

Abstract
NOD.H-2h4 mice develop spontaneous autoimmune thyroiditis (SAT) with chronic inflammation of thyroids by T and B cells. B-cell deficient (B–/–) mice are resistant to SAT but develop SAT if regulatory T (Treg) cells are transiently depleted. We established a transfer model using splenocytes from CD28–/– B–/– mice (effector cells and antigen-presenting cells) cultured with or without sorted Treg cells from Foxp3-GFP wild-type (WT) or B–/– mice. After transfer to mice lacking T cells, mice given Treg cells from B–/– mice had significantly lower SAT severity scores than mice given Treg cells from WT mice, indicating that Treg cells in B–/– mice are more effective suppressors of SAT than Treg cells in WT mice. Treg cells from B–/– mice differ from WT Treg cells in expression of CD27, tumour necrosis factor receptor (TNFR) II p75, and glucocorticoid-induced TNFR-related protein (GITR). After transient depletion using anti-CD25 or diphtheria toxin, the repopulating Treg cells in B–/– mice lack suppressor function, and expression of CD27, GITR and p75 is like that of WT Treg cells. If B–/– Treg cells develop with B cells in bone marrow chimeras, their phenotype is like that of WT Treg cells. Addition of B cells to cultures of B–/– Treg and T effector cells abrogates their suppressive function and their phenotype is like that of WT Treg cells. These results establish for the first time that Treg cells in WT and B–/– mice differ both functionally and in expression of particular cell surface markers. Both properties are altered after transient depletion and repopulation of B–/– Treg cells, and by the presence of B cells during Treg cell development or during interaction with effector T cells.
Keywords: autoimmunity, B cells, regulatory T cell
Introduction
NOD.H-2h4 mice develop autoimmune thyroiditis spontaneously (SAT),1–4 characterized by infiltration of the thyroid by T and B cells, destruction of thyroid follicles and production of antibodies to mouse thyroglobulin (MTg).1,4,5 Development of SAT is facilitated by addition of NaI to the drinking water, although many mice maintained on normal water also develop thyroid lesions and produce anti-MTg antibodies.1,2 B cells are required for the development of SAT, and anti-MTg autoantibody levels generally correlate with SAT severity scores.5 B-cell-deficient (B–/–) NOD.H-2h4 mice do not develop SAT5 and if the majority of B cells in wild-type (WT) NOD.H-2h4 mice are transiently depleted by administration of anti-IgM5 or anti-CD206 in the first month of life, SAT severity is greatly reduced in adults even though B cells have repopulated the peripheral lymphoid organs.5,6 The resistance of B–/– and B-cell-depleted WT mice to SAT was shown to be the result of the activity of CD4+ CD25+ regulatory T (Treg) cells, because these mice develop SAT if Treg cells are transiently depleted by administration of anti-CD25.7
B–/– mice are resistant to most, if not all, spontaneous autoimmune diseases.5,8–11 Consistent with our studies in SAT, B–/– mice that are resistant to other spontaneous autoimmune diseases such as diabetes and Sjögren syndrome, develop those diseases when Treg cells are transiently depleted.12,13 In contrast, the same strains of mice that have B cells spontaneously develop SAT, diabetes and Sjögren syndrome without a requirement for Treg depletion, although some WT NOD and NOD.H-2h4 mice develop more severe disease following Treg depletion.13–16 Taken together, these results suggest an important role for Treg cells in suppressing SAT in mice that lack B cells, and suggest that Treg cells in B–/– mice may have increased suppressive function that impacts their ability to develop SAT. Alternatively, Treg cells in WT and B–/– mice could have comparable in vivo suppressive function, but B cells in WT mice could limit the function of Treg cells or promote activation of effector T (Teff) cells that are more resistant to suppression.
Several previous studies have determined if Treg cells in B–/– or B-cell-depleted mice differ functionally from those in WT mice. Using the ability of Treg cells from WT and B–/– or B-cell-depleted (anti-CD20) mice to suppress T-cell proliferation in vitro as a readout, increased suppressive function of Treg cells from B-cell-depleted mice was reported by one group,17 whereas others reported that Treg cells in WT mice had comparable,18,19 or reduced20 function compared with Treg cells in B–/– or B-cell-depleted mice. With respect to in vivo function, Treg cells from WT and B–/– B6 mice showed comparable activation and migration to the central nervous system after immunization with MOG peptide to induce expeirmental autoimmune encephalomyelitis (EAE).18 In contrast, Hamel et al.17 reported that Treg cells from anti-CD20-treated BALB/c mice were more effective than Treg cells from control mice in their ability to suppress development of experimental arthritis. The basis for the differences in results in these different studies are not readily apparent, and further studies are needed to answer this important question. In addition, while many reports indicate that multiple subsets of Treg cells can be distinguished by differences in suppressive function and expression of particular cell surface markers,21–29 phenotypic differences between Treg cells in WT versus B-cell-deficient mice have previously been addressed in only one study that we know of.18
The goal of this study was to test the hypothesis that Treg cells in unmanipulated B–/– mice differ functionally and/or phenotypically from Treg cells in WT mice since the former are more effective at suppressing SAT development. Because transient depletion of Treg is permissive for SAT development in B–/– mice, these experiments also tested the hypothesis that Treg cells that repopulate B–/– mice after transient Treg depletion have reduced suppressive function that allows for the development of autoimmunity. The results demonstrate that Treg cells in WT and B–/– mice differ both functionally and phenotypically, and following transient Treg depletion, the repopulating Treg cells in B–/– mice lack suppressive function and are phenotypically different from Treg cells in unmanipulated B–/– mice.
Materials and methods
Mice
NOD.H-2h4 mice express H-2Kk, I-Ak and Db on the NOD background.30 Mice were bred and maintained in the animal facility at the University of Missouri. All animal protocols were approved by the University of Missouri Animal Care and Use Committee. B–/– NOD.H-2h4 and CD28–/–B–/– NOD.H-2h4 mice were described previously.5,31 Foxp3-GFP WT and B–/– NOD.H-2h4 mice, generated as described previously31 were used for sorting, culture and transfer of Treg cells. Foxp3-diphtheria toxin receptor (DTR) transgenic NOD mice, provided by Drs Diane Mathis and Christophe Benoist, Harvard School of Medicine,32 were crossed with WT or B–/– NOD.H-2h4 mice and F2 offspring were selected for expression of the NOD.H-2h4 MHC by flow cytometry and for DTR expression by PCR analysis of tail DNA. T-cell receptor α deficient (TCR-α–/–) NOD.H-2h4 mice were generated by crossing TCR-α–/– NOD mice with NOD.H-2h4 mice.6 TCR-α–/– NOD.H-2h4 mice were used as recipients of cultured cells.
Cell culture system
Splenocytes from CD28–/– B–/– NOD.H-2h4 mice were cultured as previously described33,34 in the presence or absence of sorted Treg cells. Briefly, splenocytes from CD28–/– B–/– NOD.H-2h4 mice were cultured for 3 days with or without sorted Treg cells (80 : 1 ratio of splenocytes to Treg cells) in the presence of 25 μg/ml mouse thyroglobulin, 10 ng/ml interleukin-12 (IL-12) and 5 ng/ml IL-2 in fetal calf serum-supplemented RPMI-1640, harvested, counted and transferred intravenously into TCR-α–/– mice (5 × 106/mouse). The 80 : 1 ratio of splenocytes to Treg cells was determined to be above the minimum number of Treg cells required to inhibit SAT development (data not shown). All recipients were given 0·08% NaI water for 8 weeks and thyroids were assessed histologically. In some experiments, B cells from TCR-α–/– NOD.H-2h4 mice were added to cultures. The ratio of CD28–/– B–/– splenocytes to TCR-α–/– B cells was 2 : 1 and the ratio of splenocytes to Treg cells was maintained at 80 : 1. For example, cultures not containing additional B cells were plated with 80 × 106 effector splenocytes plus 1 × 106 sorted Treg cells in two plates, while cultures containing added B cells were plated with 80 × 106 effector splenocytes, 40 × 106 TCR-α–/– B cells, plus 1·5 × 106 sorted Treg cells in three plates.
Treg sorting
Splenocytes from WT or B–/– Foxp3-GFP NOD.H-2h4 mice were incubated with anti-CD4 (RM4-5-allophycocyanin) for 30 min at 4°. Cells were washed and sorted using the DAKO MoFlo XDP cell sorter (Beckman Coulter Inc., Fullerton, CA) and used for cultures as described above. Alternatively, WT or B–/– Foxp3-DTR NOD.H-2h4 splenocytes were incubated with anti-CD4 (RM4-5-allophycocyanin) (eBioscience) and anti-DTR (BAF259 biotin) (R&D Systems, Minneapolis, MN) for 30 min at 4° followed by streptavidin-phycoerythrin (PE) for 30 min at 4°. The cells were then washed and sorted.
Assessment of thyroiditis
After 8 weeks, thyroids were collected and one thyroid lobe from each mouse was fixed in formalin, sectioned and stained with haematoxylin & eosin as described previously.1,5 All slides were scored independently in a blinded fashion by two individuals. Differences in interpretation were rare, but when rare differences in scoring arose, final scores were agreed upon by both scorers. Thyroid histopathology was scored for the extent of thyroid follicle destruction using a scale of 0 to 4+ as described previously.1,4,5 In brief, a score of 0 indicates a normal thyroid, and 0+ indicates a few inflammatory cells infiltrating the thyroids and/or mild follicular changes. A 1+ severity score is defined as an infiltrate of at least 125 cells in one or several foci, and a 2+ score represents 10–20 foci of cellular infiltration, each the size of several follicles, with destruction of up to one-quarter of the gland. A 3+ score indicates that one-quarter to one-half of the thyroid follicles are destroyed or replaced by infiltrating inflammatory cells, and, a score of 4+ indicates that more than one-half of the thyroid follicles are destroyed. Thyroid lesions in NOD.H-2h4 mice reach maximal severity 8 weeks after mice are given NaI water beginning at 2 months of age; lesions are chronic and remain relatively unchanged in severity for several months.1,5
Cell surface and intracellular staining
For determination of Treg numbers, spleen cells of WT or B–/– NOD.H-2h4 mice were incubated (1 × 106 cells/100 μl) with antibodies against CD4 (RM4-5-Peridinin chlorophyll protein-Cy5.5), CD25 (PC61-PE), and Foxp3 (FJK-16s-allophycocyanin) or isotype control antibody for 30 min at 4°. Cells were washed and data were collected using the DAKO CyAN flow cytometer (Glostrup, Denmark) and analysed using summit software version 5.2 (Beckman Coulter Inc., Fullerton, CA). For cell surface phenotyping, splenocytes were incubated (1 × 106 cells/100 μl) with antibodies against CD4 (RM4-5-Peridinin chlorophyll protein-Cy5.5), CD28 (E18-FITC) Foxp3 (FJK-16s-allophycocyanin), and tumour necrosis factor receptor (TNFR) II p75 (TR75-89-PE), CD27 (LB.7F9-PE), CD103 (2E7-PE) or glucocorticoid-induced TNFR-related protein (GITR; DTA-1-PE) for 30 min at 4°. For detection of intracellular CTLA4, cells were stained for surface markers, then with eBioscience FoxP3 Staining Buffer Set and incubated (1 × 106 cells/100 μl) with anti-CTLA4 (UC10-2B9-PE) Cells were washed and the data were collected and analysed as above. Antibodies were purchased from eBioscience (San Diego, CA) and BioLegend (San Diego, CA).
Transient depletion of Treg cells
For transient depletion of Treg cells using anti-CD25, mice were given 0·5 mg rat anti-mouse CD25 monoclonal antibody PC61 as previously described.7 Treg cells begin to repopulate the spleen in 7–10 days and return to pre-injection levels in B–/– mice after 5–6 weeks (data not shown). Foxp3-DTR NOD.H-2h4 mice were given 3 μg diphtheria toxin (DT) (Sigma Chemicals, St Louis, MO) intraperitoneally on two consecutive days. This regimen was found to result in almost complete splenic Treg depletion and did not negatively affect the health of the mice. Treg cells return to normal numbers by 5 days after the second DT injection in both WT and B–/– mice.
Bone marrow chimeras
Bone marrow chimeras were generated as previously described.5,35 Briefly, recipient B–/– NOD.H-2h4 mice were irradiated (1000 Gy) and reconstituted with 6 × 106 bone marrow cells from B–/– or WT Foxp3-GFP mice ± 6 × 106 bone marrow cells from TCR-α–/– NOD.H-2h4 donors as a source of precursor B cells. After 6 weeks, when peripheral lymphocytes had reconstituted the hosts, mice were given NaI in their drinking water. After 8 weeks, expression of CD27, p75, GITR and glycoprotein A repetitions predominant (GARP) on CD4+ Foxp3+ Treg was determined by flow cytometry. Chimerism of host mice was confirmed by flow cytometry.
Statistical analysis
Statistical analysis was performed using graphpad prism software version 4.0 (GraphPad Software, La Jolla, CA) with the non-parametric Mann–Whitney U-test or Student's t-test. A value of P < 0·05 was considered statistically significant.
Results
Treg cells in B–/– and WT NOD.H-2h4 mice differ in their ability to suppress SAT
To test the hypothesis that Treg in B–/– mice are more effective suppressors than those from WT mice, it was necessary to devise a protocol where Treg/Teff interactions and Treg sources and numbers could be controlled, and where the two Treg populations could interact with the same pool of Teff cells and antigen-presenting cells (APC) in the absence of B cells. This was achieved using a culture system in which populations of potential effector cells and APC were cultured before transfer to recipient mice.33 Splenocytes from CD28–/– B–/– NOD.H-2h4 mice were used as the source of Teff cells and APC because they do not have functional Treg and their effector cells are competent for development of SAT.31 It was important to exclude B cells in the cultures as they could influence the Treg/Teff T-cell interactions. WT and B–/– Foxp3-GFP NOD.H-2h4 mice, in which GFP expression is controlled by the Foxp3 promoter, were used as the source of Treg cells for most experiments. CD4+ GFP+ (Foxp3+) Treg cells were sorted, and co-cultured with CD28–/– B–/– splenocytes for 3 days. Cells were harvested and transferred intravenously (5 × 106 per recipient) to TCR-α–/– NOD.H-2h4 mice. Recipients were given NaI water, and evaluated for SAT 8 weeks later. For some experiments, sorted Treg cells from Foxp3-DTR mice were used as the source of Treg cells.
Recipients of APC/Teff cells that were cultured with Treg cells from WT mice had significantly reduced average SAT severity scores compared with those that received only Teff cells (Fig.1a; P < 0·05). However, when an equal number of Treg from B–/– mice was cultured with the same effector/APC population, SAT in recipient mice was suppressed to a much greater extent (P < 0·001). TCR-α–/– mice that did not receive T cells did not develop SAT (Fig.1a). Similar results were obtained when splenocytes from Treg-depleted CD28+/+ B–/– NOD.H-2h4 mice were used as the source of effector cells and APC (data not shown).
Figure 1.
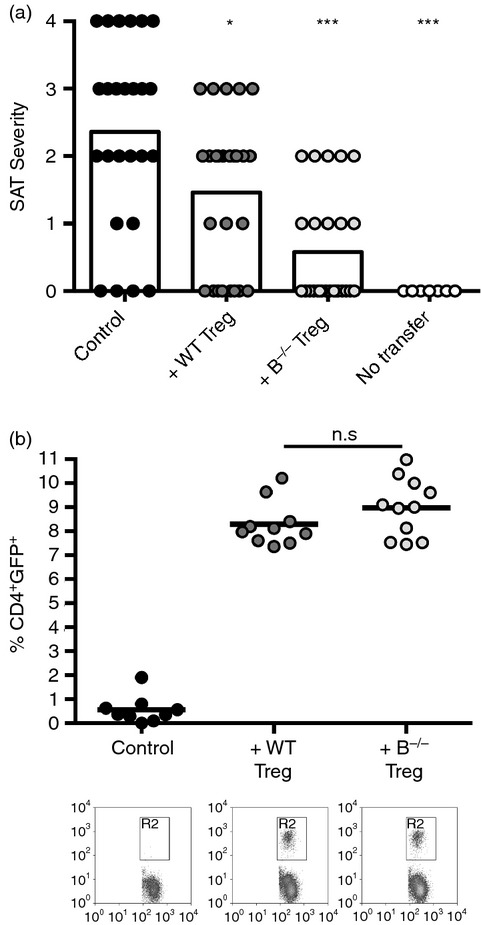
Regulatory T (Treg) cells from B-cell-deficient (B–/–) mice are more effective suppressors of spontaneous autoimmune thyroiditis (SAT) than Treg cells from wild-type (WT) mice. T-cell receptor α-deficient (TCR-α–/–) NOD.H-2h4 mice were given 5 × 106 CD28–/– B–/– splenocytes cultured in the presence or absence of sorted Foxp3+ Treg cells from naive WT or B–/– Foxp3-GFP mice. All mice were given NaI in their water, and thyroids and spleens were removed 2 months later. (a). SAT severity scores of recipient mice. Each symbol represents an individual mouse. Control mice received cultured cells without added Treg cells. SAT was suppressed more effectively by Treg cells from B–/– mice (***P < 0·001) than by Treg cells from WT mice (*P < 0·05; Mann–Whitney non-parametric test). Results are pooled from five experiments: n = 25 (Control), n = 26 (+WT Treg) and n = 26 (+B–/– Treg), and n = 7 (No transfer). (b). Percentages of CD4+ GFP+ cells in spleens of recipient mice were determined by flow cytometry. Representative flow show CD4 and FoxP3 expression on CD4+ cells. The results represent n = 9 (Control), n = 10 (+WT Treg), and n = 11 (+B–/– Treg) mice per group and are pooled from two independent experiments. Student's t-test.
To determine if the apparent differences in function of Treg cells in WT and B–/– mice could be explained by differences in survival or proliferation of Treg in recipient mice, recipient spleens were analysed for CD4 and GFP by flow cytometry. Recipients of WT or B–/– Treg had similar percentages of GFP+ CD4+ T cells (Fig.1b) (P = 0·17). The absolute numbers of GFP+ Treg cells in recipient spleens at the end of the experiment were also comparable and greater than the number of Treg cells transferred (about 2 × 104 Treg cells transferred compared with nearly 5 × 106 Treg cells per recipient spleen at the end of the experiment). Because Treg cells in WT and B–/– mice are comparable in their ability to expand and survive in recipient mice, the differences in suppression must be a result of inherent functional differences between Treg cells in WT and B–/– mice.
Treg cells in B–/– mice differ phenotypically from WT Treg cells
Differential expression of certain cell surface proteins has been associated with differences in Treg cell function.24–29,36–39 Given the differences in the ability of Treg cells from WT and B–/– mice to suppress SAT (Fig.1a), it was of interest to determine if the Treg populations differed phenotypically. To address this question, sorted CD4+ Foxp3+ Treg cells from naive WT and B–/– Foxp3GFP mice were stained and compared with respect to expression of several markers that have been used to distinguish Treg subsets in other studies. Expression of common Treg markers such as CTLA-4 was not significantly different between these two Treg populations, (Fig.2a and data not shown). The two Treg populations also did not differ in their intensity of CD25 or Foxp3 expression and both numbers and percentages of CD25+ and Foxp3+ cells were comparable (see Supporting information, Fig. S1, and data not shown). Expression of three cell surface markers, CD27, GITR and p75 was consistently reduced in CD4+ Foxp3+ Treg cells from B–/– mice compared with Treg cells of WT mice (Fig.2a,b), indicating that there are phenotypic differences that can distinguish Treg cells in B–/– and WT mice. The differences in CD27, GITR and p75 were unexpected because other reports including a recent one from our laboratory21–23,31,36 indicated that reduced expression of these molecules was associated with decreased Treg function. However, none of those studies used Treg from B–/– mice, suggesting that expression of these markers may be regulated differently in B–/– versus WT mice. The basis for this difference is unknown, but in B–/– mice it may be related to whether Treg cells are interacting with Teff cells in an inflammatory versus a non-inflammatory environment. CD27, p75 and GITR were also differentially expressed when GFP+ Foxp3+ cells were compared in recipients of WT versus B–/– Treg mice at the time thyroids were removed (Fig.2c,d), indicating that the phenotypic differences are unaffected by the recipient environment or by co-culture of Treg cells with effectors/APC.
Figure 2.

Regulatory T (Treg) cells from B-cell-deficient (B–/–) and wild-type (WT) mice can be distinguished phenotypically. (a, b). Splenocytes from naive WT or B–/– NOD.H-2h4 mice were stained for expression of CD4, Foxp3 and CD27, GITR, TNFR II p75, CD103 or intracellular CTLA-4. Plots represent the percentage (a) or mean fluorescence intensity (MFI) (b) of CD4+ Foxp3+ cells positive for the indicated marker. (n = 9 to n = 18 mice per group representative of four independent experiments.) (c, d). T-cell receptor-α-deficient (TCR-α–/–) NOD.H-2h4 mice were given 5 × 106 CD28–/– B–/– splenocytes cultured in the presence or absence of sorted Foxp3+ Treg cells from naive WT or B–/– Foxp3-GFP mice. All mice were given NaI in their water, and spleens were removed 2 months later. Recipient splenocytes were analysed for expression of CD4, Foxp3, and CD27, GITR, p75 or intracellular CTLA-4. Plots represent the percentage (c) or MFI (d) of CD4+ Foxp3+ cells positive for the indicated marker. Representative flow histograms for each marker. (n = 10 to n = 16 mice per group representative of four independent experiments.) ** P < 0·001; *** P < 0·001. Student's t-test.
As will be shown below, the reduced expression of CD27, p75 and GITR by Treg cells from B–/– compared with WT mice was a consistent finding, and differences in CD27 expression were usually greater than differences in p75 and GITR. Because the range of positive cells was much lower for p75 than for the other two markers, the y-axis for the p75 plots is different than for GITR and CD27 in Fig.2(a) and all subsequent figures. Because Treg numbers are similar in WT and B–/– NOD.H-2h4 mice6,7 (see Supporting information, Fig. S1), the differences in percentages of Treg cells expressing CD27, GITR, and p75 correspond to differences in actual numbers of Treg cells expressing these markers (data not shown). There were no significant differences in expression of these markers (see Supporting information, Fig. S2) or activation markers such as CD44 and CD62L (data not shown) by CD4+ Foxp3- non-Treg cells in WT versus B–/– mice. This indicates that reduced expression of GITR, CD27 and p75 is a property of Treg cells from B–/– mice and is not a property of all CD4+ T cells in B–/– mice.
Repopulating Treg cells from B–/– mice lack suppressive function and are phenotypically similar to WT Treg cells
B–/– NOD.H-2h4 mice are resistant to SAT,5,7 but they develop SAT when they are given anti-CD25 to transiently deplete Treg cells.7 The fact that SAT develops in B–/– mice even though Treg cells are not permanently depleted suggests that the Treg cells that repopulate after anti-CD25 depletion differ functionally from Treg cells in unmanipulated B–/– mice. To determine if transient depletion of Treg cells leads to a functional difference in the repopulating Treg cells, B–/– Foxp3-GFP NOD.H-2h4 mice were given 0·5 mg anti-CD25 or rat IgG and rested for 6 weeks until Treg cells had repopulated to normal numbers (see Supporting information, Fig. S3a). Treg cells were sorted, cultured and transferred as above. Repopulated Treg cells from B–/– mice given anti-CD25 have minimal or no suppressive function compared with Treg cells from B–/– mice given rat IgG (Fig.3a; P < 0·001). Treg cells from WT mice given anti-CD25 did not significantly differ functionally from those in unmanipulated WT mice (data not shown).
Figure 3.
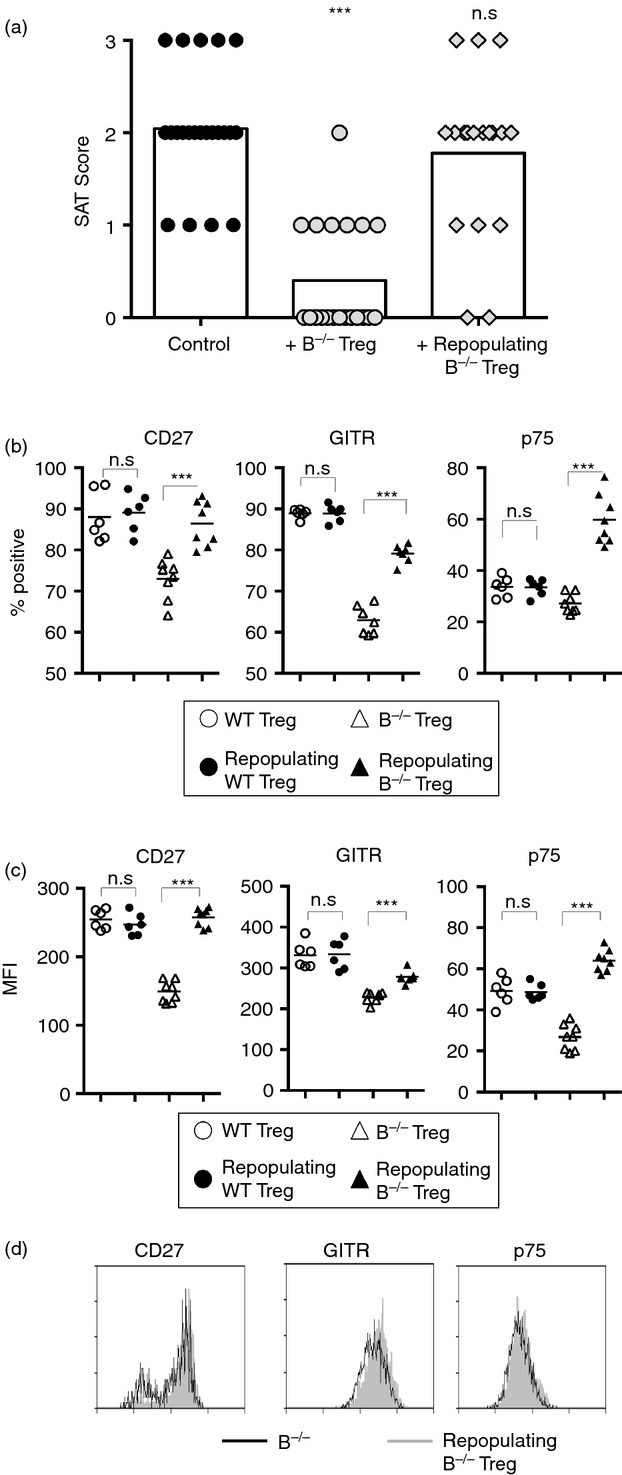
Regulatory T (Treg) cells repopulating B-cell-deficient (B–/–) mice after depletion by anti-CD25 have reduced suppressive function and altered phenotype compared with unmanipulated Treg cells. (a). Naive B–/– Foxp3-GFP NOD.H-2h4 mice were given anti-CD25 to deplete Treg cells. Six weeks later, Treg cells were sorted and cultured with CD28–/– B–/– splenocytes as described in the Materials and methods. Treg cells from B–/– mice given rat IgG were used as a control. T-cell receptor-α-deficient (TCRα–/–) NOD.H-2h4 mice were given 5 × 106 cultured CD28–/– B–/– splenocytes ± Treg cells. All mice were given NaI in their water, and thyroids and spleens were removed 8 weeks later. Spontaneous autoimmune thyroiditis (SAT) severity scores of individual mice. Control mice received cultured cells without added Treg cells. Aggregate of four experiments; n = 22(Control), n = 21(+ B–/– Treg), and n = 18 (Repopulating B–/– Treg) mice per experimental group. *** P < 0·001 compared with no Treg control. Mann–Whitney non-parametric test. (b–d). Splenocytes from naive WT and B–/– mice given anti-CD25 to deplete Treg cells and rested for 6 weeks to allow Treg repopulation were stained for the presence of CD4, Foxp3 and CD27, GITR, or TNFR II p75 and analysed by flow cytometry. Plots represent the percentage (b) or MFI (c) of CD4+ Foxp3+ cells from B–/– and WT mice positive for the indicated marker. (d) Representative flow histograms. n = 6 (WT groups) and n = 8 (B–/– groups) mice per group combined from two independent experiments. n.s. = not significant; *** P < 0·001, Student's t-test.
Because Treg cells that repopulate B–/– mice after anti-CD25-mediated Treg depletion lack suppressor function, we asked if there were phenotypic differences between repopulated and unmanipulated Treg in B–/– mice. To address this question, WT and B–/– NOD.H-2h4 mice were given anti-CD25 or rat IgG and rested for 6 weeks to allow for Treg repopulation. The repopulating Treg cells in B–/– mice given anti-CD25 differ phenotypically from those in B–/– mice given rat IgG and, importantly, have a phenotype similar to that of the less functional Treg cells from WT mice, with increased expression of CD27, p75, and GITR compared with Treg cells from rat IgG-treated B–/– mice (Fig.3b–d). Expression of these cell surface markers did not differ for repopulating Treg cells from WT mice compared with rat IgG controls (Fig.3b–d), indicating that it is primarily the source of Treg cells (B–/– versus WT) and not simply an effect of anti-CD25 that accounts for the loss of function and phenotypic changes in repopulated Treg cells from B–/– mice. These results also indicate that up-regulation of CD27, p75 and GITR on Treg cells from B–/– mice is associated with reduced Treg cell function and this can occur in the absence of B cells. These results, together with those in Fig.3(a), suggest that SAT develops in Treg-depleted B–/– mice, at least in part, because Treg cells that repopulate following transient depletion are less functional and differ phenotypically from those in unmanipulated B–/– mice.
Alternative Treg depletion strategy also results in functional and phenotypic changes in repopulating Treg cells
A significant number of CD4+ Foxp3+ Treg cells in NOD.H-2h4 mice lack CD25 and are not depleted by anti-CD25 (data not shown). Recent generation of mice in which expression of the human diphtheria toxin receptor (DTR) is driven by the Foxp3 promoter allows for the specific depletion of CD4+ Foxp3+ Treg cells regardless of CD25 expression.32 This method of Treg depletion is much shorter in duration, and provides a means to deplete both CD25+ and CD25– Foxp3+ T cells without the potential for depletion of effector cells, which can occur with anti-CD25 depletion. WT and B–/– Foxp3-DTR NOD.H-2h4 mice expressing the human DTR were therefore generated in order to determine if the reduced function of repopulating Treg cells in B–/– mice is related to the method and/or duration of Treg depletion.
Preliminary experiments indicated that two consecutive daily injections of 3 μg DT resulted in maximal depletion of splenic Foxp3+ cells 2 days later. Treg depletion was short-lived, with almost complete repopulation of Foxp3+ cells 5 days later (see Supporting information, Fig. S3b). Despite the short duration of Treg depletion, DT-treated B–/– Foxp3-DTR NOD.H-2h4 mice develop SAT, whereas non-treated B–/– Foxp3DTR mice are resistant (see Supporting information, Fig. S3c). SAT in DT-treated B–/– Foxp3DTR mice is comparable in severity to that of either Foxp3 DTR (data not shown) or Foxp3 GFP B–/– NOD.H-2h4 mice given anti-CD25.7
To determine if DT-mediated Treg depletion influenced the ability of Treg cells from B–/– mice to suppress SAT, B–/– Foxp3-DTR NOD.H-2h4 mice were treated with DT. Foxp3+ Treg were allowed to repopulate for 2 weeks, sorted based on expression of CD4 and DTR, cultured and transferred as above. Treg cells from DT-treated B–/– mice were unable to suppress SAT development, whereas those from untreated B–/– mice were highly suppressive (Fig.4a).
Figure 4.
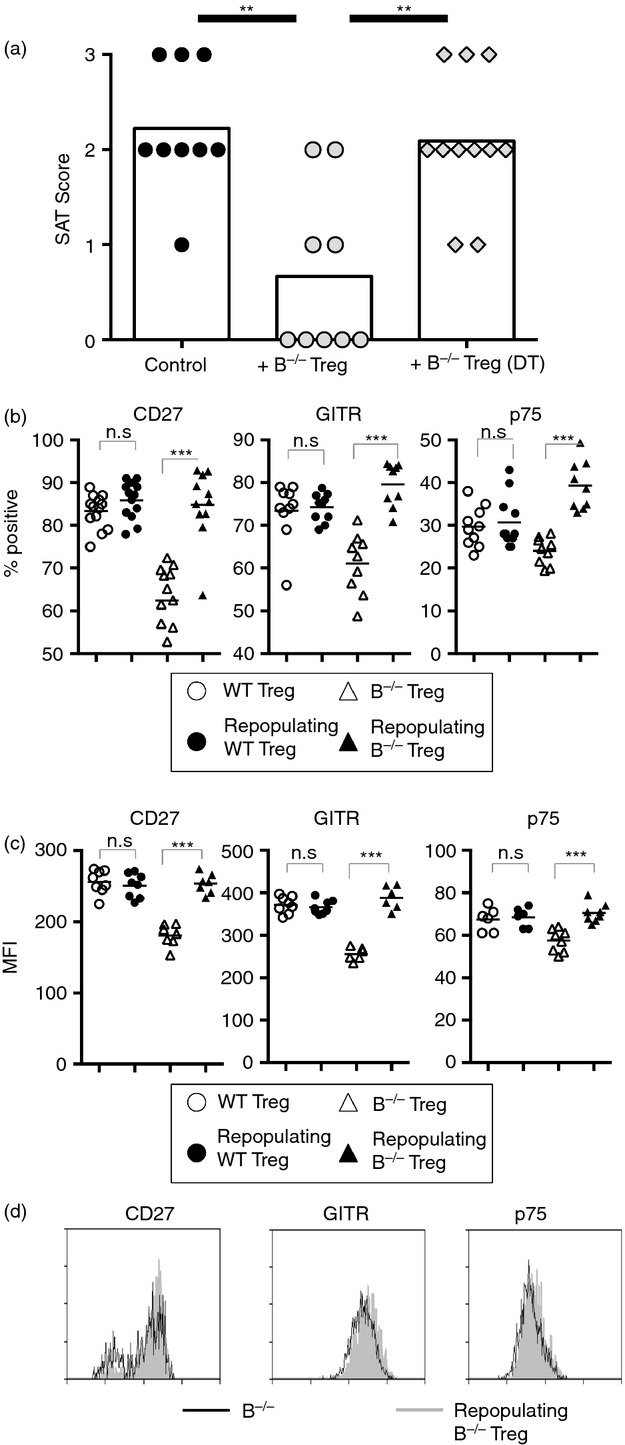
Regulatory T (Treg) cells that repopulate B-cell deficient (B–/–) mice after depletion by diphtheria toxin (DT) have reduced suppressor function and differ phenotypically from Treg cells in unmanipulated B–/– Foxp3-DTR mice. (a) Naive B–/– Foxp3-DTR NOD.H-2h4 mice were depleted of Treg cells using DT. After 6 days to allow Treg repopulation, Treg cells were sorted from DT-treated or untreated controls based on expression of CD4 and DTR, and cultured with CD28–/– B–/– splenocytes as described in Materials and methods. T-cell receptor-α-deficient (TCRα–/–) NOD.H-2h4 mice were given 5 × 106 cultured CD28–/– B–/– splenocytes ± Treg cells. All mice were given NaI in their water at the time of transfer, and thyroids and spleens were removed 8 weeks later. Control mice received cultured cells without added Treg cells. (n = 9 to n = 11 mice per group pooled from three independent experiments) **P < 0·01 compared with control. Mann–Whitney non-parametric test. (b to d). B–/– or WT Foxp3-DTR NOD.H-2h4 mice were depleted of Treg cells using DT. After Treg repopulation, splenocytes were analysed for expression of CD4, Foxp3, and CD27, TNFR II p75, or GITR by flow cytometry. Plots represent the percentage (b) or MFI (c) of CD4+ Foxp3+ cells from B–/– and WT mice positive for the indicated marker. (d) Representative flow histograms. n = 11 to n = 13 mice per group pooled from three independent experiments; **P < 0·01, ***P < 0·001, Student's t-test.
To determine if DT-mediated Treg depletion results in phenotypic changes in repopulating Treg cells, naive WT and B–/– Foxp3-DTR NOD.H-2h4 mice were depleted of Treg cells using DT. Following Treg repopulation, expression of CD27, p75 and GITR was increased on repopulating Treg cells in DT-treated B–/– mice (Fig.4b–d), whereas DT treatment had little effect on the function (data not shown) or expression of cell surface markers of WT Treg cells (Fig.4b–d). These results indicate that CD4+ Foxp3+ Treg cells that repopulate B–/– mice after transient Treg depletion differ functionally from Treg cells in unmanipulated B–/– mice, because they can no longer suppress SAT. Importantly, the repopulating Treg cells are similar to Treg cells of WT mice both in their function (data not shown) and expression of GITR, p75 and CD27 (Fig.4b–d). These results indicate that even a brief depletion of Treg cells is sufficient to lead to functional and phenotypic changes in the repopulating Treg cells in B–/– NOD.H-2h4 mice, whereas Treg depletion has little effect on the function or phenotype of Treg cells in WT NOD.H-2h4 mice.
Treg cells continue to function after transfer to recipients
The use of Treg from Foxp3-DTR NOD.H-2h4 mice in this culture and transfer system allows for depletion of the cultured Treg cells after transfer to recipient mice. To determine if the interaction between Treg and Teff/APC in culture is sufficient for suppression of SAT in recipient mice or if Treg cells are also required after transfer, Treg cells from unmanipulated WT or B–/– Foxp3-DTR NOD.H-2h4 mice were sorted, cultured and transferred as before. Groups of mice were given DT to deplete Treg cells 2 days or 1 month after transfer. Groups that received Treg cells from non-DT treated mice and one that received cultured cells without added Treg cells were included as controls. When Treg cells were depleted 2 days after cell transfer, suppression was almost completely abolished (see Supporting information, Fig. S4a), indicating that Treg continue to perform an important function after Treg and Teff cells are transferred to recipient mice. When Treg cells were depleted 1 month after T-cell transfer, mice developed more severe SAT than if Treg cells were present for the entire 2 months of the experiment, but SAT severity was partially reduced (see Supporting information, Fig. S4a). These results suggest that Treg cells are required during the initial development of SAT in recipient mice, but the requirement for the continued presence of Treg cells is reduced 1 month after cell transfer. Indeed, by this time the Treg cells were no more functional than Treg cells from WT mice (see Supporting information, Fig. S4a). It is important to note that because the recipients are TCR-α–/– mice, no new Treg cells are generated after DT treatment, and Treg depletion was complete (see Supporting information, Fig. S4b).
Treg cells that develop in the presence of B-cell precursors are phenotypically similar to WT Treg cells
Our previous studies showed that when T cells from B–/– mice develop from bone marrow precursors in the presence of B cells, B–/– mice develop SAT,5 suggesting that Treg cells in B–/– mice are less functional when B cells are present. Bone marrow chimeras were generated to determine if Treg cells from B–/– mice that develop in the presence of B cells differ phenotypically from B–/– Treg cells that develop without B cells. Lethally irradiated B–/– mice were reconstituted with bone marrow from WT or B–/– Foxp3-GFP mice with or without bone marrow from TCR-α–/– NOD.H-2h4 mice (the latter to serve as a source of B cells). After 6 weeks, mice were given NaI water, and 8 weeks later, splenocytes were stained for expression of CD4, CD8, B220, Foxp3, CD27, p75, GITR and GARP. Recipients of bone marrow from B–/– mice had no B220+ cells, while recipients of WT bone marrow or B–/– or WT bone marrow together with a source of B-cell precursors all had similar percentages of B and T cells (Fig.5a). Treg cells from B–/– mice that developed in the absence of B cells (B–/–) had lower expression of CD27, GITR and p75 compared with Treg cells that developed from WT bone marrow. In contrast, when Treg cells from B–/– mice developed in the presence of B cells (B–/– + TCR-α–/–), expression of CD27, p75 and GITR was higher and similar to that of Treg cells from WT mice. Expression of GARP was comparable for all groups regardless of the presence of B cells (Fig.5b). Expression of these surface markers was not altered in mice given WT Foxp3-GFP and TCRα–/– bone marrow compared with mice given only WT Foxp3-GFP bone marrow (Fig.5b). These results indicate that Treg cells from B–/– mice that develop in the presence of B-cell precursors are phenotypically similar to Treg cells from WT mice.
Figure 5.
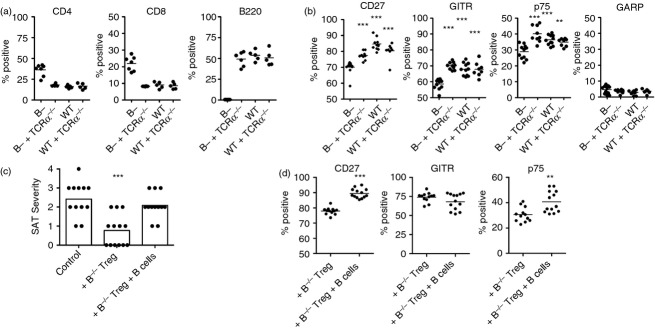
The presence of B cells during regulatory T (Treg) cell development or in culture results in phenotypic and functional changes in Treg cells from B-cell deficient (B–/–) mice. (a, b). B–/– NOD.H-2h4 mice were irradiated (1000 Gy) and given bone marrow from wild-type (WT) or B–/– Foxp3-GFP NOD.H-2h4 mice ± bone marrow from T-cell receptor-α-deficient (TCRα–/–) NOD.H-2h4 mice to provide a source of B cells. After 6 weeks, mice were given NaI water, and splenocytes were analysed 8 weeks later for expression of CD4, B220, Foxp3 and CD27, p75, GITR, or GARP. (a). Results indicate the percentage of cells expressing CD4, CD8 or B220. (b). Percentage of CD4+ Foxp3+ cells expressing the indicated marker. n = 11 (B–/–), n = 9 (B–/– + TCRα–/–), n = 11 (WT), and n = 8 (WT + TCRα–/–) combined from two separate experiments. ***P < 0·001 compared with B–/– group, Student's t-test. (c, d). TCRα–/– NOD.H-2h4 mice were given 5 × 106 cultured CD28–/–B–/– splenocytes ± Treg and ± B cells from TCRα–/– mice. Treg group cultures received sorted Treg cells from B–/– Foxp3-GFP mice. All mice were given NaI in their water at the time of transfer, and thyroids and spleens were removed 8 weeks later. (c) Spontaneous autoimmune thyroiditis (SAT) severity scores of individual mice. (d) Percentages of CD4+ Foxp3+ GFP+ cells in recipient spleens expressing CD27, TNFR II p75, or GITR. Control mice received cultured cells without added Treg cells. (***P < 0·001 compared with no Treg control. Mann–Whitney non-parametric test for SAT). **P < 0·01; *** P < 0·001; Student's t-test for flow cytometry data. n = 12 (Control), n = 13 (+ B–/– Treg) and n = 12 (+ B–/– Treg + B cells). Aggregate of two experiments.
The function and phenotype of B–/– Treg cells is altered by adding B cells to Treg/Teff cultures
Given that the presence of B cells during Treg development in bone marrow chimeras resulted in an altered phenotype of Treg cells in B–/– mice (Fig.5b), we asked whether adding B cells in vitro to the cultures of Treg and Teff cells would influence the phenotype and/or function of Treg cells from B–/– mice. To address this question, sorted Treg cells from B–/– Foxp3-GFP mice were co-cultured with splenocytes from CD28–/–B–/– mice as in previous experiments. B cells from TCR-α–/– mice were added to half of the cultures containing Treg cells, and a control group had no added Treg or B cells. Cells (5 × 106 per recipient) were transferred to TCR-α–/– NOD.H-2h4 mice, and recipients were given NaI water. As in previous experiments, SAT severity in recipient mice was reduced when Treg cells from B–/– mice were added to cultures in the absence of B cells (Fig.5c). However, when B cells were added to the Treg/Teff cultures, SAT severity in the recipients was comparable to that of controls not given Treg cells. Addition of B cells to Treg cultures also resulted in phenotypic changes of the donor Treg cells in recipient spleens, since GFP+ Treg cells from cultures containing B cells had significantly increased expression of CD27 and TNFR II p75 compared with Treg that were cultured in the absence of B cells (Fig.5d). It is not known why GITR expression was apparently unaffected by the addition of B cells to culture. Together, these results directly demonstrate that B cells can interfere with Treg suppressive function and lead to alterations in Treg phenotype. Interestingly, the function and phenotype of Treg cells was not influenced by the B cells that were present in all recipient mice. Apparently the phenotype and function of the Treg cells were fixed when Treg and Teff cells interacted before transfer of T cells to recipient mice. These results are consistent with those of our previous studies, which indicate that B cells can interfere with Treg function and phenotype when Treg and Teff cells initially interact with one another, but after this interaction has occurred, addition of B cells has no apparent effect on Treg function.5,6
Discussion
Autoreactive T cells that escape negative selection in the thymus are not usually activated in the periphery because they are inhibited by CD4+ Foxp3+ Treg cells.40–42 In autoimmune disease, this regulation is ineffective and self-reactive T cells become activated. B-cell-deficient mice are resistant to several autoimmune diseases including diabetes, systemic lupus erythematosus, Sjögren syndrome, arthritis and SAT.5,7–11,43–46 Previous studies from our laboratory showed that while unmanipulated B–/– NOD.H-2h4 mice are resistant to SAT, they develop SAT after transient depletion of Treg cells.5,7 NOD mice spontaneously develop diabetes, thyroiditis and Sjögren syndrome, but these diseases do not develop in B–/– NOD mice.13,19 However, after transient depletion of Treg cells, B–/– NOD mice develop diabetes, thyroiditis and Sjögren syndrome comparable in incidence and severity to that of age-matched WT NOD mice.13 In addition, depletion of B cells in WT NOD.H-2 h4, NOD or BALB/c mice by anti-CD20 inhibits development of SAT, diabetes and arthritis, but anti-CD20 treated mice develop the diseases if Treg cells are transiently depleted.13,17,19,47 Therefore, in several different models, normally resistant mice with few or no B cells develop autoimmune disease when Treg cells are transiently depleted. Because B–/– mice generally have comparable or reduced numbers of CD4+ Foxp3+ Treg cells compared with their WT counterparts,7,18,48–50 the resistance of B–/– mice to certain autoimmune diseases is not due to increased Treg numbers. The fact that Treg cells in B–/– mice able to keep autoreactive T cells silenced, whereas Treg cells in the corresponding WT mice are less effective at inhibiting autoimmunity, suggests that Treg cells in WT and B–/– mice may be functionally different.
Here, we demonstrate that Treg cells in B–/– NOD.H-2h4 mice are functionally superior to Treg cells from WT mice in their ability to suppress SAT. The demonstration that Treg cells in B–/– mice are functionally more effective suppressors could explain, at least in part, why B–/– mice are resistant to development of most spontaneous autoimmune diseases. To our knowledge, these results are the first to directly demonstrate the functional superiority of Treg cells from B–/– mice over Treg cells from WT mice with respect to their ability to regulate a spontaneous autoimmune disease. Consistent with our results, studies in a model of experimental arthritis suggested that Treg cells from B-cell-depleted mice were more effective at suppressing arthritis than those from untreated mice.17 In contrast, in a model of EAE induced by immunization with MOG peptide,18 Treg cells in B–/– B6 mice become activated and accumulate in the central nervous system similarly to Treg cells in WT B6 mice, suggesting that Treg cells in WT and B–/– mice have comparable functions in regulating EAE. A major difference between the EAE model18 and the SAT and experimental arthritis models is that B cells are not required for the development of EAE induced by MOG peptide,18,48 whereas B cells are essential for the development of both SAT and experimental arthritis.5,10 In EAE, Treg cells in B–/– mice are functioning under inflammatory conditions, whereas in SAT and arthritis, inflammation in B–/– mice is greatly reduced.
The results of this study also demonstrate that Treg cells that repopulate B–/– mice after transient Treg cell depletion have greatly diminished suppressive function and can no longer suppress SAT. These results provide an explanation for many earlier results indicating that Treg cells do not have to be permanently depleted for B–/– mice to develop an autoimmune disease,7,13,17,19,47 or to maintain the increases in immune responses that develop after Treg depletion.51–53 Our finding that Treg cells that repopulate after depletion are no longer capable of suppression could have implications for Treg depletion therapies that are used for treatment of cancer and other diseases where Treg activity is detrimental.51,52,54–57 Indeed, because Treg depletion is transient in Foxp3-DTR mice, it is likely that long-term control of tumours and virus-specific CD8 cells reported in some studies was maintained after Treg repopulation.51–53 Although our studies demonstrated a functional difference in repopulating versus unmanipulated Treg cells only in mice lacking B cells, similar results might be seen when B cells are present, particularly when non-B cells are functioning as the APC. Although autoimmunity could be an unwanted side effect of Treg depletion in patients with cancer, this was not observed in the studies reported so far.51,52,54,57
Our early studies and those of others showed that T cells from B–/– mice that develop from bone marrow precursors in the presence of B cells can function as effector cells for SAT and diabetes, whereas T cells from B–/– mice that develop in the absence of B cells do not have effector function.5,11 The results in Fig.5(b) extend those results by showing that Treg cells from B–/– mice acquire a phenotype similar to that of WT Treg cells when they develop with B cells. The ability of B cells to influence Treg function and phenotype was also evident when B cells were added to cultures of Treg and Teff cells (Fig.5c,d). These latter results were initially unexpected, because our earlier studies indicated that B cells promoted SAT development in B–/– or B-cell-depleted mice only if they were present early in life.5,6 In the experiments shown in Fig.5(c,d), B cells were added directly to Treg and Teff cells in a culture dish, so the initial Treg–Teff interactions occurred in the presence of B cells. In contrast, exposure of Treg cells to the B cells in the recipients after this interaction had occurred had no effect. These latter results are consistent with our earlier report,5,6 and indicate that B cells can directly interfere with Treg/Teff interactions during a limited window of time when Treg and Teff cells initially interact with one another.
B cells have both positive and negative effects on immune responses and autoimmune diseases.47–49,58–60 B cells can induce tolerance in CD4+ T cells, primarily by producing IL-10 or other suppressive cytokines.47,49,58,61–63 B cells also function as regulatory cells, whereby a specialized subset of IL-10-producing B cells can inhibit immune responses and autoimmune diseases.47,48,58,60,63–65 However, in autoimmune diseases such as SAT that require B cells for their development, B cells function primarily as APC to initiate disease.5,9,11,17,19,43,66,67 Our studies indicate that another mechanism by which B cells can promote development of SAT is by interfering with the function of Treg cells that develop to inhibit the disease. These results may explain why Treg cells in WT NOD.H-2h4 mice are less functional, because SAT develops when B cells are present, but not when they are absent. The ability of B cells to inhibit Treg function in B–/– mice is associated with phenotypic changes in the Treg cells (Fig.5 b,d), and they become phenotypically similar to Treg cells in WT mice.
The precise mechanisms by which B cells interfere with Treg functions and lead to alterations in Treg phenotype are not yet known. Interaction of GITR with its ligand can interfere with or suppress Treg function in some models,59,68,69 but B cells increase Treg activity in other models,49,50,63 including their ability to suppress EAE through GITR/GITRL interactions.48 These contrasting reports indicate that B cells can have both positive and negative effects on immune responses through multiple mechanisms.47,58,60,63 It is likely that B cells express a particular molecule(s) that interacts with a receptor either on Treg or on Teff cells, resulting in loss of Treg function and alterations in Treg phenotype. Additional studies will be required to determine what molecules are involved and whether the reduced Treg function that can occur when B cells are present in vitro is due to interaction of the B cells with Treg cells, Teff cells or both cell types. We also do not know if the phenotypic differences in Treg cells from WT and B–/– mice are directly responsible for their functional differences or whether some other process such the environment, e.g. inflammatory versus non-inflammatory, primarily dictates the phenotypic changes that occur when Treg cells are less suppressive.
Two recent reports indicate that the Foxp3-GFP reporter construct in some of the mice used for these experiments can lead to altered Treg function in autoimmune-prone strains of mice such as NOD and K/BxN.70,71 This construct had little, if any, effect on development of SAT in NOD.H-2h4 mice since B–/– Foxp3GFP NOD.H-2h4 mice, like other B–/– NOD.H-2h4 mice, are resistant to SAT, and they develop SAT following transient Treg depletion. In addition, SAT in WT Foxp3GFP NOD.H-2h4 mice is comparable in incidence and severity to that of WT NOD.H-2h4 mice that do not express GFP (our unpublished results). Moreover, all of our results were comparable in the experiments using FoxP3-DTR mice, which use a different construct (Figs3 and 4).
Together, the results of this study demonstrate that a lack of B cells in NOD.H-2h4 mice leads to the generation of Treg cells that have a greater ability to suppress SAT compared with Treg cells of mice that have B cells. Treg function in B–/– mice decreases in the presence of B cells and can be altered by transient Treg depletion followed by Treg repopulation. These studies provide an explanation for earlier results from several laboratories demonstrating that B–/– mice are resistant to many spontaneous autoimmune diseases but develop the disease when Treg cells are depleted only transiently. Our results suggest that there is an interaction between B cells, Teff cells and developing Treg cells that allows for greater regulatory function in Treg cells that suppress spontaneous autoimmune diseases when B cells are absent, possibly through interactions of the TNF receptor superfamily members CD27, p75, and GITR expressed on Treg cells with their ligands expressed by APC.
Acknowledgments
This work was supported by National Institute of Health grant RO1 AI076935 from the National Institute of Allergy and Infectious Disease and by the Lottie Caroline Hardy Trust.
Glossary
Abbreviations:
- B–/–
B-cell-deficient
- DT
diphtheria toxin
- DTR
diphtheria toxin receptor
- GITR
glucocorticoid-induced tumour necrosis factor-related protein
- SAT
spontaneous autoimmune thyroiditis
Author contribution
JSE designed and performed experiments and prepared the manuscript, HBM designed experiments and prepared the manuscript.
Disclosures
The authors declare no financial or commercial conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Wild-type (WT) and B-cell deficient (B–/–) mice have similar numbers of CD4+ Foxp3+ regulatory T (Treg) cells and CD4+ Foxp3+ CD25+ Treg cells.
Figure S2. Expression of CD27, TNFR II p75, and GITR by CD4+ Foxp3– non-regulatory T (Treg) cells in wild-type (WT) and B-cell deficient (B–/–) mice.
Figure S3. Duration of regulatory T (Treg) cell depletion by anti-CD25 and diphtheria toxin (DT).
Figure S4. Regulatory T (Treg) cells continue to exert their suppressive function after transfer to recipient mice.
References
- Braley-Mullen H, Sharp GC, Medling B, Tang H. Spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Autoimmun. 1999;12:157–65. doi: 10.1006/jaut.1999.0272. [DOI] [PubMed] [Google Scholar]
- Rasooly L, Burek CL, Rose NR. Iodine-induced autoimmune thyroiditis in NOD-H-2h4 mice. Clin Immunol Immunopathol. 1996;81:287–92. doi: 10.1006/clin.1996.0191. [DOI] [PubMed] [Google Scholar]
- Verma S, Hutchings P, Guo J, McLachlan S, Rapoport B, Cooke A. Role of MHC class I expression and CD8+ T cells in the evolution of iodine-induced thyroiditis in NOD-H2(h4) and NOD mice. Eur J Immunol. 2000;30:1191–202. doi: 10.1002/(SICI)1521-4141(200004)30:4<1191::AID-IMMU1191>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Yu S, Medling B, Yagita H, Braley-Mullen H. Characteristics of inflammatory cells in spontaneous autoimmune thyroiditis of NOD.H-2h4 mice. J Autoimmun. 2001;16:37–46. doi: 10.1006/jaut.2000.0458. [DOI] [PubMed] [Google Scholar]
- Braley-Mullen H, Yu S. Early requirement for B cells for development of spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2000;165:7262–9. doi: 10.4049/jimmunol.165.12.7262. [DOI] [PubMed] [Google Scholar]
- Yu S, Ellis JS, Dunn R, Kehry MR, Braley-Mullen H. Transient depletion of B cells in young mice results in activation of regulatory T cells that inhibit development of autoimmune disease in adults. Int Immunol. 2012;24:233–42. doi: 10.1093/intimm/dxs003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Maiti PK, Dyson M, Jain R, Braley-Mullen H. B cell-deficient NOD.H-2h4 mice have CD4+ CD25+ T regulatory cells that inhibit the development of spontaneous autoimmune thyroiditis. J Exp Med. 2006;203:349–58. doi: 10.1084/jem.20051438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan OT, Madaio MP, Shlomchik MJ. The central and multiple roles of B cells in lupus pathogenesis. Immunol Rev. 1999;169:107–21. doi: 10.1111/j.1600-065x.1999.tb01310.x. [DOI] [PubMed] [Google Scholar]
- Noorchashm H, Lieu YK, Noorchashm N, et al. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol. 1999;163:743–50. [PubMed] [Google Scholar]
- O'Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J Immunol. 2005;174:3781–8. doi: 10.4049/jimmunol.174.6.3781. [DOI] [PubMed] [Google Scholar]
- Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–8. [PubMed] [Google Scholar]
- Marino E, Villanueva J, Walters S, Liuwantara D, Mackay F, Grey ST. CD4+ CD25+ T-cells control autoimmunity in the absence of B-cells. Diabetes. 2009;58:1568–77. doi: 10.2337/db08-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JS, Wan X, Braley-Mullen H. Transient depletion of CD4+ CD25+ regulatory T cells results in multiple autoimmune diseases in wild-type and B-cell-deficient NOD mice. Immunology. 2013;139:179–86. doi: 10.1111/imm.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie I, Abiru N, Sakamoto H, Iwakura Y, Nagayama Y. Induction of autoimmune thyroiditis by depletion of CD4+ CD25+ regulatory T cells in thyroiditis-resistant IL-17, but not interferon-γ receptor, knockout nonobese diabetic-H2h4 mice. Endocrinology. 2011;152:4448–54. doi: 10.1210/en.2011-1356. [DOI] [PubMed] [Google Scholar]
- Nagayama Y, Horie I, Saitoh O, Nakahara M, Abiru N. CD4+ CD25+ naturally occurring regulatory T cells and not lymphopenia play a role in the pathogenesis of iodide-induced autoimmune thyroiditis in NOD-H2h4 mice. J Autoimmun. 2007;29:195–202. doi: 10.1016/j.jaut.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Nakahara M, Nagayama Y, Ichikawa T, Yu L, Eisenbarth GS, Abiru N. The effect of regulatory T-cell depletion on the spectrum of organ-specific autoimmune diseases in nonobese diabetic mice at different ages. Autoimmunity. 2011;44:504–10. doi: 10.3109/08916934.2010.548839. [DOI] [PubMed] [Google Scholar]
- Hamel KM, Cao Y, Ashaye S, Wang Y, Dunn R, Kehry MR, Glant TT, Finnegan A. B cell depletion enhances T regulatory cell activity essential in the suppression of arthritis. J Immunol. 2011;187:4900–6. doi: 10.4049/jimmunol.1101844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehlig K, Shen P, Lampropoulou V, et al. Activation of CD4+ Foxp3+ regulatory T cells proceeds normally in the absence of B cells during EAE. Eur J Immunol. 2012;42:1164–73. doi: 10.1002/eji.201142242. [DOI] [PubMed] [Google Scholar]
- Serreze DV, Chapman HD, Niens M, et al. Loss of intra-islet CD20 expression may complicate efficacy of B-cell-directed type 1 diabetes therapies. Diabetes. 2011;60:2914–21. doi: 10.2337/db11-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Peng J, Tai N, Hu C, Zhou Z, Wong FS, Wen L. The dual effects of B cell depletion on antigen-specific T cells in BDC2.5NOD mice. J Immunol. 2012;188:4747–58. doi: 10.4049/jimmunol.1103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+ CD25+ FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol. 2008;180:6467–71. doi: 10.4049/jimmunol.180.10.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4+ CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, Sallusto F. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med. 2005;201:1793–803. doi: 10.1084/jem.20050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevach EM. From vanilla to 28 flavors: multiple varieties of T regulatory cells. Immunity. 2006;25:195–201. doi: 10.1016/j.immuni.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Kuczma M, Pawlikowska I, Kopij M, Podolsky R, Rempala GA, Kraj P. TCR repertoire and Foxp3 expression define functionally distinct subsets of CD4+ regulatory T cells. J Immunol. 2009;183:3118–29. doi: 10.4049/jimmunol.0900514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2011;11:119–30. doi: 10.1038/nri2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Oppenheim JJ. Resolving the identity myth: key markers of functional CD4+ FoxP3+ regulatory T cells. Int Immunopharmacol. 2011;11:1489–96. doi: 10.1016/j.intimp.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med. 2013;210:257–68. doi: 10.1084/jem.20121525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14:154–65. doi: 10.1038/nri3605. [DOI] [PubMed] [Google Scholar]
- Podolin PL, Pressey A, DeLarato NH, Fischer PA, Peterson LB, Wicker LS. I-E+ nonobese diabetic mice develop insulitis and diabetes. J Exp Med. 1993;178:793–803. doi: 10.1084/jem.178.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JS, Hong SH, Zaghouani H, Braley-Mullen H. Reduced effectiveness of CD4+ Foxp3+ regulatory T cells in CD28-deficient NOD.H-2h4 mice leads to increased severity of spontaneous autoimmune thyroiditis. J Immunol. 2013;191:4940–9. doi: 10.4049/jimmunol.1301253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity. 2009;31:654–64. doi: 10.1016/j.immuni.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Fang Y, Sharav T, Sharp GC, Braley-Mullen H. CD8+ T cells induce thyroid epithelial cell hyperplasia and fibrosis. J Immunol. 2011;186:2655–62. doi: 10.4049/jimmunol.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braley-Mullen H, Johnson M, Sharp GC, Kyriakos M. Induction of experimental autoimmune thyroiditis in mice with in vitro activated splenic T cells. Cell Immunol. 1985;93:132–43. doi: 10.1016/0008-8749(85)90394-6. [DOI] [PubMed] [Google Scholar]
- Yu S, Sharp GC, Braley-Mullen H. Thyrocytes responding to IFN-γ are essential for development of lymphocytic spontaneous autoimmune thyroiditis and inhibition of thyrocyte hyperplasia. J Immunol. 2006;176:1259–65. doi: 10.4049/jimmunol.176.2.1259. [DOI] [PubMed] [Google Scholar]
- Chen X, Oppenheim JJ. The phenotypic and functional consequences of tumour necrosis factor receptor type 2 expression on CD4+ FoxP3+ regulatory T cells. Immunology. 2011;133:426–33. doi: 10.1111/j.1365-2567.2011.03460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor FoxP3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, Rajan TV, Clark RB. Natural but not inducible regulatory T cells require TNF-α signaling for in vivo function. J Immunol. 2011;186:6779–87. doi: 10.4049/jimmunol.1003868. [DOI] [PubMed] [Google Scholar]
- Tsakiri N, Papadopoulos D, Denis MC, Mitsikostas DD, Kollias G. TNFR2 on non-haematopoietic cells is required for Foxp3+ Treg-cell function and disease suppression in EAE. Eur J Immunol. 2012;42:403–12. doi: 10.1002/eji.201141659. [DOI] [PubMed] [Google Scholar]
- Bour-Jordan H, Bluestone JA. Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol Rev. 2009;229:41–66. doi: 10.1111/j.1600-065X.2009.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–25. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samy ET, Setiady YY, Ohno K, Pramoonjago P, Sharp C, Tung KS. The role of physiological self-antigen in the acquisition and maintenance of regulatory T-cell function. Immunol Rev. 2006;212:170–84. doi: 10.1111/j.0105-2896.2006.00404.x. [DOI] [PubMed] [Google Scholar]
- Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639–48. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FS, Wen L, Tang M, et al. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes. 2004;53:2581–7. doi: 10.2337/diabetes.53.10.2581. [DOI] [PubMed] [Google Scholar]
- Hayakawa I, Tedder TF, Zhuang Y. B-lymphocyte depletion ameliorates Sjogren's syndrome in Id3 knockout mice. Immunology. 2007;122:73–9. doi: 10.1111/j.1365-2567.2007.02614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson L, Jirholt J, Holmdahl R, Jansson L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA) Clin Exp Immunol. 1998;111:521–6. doi: 10.1046/j.1365-2249.1998.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol. 2010;10:236–47. doi: 10.1038/nri2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188:3188–98. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Qiao L. Resting B cells expand a CD4+ CD25+ Foxp3+ Treg population via TGF-β3. Eur J Immunol. 2008;38:2488–98. doi: 10.1002/eji.200838201. [DOI] [PubMed] [Google Scholar]
- Sun JB, Flach CF, Czerkinsky C, Holmgren J. B lymphocytes promote expansion of regulatory T cells in oral tolerance: powerful induction by antigen coupled to cholera toxin B subunit. J Immunol. 2008;181:8278–87. doi: 10.4049/jimmunol.181.12.8278. [DOI] [PubMed] [Google Scholar]
- Klages K, Mayer CT, Lahl K, et al. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. 2010;70:7788–99. doi: 10.1158/0008-5472.CAN-10-1736. [DOI] [PubMed] [Google Scholar]
- Teng MW, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res. 2010;70:7800–9. doi: 10.1158/0008-5472.CAN-10-1681. [DOI] [PubMed] [Google Scholar]
- Dietze KK, Zelinskyy G, Gibbert K, et al. Transient depletion of regulatory T cells in transgenic mice reactivates virus-specific CD8+ T cells and reduces chronic retroviral set points. Proc Natl Acad Sci U S A. 2011;108:2420–5. doi: 10.1073/pnas.1015148108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res. 2011;71:6915–20. doi: 10.1158/0008-5472.CAN-11-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–32. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kamigaki T, Yamashita K, et al. Enhancement of anti-tumor immunity by high levels of Th1 and Th17 with a combination of dendritic cell fusion hybrids and regulatory T cell depletion in pancreatic cancer. Oncol Rep. 2009;22:337–43. [PubMed] [Google Scholar]
- Rech AJ, Mick R, Martin S, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4:134ra62. doi: 10.1126/scitranslmed.3003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalampokis I, Yoshizaki A, Tedder TF. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res Ther. 2013;15(Suppl 1):S1. doi: 10.1186/ar3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson TS, Bamias G, Naganuma M, et al. Expanded B cell population blocks regulatory T cells and exacerbates ileitis in a murine model of Crohn disease. J Clin Invest. 2004;114:389–98. doi: 10.1172/JCI20855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–30. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992;258:1156–9. doi: 10.1126/science.1439825. [DOI] [PubMed] [Google Scholar]
- Murray SE, Toren KG, Parker DC. Peripheral CD4+ T-cell tolerance is induced in vivo by rare antigen-bearing B cells in follicular, marginal zone, and B-1 subsets. Eur J Immunol. 2013;43:1818–27. doi: 10.1002/eji.201242784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri C, Blair PA. Regulatory B cells in autoimmunity: developments and controversies. Nat Rev Rheumatol. 2010;6:636–43. doi: 10.1038/nrrheum.2010.140. [DOI] [PubMed] [Google Scholar]
- Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–50. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16:219–30. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- Hong SH, Braley-Mullen H. Follicular B cells in thyroids of mice with spontaneous autoimmune thyroiditis contribute to disease pathogenesis and are targets of anti-CD20 antibody therapy. J Immunol. 2014;192:897–905. doi: 10.4049/jimmunol.1301628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Harvey BP, Gee RJ, Shlomchik MJ, Mamula MJ. B cells drive early T cell autoimmunity in vivo prior to dendritic cell-mediated autoantigen presentation. J Immunol. 2006;177:4481–7. doi: 10.4049/jimmunol.177.7.4481. [DOI] [PubMed] [Google Scholar]
- Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM. Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol. 2013;43:2421–9. doi: 10.1002/eji.201343451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji HB, Liao G, Faubion WA, Abadia-Molina AC, Cozzo C, Laroux FS, Caton A, Terhorst C. Cutting edge: the natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J Immunol. 2004;172:5823–7. doi: 10.4049/jimmunol.172.10.5823. [DOI] [PubMed] [Google Scholar]
- Bettini ML, Pan F, Bettini M, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36:717–30. doi: 10.1016/j.immuni.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darce J, Rudra D, Li L, Nishio J, Cipolletta D, Rudensky AY, Mathis D, Benoist C. An N-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity. 2012;36:731–41. doi: 10.1016/j.immuni.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Wild-type (WT) and B-cell deficient (B–/–) mice have similar numbers of CD4+ Foxp3+ regulatory T (Treg) cells and CD4+ Foxp3+ CD25+ Treg cells.
Figure S2. Expression of CD27, TNFR II p75, and GITR by CD4+ Foxp3– non-regulatory T (Treg) cells in wild-type (WT) and B-cell deficient (B–/–) mice.
Figure S3. Duration of regulatory T (Treg) cell depletion by anti-CD25 and diphtheria toxin (DT).
Figure S4. Regulatory T (Treg) cells continue to exert their suppressive function after transfer to recipient mice.