performed with 1 μg total RNA and poly-A based iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed with Quantitect Primer Assays (Qiagen) and iQ SYBR Green Supermix (Bio-Rad) using a Roche LightCycler 480 System; all samples were normalized to β-actin.
RNA-seq
For RNA-seq analysis, the overexpression and withdrawal groups both consisted of three mice with subcutaneous tumors that were established from enlarged spleens of diseased donor mir-155LSLtTA mice (Extended Data Fig. 9a). The overexpression and withdrawal mice were paired such that each of the three pairs was from a separate donor littermate. Tissue was harvested once tumors reached a volume of 1 cm3; for mice in the miR-155 withdrawal group, DOX was administered for 16 hours before tissue harvest. As described in the transplant methods, tumor tissue was dispersed into a single cell suspension and red blood cells were lysed. Total RNA was extracted from the remaining cells using the hybrid Trizol and RNeasy protocol described in the qPCR methods. High quality total RNA (Agilent 2100 Bioanalyzer RIN value greater than 7) was sent to Expression Analysis for library preparation, Illumina TruSeq mRNA sequencing (50 bp paired end, 25 million reads per sample), alignment to the mouse genome (greater than 80% aligned to the NCBI37/mm9 assembly), and counts of the number of gene-mapped fragments given the maximum likelihood abundances. DESeq was used to first estimate size factors (i.e. normalize samples by their respective sizes) and dispersions (i.e. variance between samples), and then identify differentially expressed genes (Supplementary Table 1). Heatmaps were generated using variance stabilizing transformations of the count data based on a parametric fit to the overall mean-dispersions.
KEGG analysis
Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov) was used to identify the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were enriched in the genes that were both upregulated in response to miR-155 withdrawal and had a false discovery rate (FDR) less than 0.05. Enriched KEGG pathways had a minimum count threshold of 2 and a modified Fisher Exact P-value for gene enrichment less than 0.05.
Extended Data
Extended Data Figure 1. Distribution of pHLIP to the renal system and lymph node metastases.

a, Intravenous injection of A750-pHLIP distributes to the (white arrow) kidneys and (blue arrow) tumor in a representative mir-155LSLtTA subcutaneous flank model (n=3); time points indicate hours after a single injection of A750-pHLIP. Previous reports have observed systemic distribution of pHLIP to kidneys in other mouse models15. Similarly, we speculate that the increased uptake of pHLIP peptide in the kidneys is due to excretion and increased acidity of renal tubule cells. Initially kidneys are highly enriched for pHLIP, which is gradually excreted while pHLIP shows a more steady accumulation in the tumor. b, Representative example showing A750-pHLIP distribution to the (white arrow) bladder and (yellow arrow) enlarged axillary lymph node 36 hours after intravenous administration into mir-155LSLtTA mice with lymphadenopathy (n=3). c, In addition to distributing to the (white arrow) primary mir-155LSLtTA flank tumor and (red arrow) kidneys, A750-pHLIP distributes to (black arrows) enlarged lymph nodes that resulted from metastatic spread; intravital fluorescence of A750-pHLIP was detected 48 hours after intravenous injection into nude transplant mice with conspicuous lymphadenopathy (shown is a representative animal from n=3).
Extended Data Figure 2. Assessment of pHLIP-PNA conjugation and activity.

a, HPLC elution profiles of (top) free PNA, (middle) reaction mixture of PNA and pHLIP-C(Npys), and (bottom) purifed pHLIP-PNA incubated in DTT. HPLC was used to purify pHLIP-PNA (black arrow). Shown is the fluorescence detection of TAMRA (ex/em: 540/575) which was conjugated to the PNA; samples were also detected by absorbance at 260 and 280 nm (data not shown). b, Tricine SDS-PAGE evaluation of pHLIP-PNA conjugation. Gel was visualized by (top) TAMRA fluoresence to detect labeled PNA and (bottom) Coomassie stain to detect both PNA and peptide. c, Gelshift analysis of pHLIP-antimiR-155 binding to miR-155 and disulfide reduction in the presence of DTT. d, High magnification confocal projections of A549 cells incubated with labeled pHLIP-antimiR (against control miR-182); scale bars represent 7.5 μm. The diffuse intracellular fluorescence is indicative of freely distributed antimiR throughout the cytosol—note that the presence of marginal punctate fluorescence at both pH levels suggests that endocytosis is probably an additional mode of cell entry. e, Toledo DLBCL lymphocytes were incubated with labeled pHLIP-anti155 at pH 6.2; fluorescence of a representative live cell is overlayed on a bright field micrograph; scale bars represent 2 μm. f, Flow cytometry analysis of Toledo cells incubated with labeled pHLIP-anti155; cell association was dependent on dose (top, pH 6.2) and pH (bottom, 500 nM dose). g, Inhibition of miR-155 demonstrated by derepression of a miR-155 dual luciferase sensor in KB cells. h, Inhibition of miR-21 demonstrated by desuppression of luciferase expression in A549 cells transfected with a Renilla luciferase sensor. Data are shown as mean ± s.d., with n = 3; statistical analysis performed with two-tailed Student’s t-test; two asterisks, P < 0.01; three asterisks, P < 0.001.
Extended Data Figure 3. Pathology of the mir-155LSLtTA model of oncomiR addiction.

a, Organomegaly in representative diseased mir-155LSLtTA mice: (top) conspicuous lymphadenopathy seen in the (black arrow) cervical and (white arrow) brachial lymph nodes; (middle) enlarged exposed (white arrows) cervical and (black arrows) axillary lymph nodes; and (bottom) enlarged (black arrows) spleen. b, Histopathology of mir-155LSLtTA mice: H&E stain of an enlarged spleen shows expansion of the white pulp by a nodular, neoplastic infiltrate; staining of the spleen shows CD20+ and CD10+ B cells of follicular center origin. Analysis of enlarged lymph nodes indicates DLBCL with lymph node architecture effaced by a confluent population of B220+ neoplastic lymphocytes and a Ki-67 proliferative index at nearly 100%, n=5. c, Tumor regression due to DOX-induced miR-155 withdrawal in a subcutaneous tumor model established from transplanted splenic mir-155LSLtTA lymphocytes; time points indicate hours after initial administration of DOX. With a cancer phenotype that is relevant to human disease yet can be modulated by miRNA silencing, this is an excellent model for evaluating miR-155-targeted therapies.
Extended Data Figure 4. Experimental schematics for mouse tumor studies.

a, Workflow for treatment of the mir-155LSLtTA subcutaneous flank model for the early endpoint and survival studies; Day 1 indicates time of first injection. For the “early treatment” experiments in Figure 3a,b,d–f,h and Extended Figure 5b,c mice were treated on days 1 and 2 with pHLIP-anti155, mock buffer, pHLIP-antiscr and anti155 only; fed DOX starting on day 3; or treated with CHOP regimen on days 2–4. For survival experiments in Figure 3c,g and Extended Figure 5a mice were treated on days 1–3 with pHLIP-anti155, LNA against miR-155, and mock buffer. b, Workflow for investgation of the mir-155LSLtTA model of lymphoma for the biodistribution and miR-155 silencing studies. For experiments in Figure 4a and Extended Data Figure 8a,b mice were treated on day 1 with pHLIP-anti155, anti155 only, and mock buffer. For experiments in Figure 4b–d,h and Extended Data Figure 8c–g mice were treated on day 1 and day 3 with pHLIP-anti155, pHLIP-antiscr, and mock buffer; or fed DOX 16 hours before harvest.
Extended Data Figure 5. Administration of pHLIP-anti155 to mice with subcutaneous lymphoma flank tumors.

a, Fold change in tumor size in response to miR-155 withdrawal and CHOP treatment (n=3); arrow represents initiation of DOX treatment (n=3, food pellets enriched with DOX at 2.3 gm/kg, Bio-Serv), white triangle represents CHOP treatment (systemic injection of Cyclophosphamide at 40 mg/kg, Doxorubicin at 3.3 mg/kg, and Vincristine at 0.5mg/kg; oral gavage of Prednisone at 0.2 mg/kg), gray triangles represent maintenance administration of Prednisone. b, Tumor growth response to systemically administered antimiR treatment; symbols represent intravenous injections of 2 (arrowhead) or 1 (arrow) mg/kg of pHLIP-conjugated antimiR-155, molar equivalent of phosphorothioated antimiR-155 LNA, or mock delivery solution; n = 5, data are shown as mean ± s.e.; statistical comparison of pHLIP-anti155 to LNA performed with two-way ANOVA; three asterisks, P < 0.001, four asterisks, P < 0.0001. c, Representative histologic analysis of kidneys (H&E, 100x magnification) harvested from early endpoint study, in which all of the mice from Figure 3a and Extended Data Figure 5a were sacrificed at the same time for analysis. Kidney sections reveal an absence of microscopic changes in treated animals (pHLIP-anti155) that would be indicative of renal toxicity (compare with normal renal sections in mock control). d, Representative pHLIP-antiscr-treated mouse (top) with primary flank tumor (white arrow) and enlarged inguinal lymph node (black arrow) compared to an untreated mouse with no tumor (bottom). e, Measurement of circulating lymphocytes in blood collected at time of sacrifice in early endpoint study; dotted line denotes average level in nude mice with no tumor. f, Although pHLIP interacts with the outer leaflet of lipid membranes, no significant change in red blood cell (RBC) levels was detected after intraveous treatment of mice with subcutaneous mir-155LSLtTA transplant tumors. This supports the specificity of pHLIP treatments on cells of tumor origin since pHLIP-antimiR treatments affect the levels of circulating lymphocytes (Extended Data Fig. 5e); data are shown as mean ± s.d.
Extended Data Figure 6. Toxicology assessment of intravenously administered pHLIP-anti155 to C57BL/6J mice.

a, Serum-based clinical chemistry evaluation of systemic toxicity with focus on liver and kidney function; dosing schedule consisted of injections of 2 mg/kg of pHLIP-anti155 (and equamolar dose of LNA) on Day 10 and 12, as well as 1 mg/kg on Day 11. Blood samples were serially harvested retro-orbitally on Day 0 (10 days before start of treatment), as well as 1 day and 14 days after treatment. b, Circulating white blood cell count collected 14 days after treatment. c, Mouse mass throughout duration of the study. d, Organ mass normalized to total body mass at time of harvest. a–d, For all analyses mock n = 4; pHLIP-anti155 n = 5, and LNA n = 5; dotted lines indicate typical wild type values for C57BL/6J mice.
Extended Data Figure 7. Administration of pHLIP-anti155 to mice with KB oral squamous cell carcinoma xenograft tumors.

a, Intravenous injection of pHLIP-anti155 (**) and phosphorothioated LNA against miR-155 (*) significantly enhanced survival compared to mock buffer treatment; n = 4 for all groups; arrowheads indicate injections of 2 mg/kg (or molar equivalent for LNA). Survival cutoff criteria included tumor volume greater than 1 cm3 or compassionate euthanisia, which was mandated for three mock-treated mice with ulcerated tumors. b, Fold change in tumor size in response to treatment; measurements were plotted until the mock negative control group was euthanized. c, Tumor bioluminescence in response to treatment; Day 8 represents luciferase activity before first injection. d, Representative images of tumor bioluminescence. Data are shown as mean ± s.e.; statistical analysis performed with (a) Mantel-Cox analysis or (c) two-tailed Student’s t-test, asterisk, P < 0.05; two asterisks, P < 0.01.
Extended Data Figure 8. Administration of pHLIP-anti155 to mir-155LSLtTA mice with lymphoma.

a, Quantification of liver distribution of TAMRA-labeled PNA delivered with and without conjugation to pHLIP; ImageJ was used to measure fluorescence from five confocal sections per mouse liver, n = 3 mice per group. b, Visualization of whole liver fluorescence after antimiR administration; pHLIP-anti155 liver fluorescence is similar to the autofluorescence seen in the mock group. c, Lymph node tumor burden (A = axillary, B = brachial, C = cervical, and I = inguinal lymph nodes); in these specific images taken from diseased littermates, pHLIP-antiscr-treated mice had a more than 3-fold larger aggregate lymph node mass (3.179 g) than pHLIP-anti155-treated mice (1.006 g). d,e, Size of harvested (d) spleens (n = 4) and (e) lymph nodes (axillary, brachial, cervical, and inguinal; n = 5) with respect to wild type; n < 6 (i.e. total number of treated mice) due to size data not collected. f,g, TUNEL analysis of treated cervical lymph nodes of mir-155LSLtTA mice (n = 6). h, Percent of white pulp in treated spleens; n = 6. i, Measurement of lymphocyte infiltration into liver; n = 6. j, Low magnification H&E images of livers from Fig. 4d. k, Flow cytometry analysis of B220-positive cells comprising the spleens of treated mice; B220 is typically a marker for B cells, though varied expression is seen on some T cells, natural killer cells, and macrophages, n=4. l, Representative H&E image of healthy kidneys from pHLIP-anti155-treated mice; n=6. Data are shown as mean ± s.d. (a, d, e, g, h) or mean ± s.e. (i); statistical analysis performed with two-tailed Student’s t-test; two asterisks, P < 0.01; four asterisks, P < 0.0001.
Extended Data Figure 9. Differential gene expression analysis of miR-155 withdrawal.

a, Experimental design for RNA-seq analysis of miR-155 addicted tumors compared to tumors undergoing miR-155 withdrawal and tumor regression. b, RNA-seq differential gene expression analysis of three independent tumors that overexpress miR-155 in comparison to three independent tumors undergoing DOX-induced miR-155 withdrawal; shown are all differentially expressed genes with FDR < 0.05; rows are clustered by euclidean distance measure. c, KEGG pathway analysis of significantly upregulated genes after miR-155 withdrawal. d, Selection of potential miR-155 targets involved in tumor regression. Intersection of genes (Group I) that are both predicted miR-155 targets (Supplementary Table 2) and overexpressed after miR-155 withdrawal from mir-155LSLtTA tumors (Supplementary Table 1) with genes inferred from three separate miR-155 target analyses. (Group II) Xu et al. used RNA-seq to compare Mutu I B cells that overexpress miR-155 with cells transformed with a control vector36. (Group III) Gottwein et al. identified shared targets between miR-155 and a viral orthologue, miR-K12-1125. (Group IV) Loeb et al. used HITS-CLIP to identify miR-155 targets without perfect seed matches in T cells37. e, qPCR determination of gene expression levels in Toledo cells treated for 48 hours with 500 nM pHLIP-anti155 at pH 6.2; data are shown as mean ± s.d., n = 3; statistical analysis performed with two-tailed Student’s t-test, asterisk, P < 0.05.
Extended Data Figure 10. Expression levels of putative targets in response to miR-155 silencing in mir-155LSLtTA mice.

qPCR validation of potential miR-155 targets involved in tumor regression using mir-155LSLtTA mice with conspicous lymphadenopathy treated with (black bars) DOX for 16 hours compared to (white bars) untreated mice with lymphadenopathy; all samples are normalized to β-actin, n=3. Genes were selected based on criteria described in Supplementary Table 3. As shown in Fig. 4F, both Bach1 and Mafb have utility as biomarkers for miR-155 withdrawal-induced tumor regression.
Supplementary Material
Acknowledgments
We thank M. Bosenberg, Y. Dang, A. Karabadzhak, and J. Zhou for discussions and suggestions; R. Ardito, M. Bonk, K. Card, D. Caruso, D. Jenci, D. Laliberte, W. Nazzaro, N. Santiago, and S. Wilson for rodent services; A. Brooks for tissue pathology services; Antech Diagnostics for CBC analysis; E. Aronesty, B. Cooper, and E. Norris at Expression Analysis for RNA-seq services, as well as J. Deacon, A. Kasinski, J. Sawyer, and C. Stahlhut for critical reading of the manuscript. C.J.C. is the recipient of a Ruth L. Kirschstein Postdoctoral Fellowship from the NCI/NIH (F32CA174247). Our work has been supported by grants from the NCI/NIH (R01CA131301), the NHLBI/NIH (R01HL085416), the NIGMS/NIH (R01GM073857), the NIEHS/NIH (R01ES005775), the NCI/NIH (R01CA148996), the NIBIB/NIH (R01EB000487), the NHLBI/NIH (2T32HL007974), and pilot grants from the Yale Comprehensive Cancer Center.
Footnotes
SUPPLEMENTARY INFORMATION is linked to the online version of the paper at www.nature.com/nature.
AUTHOR CONTRIBUTIONS C.J.C., R.B., F.B., A.S., P.M.G., D.M.E., W.M.S., and F.J.S. designed the research; C.J.C. performed the research; R.B. synthesized the PNA; I.A.B. and C.J.C. developed and maintained the rodent colonies; C.J.C., Z.P., and C.L. performed the bioinformatics analysis; D.T.B. performed the pathological analysis; C.J.C, R.B., D.B., P.M.G., D.M.E., W.M.S., and F.J.S. analyzed the data and wrote the paper.
Gene expression data have been deposited in the Genome Expression Omnibus (GEO) under accession number GSE61851.
The authors declare no conflict of interest.
References
- 1.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 2.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 3.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 4.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467:86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- 5.Babar IA, et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci USA. 2012;109:E1695–E1704. doi: 10.1073/pnas.1201516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3:1. doi: 10.1186/1758-907X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849–864. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng CJ, Saltzman WM, Slack FJ. Canonical and Non-Canonical Barriers Facing AntimiR Cancer Therapeutics. Curr Med Chem. 2013;20:3582–3593. doi: 10.2174/0929867311320290004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elmen J, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 10.Krutzfeldt J, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 11.White PJ, Anastasopoulos F, Pouton CW, Boyd BJ. Overcoming biological barriers to in vivo efficacy of antisense oligonucleotides. Expert Rev Mol Med. 2009;11:e10. doi: 10.1017/S1462399409001021. [DOI] [PubMed] [Google Scholar]
- 12.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–6465. [PubMed] [Google Scholar]
- 13.Reshetnyak YK, Andreev OA, Lehnert U, Engelman DM. Translocation of molecules into cells by pH-dependent insertion of a transmembrane helix. Proc Natl Acad Sci USA. 2006;103:6460–6465. doi: 10.1073/pnas.0601463103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thevenin D, An M, Engelman DM. pHLIP-mediated translocation of membrane impermeable molecules into cells. Chem Biol. 2009;16:754–762. doi: 10.1016/j.chembiol.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reshetnyak YK, et al. Measuring tumor aggressiveness and targeting metastatic lesions with fluorescent pHLIP. Mol Imaging Biol. 2011;13:1146–1156. doi: 10.1007/s11307-010-0457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen PE, Egholm M, Buchardt O. Peptide nucleic acid (PNA). A DNA mimic with a peptide backbone. Bioconjugate Chem. 1994;5:3–7. doi: 10.1021/bc00025a001. [DOI] [PubMed] [Google Scholar]
- 17.Østergaard H, Tachibana C, Winther JR. Monitoring disulfide bond formation in the eukaryotic cytosol. J Cell Biol. 2004;166:337–345. doi: 10.1083/jcb.200402120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rai D, Karanti S, Jung I, Dahia PLM, Aguiar RCT. Coordinated expression of microRNA-155 and predicted target genes in diffuse large B-cell lymphoma. Cancer Genet Cytogenet. 2008;181:8–15. doi: 10.1016/j.cancergencyto.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rather MI, Nagashri MN, Swamy SS, Gopinath KS, Kumar A. Oncogenic microRNA-155 down-regulates tumor suppressor CDC73 and promotes oral squamous cell carcinoma cell proliferation: implications for cancer therapeutics. J Biol Chem. 2013;288:608–618. doi: 10.1074/jbc.M112.425736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costinean S, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in Eμ-miR155 transgenic mice. Proc Natl Acad Sci USA. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandhu SK, Croce CM, Garzon R. Micro-RNA expression and function in lymphomas. Adv Hematol. 2011;2011:1–12. doi: 10.1155/2011/347137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng CJ, Slack FJ. The duality of oncomiR addiction in the maintenance and treatment of cancer. Cancer J. 2012;18:232–237. doi: 10.1097/PPO.0b013e318258b75b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeVita Vincent T. DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology. 9. Lippincott Williams & Wilkins; Philadelphia, Pennsylvania: 2011. [Google Scholar]
- 24.Zhang Y, et al. LNA-mediated anti–miR-155 silencing in low-grade B-cell lymphomas. Blood. 2012;120:1678–1686. doi: 10.1182/blood-2012-02-410647. [DOI] [PubMed] [Google Scholar]
- 25.Gottwein E, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dweep H, Sticht C, Pandey P, Gretz N. miRWalk--database: prediction of possible miRNA binding sites by ‘walking’ the genes of three genomes. J Biomed Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Li S, et al. microRNA-155 silencing inhibits proliferation and migration and induces apoptosis by upregulating BACH1 in renal cancer cells. Mol Med Report. 2012;5:949–954. doi: 10.3892/mmr.2012.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brognara E, et al. Uptake by human glioma cell lines and biological effects of a peptide-nucleic acids targeting miR-221. J Neurooncol. 2014;118:19–28. doi: 10.1007/s11060-014-1405-6. [DOI] [PubMed] [Google Scholar]
- 29.Wang YZ, et al. Delivery of an miR155 inhibitor by anti-CD20 single-chain antibody into B cells reduces the acetylcholine receptor-specific autoantibodies and ameliorates experimental autoimmune myasthenia gravis. Clin Exp Immunol. 2014;176:207–221. doi: 10.1111/cei.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christensen L, et al. Solid-Phase synthesis of peptide nucleic acids. J Pept Sci. 1995;1:175–183. doi: 10.1002/psc.310010304. [DOI] [PubMed] [Google Scholar]
- 31.Obad S, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011;43:371–378. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sahu B, et al. Synthesis and characterization of conformationally preorganized, (R)-diethylene glycol-containing γ-peptide nucleic acids with superior hybridization properties and water solubility. J Org Chem. 2011;76:5614–5627. doi: 10.1021/jo200482d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fabani MM, et al. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010;38:4466–4475. doi: 10.1093/nar/gkq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiraishi T, Nielsen PE. Peptide nucleic acid (PNA) cell penetrating peptide (CPP) conjugates as carriers for cellular delivery of antisense oligomers. Artif DNA PNA XNA. 2011;2:90–99. doi: 10.4161/adna.18739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fabbri E, et al. miRNA therapeutics: delivery and biological activity of peptide nucleic acids targeting miRNAs. Epigenomics. 2011;3:733–745. doi: 10.2217/epi.11.90. [DOI] [PubMed] [Google Scholar]
- 36.Xu G, et al. Transcriptome and targetome analysis in MIR155 expressing cells using RNA-seq. RNA. 2010;16:1610–1622. doi: 10.1261/rna.2194910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loeb GB, et al. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell. 2012;48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.