

Abstract
Over the past decade, two independent technologies have emerged and been widely adopted by the neuroscience community for remotely controlling neuronal activity: optogenetics which utilize engineered channelrhodopsin and other opsins, and chemogenetics which utilize engineered G protein-coupled receptors (Designer Receptors Exclusively Activated by Designer Drugs (DREADDs)) and other orthologous ligand–receptor pairs. Using directed molecular evolution, two types of DREADDs derived from human muscarinic acetylcholine receptors have been developed: hM3Dq which activates neuronal firing, and hM4Di which inhibits neuronal firing. Importantly, these DREADDs were not activated by the native ligand acetylcholine (ACh), but selectively activated by clozapine N-oxide (CNO), a pharmacologically inert ligand. CNO has been used extensively in rodent models to activate DREADDs, and although CNO is not subject to significant metabolic transformation in mice, a small fraction of CNO is apparently metabolized to clozapine in humans and guinea pigs, lessening the translational potential of DREADDs. To effectively translate the DREADD technology, the next generation of DREADD agonists are needed and a thorough understanding of structure–activity relationships (SARs) of DREADDs is required for developing such ligands. We therefore conducted the first SAR studies of hM3Dq. We explored multiple regions of the scaffold represented by CNO, identified interesting SAR trends, and discovered several compounds that are very potent hM3Dq agonists but do not activate the native human M3 receptor (hM3). We also discovered that the approved drug perlapine is a novel hM3Dq agonist with >10 000-fold selectivity for hM3Dq over hM3.
Keywords: DREADD, CNO, hM3Dq, SAR, neuronal activation, perlapine
To elucidate how neuronal ensembles interactively encode higher brain processes, new and improved methods for both recording and manipulating neuronal activity will be required.1,2 The ability to selectively modulate the activity of defined neuronal populations and to elucidate the behavioral consequences of this selective neuronal modulation affords powerful approaches for studying mammalian brain function in health and disease. Historically, important methods include Wilder Penfield’s pioneering studies of focal electrical stimulation of the human cortex.3 The development of the optogenetics technology pioneered by Diesseroth and colleagues to visualize and activate neuronal activity with exquisite temporal resolution using engineered channelrhodopsin4,5 and other opsins6 has provided an expanding toolbox for decoding the neuronal correlates of brain function.4,6−10 More recently, Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) have been developed as a powerful chemogenetics technology for remotely controlling neuronal activity11,12 and have been widely adopted by the neuroscience and greater biological communities.13−18
DREADDs, first revealed in 2005,5 were developed using directed molecular evolution of human muscarinic acetylcholine receptors.11,12 After multiple rounds of random mutagenesis, DREADDs derived from the human muscarinic acetylcholine M3 receptors (hM3Dq) to be insensitive to the endogenous ligand acetylcholine (ACh) but potently and selectively activated by the pharmacologically inert clozapine N-oxide (CNO) were discovered. Importantly, CNO lacks appreciable affinity (Ki > 1 μM) for all relevant native CNS (central nervous system) targets.11,19 The DREADDs have no detectable constitutive activity in vitro11 and, thus, provide an attractive orthologous receptor-effector chemogenetic platform for modifying neuronal activity remotely with minimal invasiveness. In addition to hM3Dq, which activates neuronal firing upon the CNO stimulation in part by depolarization and elevation of intracellular calcium levels,12 hM4Di was developed from human muscarinic acetylcholine M4 receptors for inhibiting neuronal firing via activation of G-protein inwardly rectifying potassium (GIRK) channels.11 Since the introduction of the DREADD technology, a large number of papers have independently validated the utility of excitatory and inhibitory DREADDs.12,21−34 In addition, no effect related to the ectopic expression of hM3Dq or hM4Di has been observed.
In addition to being pharmacologically inert, CNO, the “chemical switch” of this chemogenetic approach, is orally bioavailability and CNS penetrant21,35−37 and is not subject to significant metabolic transformation in mice and rats. However, a small fraction of CNO is apparently metabolized to clozapine in humans, nonhuman primates and guinea pigs.36,39,41 Because clozapine modulates the activity of many native CNS receptors,42 thus interfering with the selective activation of the DREADDs in defined neuronal populations, the “back-metabolism” issue presents a hurdle for translating the DREADD technology forward. To ultimately develop the next generation of DREADD ligands that can selectively activate defined neuronal populations in primates including human, a thorough understanding of structure–activity relationships (SARs) of DREADDs is needed. To date, no SAR studies have been reported for any DREADDs including hM3Dq and hM4Di.
Here, we report the first SAR studies of hM3Dq. We extensively explored multiple regions of the scaffold represented by CNO, which resulted in the discovery of compounds 13 and 21 that are very potent hM3Dq agonists but do not activate the native human M3 receptor (hM3). We describe the design, synthesis, and pharmacological evaluation of new CNO analogues and discuss the interesting SAR trends revealed from the studies. We also report the discovery that perlapine, a hypnotic agent first reported in 1966,43−45 is a novel, potent, and selective agonist of hM3Dq (>10 000-fold selective for hM3Dq over hM3).
Results and Discussion
Design and Synthesis
To understand the SAR of CNO analogues as hM3Dq agonists, we explored several regions of the CNO scaffold. In particular, we focused on investigating the R1 and R2 substituents as well as modifications to the piperazine ring (highlighted in red in Figure 1). We also studied whether the chloro group (R3, highlighted in blue in Figure 1) on the tricyclic core is required and whether a different tricyclic core (e.g., the core of the perlapine scaffold, see below) can be tolerated.
Figure 1.
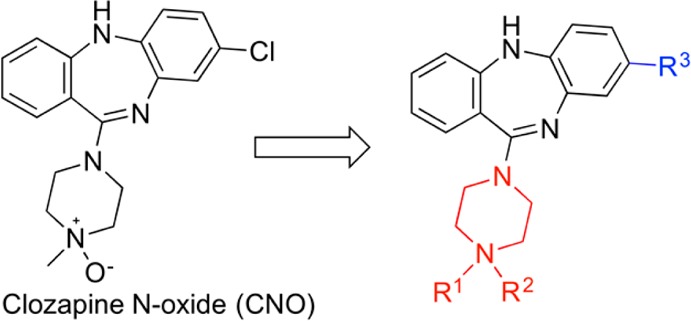
SAR studies of the CNO scaffold.
We first explored the size of the alkyl group (R1) on the N4′ position of the CNO scaffold and synthesized a set of close analogues as outlined in Scheme 1. The commercially available 2-((2-amino-4-chlorophenyl)amino)benzoic acid (1) was refluxed in xylene for 48 h to give the cyclized compound 2, which was then treated with POCl3 and N,N-dimethylaniline in toluene at 95 °C for 2 h to afford the chloride 3. Compound 3 was reacted with various N-alkylpiperazines in toluene at 120 °C for 2 h to yield compounds 4a–4d, which were then converted to their corresponding N-oxides 5a–5d by treating with meta-chloroperoxybenzoic acid (mCPBA) at room temperature in CH2Cl2 for 10 min. The synthetic route to compounds 4a – 4d was described previously.46,47
Scheme 1. Synthesis of N4′-alkyl Substituted CNO Analogues.

Reagents and conditions: (a) xylene, reflux, 48 h, 95% yield; (b) POCl3, N,N-dimethylaniline, toluene, 95 °C, 2 h, 67% yield; (c) N-alkylpiperazines, toluene, 120 °C, 2 h, 69–80% yield; and (d) mCPBA, CH2Cl2, rt, 10 min, 65–75% yield.
We next synthesized the analogues outlined in Scheme 2 to determine whether the positive charge of CNO is required for activating hM3Dq. For example, compound 6, which contains a quaternary ammonium moiety, has a permanent positive charge, while compounds 7, 9, 12, and 14 do not possess a basic amino group and therefore do not contain a positive charged group. As illustrated in Scheme 2, compound 4a (clozapine) was converted to the quaternary ammonium iodide 6 by stirring overnight with CH3I in acetone at room temperature. Compound 3 was treated with piperazin-2-one at 99 °C overnight in the 1:1 mixture of 1,4-dioxane and ethanol to give compound 7. Similarly, compound 9 was produced by treating compound 3 with commercially available 1,3,8-triazaspiro[4.5]decane-2,4-dione (8) in the 2:1 mixture of 1,4-dioxane and N,N-dimethylformamide (DMF) at 130 °C for 24 h. Hydrolysis of compound 9 using 0.5 N aqueous NaOH solution in 1,2-dimethoxyethane under microwave irradiation afforded compound 10. Likewise, compound 11 was prepared from compound 3 and piperazine in toluene at 120 °C for 2 h. Acetylation of compound 11 by AcCl in CH2Cl2 at 0 °C in the presence of triethylamine (TEA) yielded compound 12, which was subsequently reduced to the deuterated compound 13 using LiAlD4 under reflux conditions, followed by quenching with CD3OD at 0 °C. In addition, compound 11 was reacted with MsCl in CH2Cl2 at 0 °C in the presence of diisopropylethylamine (DIPEA) to give the methylsulfonamide 14.
Scheme 2. Synthesis of Compounds 6, 7, and 9–14.

Reagents and conditions: (a) CH3I, acetone, rt, overnight, 55% yield; (b) 2-oxypiperazine, 1,4-dioxane/ethanol 1:1, 99 °C, overnight, 65% yield; (c) 1,3,8-triazaspiro[4.5]decane-2,4-dione (8), 1,4-dioxane/DMF (2:1), 130 °C, 24 h, 66% yield; (d) 1,2-dimethoxyethane, 0.5 N NaOH, microwave, 150 °C, 10 min, 16% yield; (e) piperazine, toluene, 120 °C, 2 h, 69% yield; (f) AcCl, TEA, CH2Cl2, 0 °C, 1 h, 86% yield; (g) (1) LiAlD4, THF, N2, reflux, 2h, (2) CD3OD, 0 °C, (3) NH4OH, 0 °C, 84% yield; (h) MsCl, DIPEA, CH2Cl2, 0 °C, 1 h, 93% yield.
To determine whether the 8-Cl group on the tricyclic core is required to activate hM3Dq, we prepared compounds 21–23 according to the synthetic route outlined in Scheme 3.48 The commercially available 2-aminobenzoic acid (15) and 2-nitrophenyl iodide (16) were subjected to Ullmann coupling conditions49 to afford the aniline 17. Reduction of the nitro moiety of compound 17 yielded compound 18, which was refluxed in xylene to generate the benzodiazepine 19. Treatment of compound 19 with POCl3 provided the chloride 20, which was then displaced with piperazine to afford compound 21. Similarly, compound 22 was prepared by the displacement reaction of the chloride 20 with 1-ethylpiperazine in toluene under reflux conditions. In addition, the oxidation of compound 22 by mCPBA in CH2Cl2 afforded the N-oxide 23.
Scheme 3. Synthesis of Compounds 22–24.

Reagents and conditions: (a) K2CO3, Cu, 3-methylbutan-1-ol, reflux, 4 h; (b) NaS2O4, NH4OH/H2O 3:2, 80 °C, 30 min, 75% yield in two steps; (c) xylene, reflux, Dean–Stark conditions, 48 h, 96% yield; (d) POCl3, N,N-dimethylaniline, toluene, reflux, 3 h, 52% yield; (e) piperazine, toluene, reflux, overnight, 63% yield; (f) 1-ethylpiperazine, toluene, reflux, 2 h, 72% yield; and (g) mCPBA, CH2Cl2, rt, 10 min, 78% yield.
Biological Evaluation
The newly synthesized compounds were evaluated in the hM3Dq and hM3 Ca2+ mobilization fluorometric imaging plate reader (FLIPRTETRA) assays according to the protocols reported previously.11,50,51 Agonist activities of these compounds in the hM3Dq and hM3 functional assays are summarized in Table 1.
Table 1. Agonist Activities of New Compounds in hM3Dq and hM3 FLIPR Assaysa.
| hM3Dq |
hM3 |
|||
|---|---|---|---|---|
| compd | EC50 (nM) | Emax (relative to CNO) | EC50 (nM) | Emax (relative to acetylcholine) |
| 4a | 1.1 | 95 | 360 | 88 |
| 4b | 7.0 | 91 | >30 000 | NA |
| 4c | 13 | 45 | >30 000 | NA |
| 4d | 71 | 50 | >30 000 | NA |
| 5a | 6.0 | 100 | >30 000 | NA |
| 5b | 19 | 50 | >30 000 | NA |
| 5c | 190 | 45 | >30 000 | NA |
| 5d | 740 | 79 | >30 000 | NA |
| 6 | 0.069 | 100 | 9.5 | 92 |
| 7 | >30 000 | NA | >30 000 | NA |
| 9 | >30 000 | NA | >30 000 | NA |
| 10 | >30 000 | NA | >30 000 | NA |
| 11 | 2.1 | 95 | 490 | 86 |
| 12 | >30 000 | NA | >30 000 | NA |
| 13 | 9.6 | 86 | >30 000 | NA |
| 14 | >30 000 | NA | >30 000 | NA |
| 21 | 1.7 | 100 | NA | ∼20 |
| 22 | 1.3 | 81 | >30 000 | NA |
| 23 | 220 | 59 | >30 000 | NA |
EC50 values are the average of at least two duplicate experiments with standard deviation (SD) values that are 3-fold less than the average. NA: not applicable.
For the size of the N-alkyl group in compounds 4a–4d and 5a–5d, we observed a clear trend showing that the longer and/or bulkier the N-alkyl group, the weaker the compounds’ potency for hM3Dq. The replacement of the methyl group in compounds 4a and 5a with the n-propyl group in compounds 4d and 5d resulted in a potency decrease of approximately 70- and 100-fold, respectively. In addition to the loss in potency, the compounds with a longer or bulkier N-alkyl group (e.g., compounds 4b–4d and 5b–5d) in general displayed lower agonist efficacy for hM3Dq and became partial agonists of hM3Dq rather than full agonists as seen for compounds 4a and 5a. Interestingly, compounds 4c and 5c, which contain an i-propyl group, were more potent than compounds 4d and 5d, which contain a n-propyl group, suggesting that the length of the N-alkyl group plays a more significant role than the bulkiness of the N-alkyl group in reducing agonist potency. We were also pleased to find that compounds 4b–4d and 5b–5d did not display any agonist activity (EC50 > 30 000 nM) for the native human M3 receptor (hM3), in contrast to compound 4a (clozapine), which was a hM3 agonist with sub-μM potency. In addition, compounds 4a–4d were in general more potent than their corresponding N-oxides 5a–5d at activating hM3Dq, suggesting that the negative charge on the N-oxides is not only not required for activating hM3Dq, but also reduces agonist potency.
The quaternary ammonium salt 6 was an extremely potent full agonist of hM3Dq with an EC50 value of 69 pM and about 15-fold more potent than compound 4a (clozapine). However, compound 6 was also a potent full agonist of hM3 (EC50 = 9.5 nM, Emax = 92) even though it achieves >100-fold higher potency for hM3Dq over hM3. On the other hand, compounds 7, 12, and 14, which do not contain a basic amino group or a group with permanent positive charge, did not display any agonist activity for hM3Dq. As expected, these compounds did not activate hM3 either. Taken together, these results suggest that either a basic amino group as in compounds 4a and 4b or a group with permanent positive charge as in compounds 5a and 6 is required to retain hM3Dq agonist activity. In addition, compound 9 that contains a hydantoin moiety and compound 10 that contains an amino acid moiety in this region did not activate hM3Dq and hM3. On the other hand, compound 11, which is the des-methyl clozapine, showed similar potency and efficacy for hM3Dq and hM3 as clozapine, suggesting that the N-methyl group is not required for activating hM3Dq. Interestingly, compound 13, which is a deuterated analogue of compound 4b, exhibited similar potency and efficacy (EC50 = 9.6 nM, Emax = 86%) for hM3Dq as compounds 4b and 5a (CNO) (Figure 2). Importantly, compound 13 did not display any agonist activity for hM3. Because compound 13 contains an α,α-dideutero ethyl group, it is likely that the N-dealkylation, the major metabolic pathway that converts clozapine to des-methyl clozapine,36,39 will be significantly reduced on the basis of the well-documented primary kinetic isotope effect52 in similar systems.53,54
Figure 2.
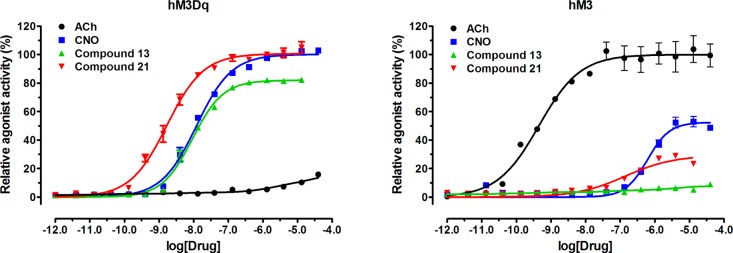
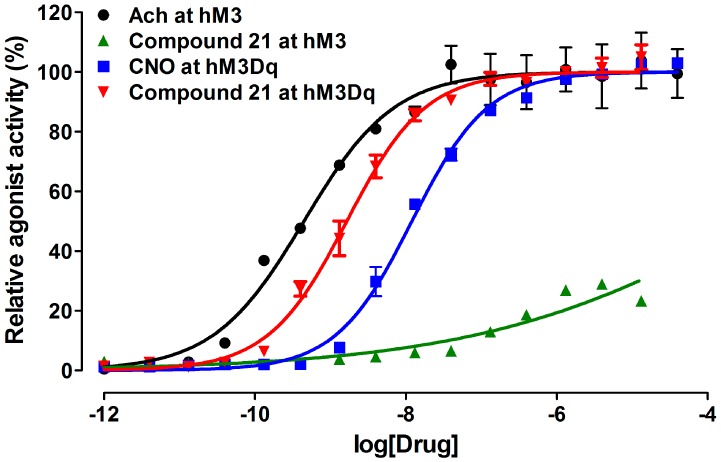
Compounds 13 and 21 are potent hM3Dq agonists and do not activate hM3 being similar to compound 5a (CNO). The endogenous ligand acetylcholine (ACh), on the other hand, is a potent hM3 agonist and does not activate hM3Dq.
We were also pleased to find that the 8-chloro group was not required to maintain high agonist potency and efficacy for hM3Dq. In particular, compound 21 was a potent full agonist (EC50 = 1.7 nM, Emax = 100%) of hM3Dq (Figure 2). In contrast to compound 11, a full hM3 agonist with sub-micromolar potency, compound 21 displayed little agonist activity for hM3 (Emax = ∼20%). In addition, compound 22 was found to be a potent hM3Dq agonist (EC50 = 1.3 nM, Emax = 81%), which was more potent than the corresponding chloro analogue, compound 4b (EC50 = 7.0 nM, Emax = 91%). On the other hand, the N-oxide 23 (EC50 = 220 nM, Emax = 59%) was about 10-fold less potent for hM3Dq than compound 5b, the corresponding chloro analog (EC50 = 19 nM, Emax = 50%). Similar to compound 21, both compounds 22 and 23 did not exhibit any agonist activity for hM3.
We next selected a subset of the above hM3Dq agonists that are inactive against hM3 and assessed their binding affinities to other aminergic GPCRs. Because compound 4a (clozapine) showed high binding affinities to 5HT2A and 5HT2C serotonin, α1A adrenergic, and H1 histamine receptors with Ki values of 5.4, 9.4, 1.6, and 1.1 nM, respectively (Table 2), we tested compounds 4b, 4c, 5b, 5c, 13, and 21 in 5HT2A, 5HT2C, α1A, and H1 radioligand binding assays. The assay results are summarized in Table 2.
Table 2. Binding Affinities of Selected hM3Dq Agonists to Other GPCRsa.
|
Ki (nM) |
||||
|---|---|---|---|---|
| compd | 5HT2A | 5HT2C | α1A | H1 |
| 4a | 5.4 | 9.4 | 1.6 | 1.1 |
| 4b | 29 | 24 | 46 | 1.9 |
| 4c | 16 | 17 | 37 | 4.6 |
| 5b | 1900 | 5100 | >10 000 | 160 |
| 5c | 5200 | 6700 | 320 | 6200 |
| 13 | 71 | 280 | 67 | 5.0 |
| 21 | 66 | 170 | 280 | 6.0 |
Ki values are the average of at least 2 duplicate experiments with standard deviation (SD) values that are 3-fold less than the average.
Compounds 4b and 4c had reduced binding affinities to 5HT2A, 5HT2C, and α1A (Ki = 16–46 nM) compared with compound 4a (clozapine), but retained high binding affinities to H1 (Ki < 5.0 nM). On the other hand, the N-oxide 5b displayed weak binding affinities for 5HT2A, 5HT2C, and α1A (Ki > 1000 nM) and was about 8-fold selective for hM3Dq over H1, while the N-oxide 5c displayed poor binding affinities to 5HT2A, 5HT2C, and H1 (Ki > 5000 nM) but was only about 2-fold selective for hM3Dq over α1A. Interestingly, compound 13, a deuterated analogue of compound 4b, exhibited reduced binding affinities to all four receptors compared with compound 4b. Compound 13 was selective for hM3Dq over 5HT2A (7-fold), 5HT2C (29-fold), and α1A (7-fold), but was not selective over H1. We were pleased to find that compound 21 displayed much improved selectivity compared with compound 4a (clozapine). In addition to being inactive at hM3, compound 21, a potent full agonist of hM3Dq (EC50 = 1.7 nM), was 40-fold selective over 5HT2A, 100-fold selective over 5HT2C, and 165-fold selective over α1A. Although it was only 3.5-fold selective for hM3Dq over H1, the overall selectivity profile of compound 21 is significantly better than compound 4a (clozapine).
Lastly, to identify an alternative compound that might activate hM3Dq, we conducted a screen of the commercially available Library Of Pharmaceutically Active Compounds (LOPAC; N = 1280 compounds) and Prestwick Chemical Library (N = 1280 compounds) using the hM3Dq FLIPR assay. From this screen, we discovered perlapine as a novel, potent agonist of hM3Dq (Figure 3). Importantly, perlapine was >10 000-fold selective for hM3Dq over hM3. Interestingly, perlapine contains a different tricyclic core in comparison with CNO. The high hM3Dq potency of perlapine suggests that the benzodiazepine tricyclic core of the CNO (compound 5a) scaffold is not required for maintaining high hM3Dq agonist activity.
Figure 3.
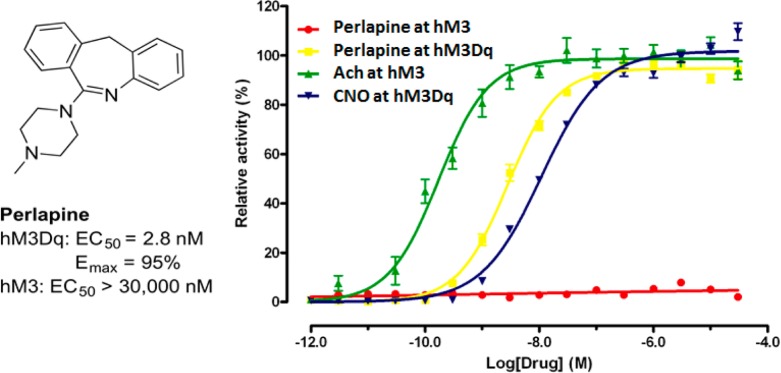
Perlapine is a potent full agonist of hM3Dq and does not activate hM3. CNO (compound 5a) was used as a positive control in the hM3Dq FLIPR assay, and acetylcholine (ACh) was used as a positive control in the hM3 FLIPR assay.
Conclusion
In summary, we conducted the first SAR studies for hM3Dq, a chemogenetic platform for activating neuronal firing, by the design, synthesis, and pharmacological evaluation of new CNO analogues. We explored multiple regions of the CNO scaffold and observed the following interesting SAR trends: (1) a longer or bulkier N-alkyl group as in compounds 4c, 4d, 5c, and 5d reduces both potency and efficacy for hM3Dq; (2) a basic amino group as in compounds 4a and 4b or a permanent positive charge group as in compounds 5a and 6 is required to retain hM3Dq agonist activity; (3) the negative change on the N-oxides such as 5a–5d reduces hM3Dq agonist potency; (4) the 8-chloro group is not required to maintain high agonist potency and efficacy for hM3Dq; and (5) modifications to the benzodiazepine tricyclic core of CNO is tolerated. From these SAR studies, we discovered several compounds such as 13 and 21, which are very potent full agonists of hM3Dq but do not activate the native human M3 receptor (hM3). In addition, the selectivity of compound 21 against a number of aminergic GPCRs is significantly improved compared with clozapine. Furthermore, we discovered perlapine as a novel, potent hM3Dq agonist, which is >10 000-fold selective for hM3Dq over hM3. These SAR studies lay the foundation for developing the next generation of DREADD ligands that can selectively activate defined neuronal populations in primates.
Methods
Chemistry
General Methods
HPLC spectra of all compounds were acquired from an Agilent 6110 Series system with UV detector set to at 220 nm. Samples were injected (5 μL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 μM, C18 column at room temperature. A linear gradient from 10% to 100% B (MeOH + 0.1% acetic acid) in 5.0 min was followed by pumping 100% B for another 2 min with A being H2O + 0.1% acetic acid. The flow rate was 1.0 mL/min. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an electrospray ionization (ESI) source. HRMS analysis was conducted on an Agilent Technologies G1969A high-resolution API-TOF mass spectrometer attached to an Agilent Technologies 1200 HPLC system. Samples were ionized by electrospray ionization (ESI) in positive mode. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury spectrometer with 400 MHz for proton (1H NMR) and 100 MHz for carbon (13C NMR); chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to at 220 nm. Samples were injected onto a Phenomenex Luna 75 × 30 mm, 5 μM, C18 column at room temperature. The flow rate was 30 mL/min. Different linear gradient for different compounds were used with A being H2O + 0.5% TFA and B being MeOH.
Compounds 2,46,473,46,474a,46,474b,46 and 5a(35,55) were prepared according to the procedures described previously.
8-Chloro-11-(4-isopropylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine (4c)
A solution of 8,11-dichloro-5H-dibenzo[b,e][1,4]diazepine (3, 0.397 g, 1.44 mmol) and 1-isopropylpiperazine (1 g, 7.799 mmol) in 1,4-dioxane (20 mL) was stirred overnight at 120 °C. After cooling down, the reaction mixture was concentrated and the residue was dissolved with 50 mL of EtOAc. The resulting solution was washed with 30 mL of aqueous NaHCO3. The organic layer was dried over Na2SO4, and the filtrate was concentrated and the residue was purified by flash column chromatography with 5–10% MeOH in CH2Cl2 to give the desired product 4c (0.410 g) in 80% yield: 1H NMR (400 MHz, CDCl3) δ 7.38–7.16 (m, 2H), 7.04 (d, J = 1.5 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.88–6.72 (m, 2H), 6.58 (d, J = 8.3 Hz, 1H), 4.86 (s, 1H), 3.46 (br, s, 4H), 2.70 (quin, J = 6.5 Hz, 1H), 2.58 (br s, 4H), 1.06 (d, J = 6.5 Hz, 6H). 13C NMR (101 MHz,, CDCl3) δ 162.84, 152.90, 142.13, 140.59, 132.00, 130.55, 129.24, 126.94, 123.69, 123.18, 123.13, 120.25, 120.15, 54.80 (2C), 48.95 (2C), 47.77, 18.80 (2C). HPLC purity 100%, RT 4.099 min. MS (ESI) 355.2 [M + H]+. HRMS (ESI) calcd for C20H24ClN4+ [M + H]+: 355.1689. Found: 355.1693.
8-Chloro-11-(4-propylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine (4d)
Compound 4d (0.380 g, 70% yield) was prepared via the same procedure as preparing compound 4c from 8,11-dichloro-5H-dibenzo[b,e][1,4]diazepine (3, 0.400 g, 1.52 mmol), 1-propylpiperazine HCl salt (0.500 g, 3.04 mmol), and DIPEA (2 mL, 11.48 mmol) in 1,4-dioxane (20 mL). 1H NMR (400 MHz, CDCl3) δ 7.31–7.19 (m, 2H), 7.04 (d, J = 2.3 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.79 (dd, J = 8.2, 2.0 Hz, 2H), 6.58 (d, J = 8.3 Hz, 1H), 4.86 (s, 1H), 3.45 (br s, 4H), 2.50 (br s, 4H), 2.33 (t, J = 7.6 Hz, 2H), 1.52 (sixtet, J = 7.6 Hz, 2H), 0.90 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 162.93, 152.90, 142.08, 140.59, 132.03, 130.52, 129.25, 126.96, 123.69, 123.21, 123.20, 120.26, 120.16, 60.93 (2C), 53.40 (2C), 47.47, 20.21, 12.18. HPLC purity 100%, RT 4.113 min. MS (ESI) 355.2 [M + H]+. HRMS (ESI) calcd for C20H24ClN4+ [M + H]+: 355.1689. Found: 355.1687.
4-(8-Chloro-5H-dibenzo[b,e][1,4]diazepin-11-yl)-1-ethylpiperazine N-oxide (5b)
A solution of compound 4b (0.100 g, 0.293 mmol) in CH2Cl2 (5 mL) was treated with mCPBA (0.064 g, 0.371 mmol) at room temperature. After 10 min, the reaction was completed. The resulting mixture was concentrated and purified by flash column chromatography with 5–15% C (2% NH4OH in MeOH) in CH2Cl2 to give the desired N-oxide compound 5b (0.073 g,) in 70% yield: 1H NMR (400 MHz, MeOH-d4) δ 7.39–7.34 (m, 1H), 7.32 (dd, J = 7.8, 1.2 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 2.3 Hz, 1H), 6.87 (dd, J = 8.4, 2.4 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 3.88 (br s, 2H), 3.74 (t, J = 12.1 Hz, 2H), 3.55–3.43 (m, 2H), 3.367 (q, J = 7.2 Hz, 2H), 3.19–3.09 (m, 2H), 1.38 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 162.06, 153.18, 141.54, 140.62, 132.57, 130.29, 129.35, 126.98, 123.90, 123.56, 122.96, 120.45, 120.43, 66.73 (2C), 63.23 (2C), 42.23, 7.69. HPLC purity 100%, RT 4.394 min. MS (ESI) 357.2 [M + H]+. HRMS (ESI) calcd for C19H22ClN4O+ [M + H]+: 357.1482. Found: 357.1481.
4-(8-Chloro-5H-dibenzo[b,e][1,4]diazepin-11-yl)-1-isopropylpiperazine N-oxide (5c)
Compound 5c (0.068 g, 65% yield) was prepared similarly as 5b from compound 4c (0.100 g, 0.282 mmol) and mCPBA (0.063 g, 0.365 mmol) in CH2Cl2 (5 mL). 1H NMR (400 MHz, MeOH-d4) δ 7.39–7.30 (m, 2H), 7.05 (t, J = 7.5 Hz, 1H), 7.01 (d, J = 7.9 Hz, 1H), 6.97 (d, J = 2.3 Hz, 1H), 6.87 (dd, J = 8.4, 2.4 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 3.90 (br s, 2H), 3.73 (t, J = 11.9 Hz, 1H), 3.57–3.40 (m, 3H), 3.19–3.12 (m, 2H), 1.38 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 162.02, 153.15, 141.64, 140.61, 132.51, 130.31, 129.38, 126.97, 123.81, 123.55, 123.01, 120.43, 120.39, 70.99 (2C), 60.41 (2C), 42.27, 16.54 (2C). HPLC purity 100%, RT 4.305 min. MS (ESI) 371.2 [M + H]+. HRMS (ESI) calcd for C20H24ClN4O+ [M + H]+: 371.1639. Found: 371.1642.
4-(8-Chloro-5H-dibenzo[b,e][1,4]diazepin-11-yl)-1-propylpiperazine N-oxide (5d)
Compound 5d (0.075 g, 72% yield) was prepared using the same procedure as preparing 5b from compound 4d (0.100 g, 0.282 mmol) and mCPBA (0.063 g, 0.365 mmol) in CH2Cl2 (5 mL). 1H NMR (400 MHz, MeOH-d4) δ 7.39–7.34 (m, 1H), 7.32 (dd, J = 7.8, 1.2 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 2.3 Hz, 1H), 6.87 (dd, J = 8.4, 2.4 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 3.86 (br s, 2H), 3.74 (t, J = 11.9 Hz, 3H), 3.57–3.44 (m, 2H), 3.29–3.21 (m, 2H), 3.20–3.10 (m, 2H), 2.00–1.84 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, MeOH-d4) δ 164.31, 155.62, 143.36, 142.96, 133.85, 131.29, 129.72, 127.45, 124.99, 124.29, 124.04, 121.68, 121.53, 73.69 (2C), 64.26 (2C), 43.23, 16.42, 11.37. HPLC purity 100%, RT 4.439 min. MS (ESI) 371.2 [M + H]+. HRMS (ESI) calcd for C20H24ClN4O+ [M + H]+: 371.1639. Found: 371.1640.
4-(8-Chloro-5H-dibenzo[b,e][1,4]diazepin-11-yl)-1,1-dimethylpiperazin-1-ium iodide (6)
A solution of 4a (0.100 g, 0.306 mmol) in acetone (5 mL) was treated with methyl iodide (21 μL, 0.337 mmol) at room temperature. After overnight, the resulting mixture was concentrated, dissolved in 1 mL of MeOH, and diluted with 10 mL of distilled water. The aqueous solution was washed with EtOAc (2 × 10 mL) and concentrated. The residue was purified by preparative HPLC to give the compound 6 (0.079 g) in 55% yield: 1H NMR (400 MHz, MeOH-d4) δ 7.57 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 7.9 Hz, 1H), 7.26 (d, J = 2.3 Hz, 1H), 7.27–7.13 (m, 3H), 7.00 (d, J = 8.5 Hz, 1H), 4.03 (br s, 4H), 3.72 (br s, 4H), 3.33 (s, 6H). 13C NMR (101 MHz, MeOH-d4) δ 166.47, 156.55, 145.82, 136.58, 135.26, 132.80, 130.24, 128.46, 126.86, 125.08, 122.75, 122.70, 119.64, 61.86 (2C), 52.46 (2C), 44.44 (2C). HPLC purity 100%, RT 3.893 min. MS (ESI) 341.2 [M − I]+.
4-(8-Chloro-5H-dibenzo[b,e][1,4]diazepin-11-yl)piperazin-2-one (7)
A solution of compound 3 (0.200 g, 0.760 mmol) and 2-oxypiperazine (0.152 g, 1.52 mmol) in a 1:1 mixture of 1,4-dioxane and ethanol (15 mL) was stirred overnight at 99 °C. After concentration, the residue was diluted with EtOAc (50 mL) and the solution was washed with 20 mL of aqueous NaHCO3. The organic layer was dried over Na2SO4 and the filtrate was concentrated. The residue was purified by flash column chromatography with 30–50% EtOAc in hexanes to afford the desired product (0.162 g) in 65% yield: 1H NMR (400 MHz, CDCl3) δ 7.30 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.8 Hz, 1H), 7.05 (s, 1H), 7.02 (t, J = 7.6 Hz, 1H), 6.84 (d, J = 8.3 Hz, 1H), 6.81 (d, J = 7.9 Hz, 1H), 6.60 (d, J = 8.3 Hz, 1H), 6.03 (s, 1H), 4.90 (s, 1H), 4.10 (s, 2H), 3.67 (br s, 2H), 3.50 (br s, 2H). 13C NMR (101 MHz, CDCl3) δ 168.89, 161.84, 153.15, 141.39, 140.49, 132.63, 130.04, 129.37, 127.19, 124.02, 123.52, 122.99, 120.48, 120.39, 51.47, 44.12, 41.15; HPLC purity 100%, RT 4.681 min. MS (ESI) 327.1 [M + H]+. HRMS (ESI) calcd for C17H16ClN4O+ [M + H]+: 327.1007. Found: 327.1006.
8-(8-Chloro-5H-dibenzo[b,e][1,4]diazepin-11-yl)-1,3,8-triazaspiro[4.5]decane-2,4-dione (9)
A mixture of compound 3 (0.250 g, 0.951 mmol) and commercially available compound 8 (0.200 g, 1.18 mmol) in 20 mL of a mixture of 1,4-dioxane and DMF (2:1) was heated to 130 °C for 24 h. The reaction mixture was cooled down to room temperature and concentrated. The residue was purified by flash column chromatography with 0–10% MeOH in CH2Cl2 to give the desired product 9 (0.250 g) in 66% yield: 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 7.37–7.17 (m, 2H), 7.10–7.03 (m, 2H), 7.01 (t, J = 7.5 Hz, 1H), 6.85–6.76 (m, 2H), 6.62 (d, J = 8.3 Hz, 1H), 5.06 (s, 1H), 3.90 (br s, 2H), 3.20 (br s, 2H), 2.18–2.05 (m, 2H), 1.74 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 176.84, 163.26, 156.74, 152.90, 141.62, 140.70, 132.47, 130.29, 129.42, 126.97, 123.82, 123.59, 123.43, 120.48, 62.15 (2C), 43.28, 33.19 (2C). HPLC purity 100%, RT 4.289 min. MS (ESI) 396.1 [M + H]+. HRMS (ESI) calcd for C20H19ClN5O2+ [M + H]+: 396.1222. Found: 396.1221.
4-Amino-1-(8-chloro-5H-dibenzo[b,e][1,4]diazepin-11-yl)piperidine-4-carboxylic acid (10)
A solution of compound 9 (0.100 g, 0.253 mmol) in 1,2-dimethoxyethane (5 mL) was treated with 5 mL of 0.5 N NaOH at room temperature. The resulting mixture was heated under microwave for 10 min (max power 100 W, max temperature 150 °C, max pressure 17.0 bar). After cooling down to room temperature, the reaction mixture was quenched with 10% citric acid and filtered. The filtrated was purified with preparative HPLC to afford the desired product (0.015 g) in 16% yield: 1H NMR (400 MHz, MeOH-d4) δ 7.63 (t, J = 7.7 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.34 (d, J = 19.4 Hz, 1H), 7.25 (t, J = 7.9 Hz, 3H), 7.08 (d, J = 8.6 Hz, 1H), 4.36–3.59 (m, 4H), 2.70–2.40 (m, 2H), 2.40–2.01 (m, 2H). HPLC purity 100%, RT 3.585 min. MS (ESI) 371.2 [M + H]+. HRMS (ESI) calcd for C19H20ClN4O2+ [M + H]+: 371.1269. Found: 371.1264.
Compound 11 was prepared according to the previously published procedures.46
1-(4-(8-Chloro-5H-dibenzo[b,e][1,4]diazepin-11-yl)piperazin-1-yl)ethanone (12)
To the solution of compound 10 (0.400 g, 1.28 mmol) and TEA (0.27 mL, 2.0 mmol) in CH2Cl2 was added AcCl (0.10 mL, 1.4 mmol) at 0 °C. The resulting mixture was then stirred at 0 °C for 1 h. After removing the solvents, the residue was purified by flash column chromatography with 0–5% MeOH in CH2Cl2 to give the desired product 12 (0.390 g, 1.10 mmol) in 86% yield: 1H NMR (400 MHz, CDCl3) δ 7.30 (td, J = 7.7, 1.5 Hz, 1H), 7.26–7.22 (m, 1H), 7.04 (d, J = 2.4 Hz, 1H), 7.02 (td, J = 7.6, 1.0 Hz, 1H), 6.86–6.77 (m, J = 2.4 Hz, 2H), 6.60 (d, J = 8.3 Hz, 1H), 4.89 (s, 1H), 3.67 (br s, 2H), 3.59–3.46 (m, 4H), 3.34 (br s, 2H), 2.11 (s, J = 11.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.41, 162.90, 153.15, 141.57, 140.65, 132.40, 130.20, 129.25, 126.97, 123.75, 123.39, 123.32, 120.42, 120.34, 47.80, 47.26, 46.13, 41.50, 21.62. HPLC purity 100%, RT 4.919 min. MS (ESI) 355.2 [M + H]+. HRMS (ESI) calcd for C19H20ClN4O+ [M + H]+: 355.1320. Found: 355.1315.
8-Chloro-11-[4-(1,1-dideutrioethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepine (13)
To the solution of compound 12 (0.100 g, 0.282 mmol) in 15 mL of anhydrous THF was added LiAlD4 (0.024 g, 0.572 mmol) at room temperature under N2 atmosphere. The reaction mixture was heated under reflux conditions for 2 h. The reaction was quenched with 0.1 mL of CD3OD at 0 °C. The resulting mixture was treated with 0.5 mL of NH4OH at 0 °C and filtered through Celite and the filtrate was concentrated. The residue was purified by flash column chromatography with 0–10% MeOH in CH2Cl2 to give the desired product 13 (0.081 g) in 84% yield: 1H NMR (400 MHz, CDCl3) δ 7.33–7.23 (m, 3H), 7.06 (d, J = 2.4 Hz, 1H), 7.01 (td, J = 7.6, 1.0 Hz, 1H), 6.81 (dd, J = 8.3, 2.4 Hz, 2H), 6.60 (d, J = 8.3 Hz, 1H), 4.88 (s, 1H), 3.49 (br, s, 4H), 2.54 (br, s, 4H), 1.10 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 162.90, 152.87, 142.03, 140.59, 132.02, 130.46, 129.17, 126.91, 123.60, 123.17, 120.24, 120.16, 52.87 (2C), 51.78 (q, J = 20.0 Hz), 47.44, 11.86 (2C). HPLC purity 100%, RT 4.072 min; MS (ESI) 343.2 [M+1]+. HRMS (ESI) calcd for C19H20D2ClN4+ [M + H]+: 343.1653. Found: 343.1653.
8-Chloro-11-(4-(methylsulfonyl)piperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine (14)
To a solution of compound 11 (0.102 g, 0.326 mmol) and DIPEA (87 μL, 0.907 mmol) in 10 mL of CH2Cl2, MsCl (27.8 μL, 0.359 mmol) was added at 0 °C. After 1 h, the reaction was completed. The reaction mixture was diluted with 50 mL of CH2Cl2 and washed with 10 mL of aqueous NaHCO3. The organic layer was dried over Na2SO4, and the filtrate was concentrated and the residue was purified by flash column chromatography with 50% EtOAc in hexanes to give the desired product 14 (0.118 g) in 93% yield: 1H NMR (400 MHz, CDCl3) δ 7.31 (t, J = 7.7 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 7.08–6.96 (m, 2H), 6.87–6.78 (m, 2H), 6.61 (d, J = 8.3 Hz, 1H), 4.90 (s, 1H), 3.57 (br s, 4H), 3.29 (br s, 4H), 2.79 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 162.44, 154.11, 142.23, 141.48, 132.24, 129.80, 126.69, 125.58, 122.88, 122.64, 122.40, 120.69, 120.40, 46.33, 45.14 (2C), 33.85 (2C). HPLC purity 100%, RT 4.752 min. MS (ESI) 391.1 [M + H]+. HRMS (ESI) calcd for C18H20ClN4O2S+ [M + H]+: 391.0990. Found: 391.0994.
Compounds 17–21 were prepared according to the previously reported procedures.48
11-(Piperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine (21)
1H NMR (400 MHz, MeOH-d4) δ 7.39–7.24 (m, 1H), 7.06–6.95 (m, 3H), 6.94–6.82 (m, 3H), 3.34 (br s, 1H), 2.96–2.85 (m, 2H). 13C NMR (101 MHz, MeOH-d4) δ 165.39, 156.04, 145.04, 141.55, 133.31, 131.47, 127.85, 125.34, 124.88, 124.66, 123.78, 121.26, 120.71, 46.47. HPLC purity 99%, RT 2.772 min. MS (ESI) 279.2 [M+1]+. HRMS (ESI) calcd for C17H19N4+ [M + H]+: 279.1604. Found: 279.1610.
11-(4-Ethylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine (22)
A solution of compound 20 (0.420 g, 1.84 mmol) and 1-ethylpiperazine (1.5 mL, 11.81 mmol) in toluene (20 mL) was heated under reflux conditions for 2 h. After cooling down to room temperature and concentration, the residue was purified by flash column chromatography with 0–10% MeOH in CH2Cl2 to give the desired product 22 (0.409 g) in 72% yield: 1H NMR (400 MHz, CDCl3) δ 7.30–7.18 (m, 2H), 7.06 (dd, J = 7.8, 1.5 Hz, 1H), 7.00–6.90 (m, 2H), 6.84 (tt, J = 7.3, 3.7 Hz, 1H), 6.80 (dd, J = 7.4, 1.1 Hz, 1H), 6.67 (dd, J = 7.7, 1.4 Hz, 1H), 4.89 (s, 1H), 3.45 (br s, 4H), 2.53 (br s, 4H), 2.46 (q, J = 7.2 Hz, 2H), 1.10 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 162.52, 153.35, 141.97, 140.73, 131.82, 130.54, 127.40, 124.46, 123.86, 123.82, 122.93, 120.16, 119.48, 53.06 (2C), 52.65 (2C), 47.56, 12.18. HPLC purity 100%, RT 3.122 min. MS (ESI) 307.2 [M + H]+. HRMS (ESI) calcd for C19H23N4+ [M + H]+: 307.1917. Found: 307.1918.
4-(5H-Dibenzo[b,e][1,4]diazepin-11-yl)-1-ethylpiperazine N-oxide (23)
A solution of compound 22 (0.095 g, 0.31 mmol) in CH2Cl2 (10 mL) was treated with mCPBA (0.069 g) at room temperature. After 10 min, the reaction mixture was concentrated and the residue was purified with 0–10% C (5% NH4OH in MeOH) in CH2Cl2 to afford the desired product 23 (0.078 g) in 78% yield: 1H NMR (400 MHz, MeOH-d4) δ 7.39–7.29 (m, 2H), 7.07–6.97 (m, 3H), 6.96–6.82 (m, 3H), 3.84 (br s, 2H), 3.79–3.66 (m, 2H), 3.56–3.43 (m, 2H), 3.35 (q, J = 7.0 Hz, 2H), 3.18–3.07 (m, 2H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, MeOH-d4) δ 163.66, 155.93, 144.50, 141.44, 133.64, 131.23, 127.98, 125.69, 125.05, 124.16, 124.13, 121.44, 120.80, 66.90 (2C), 63.79 (2C), 43.32, 7.70. HPLC purity 100%, RT 3.312 min. MS (ESI) 323.2 [M + H]+. HRMS (ESI) calcd for C19H23N4O+ [M + H]+: 323.1866. Found: 323.1863.
Biological Assays
hM3Dq and hM3 FLIPR assays were performed according to the protocols reported previously.11,50,51 Protocols for 5-HT2A, 5-HT2C, α1A, and H1 radioligand binding assays are available at the National Institute of Mental Health–Psychoactive Drug Screening Program Web site (http://pdsp.med.unc.edu/UNC-CH%20Protocol%20Book.pdf).
Acknowledgments
We thank Dr. H. Ümit Kaniskan for checking synthetic procedures and NMR spectra of novel compounds.
Author Contributions
∥ X.C. and H.C. contributed equally to this work. X.C., H.C., X.P.H., X.Y., and O.S. performed the experiments. J.J. and B.L.R. designed the studies. J.J., B.L.R., and X.C. wrote the manuscript.
This work was supported by the NIH Grant U01MH105892 (to B.L.R. and J.J.) and Korea NRF grant NRF-2013-R1A1A2A10009907 (to H.C.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Sternson S. M.; Roth B. L. (2014) Chemogenetic Tools to Interrogate Brain Functions. Annu. Rev. Neurosci. 37, 387–407. [DOI] [PubMed] [Google Scholar]
- Urban D. J.; Roth B. L. (2015) DREADDs (Designer Receptors Exclusively Activated by Designer Drugs): Chemogenetic Tools with Therapeutic Utility. Annu. Rev. Pharmacol. Toxicol. 55, 399–417. [DOI] [PubMed] [Google Scholar]
- Penfield W., and Jasper H. H. (1954) Epilepsy and the Functional Anatomy of the Human Brain (First ed.), Little Brown, Boston. [Google Scholar]
- Boyden E. S.; Zhang F.; Bamberg E.; Nagel G.; Deisseroth K. (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Armbruster B. N.; Roth B. L. (2005) Mining the receptorome. J. Biol. Chem. 280, 5129–5132. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Wang L. P.; Brauner M.; Liewald J. F.; Kay K.; Watzke N.; Wood P. G.; Bamberg E.; Nagel G.; Gottschalk A.; Deisseroth K. (2007) Multimodal fast optical interrogation of neural circuitry. Nature 446, 633–639. [DOI] [PubMed] [Google Scholar]
- Airan R. D.; Thompson K. R.; Fenno L. E.; Bernstein H.; Deisseroth K. (2009) Temporally precise in vivo control of intracellular signalling. Nature 458, 1025–1029. [DOI] [PubMed] [Google Scholar]
- Berndt A.; Yizhar O.; Gunaydin L. A.; Hegemann P.; Deisseroth K. (2009) Bi-stable neural state switches. Nat. Neurosci. 12, 229–234. [DOI] [PubMed] [Google Scholar]
- Li X.; Gutierrez D. V.; Hanson M. G.; Han J.; Mark M. D.; Chiel H.; Hegemann P.; Landmesser L. T.; Herlitze S. (2005) Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc. Natl. Acad. Sci. U. S. A. 102, 17816–17821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Prigge M.; Beyriere F.; Tsunoda S. P.; Mattis J.; Yizhar O.; Hegemann P.; Deisseroth K. (2008) Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat. Neurosci. 11, 631–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster B. N.; Li X.; Pausch M. H.; Herlitze S.; Roth B. L. (2007) Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U. S. A. 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander G. M.; Rogan S. C.; Abbas A. I.; Armbruster B. N.; Pei Y.; Allen J. A.; Nonneman R. J.; Hartmann J.; Moy S. S.; Nicolelis M. A.; McNamara J. O.; Roth B. L. (2009) Remote Control of Neuronal Activity in Transgenic Mice Expressing Evolved G Protein-Coupled Receptors. Neuron 63, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan S. C.; Roth B. L. (2011) Remote control of neuronal signaling. Pharmacol. Rev. 63, 291–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell M. S.; Roth B. L. (2012) Pharmacosynthetics: Reimagining the pharmacogenetic approach. Brain Res. 1511, 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings J. H.; Stuber G. D. (2014) Tools for Resolving Functional Activity and Connectivity within Intact Neural Circuits. Curr. Biol. 24, R41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. M.; Giguere P. M.; Roth B. L. (2013) DREADDs: Novel tools for drug discovery and development. Drug Discovery Today 19, 469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere P. M.; Kroeze W. K.; Roth B. L. (2014) Tuning up the right signal: Chemical and genetic approaches to study GPCR functions. Curr. Opin. Cell Biol. 27, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster B. N.; Li X.; Pausch M. H.; Herlitze S.; Roth B. L. (2007) Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U. S. A. 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray R. S.; Corcoran A. E.; Brust R. D.; Kim J. C.; Richerson G. B.; Nattie E.; Dymecki S. M. (2011) Impaired Respiratory and Body Temperature Control Upon Acute Serotonergic Neuron Inhibition. Science 333, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner A. R.; Rowland D. C.; Hwang S. Y.; Baumgaertel K.; Roth B. L.; Kentros C.; Mayford M. (2012) Generation of a synthetic memory trace. Science 335, 1513–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D.; Betley J. N.; Su H. H.; Sternson S. M. (2012) Deconstruction of a neural circuit for hunger. Nature 488, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes M. J.; Shah B. P.; Madara J. C.; Olson D. P.; Strochlic D. E.; Garfield A. S.; Vong L.; Pei H.; Watabe-Uchida M.; Uchida N.; Liberles S. D.; Lowell B. B. (2014) An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman S. J.; Olivas N. D.; Tring E.; Ikrar T.; Xu X.; Trachtenberg J. T. (2013) A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature 501, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter M. E.; Soden M. E.; Zweifel L. S.; Palmiter R. D. (2013) Genetic identification of a neural circuit that suppresses appetite. Nature 503, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrontou S.; Wong A. M.; Rau K. K.; Koerber H. R.; Anderson D. J. (2013) Genetic identification of C fibres that detect massage-like stroking of hairy skin in vivo. Nature 493, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y.; Saunders A.; Johnson C. A.; Lowell B. B.; Sabatini B. L. (2012) Recurrent network activity drives striatal synaptogenesis. Nature 485, 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S. M.; Eskenazi D.; Ishikawa M.; Wanat M. J.; Phillips P. E.; Dong Y.; Roth B. L.; Neumaier J. F. (2011) Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat. Neurosci. 14, 22–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva B. A.; Mattucci C.; Krzywkowski P.; Murana E.; Illarionova A.; Grinevich V.; Canteras N. S.; Ragozzino D.; Gross C. T. (2013) Independent hypothalamic circuits for social and predator fear. Nat. Neurosci. 16, 1731–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock R.; Shin J. H.; Kaplan A. R.; Dobi A.; Markey E.; Kramer P. F.; Gremel C. M.; Christensen C. H.; Adrover M. F.; Alvarez V. A. (2013) Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat. Neurosci. 16, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnaudeau S.; O’Neill P. K.; Bolkan S. S.; Ward R. D.; Abbas A. I.; Roth B. L.; Balsam P. D.; Gordon J. A.; Kellendonk C. (2013) Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron 77, 1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio M.; Maywood E. S.; Chesham J. E.; Loudon A. S.; Hastings M. H. (2013) A Gq-Ca2+ axis controls circuit-level encoding of circadian time in the suprachiasmatic nucleus. Neuron 78, 714–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D.; Tong Q.; Ye C.; Koda S.; Fuller P. M.; Krashes M. J.; Vong L.; Ray R. S.; Olson D. P.; Lowell B. B. (2012) GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. Cell 151, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender D.; Holschbach M.; Stocklin G. (1994) Synthesis of n.c.a. carbon-11 labelled clozapine and its major metabolite clozapine-N-oxide and comparison of their biodistribution in mice. Nucl. Med. Biol. 21, 921–925. [DOI] [PubMed] [Google Scholar]
- Chang W. H.; Lin S. K.; Lane H. Y.; Wei F. C.; Hu W. H.; Lam Y. W. F.; Jann M. W. (1998) Reversible metabolism of clozapine and clozapine N-oxide in schizophrenic patients. Prog. Neuro-Psychopharmacol. 22, 723–739. [DOI] [PubMed] [Google Scholar]
- Massey C. A.; Kim G.; Corcoran A. E.; Haynes R. L.; Paterson D. S.; Cummings K. J.; Dymecki S. M.; Richerson G. B.; Nattie E. E.; Kinney H. C.; Commons K. G. (2013) Development of brainstem 5HT(1A) receptor-binding sites in serotonin-deficient mice. J. Neurochem. 126, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jann M. W.; Lam Y. W.; Chang W. H. (1994) Rapid formation of clozapine in guinea-pigs and man following clozapine-N-oxide administration. Arch. Int. Pharmacodyn. Ther. 328, 243–250. [PubMed] [Google Scholar]
- Loffler S.; Korber J.; Nubbemeyer U.; Fehsel K. (2012) Comment on “Impaired Respiratory and Body Temperature Control Upon Acute Serotonergic Neuron Inhibition”. Science 337, 646. [DOI] [PubMed] [Google Scholar]
- Roth B. L.; Sheffler D. J.; Kroeze W. K. (2004) Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discovery 3, 353–359. [DOI] [PubMed] [Google Scholar]
- Hunziker F.; Kuenzle F.; Schmutz J. (1966) Helv. Chim. Acta 49, 1433–1439. [Google Scholar]
- Schmutz J. (1975) Neuroleptic piperazinyl-dibenzo-azepines. Chemistry and structure–activity relationships. Arzneimittelforschung 25, 712–720. [PubMed] [Google Scholar]
- Steiner G.; Franke A.; Hadicke E.; Lenke D.; Teschendorf H. J.; Hofmann H. P.; Kreiskott H.; Worstmann W. (1986) Tricyclic epines. Novel (E)- and (Z)-11H-dibenz[b,e]azepines as potential central nervous system agents. Variation of the basic side chain. J. Med. Chem. 29, 1877–1888. [DOI] [PubMed] [Google Scholar]
- Hunziker F.; Fischer E.; Schmutz J. (1967) 11-Amino-5H-Dibenzo[b,e]-1,4-diazepine 0.10. Mitteilung Uber Siebengliedrige Heterocyclen. Helv. Chim. Acta 50, 1588–1599. [Google Scholar]
- Wenthur C. J.; Lindsley C. W. (2013) Classics in Chemical Neuroscience: Clozapine. ACS Chem. Neurosci. 4, 1018–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhapure R. S.; Patil B. P.; Jadhav M. N.; Kawle L. A.; Wagh S. B. (2011) Synthesis of 11-(Piperazin-1-yl)-5H-dibenzo[b,e] [1,4]diazepine on Kilo Scale. E-J. Chem. 8, 1747–1749. [Google Scholar]
- Ullmann F.; Bielecki J. (1901) Synthesis in the Biphenyl series. (I. Announcement). Ber. Dtsch. Chem. Ges. 34, 2174–2185. [Google Scholar]
- Davies M. A.; Compton-Toth B. A.; Hufeisen S. J.; Meltzer H. Y.; Roth B. L. (2005) The highly efficacious actions of N-desmethylclozapine at muscarinic receptors are unique and not a common property of either typical or atypical antipsychotic drugs: is M1 agonism a pre-requisite for mimicking clozapine’s actions?. Psychopharmacology 178, 451–460. [DOI] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. (2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiberg K. B. (1955) The Deuterium Isotope Effect. Chem. Rev. 55, 713–743. [Google Scholar]
- Elison C.; Rapoport H.; Laursen R.; Elliott H. W. (1961) Effect of Deuteration of N-CH3 Group on Potency and Enzymatic N-Demethylation of Morphine. Science 134, 1078–1079. [DOI] [PubMed] [Google Scholar]
- Shao L.; Abolin C.; Hewitt M. C.; Koch P.; Varney M. (2006) Derivatives of tramadol for increased duration of effect. Bioorg. Med. Chem. Lett. 16, 691–694. [DOI] [PubMed] [Google Scholar]
- Korber J.; Loffler S.; Schollmeyer D.; Nubbemeyer U. (2013) Synthesis and Oxidant Properties of Phase 1 Benzepine N-Oxides of Common Antipsychotic Drugs. Synthesis 45, 2875–2887. [Google Scholar]