

Abstract
Tumor cells have a tendency to use glucose fermentation to obtain energy instead of mitochondrial oxidative phosphorylation (OXPHOS). We demonstrated that this phenotype correlated with loss of ERK5 expression and with reduced MHC class I expression. Consequently, tumor cells could evade cytotoxic T lymphocyte (CTL)-mediated immune surveillance, but also increase their sensitivity to natural killer (NK) cells. These outcomes were evaluated using two cellular models: leukemic EL4 cells and L929 transformed fibroblasts and their derived ρ° cell lines, which lack mitochondrial DNA. We have also used a L929 cell sub-line that spontaneously lost matrix attachment (L929dt), reminiscent of metastasis generation, that also downregulated MHC-I and ERK5 expression. MHC-I expression is lower in ρ° cells than in the parental cell lines, but they were equally sensitive to CTL. On the contrary, ρ° cells were more sensitive to activated NK cells than parental cells. On the other hand, L929dt cells were resistant to CTL and NK cells, showed reduced viability when forced to perform OXPHOS, and surviving cells increased MHC-I expression and became sensitive to CTL. The present results suggest that when the reduction in MHC-I levels in tumor cells due to glycolytic metabolism is partial, the increase in sensitivity to NK cells seems to predominate. However, when tumor cells completely lose MHC-I expression, the combination of treatments that increase OXPHOS with CTL-mediated immunotherapy could be a promising therapeutic approach.
Keywords: cancer immunotherapy, cytotoxic T lymphocytes, dichloroacetate, glucose metabolism, NK cells, ρ° cells
Abbreviations: CTG, cell tracker green; CTL, cytotoxic T lymphocyte; DCA, dicholoroacetate; DNA, deoxy-ribonucleic acid; ERK5, extracellular regulated kinase 5; FCS, fetal calf serum; mAb, monoclonal antibody; MACS, magnetic cell separation; MHC-I, major histocompatibility complex class I; mRNA, messenger ribonucleic acid; NK, natural killer; OXPHOS, oxidative phoshorylation; PBS, phosphate buffered saline; Poly I:C, polyinosinic: cytidilic acid; RNA, ribonucleic acid; sh, small hairpin
Introduction
During tumorigenesis, tumor cells need to obtain nutrients for their rapid growth. Early studies by Otto Warburg 1 found that, even in the presence of oxygen, most cancer cells metabolize glucose by fermentation even though it generates ATP less efficiently than the aerobic processes of OXPHOS, which takes place in mitochondria. Our previous data demonstrated that this tendency toward fermentation was related to a decrease on MHC class I expression through inactivation of the ERK5 pathway.2 Cancer cells could use this mechanism to evade immune-surveillance. In fact, the “cancer immune-editing” model proposes that the appearance of clinically detectable tumors may be the result of the proliferation of cells that have been selected to escape the immune response.3,4 We have recently shown that leukemic cells also suffer these immune-surveillance and immune-selection processes.5,6 One of the most relevant mechanisms for immune escape could be the aberrant, including total or partial loss, of MHC-I expression.4,7,8 In addition to mediate self-recognition, MHC-I present endogenously synthesized tumor antigens to CD8+ CTLs. Changes in MHC-I expression could allow tumor cells to avoid CTLs and thereby an adaptive immune response.4,7,8 Hence, an interesting possiblity, although not demonstrated experimentally, would be to treat cancer cells with drugs, such as the pyruvate dehydrogenase kinase 1 inhibitor dichloroacetate (DCA), that, on one hand, would reduce tumor growth rate,9,10 and on the other, would increase MHC-I expression allowing their sensitization to CTL.2 In this context, it has been recently demonstrated that metformin, an antidiabetic drug that induces OXPHOS in cancer cells, increases MHC-I expression in breast carcinoma tumor cell lines.11
Another lymphocyte lineage, the NK cell, has also a critical role in antitumor immunity.12-14 In particular, NK cells target tumor cells that have evaded the control of CTL by down-regulating MHC-I expression, since several of their inhibitory receptors recognize self MHC-I as ligands.15,16 Nevertheless, NK cell activating receptors recognize ligands on distressed cells, such as tumor of virus-infected cells, and the final outcome depends on the balance between activating and inhibitory signals.14,17 The abundance of NK cells in the peripheral blood and spleen may be of particular benefit in blood-borne cancers, such as leukemias/lymphomas. In fact, NK cell alloreactivity in hematological cancers improves prognosis.18-20
Murine leukemic cells with reduced ERK5 expression show low levels of MHC-I at the cell surface. This is associated with ERK5 transcriptional regulation of both chains of MHC-I, the α heavy chain and the β2-microglobulin.21 Tumor cells expressing low ERK5 levels, which are metabolically deficient to perform OXPHOS,2 activate NK cells and induced an immune response against parental tumor cells when injected in syngenic mice.21 Hence, the metabolic remodeling described in tumor cells can also sensitize them to NK cell-mediated tumor immune surveillance.
In this study we have evaluated the effect of tumor cell metabolims on tumor immune escape using two cellular models: the murine leukemia EL4 and the L929 transformed fibroblasts and their derived ρ° cell lines. ρ° cells should be considered as an extreme case of the Warburg effect, since they lack mitochondrial DNA and, as a consequence, are completely dependent on glycolysis for survival.22,23 We also describe that matrix detachment is associated with a similar glycolytic phenotype in L929 cells, resulting in loss of MHC-I expression and loss of sensitivity to CTL-mediated cytotoxicity, which can be recovered by treatments that force OXPHOS metabolism.
Results
ρ° cells down-modulate MHC-I expression compared with parental cells
EL4-ρ° cells and L929-ρ° cells were previously generated in our laboratories and their ρ° status verified by different methods, such as confirmation of the absence of mtDNA, impairment in respiration, and resistance to menadione.23,24 As previously described,2 ρ° cells, which should be considered an extreme case of the “Warburg effect," showed lower levels of MHC-I expression (Fig. 1A). EL4-ρ° did not lose completely MHC-I expression, but showed a 45% of reduction in mean fluorescence intensity (MFI). We have also used another clone of EL4 cells, called EG7, that was obtained by transfecting the ovoalbumin (OVA) gene into EL4 cells.25 EG7 cells expressed similar MHC-I levels than EL4 cells and higher than EL4-ρ° cells (Fig. 1B). Finally, L929 cells expressed similar MHC-I levels than EL4 cells and L929-ρ° showed a reduction of around 50% in MFI (Fig. 1C).
Figure 1.
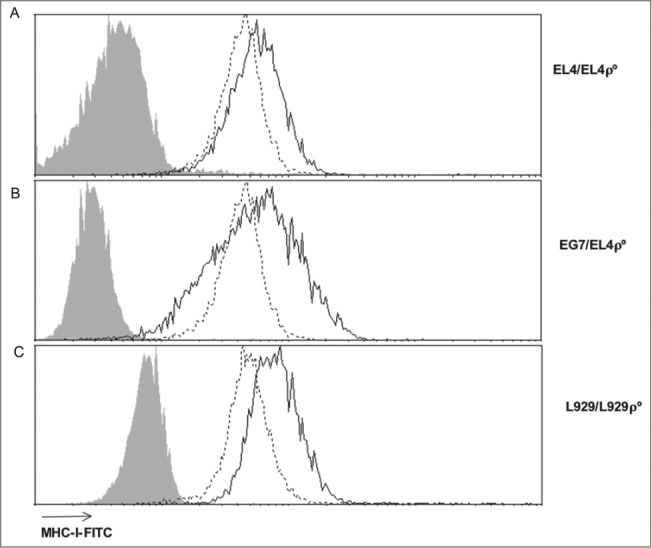
ρ° cells show low MHC-I expression. Surface MHC-I expression was analyzed by flow cytometry using FITC-labeled anti-H-2Kb (EL4, EG7 and EL4-ρ° cells) or anti-H-2Kk (L929 and L929-ρ° cells) or isotype control antibodies (gray-filled). The continuous black line represents parental cells and the discontinuous black line corresponds to ρ° cells in each case.
Lower MHC-I expression in EL4-ρ° cells did not result in reduced sensitivity to CTL but increased sensitivity to activated syngeneic NK cells
We previosuly showed that EL4-ρ° cells were equally sensitive than parental EL4 cells to granzyme B- or to primary antiviral CTL. This work also demonstrated that the apoptotic mechanisms used by cytotoxic cells to kill these tumors were not affected by the lack of OXPHOS in EL4-ρ° cells.23,26 We confirmed these results using the cells described in Fig. 1. Antiviral CTL were obtained from mice previously infected with the lympho-corio meningitis virus (LCMV), and tested on EL4 and EL4-ρ° cells pulsed with the immunodominant LCMV peptide gp33, as a standard and physiological method described in previous works.27,28 For those experiments, we used wild type C57BL/6 mice or granzyme A knockout mice, in which cytotoxicity on EL4 cells was only dependent on the perforin/granzyme B system. We have previously demontrated that the execution of this type of cell death was not dependent on mitochondrial ROS generation, that is impaired in EL4-ρ° cells.23 Both, EL4 and EL4-ρ° cells were killed by the antiviral CTL derived from granzyme A knockout mice (representative experiment of Fig. 2A) or from wt mice (data not shown). Phosphatidylserine (PS) exposure, determined by annexin-V-FITC labeling, and loss of plasma membrane integrity, determined by nuclear staining with 7-AAD, were not significantly different on both cell lines after incubation with CTL from gzmA−/− mice. The effector to target ratio in these experiments was 10 to 1. However, only 10% of the CD8+ T cells recovered from infected mice recognized the viral peptide bound to H-2Kb, 28 so the real effector to target ratio was lower, close to 1 to 1. Fig. 2B shows the summary of all experiments performed, indicating that no statistically significant difference in sensitivity to primary antiviral CTL could be observed between EL4 and EL4-ρ° cells, in spite of the difference in MHC-I expression. Similar results were also obtained using the long-term allogenic CTL clone BM3.3 (data not shown).
Figure 2.

EL4 and EL4ρ° cells were equally sensitive to CTLs. (A) CD8+ T cells were purified by MACS from the spleens of C57BL/6 granzyme A−/− mice after 8 d of infection with LCMV, labeled with cell tracker green (CTG) and tested against EL4 or EL4-ρ° cells for 2 h at an effector:target (E:T) ratio of 10:1 in the presence (+gp+CTL) or absence (−gp+CTL) of the LCMV peptide gp33. Then, target cells were gated as the CTG-negative population and, at time 0 (Ctr) or after the 2 h incubation with the CTL, plasma membrane intregrity was tested by nuclear 7-AAD incorporation and PS exposure by labeling with annexin-V-PE and flow cytometry. Numbers in the dot-plots show the percentage of cells in each quadrant. (B) Summary of all experiments performed. Antiviral CTL from gzmA−/− mice were incubated with EL4 or EL4-ρ° cells, as indicated in A. The cytolytic activity was evaluated by the percentages of annexin-V+ cells. Data showed the mean ± SD of at least three different experiments.
We next analyzed the sensitivity of these cell lines to activated NK cells. NK cells from syngeneic gzmA−/− C57BL/6 mice were activated by in vivo injection of poly I:C, and 16 h later, NK cells were purified from spleens and tested against EL4 or EL4-ρ° cells, in 2 h and 4 h assays. As shown in Fig. 3A, activated NK cells induced apoptosis in both cell lines. However, as shown in this representative experiment, EL4-ρ° cells were more sentitive than EL4 cells, especially at the higher E:T ratios used. Fig. 3B shows the summary of all experiments performed, indicating that these differences were statistically significant. We also tested activated NK cells against EG7 and EL4-ρ° cells, and observed again higher cytotoxicity on EL4-ρ° cells (Fig. 3C). This higher sensitivity to NK cells can be associated with the reduced MHC-I expression level, but it could be also related with a higher expression of ligands for NK cell activating receptors. We have tested this possibility for the ligands of NKG2D and NKp46 (NCR1), using NKG2D-Fc and NKp46-Fc chimeras, and, as shown in Fig. S1, the expression of the ligands for these activating receptors were similar in both EL4 and EL4-ρ° cells, indicating that the difference in sensitivity should be rather attributed to the reduced MHC-I expression level.
Figure 3.

EL4ρ° cells were more sensitive to NK cells. (A) C57BL/6 granzyme A−/− mice were injected with 100 μg of poly I:C and 16 h later, NK cells were purified by MACS, labeled with cell tracker green (CTG) and tested against EL4 and EL4-ρ° cells for 4 h at the effector:target ratios indicated. Then, target cells were gated as the CTG-negative population at time 0 (Ctr) or after 4 h incubation with NK cells, plasma membrane intregrity was tested by 7-AAD incorporation and PS exposure by annexin-V-PE. Numbers in the dot-plots show the percentage of cells in each quadrant. (B) Activated NK cells were tested for 2 (black symbols) or 4 h (white symbols) against EL4 (circles) or EL4-ρ° cells (squares) target cells at different E:T ratios used. Data showed the percentages of apoptosis in each experimental condition and are the mean ± SD of at least three different experiments. * p < 0.05. (C) Representative experiment as that shown in (A), using activated NK cells from gzmA−/− mice against EG7 or EL4-ρ° cells at an 10:1 E:T ratio.
In vivo cytotoxicity assays
We next used an in vivo cytotoxicity assay to validate our ex vivo results. For that, we labeled the two target cells, EG7 and EL4-ρ° cells, with different amounts of Cell Green Tracker (CGT), mix them at a 1:1 ratio, and injected into the mice peritoneum. We performed a peritoneal wash after 4 h and identified labeled cells.6,21 If the cell ratio changes after this short passage by the peritoneum means that one tumor cell is comparatively eliminated at a higher rate than the other, giving an idea on the relative in vivo clearance. The results of these experiments are not quantitative, since probably both types of cells are eliminated. As shown in the representative experiment of Fig. 4A, EL4-ρ° cells were labeled with a higher CGT concentration than EG7 cells, and mixed at an approximative 1:1 ratio at the beginning of the experiment (initial ratio). After 4 h in the peritoneum and without previous poly I:C injection, EL4-ρ° cells were spontaneously cleared at a higher rate than EG7 cells (Figs. 4A and B). When NK cells were previously activated by in vivo poly I:C injection, the relative clearance of EL4-ρ° cells with respect to EG7 cells was significantly increased (Fig. 4), in agreement with the ex-vivo experiments shown in Fig. 3.
Figure 4.

NK cells eliminated EL4ρ° cells in vivo. (A) 5 × 105 EG7 or EL4-ρ° cells were labeled with 0,5 μM (CTGlow) or 5 μM cell tracker green (CTGhigh), respectively, and mixed at a 1:1 ratio (Initial ratio). Labeled target cells were then injected i.p. in 200 μL RPMI 2% heat-inactivated FBS in gzmA−/− mice, untreated (- poly I:C), or injected 16 h before with 0.1 mg poly-IC in 0.1 mL PBS (+poly I:C). Mice were sacrificed 4 h later and peritoneal cells collected, washed in PBS, and analyzed on a FACSCalibur flow cytometer. (B) Relative in vivo clearance of EL4-ρ° cells with respect to EG7 cells in untreated (gray bar) or in gzmA−/− mice pre-treated with poly I:C (black bar). Results are the mean ± SD of 6 gzmA−/− mice for each experimental condition. (C) Wild type mice were injected either with 50 μL of rabbit control serum or with a polyclonal rabbit anti-asialo GM1 serum at days -2 and 0 before injecting the labeled cells. Then, the same experiment as in (A) was performed, using these mice injected 16 h before with poly I:C. The graphic in the right shows the relative in vivo clearance of EL4-ρ° cells with respect to EG7 cells in control mice (gray bar) or in mice pre-treated with the anti-asialo GM1 serum (black bar). Results are the mean ± SD of four mice for each experimental condition. (D) The labeling of NK1.1+ cells in splenocytes from mice untreated (black histogram) or treated with the anti-asialo GM1 serum (gray histogram) was shown in the top right panel. *, p < 0.05
To ensure that these in vivo observations were mediated by NK cells, we treated mice with an anti-asialoGM1 antiserum, a method that results in elimination of NK cells.21,29 As shown in Fig. 4C, while treatment with poly I:C in control mice resulted again in an increased relative clearance of EL4-ρ° cells, in those mice treated previously with the anti-asialoGM1 antiserum, this relative clearance was substantially reduced. There is still some relative clearance in the absence of NK cells, but this in agreement with the existence of a basal level of relative clearance in the absence of NK cell stimulaton (see Fig. 4B), that could be due to the action of other cellular population. The elimination of NK cells by the anti-asialoGM1 antiserum was confirmed by NK1.1 staining (Fig. 4D).
L929-ρ° cells become sensitive to cytotoxicity exerted by activated syngeneic NK cells
In the case of H-2k L929 cells, since the LCMV/gp33 system only works in an H-2b background, we needed to generate allogeneic C57BL/6 CTL against C3H splenocytes. We performed similar experiments as those reported in Fig. 2 using these allogeneic CTL against L929 and L929-ρ° cells and we did not find statistically significant differences in their sensitivity to these effector cells (representative experiment in the panels of Fig. 5A; summary of all the experiments perforemd in the graphic below). When testing their sensitivity to activated syngeneic (C3H) NK cells, we found that while L929 cells were not very sensitive to their cytotoxicity, L929-ρ° cells were very sensitive, at a similar level as the classical NK cell target YAC-1 (Fig. 5B, left panel).
Figure 5.

L929ρ° cells were more sensitive to NK cells. (A) Allogeneic CTL were generated by culturing C57BL/6 splenocytes with mitomycin-treated C3H splenocytes in the presence of murine IL-2 during 5 d. Aferwards, CD8+ T cells were purified by MACS, labeled with cell tracker green (CTG) and tested against L929 or L929-ρ° cells for 4 h at an effector:target (E:T) ratio of 10:1. Then, target cells were gated as the CTG-negative population at time 0 (controls) or after the 4 h incubation with the CTL. Plasma membrane intregrity was tested by nuclear 7-AAD incorporation and PS exposure by labeling with annexin-V-PE and flow cytometry, as indicated in the panels. Numbers in the figures showed the percentage of cells negative (left) or positive (right) for each parameter. The bottom graphic showed the summary of apoptosis inductin in all experiments perfromed. Data correspond to the annexin-V-PE target cell labeling after incubation with the CTL minus spontaneous labeling. The results represent the mean ± SD of at least four different experiments.(B) C3H mice were injected with 100 μg of poly I:C and 16 h later, NK cells were purified by MACS, labeled with cell tracker green (CTG) and tested against L929, L929-ρ° or YAC-1 cells for 4 h at a 10:1 E:T ratio. Target cells were gated as the CTG-negative population and PS exposure determined by labeling with annexin-V-PE and flow cytometry. Data correspond to the labeling for each target cell after incubation with the NK cells minus spontaneous labeling and represents the mean ± SD of three different experiments.
The increase in susceptibility to NK cells in ρ° cells could also be due to changes in the expression of ligands of killer activating receptors such as NKG2D and NKp46 (NCR1). We have tested this possibility and observed that the expression of NKG2D and NKp46 ligands are similar in L929 and L929ρ° cells (see Fig. 9B), suggesting that the changes in MHC-I expression should predominate in the functional outcome observed.
Figure 8.

DCA effects on MHC-I expression in EL4 cells and sensitivity to CTL and NK cells. (A) EL4 cells were cultured during 3 d in medium containing 25 mM glucose in the absence (black histogram) or in the presence of 15 mM DCA (pointed histogram). MHC-I expression was analyzed by flow cytometry as indicated in the legend of Fig. 1. The black histogram corresponds to the labeling by a control Ig. (B) Antiviral CD8+ T cells were tested against EL4 cells supplemented or not with DCA, as indicated in the legend of Fig. 3. The percentages of Annexin-V+ cells are shown, at the basal level (white bar), or after the 2 h incubation with the antiviral CTL from wild type mice (black bar) Results were the mean ± SD of three different experiments. (C) NK cell-mediated cytotoxicity was tested on EL4 cells supplemented or not with DCA, as indicated in the legend of Fig. 4. The percentages of Annexin-V+ cells are shown, at the basal level (white bar), or after the 4 h incubation with activated NK cells from wt mice (black bar). Results are the mean ± SD of four different experiments. (D) 5 × 105 EL4 cells cultured during 3 d in medium containing 25 mM glucose in the presence of 15 mM DCA were labeled with 0, 5 μM (CTGlow) and those cultured in the absence of DCA were labeled with 5 μM cell tracker green (CTGhigh), respectively, and mixed at a 1:1 ratio (Control. Time 0). Labeled target cells were then injected i.p. in 200 μL RPMI 2% heat-inactivated FBS in wild type mice injected 16 h before with 0.1 mg poly-IC in 0.1 mL PBS (+poly I:C). Mice were sacrificed 4 h later and peritoneal cells collected, washed in PBS, and analyzed on a FACSCalibur flow cytometer.
Characterization of L929dt cells. Resistance to CTL and reversal by DCA
Matrix detachment has been associated with cellular energy stress, and metabolic adaptations have been described for anchorage-independent cell growth, including activation of the AMPK pathway.30 These adaptations are characteristic of aggressive tumors and crucial for metastasis establishment.30 We have generated a L929 sub-line, termed L929dt for “dettached," by collecting cells that completely and spontaneously dettached from the culture dish, and seeding and passaging them in the same conditions as parental L929 cells. These cells never re-acquired the adherent phenotype. In their initial characterization, we found that L929dt cells had completely lost MHC-I expression (Fig. 6A). This was associated, as expected, with resistance to allogenic CTL-induced cell death as compared with parental L929 cells (Fig. 6B).
Figure 6.

L929dt cells lose MHC-I and ERK5 expression and are resistant to CTL and menadione. (A) MHC-I expression in L929 and L929dt cells was determined as indicated in Fig. 1. (B) L929, L929dt or L929dt cells supplemented or with 15 mM DCA were incubated with allogeneic CTL and cytotoxicity was tested as indicated in Fig. 6A. Histograms showed cell death measured by 7-AAD incorporation (left) and annexin-V+ binding (right). Results represent mean ± SD of at least two different experiments for each experimental condition. ***, p < 0.01; *, p < 0.05. (C) L929, L929dt or L929-ρ° cells were incubated during 24 h with the indicated concentrations of menadione and cell death was determined by Trypan blue staining. (D) ERK5 expression was determined by immunoblot on extracts from L929, L929dt or L929-ρ° cells, as indicated. β-actin immunobloting was used as a loading control.
The absence of a functional mitochondrial electron transport chain (ECT) in ρ° cells can be demonstrated by their resistance to the toxicity of menadione, a ketone that displaces ubiquinone from the ECT and kills cells through massive mitochondrial superoxide anion generation.23 This was demonstrated in our previous work for EL4 and EL4-ρ° cells 23 and is shown for L929-ρ° cells in Fig. 6C, which, contrary to their respective parental cells, were not sensitive to menadione toxicity. Interestingly, L929dt cells were also resistant to menadione-induced cell death, indicating the presence of a mitochondrial disfunction in these cells (Fig. 6C). Since MHC-I expression was regulated by OXPHOS-dependent ERK5 expression and activation,2,21 we also tested ERK5 expression by immunoblot in L929, L929-ρ° and L929dt cells. Correlating with their complete loss of MHC-I expression, ERK5 expression was abrogated only in L929dt cells (Fig. 6D).
DCA, an inhibitor of pyruvate dehydrogenase kinase (PDK1), forces cells to use OXPHOS to generate ATP.2,9 In EL4 cells, DCA decreased growth, while in the case of EL4-ρ° cells, DCA completely abrogated it (Fig. 7A). In addition, while DCA did not induce EL4 cell death during the 72 h of incubation, it induced death of ρ° cells, in agreement with their complete dependence on glycolysis for survival (Fig. 7B, left panel). Strikingly, L929dt cells were sensitive to DCA-induced cell death in a dose-dependent manner, while L929 were resistant, indicating that L929dt cells are highly glycolytic (Fig. 7B, right panel). An alternative treatment to force tumor cells to perform OXPHOS is depleting glucose from the culture media and supplement it with pyruvate and malate as metabolites to support energy production.2 This treatment is even more toxic for L929dt cells than DCA (Fig. 7B, right panel), showing that DCA-mediated effects were mainly dependent on a metabolic switch.
Figure 7.

Changing tumor cell metabolism sensitize them to CTL. (A) Cell growth of EL4 and EL4-ρ° cells in the presence or absence of 15 mM DCA was determined by counting the number of viable cells during 4 d of culture and the results expressed as the ratio between the number of cells at a given time (N(t)) and the initial number of cells (N(o)). (B) EL4 and EL4-ρ° cells (left graphic) or L929 and L929dt cells (right graphic) were incubated during 72 h with the indicated concentrations of DCA or in the absence of glucose and in the presence of 12.5 mM pyruvate/malate. Cell death was determined by Trypan blue exclusion. (C) L929dt cells were incubated during 72 h in the absence or presence of the indicated concentrations of DCA or with pyruvate/malate in the absence of glucose, viable cells gated by flow cytometry, and MHC-I expression determined as indicated in Fig. 1. In each panel, the black histogram represents the labeling with a control Ig and the gray histogram the labeling with the anti-MHC-I antibody.
DCA, through OXPHOS metabolism and ERK5 activation, also results in the increase in MHC-I expression,2 an approach that could be useful to increase tumor sensitivity to CTL. To confirm this possibility, we performed experiments using EL4 cells treated with DCA. DCA incubation for 72 h did result in an increase in MHC-I expression in EL4 cells (Fig. 8A), but this did not result in an increase in sensitivity to antiviral CTL (Fig. 8B). However, this increase in MHC-I expression did result in a partial reduction in sensitivity to activated syngeneic NK cells, that was statistically significant (Fig. 8C). On the other hand, DCA did not affect the expression of NKG2D or NKp46 ligands on EL4 cells ( Fig. S2). The reduced sensitivity to NK cells in DCA-treated EL4 cells was confirmed by in vivo cytotoxicity experiments (Fig. 8D). These results are completely in agreement with those obtained using ρ° cells, further confirming the connection between MHC-I expression, OXPHOS and change in sensitivity to activated NK cells.
Since L929dt cells had lost completely MHC-I expression and sensitivity to CTL, this cellular model would be adequate to test if treatments that force OXPHOS and, consequently, induces MHC-I expression, recover their sensitivity to CTL. Fig. 7B showed that both DCA and Pyr/Mal treatments were toxic to these cells. However, after gating in the suriving cells, we observed an increase in MHC-I expression that correlated with increasing concentrations of DCA, with the maximal increase obtained after Pyr/Mal supplementation (Fig. 7C).
We next investigated if allogeneic CTL increased their activity against cells using OXPHOS for ATP production. Unfortunately, only in cells supplemented with the lower DCA concentration (15 mM) the basal toxicity allowed us to continue these studies. In all other cases the basal percentage of apoptotic cells was too high to produce reproductible results after CTL incubation. Although the increase in MHC-I expression was moderate (see Fig. 7C), we still observed a partial increase in sensitivity to allogeneic CTL (Fig. 6B). These results confirmed the hypothesis that the combination of treatments that increased OXPHOS metabolism with CTL-mediated immunotherapy could be a new promising therapeutic approach.10
Finally, we tested NK cells against L929dt cells, and we found that, in spite of its loss of MHC-I expression, they were not more sensitive to NK cell cytotoxicity than parental L929 cells (Fig. 9A). We then tested the expression of ligands of the NK cell activating receptors NKG2D and NKp46 in the different cell types used. The expression of ligands for NKG2D was high, while the expression of ligands for NKp46 was almost undetectable in both L929 and L929-ρ° cells (Fig. 9B), indicating, as in the case of EL4/EL4ρ° cells, that the higher sensitivity of L929-ρ° cells to NK cells should be rather attritbuted to their lower MHC-I expression. However, the situation is different in L929dt cells, in which there is also no expression of NKp46 ligands but the expression of NKG2D ligands is substantially reduced if compared with parental L929 cells (Fig. 9C), explaining their low sensitivity to NK cells.
Figure 9.

The sensitivity of L929dt cells to NK cells is not increased despite loss of MHC-I expression.(A) C3H mice were injected with 100 μg of poly I:C and 16 h later, NK cells were purified by MACS, labeled with cell tracker green (CTG) and tested against L929, L929dt or YAC-1 cells for 4 h at a 10:1 E:T ratio. Target cells were gated as the CTG-negative population and the percentage of dead cells was determined by labeling with annexin-V-PE and 7-AAD and flow cytometry. Data correspond to the labeling for each target cell after incubation with the NK cells minus spontaneous labeling and represents the mean ± SD of three different experiments. (B, C) Expression of NKG2D and NKp46 (NCR1) ligands on the surface of L929, L929-ρ° (B) and L929dt cells (C) was analyzed by flow cytometry using NKG2D-Fc and NKp46-Fc chimeras and a secondary FITC-labeled anti-human IgG mAb. The gray-filled histograms represent the labeling with the secondary mAb alone, and, in (C), the pointed-line histogram represents the labeling for L929dt cells, while the black-line histogram represents the labeling for L929 cells.
Discussion
Tumor MHC-I levels are metabolically regulated and the tendency of tumor cells to use glycolysis over mitochondrial OXPHOS (“Warburg effect”) results in fact in reduced MHC-I levels. This could result in escape from CTL control; or, on the contrary, in increased sensitivity to NK cell-mediated immunosruveillance.4 Our present results, using an extreme case of the “Warburg effect," ρ° cells, depleted of mitochondrial DNA and unable to perform OXPHOS, indicate that the increase in susceptibility to NK cells predominates in both EL4 and L929 models. This result is further validated by the use of DCA on EL4 cells, a drug that specifically targets glucose metabolism, that, through the increase in MHC-I expression due to OXPHOS usage, decreases sensitivity to NK cells but does not affect to sensitivity to CTL. This result shows the importance of NK cells in antitumor immune surveillance, and that complete elimination of OXPHOS does not seem to confer an advantage to tumor cells in the context of immune surveillance. However, our results are based on established tumors. The relative importance of tumor metabolism in immune surveillance during the first steps of tumorigenesis is not addressed in this study. However, the essential role of metabolic shift in tumorigenesis has been uncovered by mice bearing lysine to arginine mutations at three p53 acetylation sites (p533KR). The p533KR mutant is not competent for p53-mediated cell-cycle arrest, senescence and apoptosis. Mice bearing this construct do not succumb to spontaneous thymic lymphomas, while p53 null mice do. The p533KR mice retain the ability to regulate energy metabolism and reactive oxygen species (ROS) production. These findings underscore that metabolic regulation and antioxidant function activities are critical to block spontaneous tumorigenesis.31
MHC-I reduction does not decrease sensitivity to CTL-induced cell death in ρ° cells, perhaps because few MHC-I molecules are needed to activate CTL cytotoxicity.32 The effective signaling complex in CTL is formed not only by the TCR, but also by the co-receptor CD8. Both of them bind to MHC-I expressed on the surface of the target cell. It has been calculated that virtually all syngeneic TCR interactions require CD8, which enhances the affinity for the TCR ligand by one million-fold or more.33 CD8 contributes to the stabilization of the interaction, and also to signaling, by bringing p56lck to the proximity of the engaged TCRs.34 In addition, CD8 also contributes to increase the binding of CTL to target cells, because it is able to interact with non-cognate MHC-I or with nonstimulatory peptide-MHC-I complexes, especially after the CTL has been triggered by the TCR35,36 And looking from the other side, it has been demonstrated that the number of TCRs on a T cell can be reduced to 1/20th, and responses are still normal.37
However, in the case of NK cells, the alteration of MHC-I levels due to metabolic changes seems more determinant for the final outcome, at least in the extreme case of ρ° cells. NK cell-mediated cytotoxicity depends on a fine-tuned equilibrium between signals coming from activating and inhibitory receptors.14,17,38 In most cases, MHC-I is the ligand for inhibitory receptors, mostly those belonging to the family of killer cell Ig-like recetors (KIR). However, some KIRs isoforms, especially in humans, transmit activating signals. In this context, it is not strange that a small alteration of the equilibrium between activating and inhibitory signals given by a reduction or increase in MHC-I expression results in significant changes in the final outcome. As indicated above, the increase in susceptibility to NK cells in ρ° cells could also be due to changes in the expression of ligands of killer activating receptors such as NKG2D and NKp46 (NCR1), but no significant differences exist between parental and ρ° cells, suggesting that the changes in MHC-I expression should predominate in the functional outcome observed.
Activated syngenic NK cells also target the original EL4 tumor, although its relative MHC-I expression level is high. This observation is in line with our previous study, in which injection of EL4shERK5 cells, with low levels of MHC-I expression, resulted in massive activation of NK cells, which were afterwards able to protect against the original MHC-I-expressing parental EL4 tumor.21 In vivo NK cell activation by poly I:C or by injecting tumor cells increase the expression of effector molecules, such as granzyme B, and, probably, of activating receptors. Usually only virus-infected or tumor cells express the ligands for these activating receptors, suggesting the use of activated NK cells for immunotherapy.12,18– 20,39
In any case, it should be noted that the functional outcomes analyzed in this work in the EL4/EL4-ρ° and L929/L929-ρ° systems are due to a partial down-modulation of MHC-I, that still allows recognition by CTL.
Only L929dt cells completely lose MHC-I expression. Strikingly enough, matrix detachment in this subline is also associated with mitochondrial disfunction, loss of ERK5 expression and extreme glycolytic phenotype. This process is reminiscent of metastasis generation,30 and more studies will be needed to characterize the molecular relationships between matrix detachment and glycolytic phenotype, for which L929dt cells will be extremely useful. In fact, the complete loss of MHC-I expression has been described in many human cancer metastasis, called “hard lessions," and, in those cases, tumor escape is mainly due to resistance to CTL.7,8 In this case, re-establishment of MHC-I levels on the tumor surface by DCA, pyruvate/malate or by metformin treatment 2,10,11 would be beneficial. Here we show that treatments that force OXPHOS result especially toxic for cells in which MHC-I expression is lost by metabolic means. However, surviving cells increased indeed MHC-I expression and re-established sensitivity to CTL cytotoxicity. This could be relevant in the clinic where a low percentage of tumor cells that escape chemo- and/or immunotherapy could be responsible of patient's relapse many months later. It could be reasoned that if tumor cells loss MHC-I, they will become more sensitive to NK cells. However, since L929dt cells reduced also the expression of ligands for at least the activating receptor NKG2D, they do not result especially sensitive to NK cells. This observation suggest that aggressive metastasis could become also resistant to NK cells by losing the expression of ligands for those activating receptors, and this has been reported in some clinical studies.40 In addition, it has been reported that in vivo, NK cells could lack migration inside the tumor mass, especially in solid tumors already established and metastasized.41
In fact, NK cells, as members of the innate immune system, are the first line of defense against tumor establishment.42 Data from this work indicate that at this point, small changes in MHC-I expression will be important in determine tumor sensitivity to NK cell-mediated cytotoxicity. The adaptive antitumor immune response, mediated by CTL but also by CD4+ T cells, is slower, and will only take place efficiently in the presence of tumor danger signals that activate antigen-presenting cells to present tumor antigens.43,44 The tumor immunoediting model proposes that if an efficient adaptive antitumor response occurs, then there is a phase of equilibrium between the tumor and immune antitumor cells.42 It was demonstrated that this phase of equilibrium exists, and that it is mediated by CTL and CD4+ T cells, but not by NK cells.45 Hence, it seems that the lower MHC-I expression, related in tumors to their glycolytic phenotype, would allow tumors to avoid CTL control when the tumor is already established and arriving to the equilibrium stage and that the therapeutic interventions described in this work to increase MHC-I expression will be beneficial especially in this time window.
Recently, immunotherapy based in antitumor CTL stimulation was approved for prostate cancer (Sipuleucel-T) 46 and antibody-based blocking of CTL de-activation through CTLA4 or through the PD-1/PD-1L system, have given very promising results in clinical studies.47-49 Drugs that induce an increase in MHC-I expression, i.e. DCA or metformin, could be combined with these treatments or with an adoptive cell transfer by selecting T lymphocytes with antitumor activity.10 Hence, this therapy will combine metabolic drugs and immunotherapy, leading to a new approach to treat patients in relapse to standard treatments.
Materials and Methods
Cells and mice
Inbred C57BL/6 (B6) and a mouse strains deficient for gzmA (gzmA−/−) bred on the B6 background,28 were maintained at the CITA (Agrifood Research and Technology Centre of Aragon). Mice from the C3H strain (CRIFFA, Barcelona) were maintained at the Unidad Mixta de Investigación from Universidad de Zaragoza. All experimental work involving mice was performed according to FELASA guidelines under the supervision and approval of “Comite Etico para la Experimentacion Animal” (Ethics Committee for Animal Experimentation) from CITA or Universidad de Zaragoza.
The mouse tumor lines EL4, EL4ρ°, EG7, L929 and L929ρ° were used as target cells. EL4 and EG7 cells were cultured in in RPMI-1640 medium supplemented with 5% or 10% heat-inactivated FBS. The EL4ρ° cell line was cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, 4, 5 mg/mL glucose, 100 μg/mL sodium pyruvate and 50 μg/mL uridine. The L929 cell line was cultured in DMEM medium supplemented with 10% heat-inactivated FBS. The L929ρ° cell line was cultured in DMEM supplemented with 10% heat-inactivated FBS, 4, 5 mg/mL glucose, 100 μg/mL sodic piruvate and 50 μg/mL uridine. The absence of mtDNA in the ρ° cell lines was confirmed previously by qPCR.23,24
Determination of MHC-I and of NKG2D and NKp46 ligands surface expression by flow cytometry
2 × 105 cells were stained for 15 min at 4°C with anti H-2Kb-FITC, anti-H2Kk or isotype control (rabbit IgG-FITC) in 100 μL PBS 5% FBS. Both antibodies were obtained from BD Pharmigen. The expression of NKG2D ligands was performed using a murine NKG2D-human Fc chimera (R&D systems, Minneapolis, USA ) and a secondary FITC-labeled anti-human IgG. The expression of NKp46 (NCR1) ligands was performed using a human NKp46-human Fc chimera, kindly provided by Dr. Miguel López-Botet (IMIM, Barcelona) and a secondary FITC-labeled anti-human IgG. The cross-reactivity of human NKp46 with murine ligands was described previously.50 Cells were washed and analyzed on by FACSCalibur flow cytometer (BD Biosciences) using CellQuestPro software (BD Biosciences).
Ex vivo cytotoxicity assays using CTL or activated NK cells
Antiviral CTL were generated by infection with 105 pfu LCMV-WE i.p. according to established protocols.27,28 On day eight after infection, spleen CTL were isolated by positive selection using anti-CD8α magnetic beads (Miltenyi Biotec). Purified CTL cells were labeled with 1 μM Cell Tracker Green (CTG) (Invitogen) according to the manufacturer´s instructions, and target cells were pre-treated with the LCMV-immunodominant peptide gp33. Once isolated and labeled, CTL and target cells were incubated in RPMI 5% heat-inactivated FBS in a 96-wells plate at the corresponding E:T ratios in a final volume of 200 μL. After incubating for the specific time in each experiment at 37°C, different apoptotic parameters were tested in the target population (CTG negative cells) by fluorescence activated cell sorting with a FACSCalibur (BD) and CellQuest software. PS exposure was analyzed using annexin V-DY634 and DNA damage was measured using 7AAD, both from Immunostep.
For the generation of allogenic CTL, spleen cells from C3H mice (stimulatory cells) were treated with 25 μg/mL mitomycin C for 25 min at 37°C. After three washing steps with RPMI medium supplemented with 5% heat-inactivated FBS, cell were mixed with spleen cells from C57BL/6 (effector cells) in 40 mL of MEM supplemented with 10% heat-inactivated FBS, β-mercaptoethanol and 10% X63Ag8 cell line supernatant containig murine IL-2, and incubated at 37°C for 4 d After that, CD8+ cells were isolated and labeled, incubated with target cells and cell death analyzed as indicated above.
For the generation of activated NK cells, mice were injected i.p. with 0.1 mg poly-IC (Sigma) in 0.1 mL PBS 16 h before the assays. Then, spleen NK cells were isolated by positive selection using anti-DX5 magnetic beads (Miltenyi Biotec). Purified NK cells were labeled with 1 μM CTG (Invitogen) according to the manufacturer´s instructions. Once isolated and labeled, NK cells and target cells were incubated in RPMI 5% heat-inactivated FBS in a V-bottom 96-wells plate at the corresponding E:T ratios in a final volume of 200 μL. After incubating for the specific time in each experiment at 37°C, different apoptotic parameters were tested in the target population (CTG negative cells) by fluorescence activated cell sorting with a FACSCalibur (BD) and CellQuest software. PS exposure was analyzed using annexin V-PE and DNA damage was measured using 7AAD, both from Immunostep.
In vivo cytotoxicity assays
For in vivo cytotoxicity assays, 5 × 105 cells were labeled with 0,5 μM CTG (CTGlow) or 5 μM CTG (CTGhigh). Labeled target cells were injected i.p. in 200 μL RPMI 2% heat-inactivated FBS in untreated mice or in mice previously injected (−16 h) i.p. with 0.1 mg poly-IC in 0.1 mL PBS. Mice were sacrificed 4 h later and peritoneal cells were collected, washed in PBS, and analyzed on a FACSCalibur flow cytometer (BD Biosciences). In some experiments mice were injected with 50 μL of polyclonal rabbit anti-asialo GM1 serum (Wako Chemicals) or the same amount of rabbit serum control at days -2 and 0 before injecting labeled cells. Peritoneal exudates cells were obtained 4 h later and CTG+ cells were analyzed by flow cytometry.
Supplementation with DCA or pyruvate/malate
In certain experiments, cells were incubated in RPMI 1640 that contains 2 mM glutamine and 25 mM glucose, with or without DCA (Sigma) at diferente concentrations, between 15 and 25 mM. In other experiments, cells were cultured in glucose-free RPMI-1640 medium (PAN Biotech) supplemented with 4 mM glutamine plus 12.5 mM each of sodium pyruvate and malate. Cells were cultured in these mediums for 3 d, cell growth and cell death was controlled by counting of Trypan blue negative and positive cells, and after that, MHC-I surface expression and ex vivo and in vivo cytotoxicity assays were carried out as indicated above.
ERK5 immunoblotting
106 cells were washed in PBS and lysed in a buffer containing 1% Triton X-100 during 15 min on ice. Soluble protein fractions were recovered by centrifugation, separated by SDS polyacrylamide gel electrophoresis (14%) and transferred onto PVDF membranes. ERK5 expression was analized by using a rabbit anti-ERK5 antibody (Cell Signaling, Beverly, USA), incubated overnight at 4°C and 1/500 dilution. Then, after a washing step, blots were incubated during 1 h at room temperature with secondary anti-rabbit antibody conjugated with HRP and revealed by chemilumiscence (Amerhsam ECL kit).
Stastitical analysis
Results are shown as average ± SD and statistical significance was evaluated using the Student t test. Differences were not considered as significant if p values were ≥ 0.05.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. Miguel López-Botet, Pompeu Fabra University/IMIM, Barcelona, for the kind gift of the human NKp46 chimera.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the collaborative projects CliNK SOE2/P1/E341 from Sudoe/Interreg IV B (EU) and Grant CTPP5/12 from “Communauté de Travail des Pyrénées” (CTP); by grants SAF2010–15341 to AA, SAF2011-2539 to JP, and SAF2009-08007 to JAE from Ministerio de Economía y Competitividad (MEC, Spain); by grant EU-ITN (MEET-317433) to JAE; and by the program “Chercheur d’avenir” from the Region Languedoc-Rousillon, the Association pour la Recherche contre le Cancer and the Fondation pour la Recherche Medicale to MV and by Gobierno de Aragón/Fondo Social Europeo. The CNIC is supported by the Ministerio de Economía y Competitividad and the Pro-CNIC Foundation. EC was supported by an FPI fellowship from MEC associated to SAF2010-15341 grant.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Warburg O. On respiratory impairment in cancer cells. Science 1956; 124:269y-70; PMID: [PubMed] [Google Scholar]
- 2. Charni S, de Bettignies G, Rathore MG, Aguiló JI, van den Elsen PJ, Haouzi D, Hipskind RA, Enriquez JA, Sanchez-Beato M, Pardo J, et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. J Immunol 2010; 185:3498-503; PMID:; http://dx.doi.org/ 10.4049/jimmunol.1001250 [DOI] [PubMed] [Google Scholar]
- 3. Dunn C, Wiltshire C, MacLaren A, Gillespie DAF. Molecular mechanism and biological function of JNK signaling via the c-Jun transcription factor. Cell Signal 2002; 14:585-93; PMID:; http://dx.doi.org/ 10.1016/S0898-6568(01)00275-3 [DOI] [PubMed] [Google Scholar]
- 4. Villalba M, Rathore MG, López-Royuela N, Krzywinska E, Garaude J, Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol 2013; 45:106-13; PMID:; http://dx.doi.org/ 10.1016/j.biocel.2012.04.024 [DOI] [PubMed] [Google Scholar]
- 5. Garaude J, Kaminski S, Charni S, Aguiló JI, Jacquet C, Plays M, Rodriguez F, Hernández J, Hipskind RA, Anel A, et al. Impaired anti-leukemic immune response in PKCq-deficient mice. Mol Immunol 2008; 45:3463-9; PMID:; http://dx.doi.org/ 10.1016/j.molimm.2008.03.016 [DOI] [PubMed] [Google Scholar]
- 6. Aguiló JI, Garaude J, Pardo J, Villalba M, Anel A. Protein kinase C-q is required for NK cell activation and in vivo control of tumor progression. J Immunol 2009; 182:1972-81; PMID:; http://dx.doi.org/ 10.4049/jimmunol.0801820 [DOI] [PubMed] [Google Scholar]
- 7. Aptsiauri N, Cabrera A, García-Lora A, López-Nevot MA, Ruiz-Cabello F, Garrido F. MHC class I antigens and immune surveillance in transformed cells. Int Rev Cytol 2007; 256:139-89; PMID:; http://dx.doi.org/ 10.1016/S0074-7696(07)56005-5 [DOI] [PubMed] [Google Scholar]
- 8. Garrido F, Algarra I, García-Lora AM. The escape of cancer from T lymphocytes: immunoselection of MHC class I loss variants harboring structural-irreversible “hard” lesions. Cancer Immunol Immunother 2010; 59:1601-6; PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010; 12:31ra4; PMID: [DOI] [PubMed] [Google Scholar]
- 10. Villalba M, López-Royuela N, Krzywinska E, Rathore M, Hipskind R, Haouas H, Allende-Vega N. Chemical metabolic inhibitors for the treatment of blood-borne cancers. Anticancer Agents Med Chem 2014; 14:223-32; PMID:; http://dx.doi.org/ 10.2174/18715206113136660374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oliveras-Ferraros C, Cufí S, Vázquez-Martín A, Menendez OJ, Bosch-Barrerra J, Martín-Castillo B, Joven J, Menendez JA. Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle 2012; 11:865-70; PMID:; http://dx.doi.org/ 10.4161/cc.11.5.19252 [DOI] [PubMed] [Google Scholar]
- 12. Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell–directed therapies: moving from unexpected results to successful strategies. Nat Immunol 2008; 9:486-94; PMID:; http://dx.doi.org/ 10.1038/ni1580 [DOI] [PubMed] [Google Scholar]
- 13. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol 2008; 9:503-10; PMID:; http://dx.doi.org/ 10.1038/ni1582 [DOI] [PubMed] [Google Scholar]
- 14. Anel A, Aguiló JI, Catalán E, Garaude J, Rathore MG, Pardo J, Villalba M. Protein kinase C-q (PKC-q) in natural killer cell function and anti-tumor immunity. Front Immunol 2012; 3:187; PMID:; http://dx.doi.org/ 10.3389/fimmu.2012.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van den Broek MF, Kägi D, Zinkernagel RM, Hengartner H. Perforin dependence of natural killer cell-mediated tumor control in vivo. Eur J Immunol 1995; 25:3514-6; PMID:; http://dx.doi.org/ 10.1002/eji.1830251246 [DOI] [PubMed] [Google Scholar]
- 16. Pardo J, Balkow S, Anel A, Simon MM. Granzymes are critically involved in NK-mediated control of RMA-S tumor growth in vivo. Eur J Immunol 2002; 32:2881-6; PMID:; http://dx.doi.org/ 10.1002/1521-4141(2002010)32:10%3c2881::AID-IMMU2881%3e3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- 17. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008; 9:495-502; PMID:; http://dx.doi.org/ 10.1038/ni1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, Stern M, Pende D, Perruccio K, Burchielli E, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood 2007; 110:433-40; PMID:; http://dx.doi.org/ 10.1182/blood-2006-07-038687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stern M, Ruggeri L, Mancusi A, Bernardo ME, de Angelis C, Bucher C, Locatelli F, Aversa F, Velardi A. Survival after T cell-depleted haploidentical stem cell transplantation is improved using the mother as donor. Blood 2008; 112:2990-5; PMID:; http://dx.doi.org/ 10.1182/blood-2008-01-135285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Willemze R, Rodrigues CA, Labopin M, Sanz G, Michel G, Socie G, Rio B, Sirvent A, Renaud M, Madero L, et al. KIR-ligand incompatibility in the graft-versus-host direction improves outcomes after umbilical cord blood transplantation for acute leukemia. Leukemia 2009; 23:492-500; PMID:; http://dx.doi.org/ 10.1038/leu.2008.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Charni S, Aguiló JI, Garaude J, de Bettignies G, Jacquet C, Hipskind RA, Singer D, Anel A, Villalba M. ERK5 knockdown generates mouse leukemia cells with low MHC class I levels that activate NK cells and block tumorigenesis. J Immunol 2009; 182:3398-405; PMID:; http://dx.doi.org/ 10.4049/jimmunol.0803006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gamen S, Anel A, Montoya J, Marzo I, Piñeiro A, Naval J. mtDNA depleted U937 cells are sensitive to TNF- and Fas-induced cytotoxicity. FEBS Lett 1995; 376:15-8; PMID:; http://dx.doi.org/ 10.1016/0014-5793(95)01236-1 [DOI] [PubMed] [Google Scholar]
- 23. Aguiló JI, Anel A, Catalán E, Sebastián A, Acín-Pérez R, Naval J, Wallich R, Simon MM, Pardo J. Granzyme B of cytotoxic T cells induces extramitochondrial reactive oxygen species production via caspase-dependent NADPH oxidase activation. Immunol Cell Biol 2010; 88:545-54; PMID:; http://dx.doi.org/ 10.1038/icb.2010.5 [DOI] [PubMed] [Google Scholar]
- 24. Acín-Pérez R, Bayona-Bafaluy M, Bueno M, Machicado C, Fernández-Silva P, Pérez-Martos A, Montoya J, López-Pérez M, Sancho J, Enríquez J. An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum Mol Genet 2003; 12:329-39; PMID:; http://dx.doi.org/ 10.1093/hmg/ddg021 [DOI] [PubMed] [Google Scholar]
- 25. Brossart P, Goldrath AW, Butz EA, Martin S, Bevan MJ. Virus-mediated delivery of antigenic epitopes into dendritic cells as a means to induce CTL. J Immunol 1997; 158:3270-6; PMID: [PubMed] [Google Scholar]
- 26. Christensen ME, Waterhouse NJ. Mechanisms of CTL cytotoxicity. A reactive response to granzyme B. Immunol Cell Biol 2010; 88:500-1; PMID:; http://dx.doi.org/ 10.1038/icb.2010.23 [DOI] [PubMed] [Google Scholar]
- 27. Pardo J, Balkow S, Anel A, Simon MM. The differential contribution of granzyme A and granzyme B in CTL-mediated apoptosis is determined by the quality of target cells. Eur J Immunol 2002; 32:1980-5; PMID:; http://dx.doi.org/ 10.1002/1521-4141(200207)32:7%3c1980::AID-IMMU1980%3e3.0.CO;2-Z [DOI] [PubMed] [Google Scholar]
- 28. Pardo J, Bosque A, Brehm R, Wallich R, Naval J, Müllbacher A, Anel A, Simon MM. Apoptotic pathways are selectively activated by granzyme A and/or granzyme B in CTL-mediated target cell lysis. J Cell Biol 2004; 167:457-68; PMID:; http://dx.doi.org/ 10.1083/jcb.200406115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kasai M, Yoneda T, Habu S, Maruyama Y, Okumura K, Tokunaga T. In vivo effect of anti-asialo GM1 antibody on natural killer activity. Nature 1981; 291:334-5; PMID:; http://dx.doi.org/ 10.1038/291334a0 [DOI] [PubMed] [Google Scholar]
- 30. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumor cell survival during enery stress. Nature 2012; 485:661-5; PMID:; http://dx.doi.org/ 10.1038/nature11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149:1269-83; PMID:; http://dx.doi.org/ 10.1016/j.cell.2012.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, Peixoto A, Flynn M, Senman B, Junt T, et al. In vivo imaging of T cell priming. Sci Signal 2008; 1:pt2; PMID:; http://dx.doi.org/ 10.1126/stke.112pt2 [DOI] [PubMed] [Google Scholar]
- 33. Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity 2003; 18:255-64; PMID:; http://dx.doi.org/ 10.1016/S1074-7613(03)00019-0 [DOI] [PubMed] [Google Scholar]
- 34. Veillette A, Bookman MA, Horak EM, Bolen JB. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell 1988; 55:301-11; PMID:; http://dx.doi.org/ 10.1016/0092-8674(88)90053-0 [DOI] [PubMed] [Google Scholar]
- 35. Yachi PP, Ampudia J, Gascoigne NR, Zal T. Nonstimulatory peptides contribute to antigeninduced CD8–T cell receptor interaction at the immunological synapse. Nat Immunol 2005; 6:785-92; PMID:; http://dx.doi.org/ 10.1038/ni1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Rourke AM, Rogers J, Mescher MF. Activated CD8 binding to class I protein mediated by the T-cell receptor results in signalling. Nature 1990; 346:187-9; PMID:; http://dx.doi.org/ 10.1038/346187a0 [DOI] [PubMed] [Google Scholar]
- 37. Labrecque N, Whitfield LS, Obst R, Waltzinger C, Benoist C, Mathis D. How much TCR does a T cell need? Immunity 2001; 15:71-82; PMID:; http://dx.doi.org/ 10.1016/S1074-7613(01)00170-4 [DOI] [PubMed] [Google Scholar]
- 38. López-Botet M, Bellón T, Llano M, Navarro F, García P, de Miguel M. Paired inhibitory and triggering NK cell receptors for HLA class I molecules. Hum Immunol 2000; 61:7-17; PMID:; http://dx.doi.org/ 10.1016/S0198-8859(99)00161-5 [DOI] [PubMed] [Google Scholar]
- 39. Cho D, Campana D. Expansion and activation of natural killer cells for cancer immunotherapy. Korean J Lab Med 2009; 29:89-96; PMID:; http://dx.doi.org/ 10.3343/kjlm.2009.29.2.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGilvray R, Eagle R, Watson N, Al-Attar A, Ball G, Jafferji I, Trowsdale J, Durrant L. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res 2009; 15:6993-7002; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mamessier E, Pradel LC, Thibult ML, Drevet C, Zouine A, Jacquemier J, Houvenaeghel G, Bertucci F, Birnbaum D, Olive D. Peripheral blood NK cells from breast cancer patients are tumor-induced composite subsets. J Immunol 2013; 190:2424-36; PMID:; http://dx.doi.org/ 10.4049/jimmunol.1200140 [DOI] [PubMed] [Google Scholar]
- 42. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002; 3:991-8; PMID:; http://dx.doi.org/ 10.1038/ni1102-991 [DOI] [PubMed] [Google Scholar]
- 43. Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, Hemmi S, Hengartner H, Zinkernagel RM. Roles of tumor localization, second signals and cross priming in cytotoxic T-cell induction. Nature 2001; 411:1058-64; PMID:; http://dx.doi.org/ 10.1038/35082583 [DOI] [PubMed] [Google Scholar]
- 44. Pardoll D. T cells and tumors. Nature 2001; 411:1010-2; PMID: [DOI] [PubMed] [Google Scholar]
- 45. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007; 450:903-7; PMID:; http://dx.doi.org/ 10.1038/nature06309 [DOI] [PubMed] [Google Scholar]
- 46. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Eng J Med 2010; 363:411-22; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1001294 [DOI] [PubMed] [Google Scholar]
- 47. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Eng J Med 2010; 363:711-23; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ribas A. Tumor immunotherapy directed at PD-1. New Eng J Med 2012; 366:2517-9; PMID: [DOI] [PubMed] [Google Scholar]
- 49. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of Anti–PD-1 antibody in cancer. New Eng J Med 2012; 366:2443-54; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Halfteck GG, Elboim M, Gur C, Achdout H, Ghadially H, Mandelboim O. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J Immunol 2009; 182:2221-30; PMID:; http://dx.doi.org/ 10.4049/jimmunol.0801878 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.