

Abstract
Macrophages have been at the heart of immune research for over a century and are an integral component of innate immunity. Macrophages are often viewed as terminally differentiated monocytic phagocytes. They infiltrate tissues during inflammation, and form polarized populations that perform pro-inflammatory or anti-inflammatory functions. Tissue-resident macrophages were regarded as differentiated monocytes, which seed the tissues to perform immune sentinel and homeostatic functions. However, tissue-resident macrophages are not a homogeneous population, but are in fact a grouping of cells with similar functions and phenotypes. In the last decade, it has been revealed that many of these cells are not terminally differentiated and, in most cases, are not derived from haematopoiesis in the adult. Recent research has highlighted that tissue-resident macrophages cannot be grouped into simple polarized categories, especially in vivo, when they are exposed to complex signalling events. It has now been demonstrated that the tissue environment itself is a major controller of macrophage phenotype, and can influence the expression of many genes regardless of origin. This is consistent with the concept that cells within different tissues have diverse responses in inflammation. There is still a mountain to climb in the field, as it evolves to encompass not only tissue-resident macrophage diversity, but also categorization of specific tissue environments and the plasticity of macrophages themselves. This knowledge provides a new perspective on therapeutic strategies, as macrophage subsets can potentially be manipulated to control the inflammatory environment in a tissue-specific manner.
Keywords: environmental programming, Gata6, tissue-resident macrophages
Introduction
Élie Metchnikoff first described macrophages in 1893 in his observations of phagocytosis during tissue inflammation.1 Metchnikoff observed microphages (neutrophils) and larger ‘macrophages’ consuming pathogens during inflammation, and developed the well-known theory of immunity by phagocytosis.2 Thereafter, the macrophage was labelled as a tissue-residing cell that could eat and kill infectious pathogens, thereby contributing to frontline immunity. Macrophages were eventually incorporated into the reticulo-endothelial system in 1924, implying that they originate from, and reside and renew within, that tissue.3 In 1968, Ralf van Furth, Zanvil Cohn and colleagues formulated the mononuclear phagocyte system: the origin of all macrophages is terminal differentiation from blood monocytes.4 There was significant evidence to the contrary at the time; however, due to the technological constraints of experiments, the mononuclear phagocyte system became the prevailing model for macrophage origin. As a result, it was generally assumed that all macrophages had the same monocytic/myeloid progenitor. Therefore, a large proportion of the subsequent research was steered toward the functional characteristics of monocyte-derived or bone-marrow-derived macrophages. This included the discovery of classically5 and alternatively activated6 macrophage phenotypes, which ultimately led to the ‘M1/M2’ nomenclature, respectively.7 Hence, traditionally, macrophages were accepted as phagocytic, terminally differentiated blood monocyte-derived cells, which can be activated by classic or alternative ligands to form polarized populations. We will review some recent advances in the field of macrophage biology and highlight how this impacts on this dogma.
Dual origins of tissue macrophages
In 1924 Ludwig Aeschoff labelled macrophages as tissue phagocytes,3 which could import lithium carmine by phagocytosis. This theory was ultimately flawed as lithium carmine could also be imported by pinocytosis (cell drinking) in non-phagocytes.8 In 1925 the findings of Florence Sabin showed that a proportion of these cells can be derived from the blood,9 and in 1968 this was applied to all macrophages to form the mononuclear phagocyte system.4 At this time, there was a reasonable body of evidence that macrophages were not terminally differentiated10–12 and were persistent in tissues.13 However, the experiments used in these studies were imperfect, mainly due to the limitations of technology, and so the theories put forward struggled for attention.
Toward the end of the twentieth century a greater appreciation for macrophage heterogeneity was observed.14 This included a consensus that some macrophages could proliferate14 and existed in the embryo long before definitive haematopoiesis.15,16 This was contradictory to the mononuclear phagocyte system; but could have been considered as special cases. The observation that human Langerhans cells (a specialized tissue macrophage)17 from the donor still existed years after a double hand transplant18 provided evidence that either these cells could survive an extended length of time, or that they were replenished locally. In 2009, Chorro et al.19 showed that murine Langerhans cells were not terminally differentiated and could proliferate in situ to restore cells lost in inflammation. This result, along with similar observations in brain microglia20 led to related observations of in vivo proliferation, for example: in peritoneal macrophages,21,22 pleural macrophages,22 alveolar macrophages,23 red pulp macrophages,23 adipose tissue macrophages,24 cardiac macrophages25 and macrophages within atherosclerotic lesions.26 Gentek et al.27 have recently discussed aspects of macrophage proliferation in detail.
It became evident that many tissue macrophage populations are renewed independently of the bone marrow, and therefore their origin was scrutinized. Fate-mapping studies showed that a significant number of tissue macrophages are derived from primitive macrophages existing within the yolk sac or fetal liver.28,29 These macrophages are seeded into the tissues before birth and proliferate to populate the expanding tissue with resident macrophages. Resident macrophages in organs, such as the intestine and dermis, are continually replenished from blood monocyte precursors,30–32 and monocyte-derived macrophages are a key constituent of inflammatory environments.33 A significant number of tissue macrophages can remain independent from blood monocytes; though ‘embryonic’ macrophages can be replaced by monocyte-derived cells after severe inflammation,34 or in active, ageing tissues such as the heart.35 The change in macrophage origin may result in different phenotypes or functions in that tissue. Considering the majority of our knowledge on tissue macrophages is focused on rodents and cells with limited origins, the next logical questions for the field include: what effect does origin have on macrophage function? And how do these primary rodent studies relate to human macrophages? This may be an important consideration, because mice used in research are usually 6 weeks, and at maximum 2 years, old, whereas humans live for over eight decades. So it is possible that the embryonic origin of adult macrophages in rodents is not indicative of human cell populations. However, results from rodent studies can still be mimicked in human tissues, such as the long-term independence of human Langerhans cells from monocytes.18
The peritoneal cavity: a model for tissue-specific transcriptional control
Immunological cell subsets are defined not only by their phenotype, but also by their expression of transcription factors required for specific functions. This is relatively well defined in other fields of immunity, such as T-cell biology;36 however, the transcription factor profiles of macrophage subsets are still largely unknown. There have been notable studies on tissue niche-specific transcriptional control of macrophage populations in the past, such as in the spleen.37 The haem-induced38 transcription factor Spic is required for the presence of red pulp macrophages,39 whereas Nr1h3 is necessary for all marginal zone macrophages.40 Importantly, the loss of these factors results in the loss of both the cells and their functions, and is localized to specific tissue microenvironments.39,40 Therefore, it is difficult to dissect the mechanisms controlling specific functions of these subsets, but implies that these factors are required for cell survival/development in that tissue niche.
In 2012 the immunological genome consortium identified the zinc finger transcription factor Gata6 as peritoneal tissue-resident macrophage (pResMϕ)-selective.41 In addition, they showed that MerTK and CD64 provided an alternative to F4/80 expression when identifying tissue macrophages.41 This study highlighted both the shared and distinct characteristics of tissue macrophage subsets and provided more information to discriminate them from dendritic cells.41
Other groups including our own also identified Gata6 as a select transcription factor in pResMϕ.42–44 Conditional knockout (KO) of the Gata6 gene in myeloid cells (‘Lyz2-Cre recombinase’, Gata6-floxed: which we termed Gata6-KOmye) resulted in gross phenotype changes in the common macrophage markers F4/80 and CD11b in pResMϕ. Importantly there were still pResMϕ present, albeit in reduced numbers.42–44 In vivo lentiviral manipulation of Gata6 in adult pResMϕ resulted in similar phenotype changes, indicating that Gata6 is required for cell phenotype in the adult.42 Transcriptome studies, which compared Gata6-KOmye with wild-type pResMϕ, identified a diverse range of genes altered in the absence of Gata6. Crucially, a large number of these genes were pResMϕ selective, so it was concluded that Gata6 is in part responsible for the pResMϕ-specific gene profile.42–44 These studies corroborated the same overall conclusion; however, the authors of each study provided additional insights into the function of Gata6 in pResMϕ.
Okabe and Medzhitov,43 identified that vitamin A-derived retinoic acid drives the expression of Gata6 in pResMϕ. The source of this retinoic acid is the peritoneal omental tissue. Interestingly pResMϕ of Gata6-KOmye mice accumulate in omental milky spots,43 perhaps because of their altered cell-adherent gene signature.42–44 The loss of Gata6 in pResMϕ can also have physiological consequences. Peritoneal B1 cells migrate to the gut and can secrete IgA.45 Gata6 ablation in pResMϕ resulted in a decline in immunoglobulin A production from migrating peritoneal B-1 cells. This was dependent upon transforming growth factor β2, which is secreted by pResMϕ and controlled by Gata6.43 The functional importance of this is not fully understood as B2 cells can compensate for this drop in IgA production, but it highlights a physiological role of resident macrophages, which enables us to tease apart the subtle control of tissue physiology.
Peritoneal pResMϕ proliferate at a low level to maintain homeostasis, but go through a burst in proliferation to recover lost numbers during the resolution from inflammation.21 We identified alterations in genes linked to proliferation in Gata6-KOmye pResMϕ.42 Interestingly, the proportion of Gata6-KOmye macrophages proliferating was actually higher than controls, but nuclear polyploidy was also enhanced.42 This could be either attributed to failed cytokinesis (as considered to be caused by lack of emerin in the nuclear envelope), as seen in Gata6-knockdown epithelial cells,46 or by enhanced cell fusion. Gata6-KOmye pResMϕ failed to recover their numbers after acute inflammation and those cells still present were deficient in their proliferative response.42 Therefore, Gata6 contributes toward pResMϕ persistence by regulating normal proliferative responses. Reduced pResMϕ numbers could also be explained by increased migration to omental milky spots,43 increased tissue adherence42–44 or enhanced apoptosis.
Gautier et al.44 found that loss of Gata6 results in decreased expression of the metabolic enzyme Aspa in pResMϕ. Aspa-KO pResMϕ shared characteristics such as enhanced apoptosis and lower cell numbers with Gata6-KO pResMϕ.44 However, the mechanisms for apoptosis may not be shared between the two knockouts. Genes associated with oxidative phosphorylation were also increased in Gata6-KOmye pResMϕ, which suggests enhanced mitochondrial function.44 When investigated, oxygen consumption was found to be higher in Gata6-KOmye pResMϕ, suggesting increased oxidative phosphorylation.44 However, this could also be enhanced basal oxidase activity. It has been suggested that alternative activation of macrophages can tip the metabolic balance away from glycolysis and toward mitochondrial oxidative phosphorylation.47 Therefore, the authors conclude that Gata6-KOmye pResMϕ are alternatively activated both due to increased expression of genetic markers such as Arg1, and enhanced oxygen consumption.44 However, many genes are enhanced in the absence of Gata6, so perhaps this activation state is merely a status of cell health, and failure to respond to the environment. It is also not clear whether the levels of glycolysis are also higher in Gata6-KOmye pResMϕ, and perhaps the increase in oxygen consumption is indicative of a more general metabolic activation. There has been a growing interest in metabolic control of immune cell function.47 This study44 demonstrates that the persistence of a tissue-specific macrophage population is maintained in part by selective expression of a metabolic enzyme. Aspa is generally considered to be restricted to oligodendrocytes in the nervous system.48 Hence, its presence in a macrophage population is interesting, and the unknown mechanisms behind the apoptotic consequence of Aspa deletion may additionally influence specific macrophage functions.
The Gata6 model for tissue-specific control of a macrophage population provides a valuable tool to investigate the fine-tuning of tissue-specific cellular function in vivo. A summary on the recent findings of Gata6 is shown in Fig.1. Tissue physiology is tightly regulated, and it is likely that each cell within each tissue has its own unique genetic expression profile. This enables cell survival in the tissue, but may restrict survival in other tissues. Immune cells of the blood are not necessarily exempt from such control, as one fate of monocyte-derived macrophages as well as neutrophils in tissues is apoptosis.49 However, these cells can adapt to new environments, as is evident in the ‘training’ of monocyte-derived macrophage subsets to survive and function in the intestine.31 Therefore, both cell plasticity and environmental factors play a role in the fate and persistence of cells in tissues, and a colossal effort is required to unravel these complexities.
Figure 1.
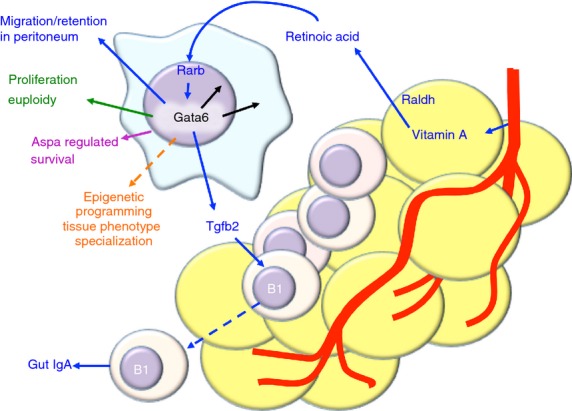
Gata6 controls peritoneal macrophage phenotype. Diagram showing recent discoveries on the control of peritoneal macrophage phenotype by Gata6. The yellow cells represent peritoneal omental tissue, B cells are shown as small circular cells, and the large globular cell is a peritoneal resident macrophage. Results from different laboratories are colour coded: green,42 blue,43 magenta44 and orange50,51.
Environmental programming of tissue-specific macrophage subsets
Two recent publications have implicated the tissue environment as the strongest factor in the determination of cell phenotype.50,51 These studies used sequencing (Seq) techniques to investigate the genetics and epigenetics of tissue-resident macrophage populations in different tissues, including Gata6-controlled pResMϕ. The authors describe enhancer landscapes, which are essentially a measure of specific histone modifications, principally histone-3 lysine-4 single methylation (H3K4me1). This modification is associated with enhancer regions in gene promoters. Areas rich in H3K4me1 highlight active gene landscapes.52 Lavin et al.51 used a combination of RNA-Seq, chromatin immuno-precipitation-Seq and assay for transposase-accessible chromatin-Seq to pinpoint enhancer landscapes coupled to gene expression and accessible chromatin. This allowed them to build a three-dimensional map of chromatin, which included whether the gene is expressed and what transcription factors are likely to be bound to that gene. This is a useful method to visualize transcription factor control of gene expression profiles in specific cell subsets. The two studies conclude that tissue-resident macrophages shared epigenetic structure and gene expression with other myeloid cells, and is modulated by master transcription factors such as PU.1.50,51 However, each tissue additionally has its own unique gene expression profiles controlled by changes in enhancer landscapes50,51 Lavin et al. transferred macrophages from the peritoneum to the lung. The transferred cells lost most of the old tissue programming, but acquired the majority of a new tissue programme specific to their new environment.51 Tissue-specific reprogramming was also evident when existing ‘embryonic’ tissue-resident macrophages were lethally irradiated and replaced by tissue-resident macrophages derived from transferred healthy bone marrow.51 This shows that the environment predominantly controls the cells’ phenotype by directing epigenetic programmes.
Gosselin et al. additionally showed the impact of selective environmental factors on gene expression of cells cultured ex vivo. Retinoic acid or transforming growth factor β1 inclusion in culture media partially restored the enhancer environments and gene expression of pResMϕ and microglia, respectively.50 This enables us to carry out ex vivo experimentation, which will be more faithful to the phenotype of specific tissue macrophages than existing culture techniques. The challenge now is to fully characterize unique tissue environments and dissect the functions of each factor. An example of such a factor is retinoic acid in the peritoneum, which drives Gata6 expression in pResMϕ and maintains the epigenetic landscape and gene expression profile,50 (Fig. 1). These studies50,51 emphasize the plasticity of macrophages and highlights that cell polarization in chronic inflammation for example, may not be irreversible, as a new environment can reprogramme the epigenetics and gene expression prolife. However, this might not be the only determiner of cell phenotype, as the expression of certain genes may still be dependent on cell origin.
These are not the first reports of macrophage environmental reprogramming; however, as evidenced by a recent report from Suzuki et al.53 They describe a model of pulmonary macrophage therapy, which is essentially the replacement of functionally deficient (Csf2rb−/−) alveolar macrophages by site-specific transfer of wild-type bone-marrow-derived macrophages. When analysed by microarray, the transferred cells acquired the majority of the phenotype associated with alveolar macrophages and persisted for at least a year. In their model, these cells are functional and restore the normal surfactant clearing function in the lungs, which is impaired in Csf2rb−/− mice. Genetically manipulated Csf2rb−/− cells with enforced Csf2rb expression were also able to re-populate the lungs of Csf2rb−/− mice with functional macrophages that restored lung homeostatic functions. This study highlights the potential for macrophage transfer and subsequent tissue-specific environmental reprogramming as a therapeutic.
Tissue-resident macrophage activation and inflammation
Inflammatory stimuli are often sensed by macrophage receptors, resulting in downstream signalling cascades that force activation of the cell. Existing dogma for macrophage activation is dedicated to M1/M2 polarization: macrophages can either be classically (‘M1’) or alternatively (‘M2’) activated. This is dependent on the stimulus and resulting phenotype of the cell.54 Classical activation of macrophages by stimuli such as lipopolysaccharide (‘M1’) through Toll-like receptor 4 has been extensively categorized, but the majority of this research has been performed in vitro on cells with limited origins. Classical activation of tissue-resident macrophages in vivo has usually been thought of as equivalent; however, it is likely that there are subtle differences, which are dependent upon environment and genetic/epigenetic programming.41,55 Although classic (‘M1’) and alternative (‘M2’) activation have been applied in the in vivo setting, such as in T helper type 1 and type 2 environments, respectively;54 the complexity of signals present will change both the activation process and outcome. For example, the phenotype of macrophages alternatively activated by interleukin-4 was reported to be different depending on cell origin.56 The authors compared thioglycollate-elicited macrophages (with bone marrow origins) to pResMϕ (with prenatal origins). The gene expression profiles were distinct, even after interleukin-4 treatment in vivo. This shows that origin may still play a role, as monocytes arriving from the blood may need time to switch environmental epigenetic programming before activation. Additionally, a transcriptomic study on resolution-phase macrophages in the peritoneum found that these cells did not resemble ‘M1’ or ‘M2’ activation phenotypes, but seemed a hybrid;57 however, at the time of these transcriptomic studies the complexity of cellular subsets present was not appreciated. We now know that some tissue macrophages can renew their population independently from monocytes during inflammation19,21,22 and have prenatal origins.28,29 Therefore, a proportion of the macrophages existing at the end of inflammation may be those present at the start. These cells have been activated and then must return to homeostasis. This aspect of tissue macrophage biology has been largely under-studied because it is often assumed that activated macrophages perish. Therefore some macrophage subsets present during the later stages of inflammation, including wound healing, may in fact be recovering tissue-resident macrophages. Recently, Newson et al. carried out a refined analysis of resolution-phase macrophages in the peritoneum.58 This study appreciated that tissue-resident macrophages survive initial acute inflammation and persist during the recovery phase. However, the authors noted that the cell environment in the peritoneum remained complicated by the co-existence of persisting tissue-resident macrophages and monocyte-derived cells, even up to 60 days after stimulus. The tissue-resident macrophages at 60 days do not mimic the naive phenotype and also maintain differential gene expression from monocyte-derived macrophages. However, this study itself shows that even acute inflammation has long-term consequences on the tissue environment and cell phenotype, with a reported second wave of leucocyte recruitment and continued abnormality in the cell milieu.58 Therefore, the time that recruited monocytes are maintained in an inflammatory lesion is poorly defined, and importantly, it is likely that the tissue environment itself may take a long time to return to homeostasis, if at all. Perhaps factors dictating tissue reprogramming are either missing or are overridden by the new inflammatory context. This work58 highlights the gaps in our understanding of macrophage function and phenotype during the resolution of acute inflammation, and further study is required to more thoroughly define the restoration of homeostasis.
The extreme heterogeneity of tissue-resident macrophages during homeostasis and inflammation shows that a macrophage cannot be just ‘M1’ or ‘M2’ when residing in a tissue. A number of reports have described the ‘M1/M2’ polarization nomenclature as being too simplistic,37,59–61 and a recent perspective has proposed a framework to standardize in vitro and ex vivo macrophage activation nomenclature.62 Although there is no new consensus model for macrophage phenotype in vivo, it has been suggested that macrophages be named by origin, and the historical nomenclature maintained for cells with an embryonic origin.17 Although cell identification is important, the difficulties associated with defining lineage should not detract from meaningful functional study of the cells. The lack of a clear model for macrophage activation phenotype in vivo results from the complexity of the environment; construction of this model would require enhanced understanding of cell-tissue interactions. Additional large-scale analyses, such as proteomics, metabolomics, genomics and epigenomics in combination with in silico algorithms will be essential to unravel further the complexity of these systems.55,63,64
One of the most studied inflammatory environments is the tumour. Tumour-associated macrophages (TAMs) are phagocytic cells with unclear origins residing in what is essentially an inflammatory tissue. These cells have been poorly categorized, as their existence and phenotype depends upon a broad range of variables.65 These variables include: tumour origin, formation, location, environment status, organism/species and individual genetics. Recently, Franklin et al.66 investigated the origin of TAMs in a murine model of breast cancer. The study found that these TAMs were derived from blood monocytes. However, this model may not be indicative of all tumours, because of the variables mentioned above. In other inflammatory environments there are known to be varying mixtures of tissue-dervied and monocyte-derived subsets; examples being encephalomyelitis, with microglia and monocyte-derived macrophages67 and dermatitis, which includes epidermal Langerhans cells.68 Regardless of the origin, macrophages are a resident of the tumour site and contribute to tumour growth. Recently, Colegio et al.69 showed that the tumour microenvironment itself controls the functions of TAMs. Tumours can produce a substantial level of lactic acid, which is scavenged by TAMs and can activate Hif1α. Hif1α is usually induced by low oxygen and drives the expression of a wide array of genes.70 The authors focused on two alternative activation genes: Arg1 and Vegf, known to be important for tumour growth and neovascularization, respectively.69 Tumour lactic acid can therefore control certain aspects of the TAM gene expression profile. Hif1α activation has been reported to shift macrophages into an enhanced glycolytic state,71 which includes increased production of lactic acid, which could further propagate the phenotype of these cells. This is an example of macrophage activation by the inflammatory environment, and because this environment can be extremely complex, methods for dissecting this need to become both more intricate and encompassing.
Development of a new paradigm
The ‘M1/M2’ system has undoubtedly been useful in studying alternate aspects of macrophage activation; however, overuse in the in vivo setting will probably hinder our progress in the understanding of how a macrophage responds to its environment, and contributes to disease pathology. A cells’ environment can become polarized with overpowering or chronic stimuli, but this is not permanent or black and white, and a change in environment will result in changes in cell phenotype in shades of grey. Therefore, the new ‘model’ is the appreciation that the local environment controls macrophage phenotype, be it in a tissue or in vitro. This may be complex but it can be applied across organisms; and although cells, mechanisms and environments may be different, animal models can be used to understand the control of tissue systems. This can then be applied to translational research contexts. When culturing cells in vitro we should update the culture conditions to closely match those of a specific tissue environment. This can be something simple, such as including retinoic acid for pResMϕ culture or transforming growth factor β1 for microglial culture,50 using physiologically relevant oxygen levels, or providing microfluidic flow72 to retain phenotype ex vivo. However, this is destined to become more advanced with increased understanding of tissue environments. Rather than standardizing basic mechanisms, perhaps this argues for an increase in the complexity of in vitro systems when culturing human cells in order to learn new aspects of cell activation in more complex environments. This would require a working knowledge of the factors specific to that environment, but it is essential for advancement of out-dated tissue culture techniques.
Concluding remarks
The field of macrophage biology, like macrophages themselves, has evolved slowly from ancient roots. However, during the last decade it has gone through a boom, with the discovery of proliferative potential, extreme heterogeneity and divergent origins. Recently it has been shown that tissue-specific transcription factors and tissue programmed epigenetics control the gene expression of resident macrophages, regulating their functions; which can then in turn impact upon the environment itself. These tissue feedback loops are probably important to maintain healthy tissue homeostasis. This environmental programming of macrophages can potentially be ‘hotwired’ in order to alter macrophage processes regardless of the environment, thus providing therapeutic benefit in inflamed tissues. Furthermore, the field of immune cell metabolism is becoming increasingly popular.47 Simple metabolites such as glucose and glutamine have been included in tissue culture media for decades; however, we are only now starting to appreciate their roles in immune cell activation and functions.47 The Gata6–retinoic acid axis in the peritoneum,43 shows how metabolites can be a key tissue factor and an area of focus for future research. Additionally, considering that metabolic enzymes are among the best-conserved proteins between organisms, it may reveal interesting analogies between animal models and human disease.
Acknowledgments
We would like to acknowledge Dr Anja Bloom for her assistance in critically reviewing the manuscript. PRT is supported by the Medical Research Council (grants MR/J002151/1 and MR/K02003X/1), and additionally supported through a Wellcome Trust Institutional Strategic Support Fund. LCD is currently a postdoctoral Visiting Fellow with the LEI, CIP, CCR, NCI. This work has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, Intramural Research Program USA and Medical Research Council UK. The content of this review does not necessarily reflect the views or policies of Cardiff University UK or the Department of Health and Human Services USA, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Glossary
Abbreviations:
- Gata6-KOmye
Lyz2-Cre recombinase: Gata6-floxed, i.e. Gata6 deletion in myeloid cells.
- H3K4me1
histone-3 lysine-4 single methylation
- KO
knockout
- pResMϕ
peritoneal tissue-resident macrophages
- Seq
sequencing
- TAM
tumour-associated macrophages
Disclosures
We have no conflicts of interest.
References
- Metchnikoff E. Leçons Sur La Pathologie Comparée De L'inflammation. Paris: Masson; 1892. [Google Scholar]
- Gordon S. Elie Metchnikoff: father of natural immunity. Eur J Immunol. 2008;38:3257–64. doi: 10.1002/eji.200838855. [DOI] [PubMed] [Google Scholar]
- Aschoff L. Das reticuloendotheliale system. Erg Inn Med Kinderheilk. 1924;26:1–119. [Google Scholar]
- van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ. 1972;46:845–52. [PMC free article] [PubMed] [Google Scholar]
- Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-γ as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158:670–89. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–92. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–6. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Lewis WH. Pinocytosis. Bull Johns Hopkins Hosp. 1931;29:17–26. [Google Scholar]
- Sabin FR, Doan CA, Cunningham RS. Discrimination of two types of phagocytic cells in the connective tissues by the supravital technique. Contrib Embryol. 1925;16:125–62. [Google Scholar]
- Parwaresch MR, Wacker HH. Origin and kinetics of resident tissue macrophages. Parabiosis studies with radiolabelled leucocytes. Cell Tissue Kinet. 1984;17:25–39. doi: 10.1111/j.1365-2184.1984.tb00565.x. [DOI] [PubMed] [Google Scholar]
- Sawyer RT, Strausbauch PH, Volkman A. Resident macrophage proliferation in mice depleted of blood monocytes by strontium-89. Lab Invest. 1982;46:165–70. [PubMed] [Google Scholar]
- Czernielewski JM, Demarchez M. Further evidence for the self-reproducing capacity of Langerhans cells in human skin. J Invest Dermatol. 1987;88:17–20. doi: 10.1111/1523-1747.ep12464659. [DOI] [PubMed] [Google Scholar]
- Melnicoff MJ, Horan PK, Breslin EW, Morahan PS. Maintenance of peritoneal macrophages in the steady state. J Leukoc Biol. 1988;44:367–75. doi: 10.1002/jlb.44.5.367. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov DA. Macrophages in the embryo and beyond: much more than just giant phagocytes. Genesis. 2008;46:447–62. doi: 10.1002/dvg.20417. [DOI] [PubMed] [Google Scholar]
- Lichanska AM, Hume DA. Origins and functions of phagocytes in the embryo. Exp Hematol. 2000;28:601–11. doi: 10.1016/s0301-472x(00)00157-0. [DOI] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14:571–8. doi: 10.1038/nri3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanitakis J, Petruzzo P, Dubernard JM. Turnover of epidermal Langerhans’ cells. N Engl J Med. 2004;351:2661–2. doi: 10.1056/NEJM200412163512523. [DOI] [PubMed] [Google Scholar]
- Chorro L, Sarde A, Li M, et al. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med. 2009;206:3089–100. doi: 10.1084/jem.20091586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–43. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Davies LC, Rosas M, Smith PJ, Fraser DJ, Jones SA, Taylor PR. A quantifiable proliferative burst of tissue macrophages restores homeostatic macrophage populations after acute inflammation. Eur J Immunol. 2011;41:2155–64. doi: 10.1002/eji.201141817. [DOI] [PubMed] [Google Scholar]
- Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, Macdonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–8. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, et al. Tissue resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano Shinya U, Cohen Jessica L, Vangala P, et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2013;19:162–71. doi: 10.1016/j.cmet.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine Kory J, Beaudin Anna E, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–72. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentek R, Molawi K, Sieweke MH. Tissue macrophage identity and self-renewal. Immunol Rev. 2014;262:56–73. doi: 10.1111/imr.12224. [DOI] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2012;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamoutounour S, Guilliams M, Montanana Sanchis F, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. 2013;39:925–38. doi: 10.1016/j.immuni.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Zigmond E, Jung S. Intestinal macrophages: well educated exceptions from the rule. Trends Immunol. 2013;34:162–8. doi: 10.1016/j.it.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Bain CC, Bravo-Blas A, Scott CL, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15:929–37. doi: 10.1038/ni.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LC, Rosas M, Jenkins SJ, et al. Distinct bone marrow-derived and tissue resident macrophage-lineages proliferate at key stages during inflammation. Nat Commun. 2013;4:1886. doi: 10.1038/ncomms2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Tacke F, Angeli V, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. 2006;7:265–73. doi: 10.1038/ni1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molawi K, Wolf Y, Kandalla PK, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. 2014;211:2151–8. doi: 10.1084/jem.20140639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15:1104–15. doi: 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue resident macrophages. Nat Immunol. 2013;14:986–95. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar M, Kohyama M, So AY, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell. 2014;156:1223–34. doi: 10.1016/j.cell.2014.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama M, Ise W, Edelson BT, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature. 2009;457:318–21. doi: 10.1038/nature07472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A-Gonzalez N, Guillen JA, Gallardo G, et al. The nuclear receptor LXRα controls the functional specialization of splenic macrophages. Nat Immunol. 2013;14:831–9. doi: 10.1038/ni.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–28. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas M, Davies LC, Giles PJ, et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science. 2014;344:645–8. doi: 10.1126/science.1251414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157:832–44. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Ivanov S, Williams JW, et al. Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J Exp Med. 2014;211:1525–31. doi: 10.1084/jem.20140570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagarasan S, Kinoshita K, Muramatsu M, Ikuta K, Honjo T. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature. 2001;413:639–43. doi: 10.1038/35098100. [DOI] [PubMed] [Google Scholar]
- Capo-chichi CD, Cai KQ, Testa JR, Godwin AK, Xu XX. Loss of GATA6 leads to nuclear deformation and aneuploidy in ovarian cancer. Mol Cell Biol. 2009;29:4766–77. doi: 10.1128/MCB.00087-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol. 2014;32:609–34. doi: 10.1146/annurev-immunol-032713-120236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namboodiri AMA, Peethambaran A, Mathew R, Sambhu PA, Hershfield J, Moffett JR, Madhavarao CN. Canavan disease and the role of N-acetylaspartate in myelin synthesis. Mol Cell Endocrinol. 2006;252:216–23. doi: 10.1016/j.mce.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Ivanov S, Lesnik P, Randolph GJ. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood. 2013;122:2714–22. doi: 10.1182/blood-2013-01-478206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159:1327–40. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–26. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Arumugam P, Sakagami T, et al. Pulmonary macrophage transplantation therapy. Nature. 2014;514:450–4. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–73. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Glass CK. Epigenomics of macrophages. Immunol Rev. 2014;262:96–112. doi: 10.1111/imr.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundra UM, Girgis NM, Ruckerl D, et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood. 2014;123:110–22. doi: 10.1182/blood-2013-08-520619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stables MJ, Shah S, Camon EB, Lovering RC, Newson J, Bystrom J, Farrow S, Gilroy DW. Transcriptomic analyses of murine resolution-phase macrophages. Blood. 2011;118:e192–208. doi: 10.1182/blood-2011-04-345330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newson J, Stables M, Karra E, et al. Resolution of acute inflammation bridges the gap between innate and adaptive immunity. Blood. 2014;124:1748–64. doi: 10.1182/blood-2014-03-562710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–88. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine Kory J, Randolph Gwendalyn J. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262:36–55. doi: 10.1111/imr.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume DA, Freeman TC. Transcriptomic analysis of mononuclear phagocyte differentiation and activation. Immunol Rev. 2014;262:74–84. doi: 10.1111/imr.12211. [DOI] [PubMed] [Google Scholar]
- Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012;4:376–89. [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–5. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy SS, Lees JG, Moalem-Taylor G. The contribution of immune and glial cell types in experimental autoimmune encephalomyelitis and multiple sclerosis. Mult Scler Int. 2014;2014:285245. doi: 10.1155/2014/285245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malissen B, Tamoutounour S, Henri S. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol. 2014;14:417–28. doi: 10.1038/nri3683. [DOI] [PubMed] [Google Scholar]
- Colegio OR, Chu N-Q, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–63. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A, Goldrath Ananda W, Nizet V, Johnson Randall S. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41:518–28. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-W, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Dominical VM, Vital DM, O'Dowd F, Saad STO, Costa FF, Conran N. In vitro microfluidic model for the study of vaso-occlusive processes. Exp Hematol. doi: 10.1016/j.exphem.2014.10.015. In press. [DOI] [PubMed] [Google Scholar]