

Abstract
Theoretical predictions show that depending on the populations of the Fe 3dxy, 3dxz, and 3dyz orbitals two possible quintet states can exist for the high-spin state of the photoswitchable model system [Fe(terpy)2]2+. The differences in the structure and molecular properties of these 5B2 and 5E quintets are very small and pose a substantial challenge for experiments to resolve them. Yet for a better understanding of the physics of this system, which can lead to the design of novel molecules with enhanced photoswitching performance, it is vital to determine which high-spin state is reached in the transitions that follow the light excitation. The quintet state can be prepared with a short laser pulse and can be studied with cutting-edge time-resolved X-ray techniques. Here we report on the application of an extended set of X-ray spectroscopy and scattering techniques applied to investigate the quintet state of [Fe(terpy)2]2+ 80 ps after light excitation. High-quality X-ray absorption, nonresonant emission, and resonant emission spectra as well as X-ray diffuse scattering data clearly reflect the formation of the high-spin state of the [Fe(terpy)2]2+ molecule; moreover, extended X-ray absorption fine structure spectroscopy resolves the Fe–ligand bond-length variations with unprecedented bond-length accuracy in time-resolved experiments. With ab initio calculations we determine why, in contrast to most related systems, one configurational mode is insufficient for the description of the low-spin (LS)–high-spin (HS) transition. We identify the electronic structure origin of the differences between the two possible quintet modes, and finally, we unambiguously identify the formed quintet state as 5E, in agreement with our theoretical expectations.
Introduction
Switchable molecular compounds have significant potential as very high-density devices in the areas of data storage systems, molecular switching, and display devices.1−3 Promising candidates include transition metal compounds, in particular octahedral FeII complexes, which can exist either in a low-spin (LS) or a high-spin (HS) state, depending on parameters such as temperature or pressure.4 Light can also induce a spin-state transition in many of these compounds,5,6 where the resulting HS state can often be stabilized under certain conditions, typically at low temperatures, thus creating a molecular switch in the “on’ position. In order to improve on the performance of these systems, it is essential to characterize the HS state thoroughly, particularly in compounds where some sort of anomaly occurs; understanding how we can modify the properties and the stability of the excited HS state can lead to developments toward applications at room temperature.
The full dynamics of the LS–HS switching has long been described by a simple and rather complete theoretical framework, suggested by A. Hauser and co-workers.7−9 This theory describes the transitions along a single configurational coordinate (SCC), typically the breathing mode of the molecule (i.e., a symmetric stretching along the Fe–ligand bonds), which was found to be valid for all complexes with monodentate, bidentate, and often even polydentate ligands. However, experiments10,11 and theory11,12 suggested that the tridentate [Fe(terpy)2]2+ (terpy: 2,2′:6′,2″-terpyridine) molecule behaves differently in several respects, the most important being that the SCC model for the HS–LS transition fails,11,12 and the switching mechanism involves (at least) two vibrational modes (configuration coordinates). In addition to this anomaly, the nature of the HS state was unclear. Due to the axial compression exerted by the terpy ligands, the symmetry is lowered, and thus the octahedral 5T2 is split into two quintet states of different symmetry with terms 5B2 and 5E. These states differ in the configuration of the t2g subshell and the molecular geometry, particularly the Fe–N bond lengths. A very recent extended X-ray absorption fine structure (EXAFS) study made an attempt to determine the structure of the populated quintet state, but instead of resolving the structure it could only address its distortion due to limited signal-to-noise conditions.13 On the basis of this it was concluded that the structure for the 5E is in somewhat better agreement with the experimental data than that of the 5B2, although this proposed structure contradicts the energetics arising from the density functional theory (DFT) models applied by these authors, which suggest 5B2 as the more stable quintet state.
In this paper, we revisit the electronic and molecular structure of the quintet state of [Fe(terpy)2]2+ in aqueous solution with advanced experimental and theoretical techniques. We investigate the reasons for and the consequences of the failure of the SCC model (and thus the necessity for a description with two modes) in the molecular geometry and electronic structure. By combining X-ray absorption and emission spectroscopies (XAS and XES, respectively) and X-ray diffuse scattering (XDS) in a single pump–probe setup at the megahertz (MHz) repetition rate,14 we acquire simultaneously time-resolved structural and electronic structural data with unprecedented quality in the picosecond time domain. We prepare the quintet state of [Fe(terpy)2]2+ with intense, 10 ps, green (532 nm) laser pulses leading to metal-to-ligand charge-transfer (MLCT) excitations that subsequently relax into the HS state on a subpicosecond time scale. We exploit XES and XAS X-ray probes to identify the formation of the quintet state and determine its lifetime; we also employ resonant X-ray emission spectroscopy (RXES) at the 1s → 3d excitations to gain further insights into the 3d states. With EXAFS and XDS we determine the structure of the molecule in the quintet state and its surrounding solvent cage. The results are compared to those obtained on photoexcited [Fe(bipy)3]2+, which has been characterized by a wide range of experimental and theoretical techniques11,12,14−27 and which follows the SCC model (the formation of its photoinduced quintet state was shown to be accompanied by a 0.2 Å elongation of all Fe–N bonds).11,18,20 Moreover, it has served to calibrate coarsely the arsenal of different X-ray tools used in this work.14 Indeed, optical tools to date suffer from the clear identification of the excited state in such compounds, as this requires an unambiguous observable. The UV–vis range is quite useful in detecting ultrafast changes, but the overlapping bands due to different excited states hampered in most cases a clear identification of the excited-state electronic and (even more) molecular structure. The X-ray tools employed here seek to fill this knowledge gap, and exploiting laser-pump–X-ray probe techniques at MHz repetition rates allows us to gain new insights into both the electronic and geometric structure changes due to its unprecedented signal quality, as we demonstrate in this work.
Experimental and Computational Methods
Experimental Details
The laser-pump X-ray-probe experiments were performed at sector 7ID-D of the Advanced Photon Source (APS) and at beamline ID26 of the European Synchrotron Radiation Facility (ESRF). Both setups utilized amplified laser systems with adjustable repetition rates in the MHz range, thus increasing the duty cycle of the pump–probe studies by orders of magnitude compared to others that use conventional amplified laser systems with kilohertz (kHz) rates. The experimental strategy combined different time-domain X-ray tools into one single setup, which enabled simultaneous measurements of X-ray diffuse scattering (XDS) together with nonresonant X-ray emission spectroscopy (XES)14 and also variants of X-ray absorption spectroscopy (XAS): (i) high-energy-resolution fluorescence detection (HERFD)28,29 and (ii) total fluorescence yield (TFY) XAS. The former was possible owing to the implementation of a secondary X-ray crystal spectrometer that energy-resolved the emitted fluorescence. This spectrometer also permitted us to record resonant XES (RXES) to better resolve the 1s pre-edge and thus exploit the improved sensitivity to the 3d orbitals (details further below). Time-resolved XDS was recorded in the forward direction up to a Q-value of around 4 Å–1 with a MHz-gateable, single-photon-counting area detector. This strategy of combined tools had been implemented and tested on the photoinduced spin transition of aqueous [Fe(bipy)3]2+.14,25 The present study applied this strategy and advanced setup to obtain complementary information about [Fe(terpy)2]2+, whose investigation is supported by the analysis of the [Fe(bipy)3]2+ data. The experiments at 7ID-D (APS) utilized 24-bunch mode (in top-up mode with a constant 102 mA ring current) with a 6.52 MHz X-ray pulse repetition rate. X-rays were monochromatized with a diamond (111) double-crystal monochromator. An amplified laser system (Duetto from Time-Bandwidth) generating 10 ps pulses at its second harmonic, 532 nm, was used for laser excitation. It was synchronized to the storage ring radio frequency system and operated at a repetition rate of 3.26 MHz, half that of the X-ray rate. The time delay between laser and X-ray pulses could be electronically adjusted in step sizes of 5 ps minimum duration. Details of this time-resolved setup are given in refs (14) and (30). The X-ray beam was focused by a pair of Kirkpatrick–Baez mirrors to 8 × 7 μm2, (H × V) on the sample, and the laser beam overlapped this spot with a larger spot size (100 × 80 μm2, H × V). The sample consisted of a free-flowing liquid jet of 13 mM aqueous [Fe(terpy)2]2+ solution (total volume 200 mL). The liquid near the nozzle output was a flat sheet, with thickness 0.1 mm, which was oriented at about 45° to both laser and X-ray beams. This geometry permitted simultaneous recording of XDS data (collected in forward scattering direction) and XES data (collected sideways at 90° scattering angle, along the X-ray polarization vector, via the analyzer crystal). For these measurements the incident X-ray probing energy was tuned to 8.2 keV, far above the Fe K edge. The XDS experiments were performed with a Pilatus 100 K detector gated and synchronized to the bunch frequency as described in our previous MHz work.14 Following masking, azimuthal integration, and scaling, difference signals were constructed and averaged for each time delay and then corrected for multiple-pump–pulse contributions as described in ref (14). Considering the jet speed (6 m/s), the 80 μm spot illuminated by the laser travels only about 2 μm before the next laser pulse arrives 307 ns later. Therefore, it is obvious that the same sample volume is hit many times in such a high repetition rate experiment. Nevertheless, the studied [Fe(terpy)2]2+ complex is very stable, and no degradation was found in any of the signals with exposure. The lifetime of the excited state is 2 orders of magnitude shorter than the separation between the X-ray pulses; therefore, the spectroscopic data are not affected by the previous excitation pulses. On the other hand, the energy transfer to the solvent leads to a temperature increase in the water, which affects the X-ray scattering data. This has been circumvented by using the scattering signal recorded at a Δt = −200 ps time delay as the background. The increased solvent temperature due to the previous laser shots is present in this water signal, and thus it serves as an appropriate reference, as explained in detail in our previous work on [Fe(bipy)3]2+.14
The Kα XES, (1s2p) RXES, and HERFD-XAS data were collected using a 10 cm diameter, spherically bent (R = 1 m), single-crystal Ge(440) wafer analyzer that focused fluorescence onto a MHz-gated scintillator coupled to a photomultiplier. A bent Si(531) crystal with similar dimensions was used to record the Kβ spectra; the energy resolution was 1 eV for both setups. For recording HERFD-XANES (X-ray absorption near-edge structure), the selected emission energy was kept constant (at the maximum of the Kα1 line), while in the case of RXES both the energy of the incident beam (Ω) and the fluorescence energy (ω) were scanned. The recorded RXES intensity is displayed on a 2D map with one axis being the incident energy and the other their difference, the energy transfer (Ω–ω).31 At the APS RXES data were obtained on aqueous [Fe(terpy)2]2+, while at the ESRF RXES data were taken on aqueous [Fe(bipy)3]2+. The experiments at ID26 (ESRF) utilized 16-bunch mode (no top-up, with decaying ring currents from 90 to 60 mA) with an X-ray repetition rate of 5.68 MHz. Laser excitation was made possible by moving our amplified femtosecond MHz laser system (Tangerine, Amplitude Systèmes) from the European XFEL to ID26. It was synchronized to one-fourth of the 16-bunch repetition rate (1.42 MHz) and to the storage ring. The laser-X-ray time delay was adjusted electronically similarly to what is done at APS. Here we focused on recording RXES planes around the pre-edge of photoexcited aqueous [Fe(bipy)3]2+ (10 mM). The incident radiation was monochromatized by a cryogenically cooled Si(111) fixed-exit double-crystal monochromator. In contrast to the APS measurements, the X-ray (and thus laser) spot size on the sample was much larger (0.6 × 0.1 mm2, H × V). Nevertheless the fractional excited-state population was not dramatically lower than at APS due to four times larger average laser power used and to the wavelength of the Tangerine at its second harmonic, 515 nm, which is absorbed more readily by [Fe(bipy)3]2+ than 532 nm due to a larger laser cross section. X-ray emission was analyzed via four spherically bent Ge(440) crystal analyzers oriented nearly orthogonal to the X-ray beam (and along its polarization vector). In addition, the dispersed X-ray emission was directed above the sample to an avalanche photodiode (Si with effective thickness 0.1 mm and 10 × 10 mm2 surface area). This vertical arrangement of Rowland circles allowed improvement of the overall energy resolution. The sample consisted of the same liquid sheet as described for the APS setup.
Theoretical Methods
In order to investigate the electronic structure of the quintet states of [Fe(terpy)2]2+ we carried out DFT calculations using the ORCA2.8 program package32,33 with the gradient-corrected (GGA) BP86 exchange-correlation functional34,35 and the hybrid B3LYP* functional36 in combination with the TZVP basis set. This approach has been proven to be rather accurate for the description of spin-state transitions in several FeII complexes.12 The geometries of the 1A1, 5B2, and 5E states were fully optimized with the BP86/TZVP method and then utilized as input for the simulation of the XDS data and for the calculations of XANES. To verify that the optimized molecular geometries corresponded to true minima on the potential energy surface, vibrational frequencies were calculated as second derivatives of the electronic energy. They were found to be positive for the 1A1 and 5E states confirming that these geometries corresponded to true minima. However, two negative and degenerate frequencies were found for the 5B2 state. Since this could indicate that the obtained 5B2 structure is a transition state, we performed single-point computations along the problematic normal modes as tests. We found that the potential well was rather flat around the minimum in these dimensions which can lead to a numerical error, resulting in negative frequencies. On the basis of this additional result, we concluded that the 5B2 geometry also corresponds to a true minimum.
In order to be able to compare the strengths of the Fe–N bonds in the different electronic states, the Mayer bond order indices, which quantify bond strengths utilizing the actual computed wave function of the molecule,37−39 were calculated.
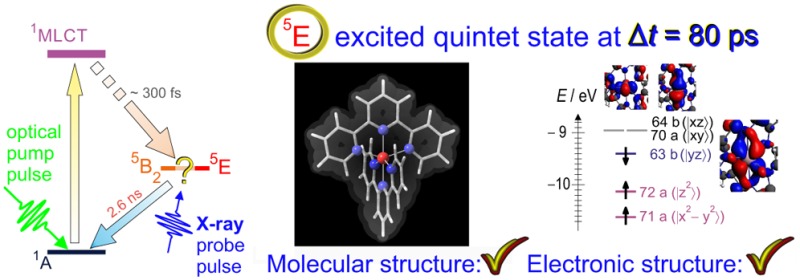
The 5E state of [Fe(terpy)2]2+ with D2d symmetry is susceptible to the Jahn–Teller (JT) effect. To estimate the contribution of this effect, geometry optimizations were repeated with the BP86/TZP method, by applying D2d and C2 point group symmetries with the ADF2013.01 code.40,41 (Note that in C2 symmetry the correct notation for the JT-distorted 5E state would be 5B, as seen in the correlation table in the Supporting Information (SI). However, in order to avoid possible confusion between 5B and 5B2 term symbols, we decided to keep the 5E notation throughout this paper.) Potential energies of excited states have been calculated using time-dependent DFT (TD-DFT) at the B3LYP*/TZVP level, which was found to perform well for describing spin-state switching for a series of FeII compounds.12 Since the ability of DFT to provide accurate spin-state energetics of FeII complexes is limited,12,42 the 5B2 and 5E potentials have been evaluated with an ab initio multireference second-order perturbation theory, the CASPT2 method (Complete Active Space Second-Order Perturbation Theory). These computations were performed with the MOLCAS7.6 program package,43−45 and their detailed description is given elsewhere.12 The potentials were calculated along two different lines, one which connects the 1A1 and 5E minima and the other the 5B2 and 5E minima. The 5B2–5E line is almost orthogonal to the 1A1–5E line (cf. Figure 9).
Figure 9.
(a) Simplified molecular-orbital diagram for the singlet ground state of [Fe(terpy)2]2+. (b) Schematic representation of the minima for the lowest singlet, lowest two triplet, and quintet states of their potential energy surfaces, along with the filling pattern of their unevenly filled t2g or eg* spin orbitals, which are most relevant to their description. On the side, surface plots present the graphical representation of the two eg antibonding orbitals of the 1A1 together with the structural changes when these antibonding orbitals are populated. The different directions of the expansions and the constraints on the ligand geometry suggest an explanation for the origin of the two modes. A hollow cross indicates the position reached in this configuration space when the coordinate changes at populating the eg*-type orbitals are added to approximate the transition to the quintet state. (c) CASPT2 potential curves of the two possible HS (5E and 5B2) states of [Fe(terpy)2]2+ along the line connecting their minima, extracted from the potential energy surface of ref (12). Their spin-up eg orbitals are both populated, and their electronic structure differs only in which of the three t2g-like nonbonding orbitals are filled with a spin-down electron.
A first-principles approach based on DFT in the generalized gradient approximation (GGA) was adopted to model the X-ray absorption near-edge spectra.46 The Fe K-edge XANES was calculated in two steps: first the charge density with a 1s core-hole on the absorbing atom was computed, and then the XANES spectrum was calculated for the electric dipole (1s → p) transitions only, using the broadening parameters given in ref (47). These two steps were performed with the PWscf48 and XSpectra49 packages, respectively, of the Quantum ESPRESSO suite of codes.50 These codes use plane-wave basis sets, pseudopotentials, and periodic boundary conditions; the [Fe(terpy)2]2+ molecule was included in a cubic cell large enough to avoid interactions between molecules belonging to neighboring cells.51,52 A 110 Ry cutoff energy was used for the plane-wave expansion; the charge density with a core-hole on the iron atom was determined at the Γ point; and a 2 × 2 × 2 Monkhorst–Pack grid was chosen for the calculation of the XAS spectrum of the LS ground state (3 × 3 × 3 for the photoinduced HS state). Spin-polarized calculations were performed in the case of the photoinduced HS state (S = 2). Norm-conserving pseudopotentials53 in the Kleinman–Bylander54 form were used, and the parameters for their generation are given in ref (52).
The absorption cross-section of RXES is given by the Kramers–Heisenberg equation55 and can be simulated using multiplet calculations based on the Ligand Field Multiplet (LFM) theory.55 The 1s2p RXES process for [Fe(terpy)2]2+ was modeled assuming electric quadrupole transitions from the initial state 1s22p63d6 to the intermediate state 1s12p63d7, followed by an electric dipole emission to the final state 1s22p53d7. The individual absorption and emission transition matrix elements were first calculated using the method developed by Thole,56 in the framework established by Cowan57 and Butler,58 and then combined in a postprocessing step. Details about these calculations can be found in refs (55), (59), (60), and (61). The calculations were performed assuming an octahedral approximation (D4h symmetry), with a crystal field parameter 10Dq = 2.0 eV for the LS state and 1.3 eV for the HS state; the transition lines were then convolved with a Gaussian, accounting for (some of) the experimental broadening (0.3 eV) and a Lorentzian accounting for the core-hole lifetime broadening (1.2 eV for the intermediate state and 0.2 eV for the final state). The Slater integrals that describe the electronic interactions were scaled down to 80% of their atomic values to account for covalency.
Results and Discussion
Formation and Decay of the Quintet State
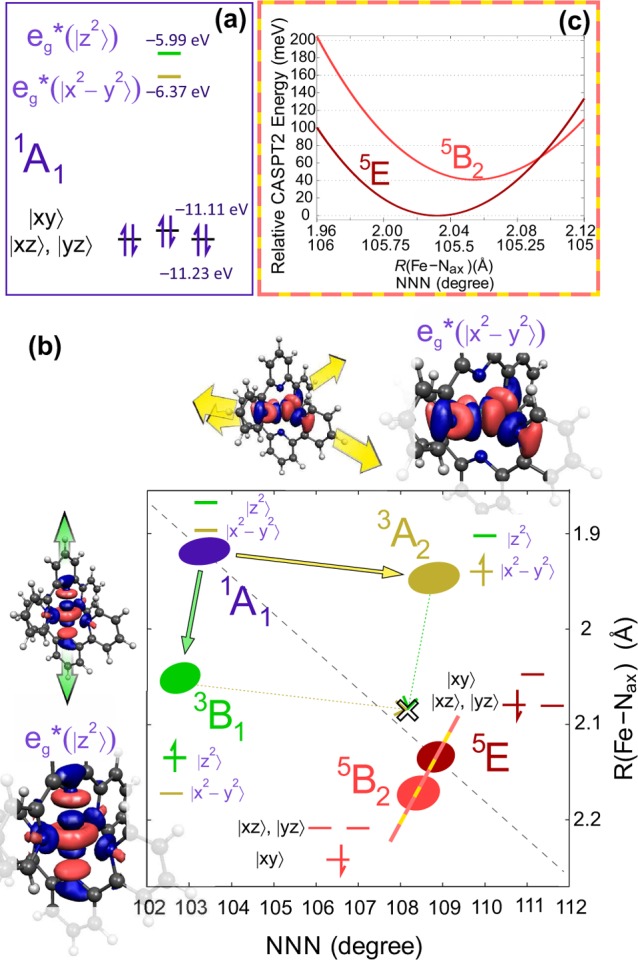
Potential energy surfaces (PESs) are the key to understanding the photophysics and photochemistry of molecular systems. It has been found recently that the PES of FeII or RuII compounds with a MN6 core (M = Fe or Ru) can be described reasonably well using DFT and TD-DFT,12,62,63 providing a means to qualitatively examine the de-excitation mechanism.63 Therefore, we have completed our previous calculations12 on [Fe(terpy)2]2+ by adding the potential energies for the singlet and triplet metal-to-ligand charge transfer (MLCT) states. The most relevant ones of them are plotted together in Figure 1 with those of the lowest singlet and quintet as well as three of the triplet metal-centered states, along a combined coordinate based on the R(Fe–Nax) bond length and the NNN angle of the donor atoms of the terpy ligands. This coordinate connects the LS and HS (5E) minima, which was found to be relevant to the description of this system.11,12 The potential energy curves show very similar structure and crossings to those of [Fe(bipy)3]2+.12,26 Therefore, similar de-excitation pathways for both complexes after the excitation into the 1MLCT may be expected. Although the metal-centered quintet (5E) crosses the 1,3MLCT band somewhat close to its minimum, the coupling between these states was found negligible in a recent theoretical work on [Fe(bipy)3]2+, and it was suggested that the system relaxes via the metal-centered triplet states before the intersystem crossing to the quintet.26 This theoretical expectation has been proven experimentally very recently by femtosecond-resolved XES.27 In [Fe(terpy)2]2+ de-excitation via the triplet states seems highly likely given that the potential curve of the 3B1 state crosses the 3MLCT band at its minimum. Strong coupling between them would make relaxation to this metal-centered triplet state rapid. Verification of this de-excitation pathway in [Fe(terpy)2]2+ requires more involved theoretical or experimental evidence; moreover, one should keep in mind that the true triplet minima do not lie on this combined coordinate, as will be obvious in the final section of this discussion. Nevertheless, based on the mentioned crossing and the almost identical structure of potential energy curves to those of [Fe(bipy)3]2+, the system is very likely to follow a decay path that includes triplets. A possible sequence is indicated by the arrows in Figure 1. From the intermediate lifetimes determined for [Fe(bipy)3]2+26,27 it is understood that relaxation to the quintet state takes place on the subpicosecond time scale, and based on the similarities and the possibly even better positioned triplet states, one can assume this is true for [Fe(terpy)2]2+ as well.
Figure 1.
DFT (1A1, 3A2, and 5E) and TD-DFT (1MLCT, 3MLCT, 3B1, and 3E) potential energy structure of [Fe(terpy)2]2+ along the line connecting the 1A1 and 5E minima; arrows indicate a possible chain of transitions for the full photocycle. The octahedral parent term symbols are also given in parentheses to aid the identification of the metal-centered states. (As the curves for the 5E and 5B2 states on this scale would almost coincide, for clarity only the first one is plotted. Also, in order to keep the figure relatively simple, from each of the 1MLCT, the 3MLCT, and the metal-centered singlet states, only the lowest-lying ones are plotted. Note also that the arrows are only schematic representations of the transitions, particularly between parallel curves, as they cannot represent the coupling by the other vibrational modes between the connected states.)
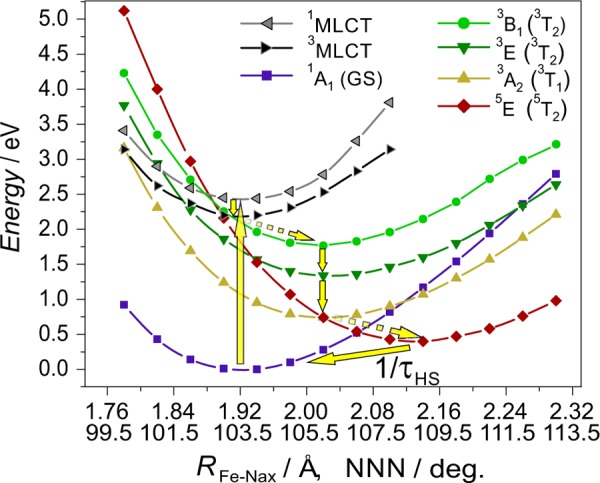
Having argued that the photocycle of [Fe(terpy)2]2+ in many respects resembles that of [Fe(bipy)3]2+, it is evident that experimental studies of the subpicosecond processes leading to the HS (quintet) state can only be achieved with femtosecond-resolved techniques. The lower temporal resolution synchrotron studies presented here probe the system after it has already reached the HS quintet state and as it decays back to the LS ground state. In order to verify the formation of the HS species and quantify its yield, XAS and XES spectra of an aqueous solution of [Fe(terpy)2]2+ were taken before and 80 ps after light excitation. The TFY-XANES spectra (Figure 2a) show significant light-induced variations, the largest ones being the intensity increase of the 7125 eV B feature and the intensity decrease of the 7142 eV D feature. These changes are similar to those observed for the thoroughly studied [Fe(bipy)3]2+ complex,14,16,20 thus confirming the formation of the photoinduced HS state. In the XES spectra (Figure 2b,c), for both the Kα and Kβ emission lines the spectral variations associated with the formation of the photoinduced HS state are evident and fully consistent with previous studies.14,24,25,64 The variations of both the Kα and the Kβ spectra are essentially identical to those of the [Fe(bipy)3]2+ complex14 which is expected since the XES line shapes of 3d transition metal ions reflect practically only the spin state. The first coordination shells of Fe in both complexes are distorted octahedra, and XES spectra are not sensitive to the other relevant differences in symmetry.
Figure 2.
Fe K-edge TFY-XANES (a), Kα (b), and Kβ (c) X-ray emission spectra of a 13 mM aqueous solution of [Fe(terpy)2]2+, with (red line) and without (blue line) laser excitation, with a time delay Δt = 80 ps for the laser-excited data. The evolution of the intensity of the B and D features, as well as the changes in the emission lines, are the signature of a change from the LS to HS configuration. The photoinduced HS fraction can be determined from XES by comparing the spectra to LS/HS references and was found to be γ = 40%.
The laser ON XAS and XES spectra (Figure 2, red line) represent the laser-excited state of the [Fe(terpy)2]2+ complex, which is a combination of both LS and HS contributions. To infer the photoinduced HS fraction (γ), we used an approach that utilizes XES spectral differences between LS and HS references, referred to as the integrated absolute difference (IAD) approach.14,25,64 The typical photoinduced HS fraction observed during our experiments at 80 ps delay was found to be γ = 40%, although at high laser power 60% could also be achieved.
The formation and decay of the photoinduced HS state can be followed by monitoring the intensity of an appropriate spectral or scattering feature at different time delays (Δt). From the XANES of [Fe(terpy)2]2+ the intensity of the B feature at 7125 eV (which presents the largest variation at the spin-state transition) was used for this purpose; its time evolution is displayed in Figure 3. The corresponding fit of the kinetics uses a model consisting of an exponential decay and a broadening by a Gaussian pulse, which is described in the SI. The lifetime of the photoinduced HS state is τHSXAS = 2.61 ± 0.01 ns. This value is fully consistent with the lifetime of τHS = 2.54 ± 0.13 ns obtained with the optical probe for aqueous [Fe(terpy)2]2+.65
Figure 3.
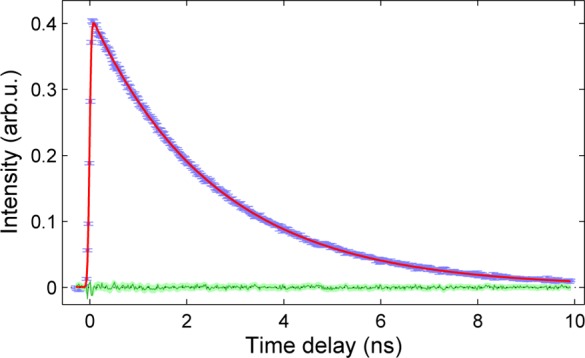
Time evolution of the intensity of the 7125 eV (B) feature of the [Fe(terpy)2]2+ XANES spectrum (Figure 2a), reflecting a quintet lifetime of τHS = 2.61 ± 0.01 ns.
Structure of the Photoinduced Quintet State
Having verified the formation of the HS state and determined its lifetime, we make an attempt at determining its structure. The theoretical molecular geometry is available from DFT calculations,11,12 and the ground state matches excellently the available experimental data, as can be seen in Table 1. For the possible quintet structures, the DFT models are very similar to each other; their corresponding Fe–N bond lengths differ at the few picometer level. This presents a great challenge to the experimental efforts that aim to determine the difference in order to identify which HS state is populated. The most distinct difference between the possible quintet states is the anisotropic distribution of Fe–N bond lengths in the first coordination shell, which is present in the case of the 5E quintet (leading to small but appreciable differences between axial and equatorial bonds), whereas it is lifted in the 5B2 state. As will be shown in the following section, the very high quality of the picosecond EXAFS spectra presented here was decisive to resolve these very small structural variations and provide unambiguous assignment of the quintet structure.
Table 1. Most Relevant Structural and Bonding Parameters of the LS and HS States of [Fe(terpy)2]2+a.
| LS (1A1) |
HS (5E) | HS (5B2) | ||
|---|---|---|---|---|
| exptl66 | calculated | |||
| R(Fe–Nax) | 1.891(5) Å | 1.886 Å | 2.103 Å | 2.159 Å |
| R(Fe–Neq) | 1.988(10) Å | 1.985 Å | 2.198 Å | 2.187 Å |
| NNN angle | 102.8(3)° | 102.6° | 108.4° | 107.2° |
| BFeNax | - | 0.65 | 0.39 | 0.33 |
| BFeNeq | - | 0.60 | 0.34 | 0.34 |
The experimental data are taken from ref (66). The calculations were carried out at the DFT BP86/TZVP level (for the 5E at the (relaxed) JT-distorted geometry). BAB denotes the Mayer bond order index, a powerful parameter quantifying the strength of chemical bonds between atoms A and B (based on MO coefficients and overlaps).37,39,67
Resolving the Quintet Structure with EXAFS Spectroscopy
We have measured the EXAFS spectra of the [Fe(terpy)2]2+ complex in its ground LS and photoexcited HS state (at a time delay of 80 ps) with outstanding statistical quality. The spectra allowed us to verify experimentally the DFT-predicted molecular geometries for both spin states. Moreover, the bond length accuracy of the HS spectrum, unprecedented in time-resolved experiments, permitted us to determine with high confidence which of the two possible quintet states (5E and 5B2) is populated after the light excitation.
The conventional curve-fitting approach of Fourier-transformed EXAFS spectra with standard software packages68 has been used. A detailed description of EXAFS data reduction procedures, fitting approach, and methods used for statistical evaluation of fit errors is available in the SI.
The Fourier-transformed (FT) EXAFS spectrum of the LS ground state is shown in Figure 4a together with the fit function. In this case we have started with the DFT-optimized 1A1 geometry as input structure and refined it during the fitting process. The inset in Figure 4a shows the corresponding k2-weighted EXAFS spectrum in k-space together with the fit curve. (The spectra in this figure appear shifted, as they have not been phase corrected.) The first peak observed in the FT-EXAFS spectrum in Figure 4a corresponds to the first coordination shell, which contains solely contributions from single-scattering paths involving axial and equatorial N atoms (Nax and Neq, respectively) and the central Fe atom. The peaks in the range between 2 and 4 Å correspond to the second and further coordination shells around the FeII ion, as well as multiple scattering paths from the first coordination shell. In the theoretical structure of the LS singlet state, the molecule is axially compressed. This leads to an anisotropic distribution of Fe–N bond lengths in the first coordination shell, with the axial Fe–N bonds about 0.1 Å smaller than the equatorial ones (cf. Table 1). The obtained EXAFS fit is in full agreement with this prediction, and we can unambiguously distinguish between the shorter axial Fe–Nax bonds and the expanded equatorial bonds. The refined experimental bond lengths are R(Fe–Nax) = 1.874 ± 0.004 Å and R(Fe–Neq) = 1.969 ± 0.004 Å, in very good agreement with the DFT-calculated bond lengths listed in Table 1.
Figure 4.
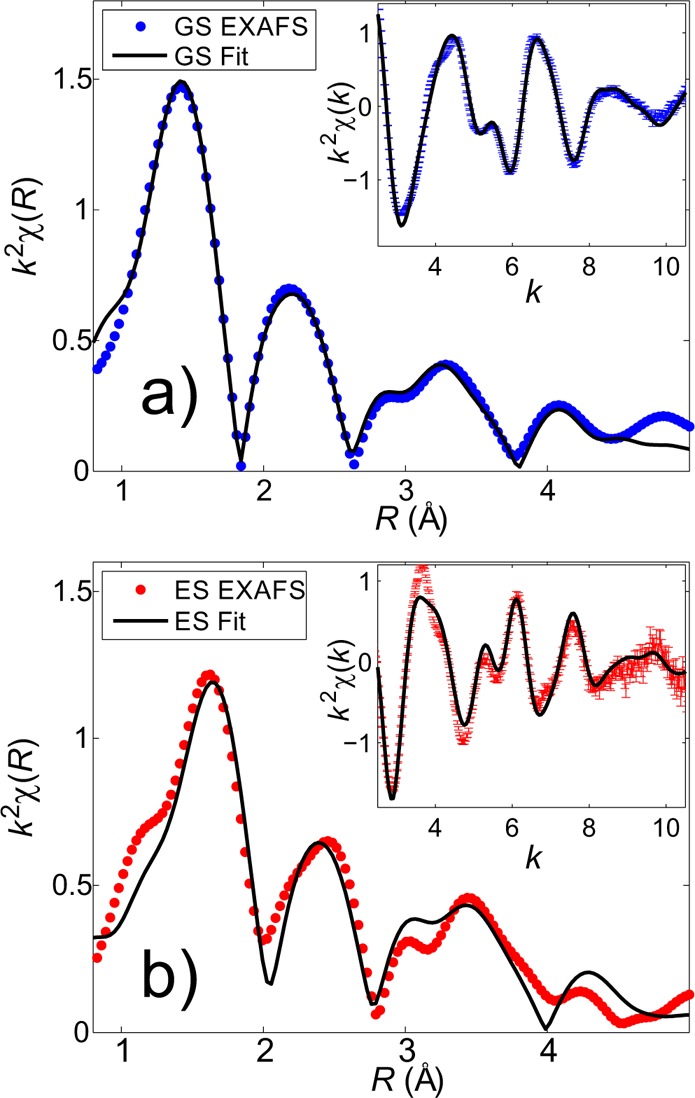
(a) EXAFS of [Fe(terpy)2]2+ in the ground state; the fitted bond lengths are R(Fe–Nax) = 1.874 ± 0.004 Å and R(Fe–Neq) = 1.969 ± 0.004 Å. (b) EXAFS spectrum of the photoexcited quintet state of [Fe(terpy)2]2+; the fitted bond lengths are R(Fe–Nax) = 2.08 ± 0.02 Å and R(Fe–Neq) = 2.20 ± 0.01 Å.
The EXAFS spectrum of the photoexcited [Fe(terpy)2]2+ molecule measured at 80 ps time delay is presented in Figure 4b. The spectrum was obtained using the data reduction method described in the SI. The expected elongation of Fe–N bonds (together with the expansion of outer-lying coordination shells) is clearly visible from the shift of all main peaks present in the FT spectrum toward higher R-values. Similarly to Figure 4a, the inset shows the photoexcited EXAFS spectrum in k-space with the corresponding best-fit result.
To ensure an unbiased determination of the excited-state structure, a range of starting parameters was used that covered the structures of the two proposed quintet states (using the corresponding theoretical models) as initial guesses. As derived from the DFT, the anisotropy affecting the Fe–N bond lengths is preserved in the HS excited state only for the 5E structure, whereas the structure of the 5B2 state has fairly similar Fe–N bond lengths for all six N atoms. Independent of the initial guess, in all cases the fit converged to a structure with substantially distinct Fe–Nax and Fe–Neq bonds lengths, clearly indicating that the anisotropic bond distribution is preserved in the photoexcited state and that we can distinguish between the two possible excited states. Furthermore, a rigorous statistical evaluation of the fit results based on χ2-analysis is discussed in detail in the SI and undoubtedly excludes the 5B2 quintet state due to the lack of any substantial differences between its Fe–N bonds within the first coordination shell and supports the 5E quintet state as the only possible structural model of the HS state. The refined Fe–N bond lengths are R(Fe–Nax) = 2.08 ± 0.02 Å and R(Fe–Neq) = 2.20 ± 0.01 Å, in excellent agreement with the theoretical model for the 5E state (cf. Table 1), which allows us to unambiguously assign its structure to the dynamically measured HS state.
It is worth mentioning that the inclusion of outer coordination shells (beyond Fe–N single scattering contributions in the 1–2 Å range in Figure 4) was possible given the superior EXAFS data quality obtained in the experiment for both the LS ground and photoexcited HS state spectra. In both cases, the statistical goodness of the fit, evaluated using the χ2 test (see the SI for more details), significantly improved when extending beyond the first coordination shell and also by taking into account some of the multiple scattering contributions from outer-lying C atoms. For both LS and HS spectra, only very minor changes to the second and third coordination shells of DFT-optimized geometries were needed for fits to converge to minimum χ2 values (further details can be found in the SI).
Structural Information from XDS
The X-ray scattering signal from a (dilute) solution usually contains only a very small contribution from the solute. However, calculating the difference signal ΔS(Q, Δt) = S(Q, Δt) – S(Q, −∞) effectively removes the otherwise dominating, but almost constant, scattering signal from the solvent. This procedure thus highlights the scattering signature of the structural dynamics of the solute and its environment, the solvent cage, and the structural changes in the surrounding bulk solvent. The analysis approach applied here is fully analogous to the one presented in ref (14), where the analogous [Fe(bipy)3]2+ compound was investigated.
Such difference signals acquired at six time delays from Δt = 100 ps to Δt = 25 ns measured on the [Fe(terpy)2]2+ solution are shown in Figure 5a. Qualitatively, the characteristic negative feature around Q = 0.8–1.2 Å–1 corresponds to the immediate (on the 100 ps time scale) appearance of the bond-elongated HS structure, and the oscillatory feature around Q = 2.2 Å–1 arises from temperature (T) and density (ρ) changes in the bulk solvent, as described in detail in ref (14) for the [Fe(bipy)3]2+ complex and in ref (69) for the PtPOP complex. Figure 5b shows the experimental signal ΔS(Q, Δt = 1 ns) as well as two simulated signals ΔSmodel = γ × ΔSSolute+Cage + ΔT × ΔSΔT + Δρ × ΔSΔρ, where γ is the photoexcitation fraction and ΔSSolute+Cage has been calculated for each of the two proposed HS structures 5E and 5B2. The ΔT × ΔSΔT and Δρ × ΔSΔρ contributions describe the changes in scattering due to bulk-solvent heating and density changes, respectively, and are determined in a separate experiment.70,71 Figure 5c displays each of these three contributions to the model difference signal at Δt = 1 ns, where the magnitudes of the individual contributions are comparable. Separate fits were carried out for each time delay and for both putative HS states, yielding essentially similar results.
Figure 5.
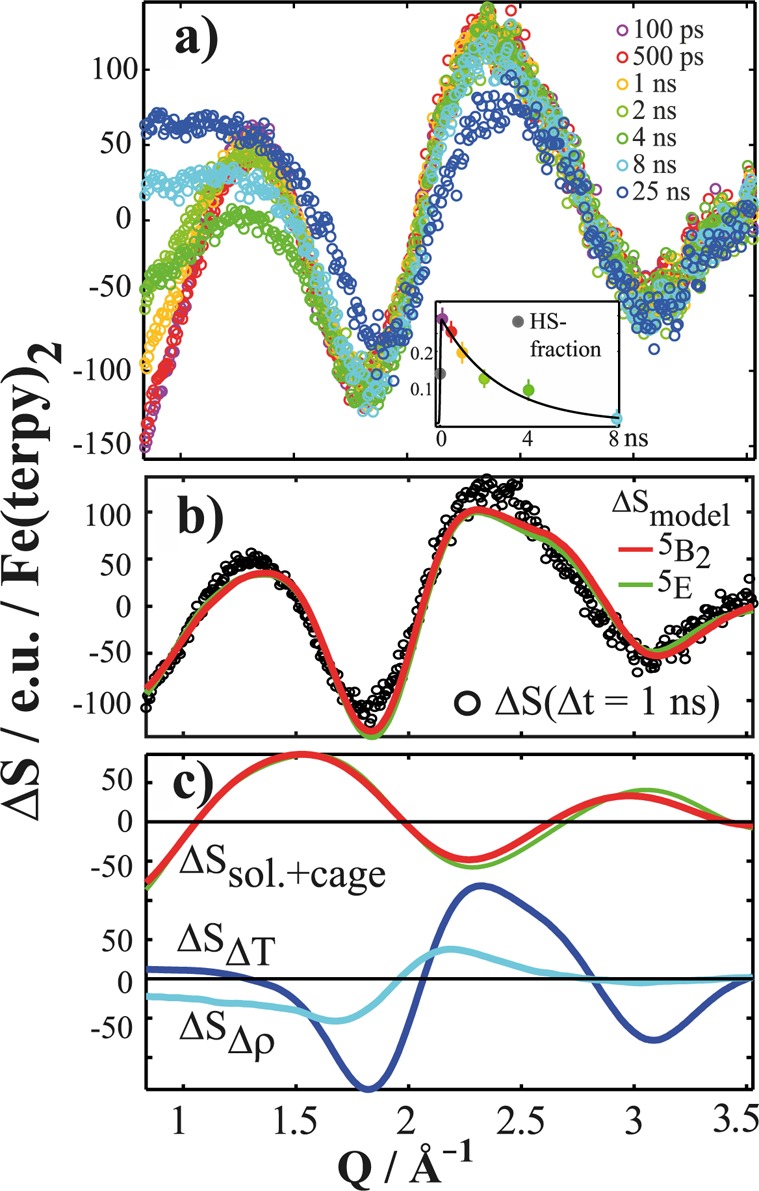
(a) Set of difference scattering signals ΔS(Q, Δt) acquired for [Fe(terpy)2]2+, color-coded according to time delay. The inset shows the evolution of the HS fraction obtained from the magnitude of the solute-related contribution to the difference scattering signal as a function of time delay. (b) ΔS(Q, Δt = 1 ns) (black circles) and the best-fit simulated difference signal for both of the suggested HS structures (red = 5B2, green = 5E). (c) Each of the three contributions to the simulated difference signal, with the two solute-related difference signals again shown in red (5B2) and green (5E). Bulk solvent contributions are shown in light Δρ × ΔSΔρ and dark blue ΔT × ΔSΔT.
The time evolution of the magnitude of γ, the photoexcited fraction for the HS state, is shown in the inset of Figure 5a. Fitting γ in analogy with the fit of the XAS signal described above, we find τHSXDS = 2.7 ± 1.5 ns, which is in reasonable agreement with the XAS-derived value; the larger uncertainty arises from the fewer time-domain data points in the XDS data set. The time evolution of ΔT and Δρ reflects the dynamics of the energy transfer to the solvent. In general, they follow what was observed in the analysis of [Fe(bipy)3]2+,14 although the density increase is somewhat smaller in magnitude in the present case (∼0.1 vs ∼0.4 kg·m–3) and is at the limit of detection. The temperature increase is also observed to be lower (∼0.3 vs ∼0.6 °C), in agreement with the lower concentration of excited-state solutes depositing energy to the solvent through the nonradiative decay processes. Finally, it is evident from Figure 5b that the expected difference signals for the two putative structures are very similar and that they fit the acquired data almost equally well.
In conclusion, the XDS results confirm the plausibility of the DFT structural models, but the present data set does not allow us to recognize the tiny differences of the quintet structures in the variations of the solute signal. Moreover, the time evolution of ΔT and Δρ provides invaluable insights into the solvent dynamics around the excited solute.
Structure and Electronic Structure from XANES
K-edge XANES relates to the p-projected unoccupied electronic density of states of the absorbing atom. Since this is largely influenced by the coordination and bonding, this technique is usually rather sensitive to structural changes around the absorbing atom. For the laser-induced quintet state of [Fe(terpy)2]2+, the population of the antibonding eg* orbitals leads to dramatic rearrangements in the molecular and electronic structure which, as has been shown in Figure 2a, are clearly evident in the near-edge region. The structural differences between the two quintet states, however, are considerably smaller, and distinguishing them with XANES is more challenging. Yet the electronic structure of the 5E and 5B2 states differs, and one expects that this would yield differences in their XANES spectra. With the objective of obtaining further insights into the nature of the quintet state while at the same time assessing the capabilities of XANES to reveal electronic and structural information when very small changes are involved, we compare experimental XANES spectra with theoretical spectra obtained from DFT calculations.
The experimental XANES spectra of the ground and laser-excited (Δt = 80 ps) states were recorded in both TFY and HERFD (taken at the maximum of the Kα1 peak) modes. These spectra are shown in Figure 6. HERFD-XANES usually leads to improvement in the energy resolution when compared to the conventional detection modes.28 In the current experiment, although the HERFD spectra appear somewhat sharper and better resolved, the effect is not dramatic. This can be explained by the fact that the relatively broad energy resolution of the spectrometer (∼0.8 eV) could not improve too much further on the good resolution obtained already in the TFY-XANES due to the narrow energy bandwidth of the incident beam (∼0.5 eV).
Figure 6.

Experimental and calculated Fe K-edge XANES spectra of (molecular) [Fe(terpy)2]2+. With the experimental data, both detection modes (TFY: thick lines and HERFD: dotted lines) are presented; for the theoretical spectrum (thin line), only the electric dipole contribution is plotted. (a) LS ground state; the theoretical spectrum was calculated using the 1A1 structure. (b) Partially excited [Fe(terpy)2]2+ (Laser ON, Δt = 80 ps); the theoretical spectrum was constructed as a linear combination of the calculated LS (60%) and HS (40%) spectra. (Given the similarity of the two theoretical HS spectra, the reconstruction was done only for the 5E structure for clarity.) (c) Photoinduced HS state of [Fe(terpy)2]2+; the plot shows the experimental spectra reconstructed from the laser ON and LS measurements (utilizing the population from XES) and the theoretical spectra calculated for both possible HS states, 5E (thin line) and 5B2 (dashed line). (d) Transient XANES obtained as the difference of the laser ON–LS (ground state) spectra.
Theoretical modeling of K-edge XANES based on the single electron picture has became reliable, and its combination with experiment grew a powerful tool in electronic structure analysis.52,72−74 The theoretical spectra were obtained with such a well-established, reliable approach that takes into account the 1s core hole, utilizing the Quantum ESPRESSO program packages.50 Electric dipole transitions from the 1s state to the unoccupied p-states were calculated, and DFT (BP86/TZVP) optimized structures for the LS and HS states were utilized. Comparing first the spectra of the LS ground state in Figure 6a it is clear that the DFT-derived spectrum agrees very well with the experiment. The energy positions and the relative intensities of the major spectral features, labeled B, C, and D, are well reproduced, including the high-energy shoulder of peak C (which is resolved only in HERFD-XANES). The pre-edge peak labeled A consists of mostly quadrupolar transitions and so is not expected to be properly accounted for by the current electric dipole calculation.
Comparisons to the laser-excited (Δt = 80 ps) experimental spectra are shown in Figure 6b, c, and d. In Figure 6b the measured laser ON spectra, which contain a mixture of LS ground state and HS excited state contributions, are compared with a superposition of LS and HS theoretical spectra. Here the calculated HS component arises solely from the 5E state. The proportion of HS contribution is taken to be 40% based on the excitation fraction derived from the XES measurement. The calculated spectrum is seen to again reproduce the key spectral features, peaks B, C, and D. For a comparison that is independent of the LS and HS population fractions, the differences between the laser ON spectra and the LS ground state spectra are shown in Figure 6d. Here the theoretical HS–LS difference, which has been scaled to match the intensity of the measured curves, can be seen to mimic the shape of the experimental difference spectra. The agreement in both of these comparisons validates the compatibility of the DFT calculated HS state structure with the experimental observations.
To test whether XANES could distinguish between the two possible HS states and perhaps confirm the EXAFS assignment, theoretical spectra for the 5E and 5B2 states were compared to experimental spectra for the HS state that have been reconstructed from the laser ON spectra assuming a 40% HS state fraction. These are shown in Figure 6c. The calculated spectra are seen to be very similar, making a definitive assignment of the experimental spectra to one or the other state difficult. Comparing the ratio of the B and C peak intensities obtained in HERFD and in the calculations, one could be tempted to interpret it as supporting the 5E model to be closer to the experiment. However, the C:D peak intensity ratios might lead to the opposite conclusion, which warns us that this approach might be very far-stretched, as the calculated intensities are apparently not accurate enough for such conclusions in the present case.
The primary electronic structure differences between the 5E and 5B2 quintets manifest themselves in the occupation of the 3d orbitals, which brings us to the discussion of the variation of the experimental pre-edge region (peaks A, A′, and A″) upon excitation. This part of the spectrum stems from 1s → 3d transitions. Being sensitive to the occupation of the 3d orbitals, this shall provide further information on the electronic structure. Indeed, as is evident from Figure 6, this region shows large variations after excitation. However, in conventional XANES these peaks are usually weak and overlapping, and they are also partly obscured by the tail of the main K edge; therefore, they are often poorly resolved. In order to overcome these difficulties and obtain better resolved spectra, we have recorded 1s2p3/2 RXES, the combination of XANES in the preedge region, and the (1s2p) XES around the Kα1 peak. This is presented in the next section.
Resolving the XANES Pre-Edges with RXES: Further Insights into the 3d Electron States
Exploring the pre-edge region of 1s XAS with RXES can reveal more details about the 3d-related part of the electronic structure. We performed 1s2p RXES on light-excited [Fe(terpy)2]2+ at a time delay of Δt = 80 ps. These data are plotted in the top row of Figure 7 next to the ground-state spectrum. The difference between the light-excited and ground-state spectra is shown along with the (fully converted) HS spectrum that was reconstructed from the difference spectrum assuming the population obtained from XES.
Figure 7.

Experimental Fe 1s2p RXES spectra, from aqueous solutions of [Fe(terpy)2]2+ (top row) and [Fe(bipy)3]2+ (bottom row), showing the ground state, the laser-excited state at 80 ps time delay, their difference, and the reconstructed HS spectra. The theoretical spectra (middle row) were obtained from ligand-field multiplet calculations in an approximate Oh local symmetry, and there the “Laser ON (partly HS)” panel was constructed by the superposition of 40% HS and 60% LS. The spectral intensity is plotted above the plane spanned by the incident energy and the difference between the incident and emitted energy, the energy transfer. The energy scaling is equal for both axes and identical for all the plots in order to aid the comparison.
FeII in (close-to) octahedral environments is expected to have a single pre-edge resonance when in the LS state and three resonances when in the HS state.55,75 This has been confirmed in several experiments including 1s2p RXES of an FeII compound at thermal spin-state switching.64 These resonances are clearly present in the spectra in Figure 7, and the increase in resolution and information content with respect to XANES is evident when these features are compared to peaks A, A′, and A″ in Figure 6. Calculated RXES spectra are shown in the middle row of Figure 7. While a single-electron approach usually cannot give an accurate description for 1s2p RXES spectra, multiplet theory can do that for both LS and HS states.55 For octahedral complexes the nature of the pre-edge transitions is electric quadrupolar due to symmetry reasons. The FeN6 cores in [Fe(terpy)2]2+ and [Fe(bipy)3]2+ are distorted octahedra but at a level of distortion that makes the quadrupolar description sufficient to model the spectra. The calculations assumed LS and HS FeII ions in octahedral crystal fields, and the results reproduced the single LS and three HS resonances as can be seen in Figure 7. Multiplet interactions in the RXES final state of the ground-state system lead to small intensity peaks that appear vertically, at different energy transfer values, in the plotted RXES spectra. In the measured LS spectrum the expected (vertical) multiplet structure is weak but obviously present near the single resonance at around 7113.5 eV. In addition, a diagonal feature is observed (at ca. Ω = 7115.5 eV incident energy, Ω–ω = 710.5 eV energy transfer) which is not present in the calculation. This feature must reflect a p-type contribution as a result of orbital mixing (i.e., covalency). In fact, this mixing is expected to bring in a dipolar character, which is seen in the XANES-calculated dipolar contribution to the pre-edges in Figure 6. Upon laser excitation two more peaks (at ca. Ω = 7113 eV, Ω–ω = 706 eV, and ca. Ω = 7116 eV, Ω–ω = 711 eV) appear, while the intensity of the main peak drops. The difference of these spectra together with the known HS population allows us to reconstruct the 1s2p RXES spectrum of the HS state of [Fe(terpy)2]2+, which shows a striking similarity to the calculated one.
We have also studied the 1s2p RXES spectra of the benchmark system [Fe(bipy)3]2+ which has a somewhat different symmetry (D3) and less geometrical constraints from the ligands as they are bidentate. The measured spectra are shown in the bottom row of Figure 7. Despite the somewhat worse energy resolution caused by a twice-larger incident bandwidth, it is apparent that the spectra are very similar to those of the [Fe(terpy)2]2+. The almost identical difference spectra indicate that the nature of the variations in the two complexes are essentially the same.
The crystal field multiplet calculations nicely reproduce the overall spectral shapes and the difference spectra. Intra-atomic electron–electron interactions together with a crystal-field splitting are therefore the main contributions to the spectra. The calculations neglect orbital mixing effects and only consider electric quadrupole transitions; as a result spectral features may be missing on the high-energy side, and the spectral intensities cannot be fully reproduced.
The chemical sensitivity of the K absorption pre-edge is very different from the main edge. While the main edge spectral shape is dominated by the coordination shells, the pre-edge reflects electron–electron interactions within the d-shell as well as crystal-field effects and orbital mixing. The striking similarity of the difference spectra in the two compounds shows that intra-atomic d–d interactions in the HS state dominate the spectral shape. The 1s2p RXES planes thus visualize the dramatic change of the electronic levels when the molecules are in their short-lived photoexcited state. However, the reasonably good description by the multiplet calculations, an ionic approach utilizing Oh symmetry, points toward the need for more involved calculations. Distinguishing between 5E and 5B2 in RXES would require full multiplet calculations including dipole and quadrupole transitions with molecular wave functions that describe the whole complex. This is currently not possible with sufficient accuracy. Angular-dependent RXES could help to identify the exact 3d configuration in single crystals;76 however, this is hardly possible in solution.
Theoretical Insights into the Electronic Structure
Although the [Fe(terpy)2]2+ molecule has already been investigated theoretically,11,12 a few details relevant to the characteristics of the quintet states have been overlooked or at least not discussed in due detail. These include the relation between the structural differences and the occupation of the 3d orbitals in the two quintet states 5E and 5B2, the effects of the Jahn–Teller (JT) distortion on the 5E, and the electronic structure origin of the two modes needed to describe the spin-state transition. Furthermore, the previous results raise disturbing questions: is it hopeless to distinguish the electronic structure differences of two possible quintets with X-ray spectroscopy? Why is the 5E state populated when almost all DFT calculations predict 5B2 as being more stable? Here we complement the referred works by performing the necessary analysis using TD-DFT and CASPT2 and provide answers to all of the above-mentioned issues.
In order to unveil the variations in the electronic structure that cause the structural and bonding differences between the 5E and 5B2 states, we compare the relevant molecular orbitals (MOs) obtained with spin-unrestricted DFT. To examine this in detail, in Figure 8 we present the highest occupied and the lowest unoccupied (Kohn–Sham) MOs. (A more extended version of this MO diagram is plotted in the SI.) Not surprisingly, these orbitals at the HOMO–LUMO gap have substantial Fe 3d character. The most interesting and relevant molecular orbitals at the gap are those with a dominant contribution from the atomic Fe dxy, dxz, and dyz orbitals of β spin (i.e., spin down). Only one spin-down electron is available for these three orbitals. Which of these orbitals the electron occupies determines the atomic and electronic structural differences.
Figure 8.

Diagram of the spin-unrestricted KS molecular orbitals for the quintet states of [Fe(terpy)2]2+ for 5E, and both the highly symmetric (D2d) and the Jahn–Teller distorted (C2) form is also shown. (Note that in a rigorous description the latter should be denoted as 5B in C2 symmetry.) The energy levels (top) as well as the 3D representation (bottom) of the most relevant MOs of the 5B2 and 5E states with relevant 3d character are shown.
In the simplest approximation of ligand-field theory, the Fe dxy, dxz, and dyz atomic orbitals are nonbonding. However, this is altered in such a covalent molecule where the atomic orbtials are mixed to some extent with ligand orbitals. In the bottom of Figure 8 3D representations show the actual composition of these MOs for the 5B2 state. Here the 5b1 orbital is populated, which still has nonbonding dxy character; the atomic orbital contribution (mixing coefficient) for the Fe dxy is 84.2%, while those for all nitrogen orbitals are below 1%. Nevertheless, it is apparent that the dxz and dyz orbitals are mixed with in-phase p-type orbitals from the axial nitrogens (58.5% Fe(3d), 6.24% N(p)). The resulting 2-fold-degenerate 32e pair of MOs have a (π-)bonding character. However, this has no effect on the properties of the 5B2 state since these MOs are unoccupied. In the 5E state (when constraining the symmetry to D2d as in ref (11)), the energy order of the 5b1 and 32e orbitals is swapped, and the 32e orbital pair is populated by a single electron. These orbitals bond to the axial nitrogens, as we just discussed, which shortens the Fe–Nax bond lengths. (This orbital mixing and the corresponding charge transfer to the ligand via the bonding is usually referred to as backbonding or back-donation.)
Without constraints on the symmetry, the JT effect removes the degeneracy of the 32e orbitals in the 5E state and lowers the symmetry to C2. The highest occupied orbital becomes the 63b, which has the main contribution from the Fe dyz atomic orbital, and thus the backbonding to the axial nitrogens remains effective. The enhanced axial bonding is clearly reflected in the Mayer bond order indices37,39,67 reported in Table 1 and provides a natural explanation for the geometry differences of the quintet states. Moreover, the energies of the levels of the quintet MOs become strikingly similar after applying the JT distortion to the 5E; the frontier orbitals have almost identical energies. The LUMO pair 64b and 70a (|xz⟩,|xy⟩) mimics the 32e (|xz⟩,|xy⟩) pair of 5B2, and the even higher-energy unoccupied orbitals show high resemblance for the 5B2 and 5E (C2) states (see SI). Such similarities of the electronic structure readily explain why the presented spectroscopy data are so similar for the two quintet states.
The question remains why the 5E state is populated, contradicting most DFT results that favor the 5B2 state as more stable.11,13 The energy difference between the quintet states is rather small, and on such a scale the DFT calculations show a large scatter that depends on the choice of the exchange-correlation functional.12 Therefore, we have performed CASPT2 calculations that provide more accurate energy differences. As can be seen in Figure 9(b), the two quintet minima are separated along a line that is almost orthogonal to the line connecting the LS and quintet states. The energy difference and the relative location of the potential wells belonging to the 5E and the 5B2 states are best appreciated along this coordinate. This is plotted in Figure 9(c). The energy difference is 41 meV with the 5E state lower in energy. Moreover, as the 5E state is expected to split due to the Jahn–Teller effect, we can expect the energy to be even lower. (Note that the JT effect only affects the angle between the planes of the two terpy ligands, which changes by 4°, while the bond lengths and other angles stay practically the same.12) The combination of the energy difference from CASPT2 and the JT energy lowering obtained from DFT suggests that the distorted 5E can be as much as 150 meV lower in energy than the 5B2 state. As the transfer to the quintet(s) takes place in the first picosecond after light excitation and the measurements are performed at a delay ≥80 ps by which time the excess energy is transferred to the solvent, we expect to find the system in the 5E, the more stable quintet state. Yet, since geometry optimization is practically impossible at the CASPT2 level on a such a large system with numerous electrons, one should keep in mind that this expectation might be biased.
Finally, we address the origin of the breakdown of the SCC model. In most octahedral FeII compounds that undergo spin-state transitions a single configuration coordinate is sufficient as a reaction coordinate, typically the breathing mode of the molecule which is proportional to the metal–ligand distance. This has been exploited in a very successful quantitative approach proposed by A. Hauser that describes well the HS–LS relaxation times for most complexes but not [Fe(terpy)2]2+.10,11 Given the slower than expected relaxations (and apparently higher barrier) found for [Fe(terpy)2]2+ in different matrices, it was suggested that the SCC-based model is not applicable to this tridentate complex, and the involvement of an extra mode, probably the ligand bending measured by the NNN angle, is required, which increases the barrier height. Many details of this have not been brought to light; in a recent paper we found that the NNN angle of the ligand and the R(Fe–Nax) bond lengths are promising candidates to describe the reaction coordinates.12
Starting with the description of the electronic structure of the 1A1 ground state, which has no unpaired electrons, we note that the spin-resctricted KS MOs have identical energies for α and β electrons, as is observed on the frontier orbitals of this state in Figure 9a. The tridentate terpyridine has geometrical constraints as the N atoms of its three linked pyridine rings in this planar ligand cannot take up ideal octahedral positions around the Fe center. Due to this, the energy of the molecular orbitals that have a dominant contribution from the 3d orbitals will have a relevant split in energy, depending on whether they are oriented parallel or orthogonal to the molecular axis that goes through the nitrogens of the central pyridine rings and the FeII ion. This splitting is small for the dxy, dxz, and dyz nonbonding or weakly backbonding orbitals but high for the dz2- and dx2-y2-based orbitals, which originate from the octahedral eg* antibonding orbitals as seen in Figure 9a. In addition to the geometrical constraints, the backbonding between the axial N and the Fe contributes to the axial distortion of the molecule as well. The stronger bonding along the axis is again clearly identified in the Mayer bond order indices, as seen in Table 1.
To reach the quintet states, both antibonding eg* orbitals need to be populated with a spin-up electron. However, it is revealing to examine what would happen to the molecular geometry if only one of these orbitals is populated. The eg(|x2–y2⟩) MO is based on the Fe dx2–y2 atomic orbital mixed with p-orbitals of the four equatorial nitrogens; therefore, populating this antibonding orbital causes an expansion in the molecule in this direction, opening up the NNN angle of the ligand, without affecting the axial R(Fe–Nax) bond length. Indeed, the transition to the 3A2 state is almost perfectly described by the NNN opening, as can be seen in Figure 9. This suggests that the opening of the NNN angle of the ligands is one of the relevant modes needed for the LS–HS transition. The eg*|z2⟩ antibonding orbital has large mixing of the Fe dz2 with the p-orbitals of the axial N atoms and smaller mixing with those of the equatorial N atoms. Therefore, the population of this MO leads to an increase in the lengths of the Fe–Nax bonds and a smaller weakening of the equatorial Fe–Neq bonds. This results in the increase of the axial R(Fe–Nax) bond length without substantial effect on the NNN angle of the ligands because the pull on the side rings at the equatorial nitrogens by the iron caused by axial elongation is compensated by the simultaneous weakening of the Fe–Neq bonds. This change corresponds to the second coordinate needed for the transition.11,12 The optimized geometry of the triplet with the populated dz2-based orbital, 3B1, differs from that of the ground state almost only in R(Fe–Nax), as shown in Figure 9. (Note that the geometry optimization of the 3B1 could only be obtained by forcing the occupation of the eg|z2⟩ MO with a symmetry constraint on the orbital populations.) Consequently, we can identify that these population changes combined with the geometrical constraints of the terpy ligand explain the need for the two-mode description of the switching and the breakdown of the SCC model in this molecule. The validity of relating the two necessary configuration coordinates to electronic structure effects is further supported if we add up the two vectors pointing from the singlet ground state to the triplet states in this coordinate system. Each of these vectors represent the structural variations caused by adding one electron to an eg* orbital. Therefore, we can expect that their sum approximates well the real quintet geometry, if the relation between structure and orbital population variations is valid. The resulting position (marked by a hollow cross in Figure 9) lies indeed fairly close to the actual positions found for the quintet states with the geometry optimizations. The mismatch is comparable to the distance between the 5E and 5B2 states, which differ in the populations of t2g-like orbitals. Accordingly, the two modes based on the opening of the NNN angle and the variation of the R(Fe–Nax) bond lengths grasp rather well the main structural changes, and these seem to clearly correlate with the populations of the eg-like orbitals. However, remaining smaller-scale variations in the electronic structure are also reflected in the molecular structure. Consequently, further refinement of these modes or involvement of further ones is needed to obtain a complete and fully satisfactory description of this system.
Conclusions
The challengingly small differences in the molecular geometry and electronic structure of the 5B2 and 5E quintets were addressed utilizing a variety of time-resolved X-ray techniques. The minuscule explorable differences provided a demanding testing ground for the techniques, enabling assessment of their ability to discern small changes that occur in short-lived transient states. The formation and decay of the high-spin state was followed with all applied techniques. XANES, XES, and RXES spectra, despite their high quality and clear variations, could not reveal which of the two quintets is realized. XDS, which captured both the light-induced conversion to HS and the behavior of the solvent in the vicinity of the molecule, was also unable to distinguish between the two quintets. Excellent quality EXAFS spectra were able to resolve the bond-length variations accurately, leading to unambiguous identification of 5E as the formed quintet state which is in agreement with our theoretical predictions. Applying theoretical calculations (DFT, TD-DFT, and CASPT2), we were able to explain the electronic structure origin of the differences in the properties of the two possible quintet states. We identified that back-bonding in the 5E state is responsible for the structural differences between the quintet states. We revealed why the 5E state is the more stable one and also checked how this state varies upon the Jahn–Teller effect. The failure of spectroscopy to distinguish the quintets was readily explained by the striking similarity of their molecular orbital diagrams. Finally, we determined why one configurational coordinate is not sufficient for the description of the LS–HS transition in [Fe(terpy)2]2+.
Due to the anomalous behavior of [Fe(terpy)2]2+ among FeII complexes that undergo photoinduced LS to HS transitions, the identification of the quintet state produced after light excitation represents a milestone. Our work should inspire studies of the dynamics on the way to the quintet state, during the first picosecond after light excitation, using X-ray free electron lasers. This will lead to a more complete understanding of the photocycle of this system, which can motivate the design of novel molecules with better photoswitching performance.
Acknowledgments
This project was supported by the European Research Council via contracts ERC-StG-259709 and ERC-AdvG-VISCHEM-226136, the ‘Lendület’ (Momentum) Program of the Hungarian Academy of Sciences, and the European XFEL. K.H., M.N., T. B. van D., K.K., and H.L. acknowledge support from the Danish National Research Foundation’s Centre for Molecular Movies and DANSCATT. C.B., W.G., A.G., A.B., and T.A. acknowledge funding by the Centre of Ultrafast Imaging (Hamburg), by the EU-funded Cluster of Research Infrastructures for Synergies in Physics (CRISP) project, and the Deutsche Forschungsgemeinschaft via SFB925. A.B., N.Z., G.V., D.C., and A.J. acknowledge the French Ministry of Foreign Affairs (MAE) and the French Ministry of Higher Education and Research (MESR) for funding through Balaton project no. 27875YK, as well as the Hungarian and French Intergovernmental S&T Cooperation Program (TET11FR-XTHEOEXP). Z.N. acknowledges support from the Bolyai Fellowship of the Hungarian Academy of Sciences. J.U. acknowledges continued funding from the Knut and Alice Wallenberg Foundation. D.C. and A.J. acknowledge GENCI (Grand Equipement National de Calcul Intensif), which allows access to the HPC resouces of IDRIS under the allocation 2013-100172. Use of the Advanced Photon Source, an Office of Science User Facility operated for U.S. Department Of Energy Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under contract no. DE-AC02-06CH11357. We are grateful to the staff of 7-ID from the APS and the staff of ID26 from the ESRF for help during experiments.
Supporting Information Available
Further details on the relation between the states in the different symmetry groups, the lifetime fitting, the EXAFS analysis, and the calculated molecular orbital diagrams. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⬢ Université Paris-Sud, ICMMO UMR CNRS 8182, Equipe de Chimie Inorganique, 91405 Orsay, France.
The authors declare no competing financial interest.
Supplementary Material
References
- Kahn O.; Martinez C. J. Spin-Transition Polymers: From Molecular Materials Toward Memory Devices. Science 1998, 279, 44–48. [Google Scholar]
- Sato O.; Tao J.; Zhang Y.-Z. Control of Magnetic Properties through External Stimuli. Angew. Chem., Int. Ed. 2007, 46, 2152–2187. [DOI] [PubMed] [Google Scholar]
- Venkataramani S.; Jana U.; Dommaschk M.; Snnichsen F. D.; Tuczek F.; Herges R. Magnetic Bistability of Molecules in Homogeneous Solution at Room Temperature. Science 2011, 331, 445–448. [DOI] [PubMed] [Google Scholar]
- Gütlich P.; Goodwin H. A.. Spin crossover - An overall perspective. Spin Crossover in Transition Metal Compounds I; Topics in Current Chemistry; Springer-Verlag: Berlin, 2004; Vol. 233, pp 1–47. [Google Scholar]
- McGarvey J.; Lawthers I. Photochemically-induced perturbation of the 1A-reversible-5T equilibrium in Fe-II complexes by pulsed laser irradiation in the metal-to-ligand charge-transfer absorption-band. J. Chem. Soc., Chem. Commun. 1982, 16, 906–907. [Google Scholar]
- Decurtins S.; Gütlich P.; Köhler C. P.; Spiering H.; Hauser A. Light-induced excited spin state trapping in a transition-metal complex: The hexa-1-propyltetrazole-iron(II) tetrafluoroborate spin-crossover system. Chem. Phys. Lett. 1984, 105, 1–4. [Google Scholar]
- Hauser A. Intersystem crossing in the [Fe(ptz)6](BF4)2 spin crossover system (ptz=1-propyltetrazole). J. Chem. Phys. 1991, 94, 2741–2748. [Google Scholar]
- Hauser A.; Vef A.; Adler P. Intersystem crossing dynamics in Fe(II) coordination compounds. J. Chem. Phys. 1991, 95, 8710–8717. [Google Scholar]
- Hauser A. Light-induced spin crossover and the high-spin →low-spin relaxation. Spin Crossover in Transition Metal Compounds II; Topics in Current Chemistry; Springer-Verlag: Berlin, 2004; Vol. 234, pp 155–198. [Google Scholar]
- Renz F.; Oshio H.; Ksenofontov V.; Waldeck M.; Spiering H.; Gütlich P. Strong Field Iron(II) Complex Converted by Light into a Long-Lived High-Spin State. Angew. Chem., Int. Ed. 2000, 39, 3699–3700. [DOI] [PubMed] [Google Scholar]
- Hauser A.; Enachescu C.; Daku M. L.; Vargas A.; Amstutz N. Low-temperature lifetimes of metastable high-spin states in spin-crossover and in low-spin iron(II) compounds: The rule and exceptions to the rule. Coord. Chem. Rev. 2006, 250, 1642–1652. [Google Scholar]
- Pápai M.; Vankó G.; de Graaf C.; Rozgonyi T. Theoretical investigation of the electronic strucure of Fe(II) complexes at spin-state transitions. J. Chem. Theory Comput. 2013, 9, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton S.; Zhang X.; Daku L. M. D.; Smeigh A. L.; Jianxin Z.; Carl-Johan W.; Attenkofer K.; Jennings G.; Kurtz C. A.; Gosztola D. J. Probing the anisotropic distortion of photoexcited spin crossover complexes with picosecond x-ray absorption spectroscopy. J. Phys. Chem. C 2014, 118, 4536–4545. [Google Scholar]
- Haldrup K.; Vankó G.; Gawelda W.; Galler A.; Doumy G.; March A. M.; Kanter E. P.; Bordage A.; Dohn A.; van Driel T. B. Guest-host interactions investigated by time-resolved x-ray spectroscopies ans scattering at MHz rates: Solvation dynamics and photoinduced spin transition in aqueous [Fe(bipy)3]2+. J. Phys. Chem. A 2012, 116, 9878–9887. [DOI] [PubMed] [Google Scholar]
- Gawelda W.; Cannizzo A.; Pham V.-T.; van Mourik F.; Bressler C.; Chergui M. Ultrafast Nonadiabatic Dynamics of [FeII(bpy)3]2+ in Solution. J. Am. Chem. Soc. 2007, 129, 8199–8206. [DOI] [PubMed] [Google Scholar]
- Bressler C.; Milne C.; Pham V.-T.; ElNahhas A.; van der Veen R. M.; Gawelda W.; Johnson S.; Beaud P.; Grolimund D.; Kaiser M. Femtosecond XANES Study of the Light-Induced Spin Crossover Dynamics in an Iron(II) Complex. Science 2009, 323, 489–492. [DOI] [PubMed] [Google Scholar]
- Bressler C.; Abela R.; Chergui M. Exploiting EXAFS and XANES for time-resolved molecular structures in liquids. Z. für Kristallogr. 2008, 223, 307–321. [Google Scholar]
- Daku L. M.; Vargas A.; Hauser A.; Fouqueau A.; Casida M. E. Assessment of Density Functionals for the High-Spin/Low-Spin Energy Difference in the Low-Spin Iron(II) Tris(2,2′-bipyridine) Complex. ChemPhysChem 2005, 6, 1393–1410. [DOI] [PubMed] [Google Scholar]
- Gawelda W.; Pham V.-T.; van der Veen R. M.; Grolimund D.; Abela R.; Chergui M.; Bressler C. Structural analysis of ultrafast extended x-ray absorption fine structure with subpicometer spatial resolution: Application to spin crossiver complexes. J. Chem. Phys. 2009, 130, 124520. [DOI] [PubMed] [Google Scholar]
- Gawelda W.; Pham V.-T.; Benfatto M.; Zaushitsyn Y.; Kaiser M.; Grolimund D.; Johnson S. L.; Abela R.; Hauser A.; Bressler C. Structural Determination of a Short-Lived Excited Iron(II) Complex by Picosecond X-Ray Absorption Spectroscopy. Phys. Rev. Lett. 2007, 98, 057401. [DOI] [PubMed] [Google Scholar]
- Bressler C.; Chergui M. Molecular Structural Dynamics Probed by Ultrafast X-Ray Absorption Spectroscopy. Annu. Rev. Phys. Chem. 2010, 61, 263–282. [DOI] [PubMed] [Google Scholar]
- Cannizzo A.; Milne C.; Consani C.; Gawelda W.; Bressler C.; van Mourik F.; Chergui M. Light-induced spin crossover in Fe(II)-based complexes: The full photocycle unraveled by ultrafast optical and x-ray spectroscopies. Coord. Chem. Rev. 2010, 254, 2677–2686. [Google Scholar]
- Daku L. M.; Hauser A. Ab Initio Molecular Dynamics Study of an Aqueous Solution of [Fe(bpy)3](Cl)2 in the Low-Spin and in the High-Spin States. J. Phys. Chem. Lett. 2010, 1, 1830–1835. [Google Scholar]
- Vankó G.; Glatzel P.; Pham V.-T.; Abela R.; Grolimund D.; Borca C. N.; Johnson S. L.; Milne C. J.; Bressler C. Picosecond Time-Resolved X-Ray Emission Spectroscopy: Ultrafast Spin-State Determination in an Iron Complex. Angew. Chem., Int. Ed. 2010, 49, 5910–5912. [DOI] [PubMed] [Google Scholar]
- Vankó G.; Bordage A.; Glatzel P.; Gallo E.; Rovezzi M.; Gawelda W.; Galler A.; Bressler C.; Doumy G.; March A. M. Spin-state studies with XES and RXIS: From static to ultrafast. J. Electron Spectrosc. Relat. Phenom. 2013, 188, 166–171. [Google Scholar]
- Sousa C.; de Graaf C.; Rudavskyi A.; Broer R.; Tatchen J.; Etinski M.; Marian C. M. Ultrafast Deactivation Mechanism of the Excited Singlet in the Light-Induced Spin Crossover of [Fe(2.2′-bipyridine)3]2+. Chem.—Eur. J. 2013, 19, 17541–17551. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Alonso-Mori R.; Bergmann U.; Bressler C.; Chollet M.; Galler A.; Gawelda W.; Hadt R. G.; Hartsock R. W.; Kroll T. Tracking excited-state charge and spin dynamics in iron coordination complexes. Nature 2014, 509, 345–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämäläinen K.; Siddons D. P.; Hastings J. B.; Berman L. E. Elimination of the inner-shell lifetime broadening in x-ray absorption spectroscopy. Phys. Rev. Lett. 1991, 67, 2850. [DOI] [PubMed] [Google Scholar]
- de Groot F. M. F.; Vankó G.; Glatzel P. The 1s x-ray absorption pre-edge structures in transition metal oxides. J. Phys.: Condens. Matter 2009, 21, 104207. [DOI] [PubMed] [Google Scholar]
- March A. M.; Stickrath A.; Doumy G.; Kanter E. P.; Krassig B.; Southworth S. H.; Attenkofer K.; Kurtz C. A.; Chen L. X.; Young L. Development of high-repetition-rate laser pump/x-ray probe methodologies for synchrotron facilities. Rev. Sci. Instrum. 2011, 82, 073110. [DOI] [PubMed] [Google Scholar]
- Glatzel P.; Bergmann U. High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes–electronic and structural information. Coord. Chem. Rev. 2005, 249, 65–95. [Google Scholar]
- Neese F. The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Neese F.ORCA, version 2.8; Max-Planck-Institut für Bioanorganische Chemie: Mülheim an der Ruhr, Germany, 2010. [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [DOI] [PubMed] [Google Scholar]
- Reiher M. Reparametrization of hybrid functionals based on energy differences of satates of different multiplicities. Theor. Chem. Acc. 2001, 107, 48–55. [Google Scholar]
- Mayer I. Charge, bond order and valence in the {AB} initio {SCF} theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar]
- Mayer I. Bond order and valence: Relations to Mulliken’s population analysis. Int. J. Quantum Chem. 1984, 26, 151–154. [Google Scholar]
- Mayer I. Bond order and valence indices: A personal account. J. Comput. Chem. 2007, 28, 204–221. [DOI] [PubMed] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar]
- Scientific Computing and Modelling ADF; version 2013.01; Vrije Universiteit, Theoretical Chemistry: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Pierloot K.; Vancoillie S. Relative energy of the high-(5T2g) and low-(1A1g) spin states of [Fe(H2O)6]2+, [Fe(NH3)6]2+, and [Fe(bpy)3]2+: CASPT2 versus density functional theory. J. Chem. Phys. 2006, 125, 124303. [DOI] [PubMed] [Google Scholar]
- Roos B. O.; Taylor P. R.; Siegbahn P. E. A complete active space {SCF} method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar]
- Andersson K.; Malmqvist P.-A.; Roos B. O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar]
- Aquilante F.; De Vico L.; Ferré N.; Ghigo G.; Malmqvist P.-Å.; Neogrády P.; Pedersen T.; Pitonak M.; Reiher M.; Roos B. MOLCAS 7: the next generation. J. Comput. Chem. 2010, 31, 224. [DOI] [PubMed] [Google Scholar]
- Taillefumier M.; Cabaret D.; Flank A.-M.; Mauri F. X-ray absorption near-edge structure calculations with the pseudopotentials: Application to the K edge in diamond and α-quartz. Phys. Rev. B 2002, 66, 195107. [Google Scholar]
- Gaudry E.; Cabaret D.; Sainctavit P.; Brouder C.; Mauri F.; Goulon J.; Rogalev A. Structural relaxations around Ti, Cr and Fe impurities in α-Al2O3 probed by X-ray absorption near-edge structure combined with first-principles calculations. J. Phys.: Condens. Matter 2005, 17, 5467–5480. [Google Scholar]
- Baroni S.; de Gironcoli S.; Dal Corso A.; Gianozzi P. Phonons and related crystal properties from density functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar]
- Gougoussis C.; Calandra M.; Seitsonen A.; Brouder C.; Shukla A.; Mauri F. Intrinsic charge transfer gap in NiO from Ni K-edge X-ray absorption spectroscopy. Phys. Rev. B 2009, 79, 045118. [Google Scholar]
- Giannozzi P.; Baroni S.; Bonini N.; Calandra M.; Car R.; Cavazzoni C.; Ceresoli D.; Chiarotti G. L.; Cococcioni M.; Dabo I. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 2009, 21, 395502. [DOI] [PubMed] [Google Scholar]
- Arfaoui M.; Cabaret D.; Longa S. D.; Seitsonen A.; Mauri F.. First-principles full-potential calculations of the Fe K pre-edge and near-edge structure in carbonmonoxy-myoglobin. AIP Conference Proceedings; 2007; pp 331–333.
- Cabaret D.; Bordage A.; Juhin A.; Arfaoui M.; Gaudry E. First-principles calculations of X-ray absorption spectra at the K-edge of 3d transition metals: an electronic structure analysis of the pre-edge. Phys. Chem. Chem. Phys. 2010, 12, 5619–5633. [DOI] [PubMed] [Google Scholar]
- Troullier N.; Martins J. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [DOI] [PubMed] [Google Scholar]
- Kleinman L.; Bylander D. Efficacious form for model pseudopotentials. Phys. Rev. Lett. 1982, 48, 1425–1428. [Google Scholar]
- de Groot F. M. F., Kotani A.. Core Level Spectroscopy of Solids; Taylor & Francis: New York, 2008. [Google Scholar]
- Thole T.; van der Laan G.; Fuggle J.; Sawatzky G.; Karnatak R.; Estava J.-M. 3d X-ray absorption lines and the 3d94fn+1multiplets of the lanthanides. Phys. Rev. B 1985, 32, 5107–5118. [DOI] [PubMed] [Google Scholar]
- Cowan R.The theory of atomic structure and spectra; University of California Press: Berkeley, 1981. [Google Scholar]
- Butler P. H.Point group symmetry: Applications, methods and tables; Plenum: New York, 1981. [Google Scholar]
- de Groot F. M. F. Multiplet effects in X-ray spectroscopy. Coord. Chem. Rev. 2005, 249, 31–63. [Google Scholar]
- van Schooneveld M. M.; Kurian R.; Juhin A.; de Groot F. M. F. Electronic structure of CoO nanocrystals and a single-crystal probed by Resonant X-ray Emission Spectroscopy. J. Phys. Chem. C 2012, 116, 15218–15230. [Google Scholar]
- Juhin A.; Brouder C.; Arrio M.-A.; Cabaret D.; Sainctavit P.; Balan E.; Bordage A.; Seitsonen A. P.; Calas G.; Eeckhout S. G. X-ray linear dichroism in cubic compounds: The case of Cr3+in MgAl2O4. Phys. Rev. B 2008, 78, 195103. [Google Scholar]
- Österman T.; Persson P. Excited state potential energy surfaces of bistridentate RuII complexes A TD-DFT study. Chem. Phys. 2012, 407, 76–82. [Google Scholar]
- Fredin L. A.; Pápai M.; Rozsályi E.; Vankó G.; Wärnmark K.; Sundström V.; Persson P. Exceptional Excited-State Lifetime of an Iron(II)-N-Heterocyclic Carbene Complex Explained. J. Phys. Chem. Lett. 2014, 5, 2066–2071. [DOI] [PubMed] [Google Scholar]
- Vankó G.; Neisius T.; Molnár G.; Renz F.; Kárpáti S.; Shukla A.; de Groot F. F. Molecular spin transitions studied with X-ray emission spectroscopy. J. Phys. Chem. B 2006, 110, 11647–11653. [DOI] [PubMed] [Google Scholar]
- Creutz C.; Chou M.; Netzel T. L.; Okumura M.; Sutin N. Lifetime, spectra and quenching of the excited states of polypiridine complexes of iron(II), ruthenium(II), and osmium(II). J. Am. Chem. Soc. 1980, 102, 1309–1319. [Google Scholar]
- Baker A. T.; Goodwin H. A. Crystal Structure of Bis(2,2′:6′,2″-terpyridine)iron(II) Bis(perchlorate) Hydrate. Aust. J. Chem. 1985, 38, 207–214. [Google Scholar]
- Bridgeman A. J.; Cavigliasso G.; Ireland L. R.; Rothery J. The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc., Dalton Trans. 2001, 2095–2108. [Google Scholar]
- Ravel B.; Newville M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [DOI] [PubMed] [Google Scholar]
- Haldrup K.; Christensen M.; Nielsen M. M. Analysis of time-resolved X-ray scattering data from solution-state systems. Acta Crystallogr., Sect. A 2010, 66, 261–260. [DOI] [PubMed] [Google Scholar]
- Cammarata M.; Lorenc M.; Kim T.; Lee J. H.; Kong Q. Y.; Pontecorvo E.; Lo Russo M.; Schiro G.; Cupane A.; Wulff M. Impulsive solvent heating probed by picosecond x-ray diffraction. J. Chem. Phys. 2006, 124, 124504. [DOI] [PubMed] [Google Scholar]
- Kjaer K. S.; van Driel T. B.; Kehres J.; Haldrup K.; Khakhulin D.; Bechgaard K.; Cammarata M.; Wulff M.; Sorensen T. J.; Nielsen M. M. Introducing a standard method for experimental determination of the solvent response in laser pump, X-ray probe time-resolved wide-angle X-ray scattering experiments on systems in solution. Phys. Chem. Chem. Phys. 2013, 15, 15003–15016. [DOI] [PubMed] [Google Scholar]
- Joly Y. X-ray absorption near-edge structure calculations beyond the muffin-tin approximation. Phys. Rev. B 2001, 63, 125120. [Google Scholar]
- Juhin A.; de Groot F.; Vankó G.; Calandra M.; Brouder C. Angular dependence of core hole screening in LiCoO2: A DFT + U calculation of the oxygen and cobalt K -edge x-ray absorption spectra. Phys. Rev. B 2010, 81, 115115. [Google Scholar]
- Rehr J. J.; Kas J. J.; Vila F. D.; Prange M. P.; Jorissen K. Parameter-free calculations of X-ray spectra with FEFF9. Phys. Chem. Chem. Phys. 2010, 12, 5503–5513. [DOI] [PubMed] [Google Scholar]
- Westre T. E.; Kennepohl P.; DeWitt J. G.; Hedman B.; Hodgson K. O.; Solomon E. I. A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes. J. Am. Chem. Soc. 1997, 119, 6297–6314. [Google Scholar]
- Brouder C. Angular dependence of X-ray absorption spectra. J. Phys.: Condens. Matter 1990, 2, 701–738. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.