

Abstract
Tumour progression is associated with immune-suppressive conditions that facilitate the escape of tumour cells from the regimen of immune cells, subsequently paralysing the host defence mechanisms. Induction of CD4+ CD25+ FoxP3+ T regulatory (Treg) cells has been implicated in the tumour immune escape mechanism, although the novel anti-cancer treatment strategies targeting Treg cells remain unknown. The focus of this study is to define the interaction between tumour and immune system, i.e. how immune tolerance starts and gradually leads to the induction of adaptive Treg cells in the tumour microenvironment. Our study identified hyperactivated mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) -signalling as a potential target for reversing Treg cell augmentation in breast cancer patients. In more mechanistic detail, pharmacological inhibitors of MEK/ERK signalling inhibited transforming growth factor-β (TGF-β) production in tumour cells that essentially blocked TGF-β-SMAD3/SMAD4-mediated induction of CD25/interleukin-2 receptor α on CD4+ T-cell surface. As a result high-affinity binding of interleukin-2 on those cells was prohibited, causing lack of Janus kinase 1 (JAK1)/JAK3-mediated signal transducer and activator of transcription 3 (STAT3)/STAT5 activation required for FoxP3 expression. Finally, for a more radical approach towards a safe MEK inhibitor, we validate the potential of multi-kinase inhibitor curcumin, especially the nano-curcumin made out of pure curcumin with greater bioavailability; in repealing tumour-shed TGF-β-induced Treg cell augmentation.
Keywords: breast cancer, janus kinase/signal transducer and activator of transcription, mitogen-activated protein kinase kinase/extracellular signal-regulated kinase, nano-curcumin, regulatory T cells, transforming growth factor-β
Introduction
Recent trends in anticancer drug development are based on identifying and then targeting precise small signalling molecules or genes that create the particular molecular abnormalities leading to generation of malignant phenotype.1 Induction and expansion of fork-head box P3 (FoxP3)-positive regulatory T (Treg) cells are associated with prognosis and progression of cancer, and have important role in suppressing tumour-specific immunity. Expansion of CD4+ CD25+ FoxP3+ Treg cells following tumour progression suppresses the activity of tumour-specific T effector cells and creates an environment that is favourable for tumour growth.2 Patients with breast cancer have an increased prevalence of Treg cells in the peripheral blood and in the tumour microenvironment.3,4 Hence, blockade of Treg cell induction can enhance immune protection from tumour-associated antigens and offer successful therapeutic interventions of cancer development and progression. Though several reports have described the significance of Treg cells in tumour development, studies to identify and target specific signalling molecules responsible for Treg cell induction are yet to be designed.
The regulatory T-cell lineage is indispensable for the induction of T-cell tolerance, which happens to be one of the mechanisms of cancer immune evasion in tumour condition.5–7 Regulatory T cells have been shown to be critical for the maintenance of immunological tolerance. The X-chromosome-encoded transcription factor FoxP3 is a lineage-specification factor required for Treg cell differentiation and function,8 which executes its multiple activities mostly through transcriptional regulation of its target genes.9 FoxP3 interacts directly with the transcription factor nuclear factor of activated T cells, and this interaction is required for FoxP3 to bind to a proximal site in the il2 promoter in vivo, to up-regulate several Treg-associated markers and to inhibit cytokine production.10 In vivo, a high-density of tumour-infiltrating FoxP3+ Treg cells has been associated with poor disease outcome in patients affected by various solid tumours.11 At least two general subsets of Treg cells exist – natural (nTreg) and adaptive/induced (iTreg), which differ in their development, antigenic specificities and mechanisms of action.12–14 The suppressive function of nTreg cells requires cell–cell contact or proximity, whereas iTreg cells exert their regulatory activity essentially by means of secreted cytokines like interleukin-10 (IL-10) and transforming growth factor-β (TGF-β).3 Treg cells kill responder T cells by a granzyme-dependent or perforin-dependent mechanism.15 Cytotoxic T-lymphocyte antigen 4-mediated cell contact-dependent suppression of Treg cells inhibits the up-regulation of CD80 and CD86 on immature dendritic cells and down-regulates the expression of CD80 and CD86 by mature dendritic cells without affecting the expression of CD40 and class II MHC.16 Treg cells also employ immunosuppression by commencing competition for growth factors thereby leading to cytokine deprivation-induced apoptosis in the target effector T cells.17 Moreover, the constitutive expression of high-affinity IL-2 receptor-α or CD25 by Treg cells gives them an initial competitive advantage for the consumption of IL-2 over naive T cells.6 Furthermore, FoxP3 is able to repress the expression of specific cytokines by interacting with phosphodiesterase 3B and the transcription factor nuclear factor-κB, the key drivers of inflammation.18
There is substantial evidence that tumour-microenvironment is characterized by abundance of factors like TGF-β, IL-6, IL-10, vascular endothelial growth factor and prostaglandin E2, which are pivotal to the development of an immunosuppressive network.19 Among these, TGF-β, produced by a diverse range of tumour cell types,20 is a critical factor in regulation of T-cell-mediated immune responses and in the induction of immune tolerance.21 When the TGF-β1-mediated inhibitory signal is abrogated by restrictive expression of dominant negative TGF-β receptor II, these mice develop unchecked T-cell proliferation and autoimmune-like diseases, documenting a TGF-β-dependent signal in T-cell activation and tolerance.22 Apart from inhibiting T-cell function TGF-β also imparts a suppressive phenotype to CD4+ T cells.23,24 The TGF-β converts CD25− CD4+ T cells into CD25+ CD4+ anergic/suppressor T cells, which not only exhibit unresponsiveness to T-cell receptor stimulation but also suppress normal CD4+ T-cell activation and cytokine production.12 SMAD family members have been identified as essential intracellular signalling components of the TGF-β super family.13 It was shown that TGF-β signalling through SMADs is required for generation of both T helper type 17 and Treg cells.14 Particularly, SMAD3/SMAD4 is involved in the induction of Treg cells, whereas SMAD2 regulates the generation of T helper type 17 cells.25,26 Although TGF-β/SMAD-mediated regulation of immune responsiveness has been validated, the mechanism as to how tumour-shed TGF-β accomplishes immunosuppression through induction of CD25 on CD4+ T cells remains to be elucidated.
Interleukin-2 has a long-established heritage as a T-cell growth factor.27,28 However, the evidence from the past few years has suggested that IL-2 is also critical for the establishment and maintenance of immune tolerance.29 The role of IL-2 in the generation and maintenance of adaptive Treg cells became clear when it was found that TGF-β, which induces CD25 on CD4+ CD25− T cells, had to be supplemented with IL-2 for induction of FoxP3.30,31 Recent studies have shown that IL-2 directly regulates the foxp3 gene in CD25+ CD4+ Treg cells.32 Janus kinase (JAK)/ signal transducer and activator of transcription (STAT)-signalling pathway plays an important role in maintaining FoxP3 status in CD3/CD28-stimulated CD4+ T cells and blockage of STAT3/STAT5 activation significantly reduces foxp3 transcription in these cells.33–35
Our study identified every sequential step, demonstrating how being derived through mitogen-activated protein kinase kinase (MEK)/extracellular singal-regulated kinase (ERK) signalling, tumour shed-TGF-β induced FoxP3+ Treg cells through SMAD3/SMAD4-directed CD25 expression and subsequent JAK/STAT activation. In addition, using several pharmacological inhibitors, we have further strengthened the candidature of MEK/ERK signalling as the potential target in reversing Treg induction in tumour condition. Most importantly, as a novel strategy to maximize the effectiveness of targeted therapies and to minimize the impact of side effects of available cytotoxic drugs, we have identified the efficacy of curcumin, when used in the form of nano-curcumin, made out of pure curcumin and with improved bioavailability, as a MEK/ERK inhibitor, in repealing Treg cell augmentation in tumour bearers.
Materials and methods
Cell culture and experiments
The present study included 24 female patients with breast cancer and 12 age/sex-matched female healthy volunteers as controls. Informed consent (IRB-1382) under the provision of ethics committee, SSKM Hospital, Kolkata, India (Approval No: Inst/IEC/306) and Human Ethics Committee, Bose Institute (Approval No: BIHEC/2010-11/2) was obtained from all patients with localized disease and from female healthy volunteers in compliance with the Helsinki Declaration (http://www.wma.net/en/30publications/10policies/b3/). Peripheral blood collected from healthy volunteers or from patients was centrifuged over Ficoll–Hypaque (GE Healthcare Life Sciences, Pittsburgh, PA) density-gradient to obtain total leucocytes. T cells were purified from total leucocytes by negative magnetic selection using a human T-cell enrichment cocktail (Stem Cell Technologies, Vancouver, BC, Canada). Cells were maintained in complete RPMI-1640 medium at 37° in a humidified incubator containing 5% CO2. Tissue from primary lesions of breast cancer was collected from patients undergoing surgical procedures to remove solid tumour mass. The inner mass of tissues was cut into small pieces of 2–4 mm, digested at 37° for 3 hr in a 1 : 1 solution of collagenase/hyaluronidase (Sigma-Aldrich, St. Louis, MO). After filtration through a 30-μm pore filter, single cells were plated in RPMI-1640 medium supplemented with 10% fetal bovine serum for overnight. The adherent cells were used for further experiments. The purity of the cells was checked flow cytometrically by CD24 and ESA-positivity or CD4 and CD25-negativity (see Supporting information, Fig. S1). After 72 hr of incubation, supernatants freed from cellular components were used in 1 : 1 ratio with RPMI-1640 medium to study the effect of tumour supernatant on T cells. These primary breast carcinoma cells were treated with 10 μm curcumin/0·5 μm nano-curcumin/10 μm U0126/100 nm SB431542/10 μm Sunitib for 90 min, excess inhibitors were washed off and cells were incubated in fresh RPMI-1640 medium. After 72 hr of incubation, supernatants freed from cellular components were used in 1 : 1 ratio with RPMI-1640 medium to study the effect of tumour supernatant on T cells. To study the role of MEK/ERK-mediated TGF-β/IL-2 signalling in tumour-induced Treg cell augmentation, CD4+ CD25− T cells (2 × 106 cells) isolated from healthy human donors were cultured with rTGF-β/rIL-2 alone or in combination with supernatant from control-/MEK-/ERK-/TGF-β-small interfering (si) RNA-transfected breast cancer cells or in the presence/absence of neutralizing anti-TGF-β/anti-IL-2 antibodies (2 μg/ml; Santa Cruz Biotechnology, Dallas, TX).
Animal tumour model
To study the role of curcumin and nano-curcumin in reversing Treg cell augmentation in vivo, BALB/c mice bearing syngeneic breast cancer cells 4T1 were used. BALB/c mice (NCLAS, Hyderabad, India) weighing 20–25 g were maintained in a temperature-controlled room with light–dark cycle. All animal experiments were performed following Principles of laboratory animal care (NIH publication No. 85–23, revised in 1985) as well as Indian laws on ‘Protection of Animals’ under the provision of the Ethics Committee for the purpose of control and supervision of experiments on animals (Reg. No. 95/99/CPCSEA; Approval No: IAEC/BI/1/2010), Bose Institute. Mice were divided into two groups including normal set (non-tumour-bearing), tumour-bearing set (which were inoculated in the mammary fat-pad with 1 × 106 exponentially grown 4T1 cells). After 14 days of 4T1 cell inoculation, tumour-bearing set was further divided into three groups: (i) untreated, (ii) curcumin and (iii) nano-curcumin treated groups. Various doses of curcumin/nano-curcumin were fed orally on alternate days. After 28 days percentage of Treg cells induced in tumour-draining lymph nodes (popliteal) and expression levels of TGF-β-signalling intermediates leading to FoxP3 expression in these cells were determined.
Flow cytometry
For the determination of Treg cells Peridinin chlorophyll protein-conjugated anti-CD4, FITC-conjugated anti-CD25 and phycoerythrin-conjugated anti-FoxP3 antibodies were used (BD Bioscience, San Jose, CA). For the analysis of cell-surface-bound TGF-β/IL-2, CD4+ T cells were incubated with FITC-anti-TGF-β/phycoerythrin-anti-IL-2 antibody (BD Bioscience) and TGF-β/IL-2 positivity was analysed flow cytometrically (FACS Versa; BD Bioscience). For the determination of intracellular TGF-β, cancer cells were first stimulated with PMA (10 ng/ml) and ionomycin (1 μm) and transport of newly synthesized cytokines from the golgi apparatus was blocked with 10 μg/ml Brefeldin A (Sigma). After incubation for 4 hr at 37° cells were washed with PBS and permeabilized with saponin. Finally, intracellular TGF-β was analysed with FITC-tagged antibody flow cytometrically. A total of 10 000 events were acquired; cells were properly gated with Simultest LeucoGATE and analysed using CellQuest Software (BD Bioscience).
Co-immunoprecipitation, Western blotting and ELISA
For Western blot analysis cell lysates were prepared in lysis buffer [20 mm Tris–HCl (pH 7·4), 100 mm NaCl, 1% nonidet P40, 0·5% deoxycholic acid and 1 mm EGTA] containing protease and phosphatase inhibitor cocktails. A total of 50 μg of protein was separated by SDS–PAGE and transferred to nitrocellulose filter paper for Western blotting using specific antibodies, e.g. anti-MEK (rabbit), anti-phospho-MEK (mouse), anti-ERK (rabbit) anti-phospho-ERK (goat), anti-SMAD1 (mouse), anti-SMAD2 (goat), anti-SMAD3 (mouse), anti-SMAD4 (mouse), anti-phospho-SMAD1 (rabbit), anti-phospho-SMAD2 (rabbit), anti-phospho-SMAD3 (rabbit), anti-CD25 (rabbit), anti-FoxP3 (mouse), anti-JAK1 (rabbit), anti-JAK2 (rabbit), anti-JAK3 (rabbit), anti-phospho-JAK1 (rabbit), anti-phospho-JAK2 (rabbit), anti-phospho-JAK3 (goat), anti-STAT1 (rabbit), anti-STAT2 (rabbit), anti-STAT3 (rabbit), anti-STAT4 (rabbit), anti-STAT5 (rabbit), anti-STAT6 (rabbit), anti-phospho-STAT1 (goat), anti-phospho-STAT2 (rabbit), anti-phospho-STAT3 (goat), anti-phospho-STAT4 (rabbit), anti-phospho- STAT5 (goat), anti-phospho-STAT6 (goat) (Santa Cruz) and anti-TGF-β (rabbit; Cell Signaling) antibodies. For the staining of phospho- and total proteins we employed parallel Western blotting using equal amounts of loading protein to avoid any cross-reactivity. For internal control we stripped the original membrane and re-probed with internal control antibody. Histone H1 (rabbit)/α-actin (mouse) (Santa Cruz) were used as internal control. For the determination of direct interaction between two proteins, a co-immunoprecipitation technique was employed. The lysates (200 μg of protein) were incubated for 4 hr at 4° with rocking with 4 μg of specific antibody, and the immune complexes were then incubated with protein A–Sepharose beads. The JAK1/JAK3-associated STAT3/STAT5 was then detected in the immunopurified complexes by Western blot analysis. Twenty per cent of the cell lysates used for co-immunoprecipitation was blotted against anti-α-actin antibody to confirm equivalent protein loading. Protein of interest was visualized by chemiluminescence. Relative protein expression was determined by densitometric quantification of semi-quantitative chemiluminescence bands obtained in Western blot relative to actin bands.3 Relative protein expression was estimated with respect to Histone H1/α-actin. Levels of TGF-β in tumour supernatants were quantified by ELISA kit (BD Pharmingen, San Diego, CA).
siRNA and transfection
Primary breast cancer or T cells were transfected with 300 pmol of ERK-/MEK-/TGF-β-/SMAD3-/SMAD4-siRNA/control-siRNA (Santa Cruz) or lipofectamine-2000 (Invitrogen, Carlsbad, CA). separately for 12 hr as described earlier.3 T cells were transduced with lentiviral STAT3/STAT5/control-shRNA (Open Biosystems, Pittsburgh, PA) in the presence of hexadimethrine bromide (Sigma) as transduction facilitator. The protein levels were estimated by Western blotting.
Real-time quantitative RT-PCR
For quantitative PCR, total RNA was prepared from 1 × 106 cells using tripure isolation reagent. The mRNA expression levels were measured by quantitative real-time PCR using a Quanti TechTM SYBR R Green real-time-PCR kit and the iCycler real-time detection system and software according to the manufacturer's instructions. Passive reference dye (ROX) was used to normalize the SYBR Green/double-stranded DNA complex signal during analysis to correct for well-to-well variation and sampling loading error. Amplification products using SYBR Green detection were checked using melting curve with iCycler software (version-3; Bio-Rad, Hercules, CA) and by 1·5% agarose gel electrophoresis to confirm the size of the DNA fragment and that single product was formed. Samples were compared using the relative (comparative) Ct method. The Ct value, which is inversely proportional to the initial template copy number, is the calculated cycle number where the fluorescence signal emitted is significantly above background levels.3 Expression level of the housekeeping gene, GAPDH, was used to normalize for variations in amount of RNA and RNA purity. The fold induction or repression by real-time RT-PCR was calculated according to the following formula: fold change = 2−ΔΔct; where ΔΔct = Δct control − Δct treatment, and Δct = target gene ct − GAPDH ct. Oligonucleotide primer sequences used in real-time quantitative RT-PCR were as follows: CD25: sense: 5′-CGTTGCTTAGGAAACTCCTGGA-3′, antisense: 5′-GCTTTCTCGATTTGTCATGGG-3′; FoxP3: sense: 5′-CAGCACATTCCCAGAGTTCCTC-3′, antisense: 5′-GCGTGTGAACCAGTGGTAGATC-3′; GAPDH: sense: 5′-GCCATCAACGACCCCTTC-3′, antisense: 5′-AGCCCCAGCCTTCTCCA-3′.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were carried out using a ChIP assay kit (Millipore, Billerica, MA) according to a modification of the manufacturer's instructions.3 To cross-link the protein–DNA complexes, 2 × 106 cells were fixed with 1% formaldehyde for 10 min at 37°. Then they were harvested and washed twice with ice-cold PBS containing protease inhibitors (1 mm PMSF, 1 μg/ml aprotinin and 1 μg/ml pepstatin A). For the remaining steps of the protein isolation, all buffers used to isolate the proteins contained protease inhibitor cocktails. Cells were added to SDS lysis buffer and incubated on ice for 10 min. Cell lysates were sonicated to shear the DNA to lengths between 200 and 1000 base pairs and then centrifuged at 15 000 g for 10 min at 4°. The sonicated cell supernatants were diluted 10-fold in ChIP dilution buffer and pre-cleared with protein A-agarose/salmon sperm DNA for 30 min at 4° with agitation. The supernatant was recovered after pelleting the agarose by centrifugation and then incubated with specific antibody against STAT3 or STAT5 overnight at 4°. The antibody–protein–DNA complexes were collected by adding protein A-agarose/salmon sperm DNA for 1 hr at 4° with rotation. Immunoprecipitated antibody–protein–DNA complexes were washed according to the described protocol. Chromatin complexes were eluted with freshly prepared extraction buffer (1% SDS, 0·1 m NaHCO3). To reverse cross-links, 5 m NaCl was added to each eluate, and then the solution was heated to 65° for 5 hr. Proteins were digested with 10 mg/ml proteinase K for 1 hr at 45°, and DNA was recovered by phenol/chloroform extraction and ethanol precipitation. DNA fragments were amplified by PCR using FoxP3-promoter: forward: 5′-CTTCCCATTCACATGGCAGGC-3′; reverse: 5′-TTGCCCTTTACGAGTCAT CTG-3′. For ChIP re-ChIP assay, cross-linking, lysis, sonication and immunoprecipitation with STAT3-specific antibodies were performed per normal ChIP assay. After the first round of immunoprecipitation, DNA–protein complexes were eluted and subjected to another round of immunoprecipitation with STAT5-specific antibodies. Following this step, the assay was continued as a routine ChIP assay.
Preparation of curcumin nano-particles
Four grams of curcumin (Sigma) was dissolved in 1 l of distilled ethanol by stirring on a magnetic stirrer at room temperature and was filtered to obtain a clear solution. The entire solution was then stirred in a high-speed homogenizer at 16 000–20 000 g rpm and an equal volume of milliQ water containing 0·1% citric acid was added over a period of 10 min when the solution (50% ethanol) became turbid with nucleation of curcumin nano-particles. Further addition of milliQ water containing 0·1% citric acid was continued with vigorous stirring (16 000–20 000 g) till the ethanol concentration became 40% (volume/volume). Then the entire suspension was immediately homogenized in a high-pressure homogenizer at 30 000–45 000 PSI for 3 hr and spray-dried immediately in a Hitachi (Hitachi High Technologies, Tokyo, Japan) spray dryer. Particle size and surface morphology of the nano-particles were determined by a high-resolution transmission electron microscope (TEM; JOEL 2100FR) and scanning electron microscope (SEM; Carl Zeiss, Jena, Germany, EM-LEO 435). For TEM, 20 μl of curcumin nano-particle suspension was adsorbed on a carbon-coated 300-mesh copper grid, air-dried, stained with 1 m uranyl acetate and then viewed under TEM. For SEM, dried nano-curcumin/curcumin was loaded on a metal stub, gold-coated under vacuum and then imaged with the SEM.
Quantitative fluorescent imaging
For quantitative fluorescent imaging, syngeneic breast cancer cells harvested from mice were allowed to adhere to poly-l-lysine-coated slides. Adhered cells were then fixed with 3% p-formaldehyde. Nuclei were labelled with 4,6-diamidino-2-phenylindole (DAPI) and cells were imaged with an Olympus disc scanning microscope (Olympus America, Center Valley, PA) fitted with an Andor EM-CCD camera (Andor Technology, Belfast, NIR, UK). Fluorescence images were quantified with ImageJ software [NIH, Bethesda, MD (http://imagej.nih.gov/ij/)].
Statistical analysis
Values are shown as standard error of mean (SEM) except where otherwise indicated. Comparison of multiple experimental groups was performed by two-way analysis of variance followed by a post-hoc Bonferroni modification of multiple comparison t-test. Data were analysed and, when appropriate, significance of the differences between mean values was determined by a Student's t-test. Results were considered significant at P ≤ 0·05.
Results
TGF-β-induced CD25 triggered FoxP3 expression in tumour-generated Treg cells
Regulatory T cells are defined by the expression of CD4, CD25 and the transcription factor FoxP3 (CD4+ CD25+ FoxP3+). However, being a nuclear protein, FoxP3 is of limited value in the isolation of Treg cells – a major reason for many functional aspects of Treg cells remains obscure. Hence, in addition to the phenotypic marker IL-2Rα chain (CD25), IL-7Rα chain (CD127) was used to characterize definitive CD4+ CD25+ CD127− Treg cells. During the course of identifying the developmental stages of these Treg-specific markers in peripheral circulation, we observed that CD25 positivity starts in CD4+ T-cell populations from as early as 12 hr of cell-free breast tumour supernatant treatment and FoxP3 expression followed CD25 (IL-2Rα) expression (Fig.1a,b). The expression of high-affinity IL-2Rα subsequently enabled the binding of trace amounts of IL-2 present in the system on the Treg surface with higher affinity (Fig.1b). Based on this result, we presumed that IL-2/IL-2R-dependent signalling might be responsible for FoxP3 expression. This result also compelled us to investigate the mechanism of Treg cell induction. In parallel, our results depicted in Fig.1(c) show that breast tumour produced and secreted abundant TGF-β, which bound to the surface of CD4+ T cells. Considering all these results, we hypothesized that tumour-shed TGF-β induced CD25 in CD4+ T cells that harboured IL-2 to elicit FoxP3 expression.
Figure 1.

Tumour cell free supernatant induces high-affinity interleukin-2 receptor CD25 and fork-head transcription factor FoxP3 in CD4+ regulatory T (Treg) cells. Human peripheral blood mononuclear cells from normal individuals were cultured with cell-free supernatant obtained from primary culture of breast tumour cells. (a) Induction of CD25 and FoxP3 in CD4+ T cells at different time-points were determined flow cytometrically. (b) Per cent CD4+ CD25+ (CD127−)/CD4+ CD25+ FoxP3+ and IL2-binding to CD4+ T-cell surface were analysed flow cytometrically at different time-points and represented graphically. (c) Intracellular and secretion levels of transforming growth factor-β (TGF-β) in normal or control-/TGF-β-siRNA-transfected human breast cancer cells were estimated by flow cytometry and by ELISA. Per cent TGF-β-binding to CD4+ T cell surface treated with normal and siRNA-transfected breast cancer cell-supernatant were analysed flow cytometrically. Values are mean ± SEM or representative of five independent experiments.
SMAD and STAT cooperatively regulated FoxP3 expression in tumour-induced Treg cells
To validate our hypothesis we next tested the involvement of TGF-β in CD25+ Treg cell augmentation. When naive (CD4+ CD25− CD127+) T cells were treated with (i) supernatants of breast cancer cells transfected with TGF-β-siRNA, or (ii) supernatants treated with TGF-β-neutralizing antibody, augmentation of CD4+ CD25+ (CD127−) Treg cells was suppressed. In a parallel experiment, TGF-β-signalling inhibitor, SB431542, also blocked such Treg cell augmentation (Fig.2a). Hence, our results signify that tumour-shed TGF-β induced CD25 in CD4+ T cells that harboured IL-2 to elicit FoxP3 expression. For cells to respond to their environment, external stimuli must be received and transmitted to the nucleus through certain signalling molecules. Hence, we next aimed to identify the signalling molecules involved in TGF-β-mediated CD25 expression and the resultant IL-2-mediated FoxP3 expression during T-cell stimulation with tumour supernatant. For this, CD4+ CD25+ (CD127−) Treg cells, induced in tumour condition, were purified and lysed for further analysis. SMAD family members have already been identified as essential intracellular signalling components of TGF-β, which are either phosphorylated and/or translocated to the nucleus. Among different isoforms of SMAD protein, our Western blot analysis showed that SMAD3 was intensely phosphorylated by ERK1/2 (Fig.2c, left panel) and phospho-SMAD3 as well as SMAD4 were translocated to the nucleus in Treg cells (Fig.2c, right panel). At this juncture, translocation of SMAD3 and SMAD4 to nucleus, before the increase in CD25 expression at both mRNA and protein levels, raised the possibility of their involvement in CD25 up-regulation (Fig.2c). Complete eradication of CD25 expression at both protein and mRNA levels in single or double knockdown studies using siRNA against SMAD3 and SMAD4 finally confirmed their role in tumour-derived TGF-β-mediated Treg development (Fig.2d).
Figure 2.
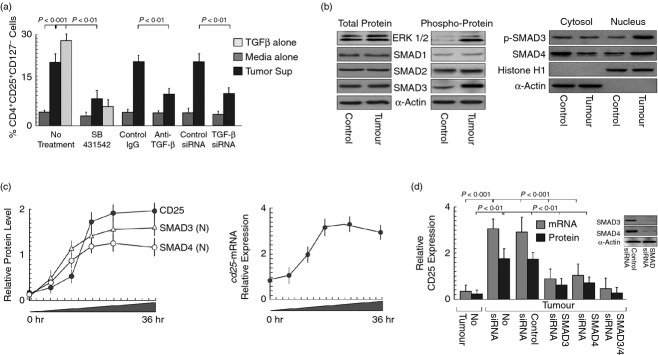
SMAD3 and SMAD4 mediated tumour-shed transforming growth factor-β (TGF-β)-induced CD25/interleukin-2 receptor α (IL-2Rα) expression on CD4+ regulatory T (Treg) cells. (a) Graphical representation of percentage of CD4+ CD25+ CD127− Treg populations in T cells cultured with recombinant-TGF-β or cell-free supernatants of control-/TGF-β-siRNA-transfected tumour cells or TGF-β neutralization antibody or TGF-β-signalling inhibitor (SB431542) supplemented cell-free tumour supernatants. (b) Western blot analysis showing total and phosphorylation status of extracellular signal-regulated kinase 1/2 (ERK1/2), SMAD1, SMAD2, SMAD3 (left panel) and nuclear translocation of phospho-SMAD3 and SMAD4 (right panel). (c) Time-dependent nuclear translocation of SMAD3/SMAD4 and induction of CD25 (left panel), as determined by semi-quantitative Western blot analysis and relative expression levels of cd25-mRNA, as determined by quantitative PCR, in tumour supernatant-treated CD4+ T cells (right panel). (d) Relative expression levels of CD25 at protein and mRNA levels were determined in SMAD3- and/or SMAD4-silenced CD4+ T cells by semi-quantitative Western blot and quantitative PCR. The house-keeping genes α-actin/GAPDH were used as internal control. Values are mean ± SEM or representative of five independent experiments.
Purified CD4+ CD25+ (CD127−) Treg cells were further analysed to discern the molecular mechanisms behind IL-2-mediated induction of FoxP3 expression. These cells exhibited increased phospho-JAK1 and phospho-JAK3 status without altering phospho-JAK2 with subsequent phosphorylation of STAT3 and STAT5 proteins among the different isoforms of STAT (Fig.3a), which coincided with FoxP3 induction in these cells. Our time-dependent study showed that when cultured with tumour supernatant, STAT3 and STAT5 hyper-activation started at 12 hr followed by FoxP3 expression at 18 hr (Fig.3b, left panel). Real-time PCR data suggest that foxp3-mRNA expression was commenced after cd25-mRNA expression (Fig.3b, right panel). Co-immunoprecipitation results depicted in Fig.3(c) showed that STAT3 was associated with JAK1 whereas STAT5 was associated with JAK3 to induce the transcription of foxp3. FoxP3 induction decreased significantly when STAT3 and STAT5 were silenced separately by RNA-interference. Interestingly, when both the isoforms were knocked-out, FoxP3 induction was completely abolished (Fig.3d, left panel). This phenomenon was also suppressed by JAK/STAT pathway inhibitor, Sunitinib (Fig.3d, left panel). All this information suggests that both the isoforms indeed contributed to FoxP3 induction and JAK/STAT signalling plays a pivotal role in Treg cell augmentation. It is well established that STAT3 and STAT5 dimerize and bind to the putative STAT-responsive elements at the responsive gene promoter region. Consistent with the presence of putative STAT-responsive elements at the foxp3 gene locus, our ChIP/Re-ChIP data demonstrated STAT3–STAT5 binding to the foxp3 promoter in these cells (Fig.3d, right panel). Interestingly, STAT3/STAT5 knock-out resulted in decreased ChIP/ReChIP signals of foxp3 promoter (Fig.3d, right panel).
Figure 3.
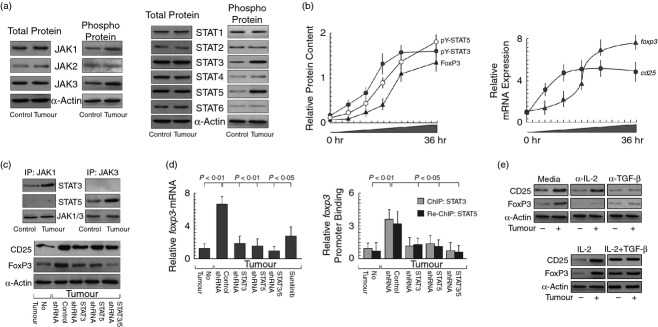
CD25/IL-2Rα-mediated FoxP3 expression through Janus kinase/signal transducer and activator of transcription (JAK/STAT) activation. (a) After incubation with tumour supernatant, CD4+ CD25+ (CD127−) regulatory T (Treg) cells were sorted and Western blot analysis for all possible form of total and phosphorylated form of JAK (left panel) and STAT (right panel) proteins was performed. (b) Time-dependent induction of pY-STAT3, pY-STAT5, FoxP3 was analysed by semi-quantitative Western blot (left panel) and relative expression level of cd25 and foxp3 mRNAs in tumour supernatant-treated CD4+ T cells were determined by quantitative PCR (right panel). (c) Co-immunoprecipitation studies revealed direct association between JAK1/STAT3 and JAK3/STAT5 in CD4+ CD25+ (CD127−) T cells (upper panel). Expression of CD25 and FoxP3 was evaluated in STAT3 and/or STAT5 siRNA transfected CD4+ CD25+ (CD127−) T cells in tumour condition (lower panel). (d) Human CD4+ T cells were transfected with STAT3- and/or STAT5-shRNA or pre-treated with Sunitinib followed by tumour supernatant incubation and relative expression levels of foxp3-mRNA were determined by quantitative PCR (right panel). ChIP re-ChIP studies were carried out to evaluate STAT3-/STAT5-associative binding at foxp3 promoter using anti-STAT3 antibodies and then by reversing the second round of immunoprecipitation with anti-STAT5 antibody of the first round immunoprecipitates followed by quantitative PCR with primers specific for the STAT-binding site of foxp3 promoter. (e) Western blot studies with interleukin-2 (IL-2)/ transforming growth factor-β (TGF-β) neutralization and recombinant-IL-2 and/or TGF-β addition clearly stated that tumour-induced CD25/FoxP3 expression in Treg cells was indeed dependent on IL-2/TGF-β. The house-keeping genes α-actin/GAPDH were used as internal control. Values are mean ± SEM or representative of five independent experiments.
Interestingly, although neutralization of IL-2 failed to bring any change in tumour supernatant-induced CD25 expression, FoxP3 induction was completely blocked (Fig.3e). In contrast, TGF-β neutralization totally inhibited up-regulation of both CD25 and FoxP3 (Fig.3e). Similarly, recombinant IL-2 alone could not induce FoxP3 in the absence of tumour supernatant whereas, when added along with TGF-β there was robust induction of FoxP3 even in the absence of tumour supernatant (Fig.3e). All of these results therefore confirmed that both SMAD3/SMAD4-induced CD25 expression and IL-2/CD25-mediated STAT3/STAT5 activation were mandatory for the conversion of CD4+ CD25− FoxP3− T cells to CD4+ CD25+ FoxP3+ definitive Treg cells.
MEK inhibitors blocked tumour-mediated induction of Treg cells by reducing TGF-β production in tumour cells
Our results signify that tumour-shed TGF-β induced CD25/IL-2Rα in CD4+ T cells that harboured IL-2 to elicit JAK/STAT-mediated foxp3 expression. Hence, we hypothesized that interference with TGF-β-signalling could be a possible way to intervene in the generation of tumour-induced Treg cells. Previously it has been shown that TGF-β production is mediated through the MEK/ERK cascade, which is hyper-activated in most of the cancer cells.36 That MEK/ERK cascade was indeed involved in TGF-β production in our system was established when primary breast cancer cells, treated with MEK inhibitor U0126/curcumin or transfected with MEK-siRNA, exhibited reduced phosphorylation and hence activation of MEK and its downstream substrate ERK1/2, which further correlated with inhibition of TGF-β expression (Fig.4a). Flow cytometric studies further confirmed that intracellular TGF-β in these cells was dependent on the MEK/ERK pathway because blocking either of them reduced TGF-β levels (Fig.4b). Similar results were obtained with secretory-TGF-β, as confirmed by ELISA (Fig.4c, left panel). In conformity with the above findings, supernatants from MEK-/ERK-inhibited breast cancer cells failed to augment CD4+ CD25+ (CD127−) Treg cells (Fig.4c, right panel). Since MEK/ERK inhibition blocked TGF-β synthesis from tumour cells, we next examined whether this effect was responsible for inhibiting the whole signalling cascade leading to FoxP3 expression in Treg cells. When CD4+ T cells were exposed to U0126-/curcumin-pretreated tumour supernatants, SMAD3 phosphorylation along with nuclear SMAD4 translocation and CD25 induction were completely diminished, which was accompanied with reduced phosphorylation of JAK1/JAK3 and STAT3/STAT5. This effect of MEK/ERK inhibition finally resulted in reduced FoxP3 expression (Fig.4d).
Figure 4.
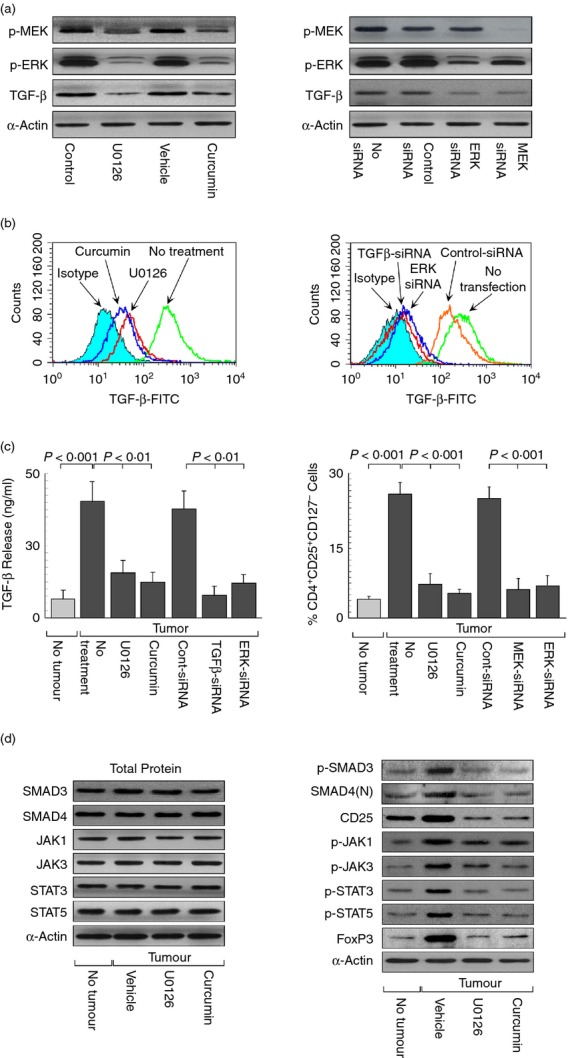
Mitogen-activated protein kinase kinase (MEK)/ extracellular signal-regulated kinase (ERK) inhibitors blocked transforming growth factor-β (TGF-β) -induced regulatory T (Treg) cell augmentation. (a) Western blot representation of phospho-MEK, phospho-ERK (p42/p44) and TGF-β patterns in untreated or U0126-/curcumin-treated and scramble control-siRNA or ERK-/MEK-siRNA-transfected breast cancer cells. (b) Histogram overlay of intracellular TGF-β levels in untreated or U0126-/curcumin-treated and untransfected or control-/TGF-β-/ERK-siRNA transfected breast cancer cells. (c) TGF-β levels in cell-free supernatant from control-/TGF-β-/ERK-siRNA transfected and U0126-/curcumin-treated breast cancer cells were determined by ELISA (left panel). Graphical representation of per cent CD4+ CD25+ (CD127−) Treg populations in T cells cultured with cell-free supernatants of U0126-/curcumin-treated and control-/MEK-/ERK-siRNA-transfected cancer cells (right panel). (d) Western blots analysis depicting the effect of U0126/curcumin on phospho-SMAD3, nuclear SMAD4, CD25 or CD25-signalling intermediates [phospho-Janus kinase (p-JAK1), p-JAK3, phospho-signal transducer and activator of transcription 3 (p-STAT3), p-STAT5] leading to FoxP3 expression in CD4+ CD25+ (CD127−) Treg cells. The housekeeping protein α-actin was used as internal control. Values are mean ± SEM or the representative of five independent experiments.
Establishing curcumin as a non-toxic immunomodulator for reversing Treg cell augmentation in tumour-bearing host
There is a usage advantage of curcumin over pharmacological MEK inhibitor U0126, as curcumin is non-toxic to normal cells (Fig.5c).37 Hence, employing curcumin to inhibit tumour-shed TGF-β, which was involved in generation of Treg cells, seemed to be a more radical approach to reverse Treg cell augmentation in the tumour microenvironment. However, in spite of its efficacy and safety, curcumin has not yet been approved as a therapeutic agent against cancer. Poor bioavailability and metabolic instability of curcumin have been highlighted as major hurdles of its use.38 Nowadays, several formulations of curcumin, which have been prepared by encapsulating curcumin into polymeric nano-particles or by trapping curcumin into dipeptide nano-particles, have been introduced to overcome the problem related to poor bioavailability. In our study, instead of using these conventional carrier-based nano-formulations that do not contain curcumin in ‘nano’ form, we have prepared nano-particles from pure curcumin. In fact, our nano-particles that contain only curcumin are less toxic in comparison to other available nano-formulations (Fig.5c). Electron microscope data (TEM and SEM) revealed that these nano-particles are homogeneous with an effective size of 200 ± 10 nm (Fig.5a) and are more efficiently internalized into target cells where these were retained for a longer time (Fig.5a,b). As a result, for a more radical therapeutic approach, we next planned to check the role of curcumin and its nano-formulation in reversing Treg cell augmentation in tumour-bearing mice. In conformity with our assumption we observed that nano-curcumin treatment significantly inhibited the induction of CD4+ CD25+ FoxP3+ Treg cells in the regional lymph nodes as well as in the tumour site of tumour-bearing mice. This nano-curcumin is 50 times more effective than curcumin alone (Fig.5d). Importantly, this nano-formulation, even at a 50 times higher dose than curcumin, showed no adverse effect in normal lymph nodes (Fig.5d). Similarly, in in vitro conditions, nano-curcumin, even at 50 times lower concentration than curcumin, blocked the signalling molecules that are responsible for TGF-β-mediated FoxP3 expression (Fig.5e).
Figure 5.
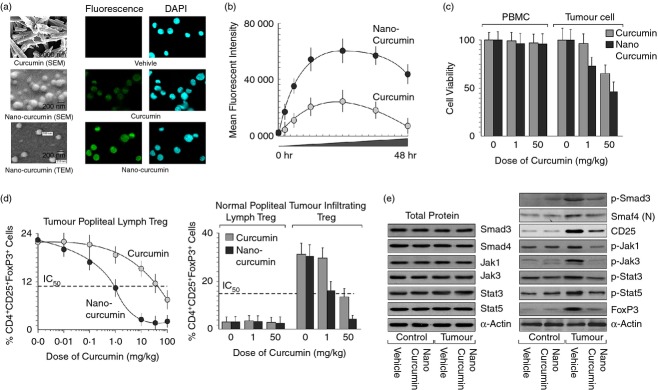
Nano-curcumin reversed regulatory T (Treg) cell augmentation in tumour-bearing mice at higher efficiency. (a) Scanning/transmission electron microscopic images of curcumin and nano-curcumin (left panel). Syngeneic breast cancer cells 4T1 were implanted into the thigh-pad of BALB/c mice and allowed to grow for 14 days. Then mice were treated with various doses of vehicle or curcumin/nano-curcumin. Green fluorescent (for curcumin)/DAPI images of 4T1 breast cancer cells isolated from vehicle/curcumin/nano-curcumin-fed mice (right panels). (b) Green fluorescent images were quantified by ImageJ software and mean fluorescent intensity was plotted against time. (c) Toxicity level of curcumin/nano-curcumin was determined by in vitro cell viability (Trypan blue dye-exclusion) assay of normal peripheral blood mononuclear cells and 4T1 cells. (d) Percentage of CD4+ CD25+ FoxP3+ Treg cells induced in tumour-draining lymph nodes (popliteal; left panel) and tumour-infiltrating lymphocytes of curcumin/nano-curcumin-treated tumour-bearing mice was determined by flow cytometry (right panel). Popliteal lymph nodes of normal mice were used as control. (e) The levels of transforming growth factor-β (TGFβ) -signalling intermediates, p-Smad3 and nuclear Smad4, leading to CD25 and p-Jak1, p-Jak3, p-Stat3, p-Stat5, leading to FoxP3 expression in these cells were determined in the presence of curcumin/nano-curcumin by Western blot. Values are mean ± SEM or the representative of five independent experiments.
Discussion
Activated through RAS-stimulated RAF activity, MEK activates a second protein kinase termed ERK. ERK regulates downstream signalling complexes of transcription factors that affect gene expression, rearrangements of the cytoskeleton and metabolism.39 ERK acts to coordinate responses to extracellular signals that result in the regulation of proliferation, differentiation, senescence and apoptosis.40 One or more activating genetic mutations of many of the components of this pathway have been found to be associated with cancers, clearly demonstrating its importance to neoplastic transformation.41 Constitutive ERK signalling results in the expression of transcriptional products, such as cyclin D1, which allow entry into the cell cycle, as well as repressing the expression of genes that inhibit proliferation.42,43 Furthermore, ERK effectors function in angiogenesis, migration, invasion and metastasis.44 Recent findings also indicated the involvement of MEK/ERK signalling in cancer cell evasion of the immune system.45 The ERK-signalling pathways differentially regulate dendritic cell maturation and modulate the initial commitment of naive helper T cells toward Th1 or Th2 subsets.46 In fact, activation of the ERK-pathway has been implicated in immune evasion by playing a role in the production of secreted immunosuppressive cytokines, such as IL-6, IL-10 and vascular endothelial growth factor.47 Adding to this knowledge, our findings identified the involvement of MEK/ERK signalling in TGF-β production from breast cancer cells, which in turn mediated the induction of Treg cells.
Although not required for generation of natural CD4+ CD25+ (CD127−) Treg cells, evidence is accumulating that TGF-β proteins are essential for the function and survival of induced Treg cells.12,14,48 Our results are in agreement with earlier reports where the addition of TGF-β to CD4+ T-cell culture markedly enhanced CD25 expression through activation of transcription factors SMAD3 and SMAD4. Because of the expression of this high-affinity receptor, these CD4+ CD25+ cells can presumably respond to physiologically low concentrations of IL-2. Interleukin-2 selectively up-regulated the expression of FoxP3 in purified CD4+ CD25+ (CD127−) T cells but not in CD4+ CD25− (CD127+) cells.49 This regulation involved the binding of activated STAT3 and STAT5 proteins to a highly conserved STAT-binding site located in the first intron of the foxp3 gene.50,51 Neutralization of IL-2 did not inhibit TGF-β-mediated enhancement of CD25 expression but completely abolished the ability of TGF-β to induce FoxP3. Our results further signify the role of tumour-derived TGF-β and IL-2 in mediating FoxP3 expression via JAK1/STAT3- and JAK3/STAT5-signalling (Fig.6).
Figure 6.
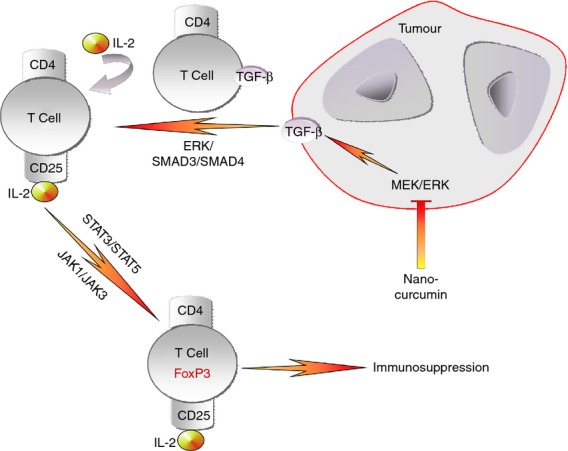
Model showing sequential events leading to induction of immunosuppressive CD4+ CD25+ FoxP3+ regulatory T (Treg) cells in breast cancer by way of transforming growth factor-β/interleukin-2 (TGF-β/IL-2) signalling pathways involving extracellular signal-regulated kinase (ERK)/SMAD and Janus kinase/signal transducer and activator of transcription (JAK/STAT).
Target-based therapies are widely considered to be the future of cancer treatment and much attention has been focused on developing inhibitors of the MEK/ERK signalling pathway.52 Among several pharmacological MEK inhibitors, PD98059 is a specific MEK inhibitor and a useful tool in studying the MEK/ERK pathway.53 In cell line studies, PD98059 treatment has been shown to reverse the malignant phenotype of mutated RAS.54 Another highly specific, non-ATP-competitive inhibitor, U0126, used in pre-clinical studies is more potent than PD98059. Both compounds, however, have limited in vivo activity in blocking phosphorylation of ERK and so were not developed further.55 Additionally, phase I studies of other MEK inhibitors have shown acceptable toxicity profiles at doses required to inhibit the target.55 Several studies from our laboratory have already acknowledged the anti-cancer and immunoprotective role of the plant polyphenol curcumin in different tumour models.56–58 Hence for a more radical therapeutic approach, we next planned to check the role of curcumin in reversing Treg cell induction as a potent MEK/ERK inhibitor. Fortunately, curcumin, like other MEK/ERK inhibitors, down-regulated TGF-β expression in breast cancer cells by inhibiting MEK-mediated ERK activation. Although, acknowledged globally as a ‘wonder drug of the future’ because of its great potential abilities to prevent chronic diseases, low bioavailability of curcumin has so far limited its medical use.59 The low bioavailability of curcumin has been attributed to its very low aqueous solubility, tendency to degrade in the gastrointestinal tract in the physiological environment, high rate of metabolism, and rapid systemic elimination.60 Nanotechnology-based novel strategies are being aggressively explored worldwide to enhance curcumin's bioavailability and to reduce the perceived toxicity because they offer several other additional benefits such as improved cellular uptake, enhanced solubility and excellent blood stability.61 In this regard, the nano-curcumin used in this study being only pure curcumin is expected not to have any toxicity because no other molecule is present in the formulation. Our results depicted that even at 50 times less concentration than normal curcumin, nano-curcumin, by inhibiting MEK/ERK, blocked TGF-β-mediated CD4+ CD25+ FoxP3+ Treg cell augmentation in tumour-bearing hosts.
Together our results explain the intricacies of MEK/ERK-mediated TGF-β-signalling in FoxP3+ Treg cell induction in the tumour microenvironment and its reversal by curcumin thereby strengthening the candidature of curcumin, particularly the nano formulation, as a pharmacologically and immunologically safe anti-cancer drug.
Acknowledgments
The authors want to thank Ms S. Das and Ms S. Samanta for technical assistance. This work was supported by grants from the Department of Biotechnology and Council for Scientific and Industrial Research Government of India.
Disclosures
There is no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Flow cytometric analysis of CD24− and ESA-positivity of breast cancer cells isolated from a patient with breast cancer.
References
- Gelmon KA, Eisenhauer EA, Harris AL, Ratain MJ, Workman P. Anticancer agents targeting signaling molecules and cancer cell environment: challenges for drug development? J Natl Cancer Inst. 1999;91:1281–7. doi: 10.1093/jnci/91.15.1281. [DOI] [PubMed] [Google Scholar]
- Beyer M, Kochanek M, Giese T, Endl E, Weihrauch MR, Knolle PA, Classen S, Schultze JL. In vivo peripheral expansion of naive CD4+CD25highFoxP3+ regulatory T cells in patients with multiple myeloma. Blood. 2006;107:3940–9. doi: 10.1182/blood-2005-09-3671. [DOI] [PubMed] [Google Scholar]
- Hossain DMS, Panda A, Manna A, et al. Novel function of FoxP3 as a co-transcription factor of STAT3 in tumor-induced Treg cells. Immunity. 2013;39:1057–69. doi: 10.1016/j.immuni.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Shen X, Li N, Li H, Zhang T, Wang F, Li Q. Increased prevalence of regulatory T cells in the tumor microenvironment and its correlation with TNM stage of hepatocellular carcinoma. J Cancer Res Clin Oncol. 2010;136:1745–54. doi: 10.1007/s00432-010-0833-8. [DOI] [PubMed] [Google Scholar]
- Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–62. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- Ramsdell F. Foxp3 and natural regulatory T cells: key to a cell lineage? Immunity. 2003;19:165–8. doi: 10.1016/s1074-7613(03)00207-3. [DOI] [PubMed] [Google Scholar]
- Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–84. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- Marson A, Kretschmer K, Frampton GM, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–5. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009;21:274–80. doi: 10.1016/j.coi.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Gao Q, Qiu SJ, Fan J, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25:2586–93. doi: 10.1200/JCO.2006.09.4565. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, Strober W. TGF-β1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol. 2004;172:834–42. doi: 10.4049/jimmunol.172.2.834. [DOI] [PubMed] [Google Scholar]
- Javelaud D, van Kempen L, Alexaki VI, Le Scolan E, Luo K, Mauviel A. Efficient TGF-β/SMAD signaling in human melanoma cells associated with high c-SKI/SnoN expression. Mol Cancer. 2011;10:1–2. doi: 10.1186/1476-4598-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Wang J, Zhang F, et al. Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells. J Immunol. 2010;184:4295–306. doi: 10.4049/jimmunol.0903418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression – a diverse arsenal for a moving target. Immunology. 2008;124:13–22. doi: 10.1111/j.1365-2567.2008.02813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated Tcells and NF-κB to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci USA. 2005;102:5138–43. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botti C, Seregni E, Ferrari L, Martinetti A, Bombardieri E. Immunosuppressive factors: role in cancer development and progression. Int J Biol Markers. 1998;13:51–69. doi: 10.1177/172460089801300201. [DOI] [PubMed] [Google Scholar]
- Seder RA, Marth T, Sieve MC, Strober W, Letterio JJ, Roberts AB, Kelsall B. Factors involved in the differentiation of TGF-β-producing cells from naïve CD4+ T cells: IL-4 and IFN-γ have opposing effects, while TGF-β positively regulates its own production. J Immunol. 1998;160:5719–28. [PubMed] [Google Scholar]
- Mesa MC, Gutiérrez L, Duarte-Rey C, Angel J, Franco MA. A TGF-β mediated regulatory mechanism modulates the T cell immune response to rotavirus in adults but not in children. Virology. 2010;399:77–86. doi: 10.1016/j.virol.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat Med. 2001;7:1118–22. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- Lan Q, Zhou X, Fan H, et al. Polyclonal CD4+Foxp3+ Treg cells induce TGFβ-dependent tolerogenic dendritic cells that suppress the murine lupus-like syndrome. J Mol Cell Biol. 2012;4:409–19. doi: 10.1093/jmcb/mjs040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroukhova M, Seguin-Devaux C, Oriss TB, Dixon-McCarthy B, Yang L, Ameredes BT, Corcoran TE, Ray A. Tolerance induced by inhaled antigen involves CD4+ T cells expressing membrane-bound TGF-β and FOXP3. J Clin Invest. 2004;114:28–38. doi: 10.1172/JCI20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Zhang Z, Chung Y, Reynolds JM, Lin X, Jetten AM, Feng XH, Dong C. Smad3 differentially regulates the induction of regulatory and inflammatory T cell differentiation. J Biol Chem. 2009;284:35283–6. doi: 10.1074/jbc.C109.078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra N, Robertson E, Kang J. SMAD2 is essential for TGFβ-mediated Th17 cell generation. J Biol Chem. 2010;285:29044–8. doi: 10.1074/jbc.C110.156745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bich-Thuy LT, Dukovich M, Peffer NJ, Fauci AS, Kehrl JH, Greene WC. Direct activation of human resting T cells by IL 2: the role of an IL 2 receptor distinct from the Tac protein. J Immunol. 1987;139:1550–6. [PubMed] [Google Scholar]
- Oppenheim JJ. IL-2: more than a T cell growth factor. J Immunol. 2007;179:1413–4. doi: 10.4049/jimmunol.179.3.1413. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Mandal D, Saha B, Sen GS, Das T, Sa G. Curcumin prevents tumor-induced T cell apoptosis through Stat-5a-mediated Bcl-2 induction. J Biol Chem. 2007;282:15954–64. doi: 10.1074/jbc.M608189200. [DOI] [PubMed] [Google Scholar]
- Lu L, Zhou X, Wang J, Zheng SG, Horwitz DA. Characterization of protective human CD4CD25 FOXP3 regulatory T cells generated with IL-2. TGF-β and retinoic acid. PLoS One. 2010;5:e15150. doi: 10.1371/journal.pone.0015150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh BB, Bhattacharya P, Gopisetty A, Sheng J, Vasu C, Prabhakar BS. IL-1β promotes TGF-β1 and IL-2 dependent Foxp3 expression in regulatory T cells. PLoS One. 2011;6:e21949. doi: 10.1371/journal.pone.0021949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhi MS, Meda F, Wang P, Samyn M, Mieli-Vergani G, Vergani D, Ma Y. Expansion and de novo generation of potentially therapeutic regulatory T cells in patients with autoimmune hepatitis. Hepatology. 2008;47:581–91. doi: 10.1002/hep.22071. [DOI] [PubMed] [Google Scholar]
- Murawski MR, Litherland SA, Clare-Salzler MJ, Davoodi-Semiromi A. Upregulation of Foxp3 expression in mouse and human Treg is IL-2/STAT5 dependent: implications for the NOD STAT5B mutation in diabetes pathogenesis. Ann N Y Acad Sci. 2006;1079:198–204. doi: 10.1196/annals.1375.031. [DOI] [PubMed] [Google Scholar]
- Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor β-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–90. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- Pallandre JR, Brillard E, Créhange G, et al. Role of STAT3 in CD4+CD25+FOXP3+ regulatory lymphocyte generation: implications in graft-versus-host disease and antitumor immunity. J Immunol. 2007;179:7593–604. doi: 10.4049/jimmunol.179.11.7593. [DOI] [PubMed] [Google Scholar]
- Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-β1-induced EMT in vitro. Neoplasia. 2004;6:603–10. doi: 10.1593/neo.04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arce F, Breckpot K, Stephenson H, Karwacz K, Ehrenstein MR, Collins M, Escors D. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 2011;63:84–95. doi: 10.1002/art.30099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berginc K, Trontelj J, Basnet NS, Kristl A. Physiological barriers to the oral delivery of curcumin. Pharmazie. 2012;67:518–24. [PubMed] [Google Scholar]
- Harrison RE, Turley EA. Active erk regulates microtubule stability in H-ras-transformed cells. Neoplasia. 2001;3:385–94. doi: 10.1038/sj.neo.7900180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- Saha S, Adhikary A, Bhattacharyya P, Das T, Sa G. Death by design: where curcumin sensitizes drug-resistant tumours. Anticancer Res. 2012;32:2567–84. [PubMed] [Google Scholar]
- Choudhuri T, Pal S, Das T, Sa G. Curcumin selectively induces apoptosis in deregulated cyclin D1 expressed cells at G2 phase of cell cycle in a p53-dependent manner. J Biol Chem. 2005;280:20059–68. doi: 10.1074/jbc.M410670200. [DOI] [PubMed] [Google Scholar]
- Wilkinson MG, Millar JB. Control of the eukaryotic cell cycle by MAP kinase signaling pathways. FASEB J. 2000;14:2147–57. doi: 10.1096/fj.00-0102rev. [DOI] [PubMed] [Google Scholar]
- Zohrabian VM, Forzani B, Chau Z, Murali R, Jhanwar-Uniyal M. Rho/ROCK and MAPK signaling pathways are involved in glioblastoma cell migration and proliferation. Anticancer Res. 2009;29:119–23. [PubMed] [Google Scholar]
- Tague SE, Muralidharan V, D'Souza-Schorey C. ADP-ribosylation factor 6 regulates tumor cell invasion through the activation of the MEK/ERK signaling pathway. Proc Natl Acad Sci USA. 2004;101:9671–6. doi: 10.1073/pnas.0403531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara T, Moroi Y, Uchi H, Furue M. Differential role of MAPK signaling in human dendritic cell maturation and Th1/Th2 engagement. J Dermatol Sci. 2006;42:1–11. doi: 10.1016/j.jdermsci.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–6. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Hossain DSM, Mohanty S, et al. Curcumin reverses T cell-mediated adaptive immune dysfunctions in tumor-bearing hosts. Cell Mol Immunol. 2010;7:306–15. doi: 10.1038/cmi.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn E, Nelson EA, Mohseni M, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–9. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Kanno Y, Kerenyi M, et al. Non redundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–75. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–6. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- Richards JD, Davé SH, Chou CH, Mamchak AA, DeFranco AL. Inhibition of the MEK/ERK signaling pathway blocks a subset of B cell responses to antigen. J Immunol. 2001;166:3855–64. doi: 10.4049/jimmunol.166.6.3855. [DOI] [PubMed] [Google Scholar]
- Hotokezaka H, Sakai E, Kanaoka K, Saito K, Matsuo K, Kitaura H, Yoshida N, Nakayama K. U0126 and PD98059, specific inhibitors of MEK, accelerated differentiation of RAW264.7 cells into osteoclast-like cells. J Biol Chem. 2002;277:47366–72. doi: 10.1074/jbc.M208284200. [DOI] [PubMed] [Google Scholar]
- Röttinger E, Besnardeau L, Lepage T. A Raf/MEK/ERK signaling pathway is required for development of the sea urchin embryo micromere lineage through phosphorylation of the transcription factor Ets. Development. 2004;131:1075–87. doi: 10.1242/dev.01000. [DOI] [PubMed] [Google Scholar]
- Sen GS, Mohanty S, Hossain DMS, et al. Curcumin enhances the efficacy of chemotherapy by tailoring p65NFκB-p300cross-talk in favor of p53-p300 in breast cancer. J Biol Chem. 2011;286:42232–47. doi: 10.1074/jbc.M111.262295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty J, Banerjee S, Ray P, et al. Gain of cellular adaptation due to prolong p53 impairment leads to functional switch-over from p53 to p73 during DNA damage in acute myeloid leukemia cells. J Biol Chem. 2010;285:33104–12. doi: 10.1074/jbc.M110.122705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain DMS, Bhattacharyya S, Das T, Sa G. Curcumin: the multi-targeted therapy for cancer regression. Front Biosci (Schol Ed) 2012;4:335–55. doi: 10.2741/272. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Mandal D, Sen GS, et al. Tumor-induced oxidative stress perturbs NFκB activity augmenting TNFα-mediated T cell death: protection by curcumin. Cancer Res. 2007;67:362–70. doi: 10.1158/0008-5472.CAN-06-2583. [DOI] [PubMed] [Google Scholar]
- Lee JC, Kinniry PA, Arguiri E, et al. Dietary curcumin increases antioxidant defenses in lung, ameliorates radiation-induced pulmonary fibrosis, and improves survival in mice. Radiat Res. 2010;173:590–601. doi: 10.1667/RR1522.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T, Sa G, Saha B, Das K. Multifocal signal modulation therapy of cancer: ancient weapon, modern targets. Mol Cell Biochem. 2010;336:85–95. doi: 10.1007/s11010-009-0269-0. [DOI] [PubMed] [Google Scholar]
- Bisht S, Feldmann G, Soni S, Ravi R, Karikar C, Maitra A, Maitra A. Polymeric nanoparticle-encapsulated curcumin (“nanocurcumin”): a novel strategy for human cancer therapy. J Nanobiotechnology. 2007;5:3. doi: 10.1186/1477-3155-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Flow cytometric analysis of CD24− and ESA-positivity of breast cancer cells isolated from a patient with breast cancer.