

Abstract
The host response to Clostridium difficile infection in antibiotic-treated mice is characterized by robust recruitment of Gr-1+ cells, increased expression of inflammatory cytokines including tumour necrosis factor-α (TNF-α), and the development of severe epithelial damage. To investigate the role of Gr-1+ cells and TNF-α during C. difficile colitis, we treated infected mice with monoclonal antibodies against Gr-1 or TNF-α. Mice were challenged with vegetative cells of C. difficile strain VPI 10463 following treatment with the third-generation cephalosporin ceftriaxone. Ceftriaxone treatment alone was associated with significant changes in cytokine expression within the colonic mucosa but not overt inflammatory histopathological changes. In comparison, C. difficile infection following ceftriaxone treatment was associated with increased expression of inflammatory cytokines and chemokines including Cxcl1, Cxcl2, Il1b, Il17f and Tnfa, as well as robust recruitment of Ly6CMid Gr-1High neutrophils and Ly6CHigh Gr-1Mid monocytes and the development of severe colonic histopathology. Anti-Gr-1 antibody treatment resulted in effective depletion of both Ly6CMid Gr-1High neutrophils and Ly6CHigh Gr-1Mid monocytes: however, we observed no protection from the development of severe pathology or reduction in expression of the pro-inflammatory cytokines Il1b, Il6, Il33 and Tnfa following anti-Gr-1 treatment. By contrast, anti-TNF-α treatment did not affect Gr-1+ cell recruitment, but was associated with increased expression of Il6 and Il1b. Additionally, Ffar2, Ffar3, Tslp, Tff and Ang4 expression was significantly reduced in anti-TNF-α-treated animals, in association with marked intestinal histopathology. These studies raise the possibility that TNF-α may play a role in restraining inflammation and protecting the epithelium during C. difficile infection.
Keywords: colitis, epithelium, microbiome, mucosal inflammation, neutrophil
Introduction
The inflammatory signals that support leucocyte recruitment, intestinal histopathology and inflammatory cytokine production during Clostridium difficile infection are still not completely understood. Recent studies have identified key roles for Myeloid Differentiation Primary Response 88, Nucleotide Binding Oligomerization Domain 1, and Caspase Recruitment Domain Containing Protein in promoting neutrophil recruitment and the production of inflammatory cytokines in response to C. difficile infection.1–3 Additionally, signalling of Toll-like receptors 4 and 5 has been shown to prevent the development of severe intestinal histopathology during C. difficile infection.4,5 However, the role of tumour necrosis factor-α (TNF-α) in promoting epithelial damage, leucocyte recruitment and inflammatory cytokine expression during C. difficile infection has yet to be investigated.
Antibiotic pre-treatment is required to infect conventional mice with C. difficile.1–4,6–11 However, antibiotic treatment is also capable of modulating inflammatory responses and immune cell function.12–16 In vitro studies have demonstrated reduced expression of inflammatory mediators from monocytes stimulated with live fungal15 or heat-killed bacterial13 cells following moxifloxacin and co-trimoxazole treatment, respectively. Macrolide antibiotics can also affect inflammatory functions of pulmonary epithelial cells and modulate TNF-α, interleukin-8 (IL-8) and Granulocyte-macrophage colony stimulating factor production by these cells.16–19 Ceftriaxone is a third-generation cephalosporin with bactericidal activity against both Gram-positive and Gram-negative bacteria.20–22 Cefoperazone, another third-generation cephalosporin, can markedly alter the composition of the intestinal microbiota and render mice susceptible to C. difficile infection.8,10 In the current study, we investigated the ability of ceftriaxone to permit C. difficile infection, and whether ceftriaxone treatment alone was sufficient to induce colonic inflammation.
Tumour necrosis factor-α promotes leucocyte recruitment and the expression of inflammatory cytokines during mucosal inflammation.23–25 TNF-α expression is significantly increased during acute C. difficile colitis,9 and macrophage TNF-α production is also enhanced by exposure to C. difficile toxins.26 Gr-1 is an epitope found on both Ly6C and Ly6G, and is expressed on neutrophils, inflammatory monocytes, and plasmacytoid dendritic cells.9,27–29 Gr-1+ cells are recruited in large numbers to the large intestine in response to C. difficile infection,1–3,9 and protect against bacterial dissemination and mortality.2,3 However, much remains unknown about the contributions of Gr-1+ cells and TNF-α in promoting intestinal histopathology, leucocyte recruitment, and the expression of inflammatory cytokines during C. difficile colitis.
Materials and methods
Bacterial culture and growth conditions
Clostridium difficile was prepared for infection as described previously.8,10 Briefly, an overnight culture of C. difficile strain VPI 10463 (ATCC 43255) was back-diluted 1 : 10 in fresh brain–heart infusion broth supplemented with 0.1% cysteine and grown for 4–6 hr. The culture was then collected, washed three times in deoxygenated PBS, and diluted to the desired dose. Mice were challenged via oral gavage. The inoculum was serially diluted and plated on brain–heart infusion supplemented with 0.1% cysteine to confirm dosage. Clostridium difficile was grown and prepared for gavage in a Coy anaerobic chamber (Coy Laboratory Products, Grass Lake, MI).
Animals and housing
C57BL/6 male mice aged 5–9 weeks at the time of antibiotic pre-treatment were used in the current study. All experiments were conducted under a protocol approved by the University Committee on Use and Care of Animals at the University of Michigan. All mice were purchased directly from Jackson Laboratories (Bar Harbor, ME) or obtained from an in-house colony founded by Jackson breeders. Mice were housed with autoclaved bedding, food and water. All animal manipulations were carried out in a laminar flow hood.
Antibiotic treatment and infection
For ceftriaxone and C. difficile infection studies, mice were treated with ceftriaxone (0·5 g/l) (Sigma, St Louis, MO) given ad libitum in their drinking water for 4 days. Antibiotic water was replaced every other day. Mice were then given a 2-day recovery period on drinking water without antibiotic before infection with C. difficile as described previously.8,10 Ceftriaxone-treated mice were given the antibiotic regimen only, and untreated animals were not manipulated at all.
For C. difficile infection studies, mice received 5·06 ± 0·31 Log10 colony-forming units vegetative C. difficile via oral gavage on day 2 (Fig.1). Clostridium difficile-infected animals were monitored for signs of severe disease (hunched posture, lethargy, weight loss exceeding 20% of baseline body weight) and were humanely killed if moribund. All surviving animals were killed on day 4 for subsequent analysis (Fig.1).
Figure 1.

(a) Experimental approach and timeline. (b–e) Effect of ceftriaxone treatment and Clostridium difficile infection on the colonic microbiota (day 4). (b) Diversity (Inverse Simpson Index) of the mucosa-associated bacterial communities of untreated, ceftriaxone-treated, or ceftriaxone-treated and C. difficile infected mice. (c–e) Relative abundance of the mucosa-associated bacterial communities of the mice in panel b. The operational taxonomic units (OTUs) were ordered by decreasing abundance of genera in the untreated group. Data are shown as mean ± SEM relative abundance. The Family and Genus-level taxonomies are displayed along the x-axis and the Phylum-level taxonomy is identified in the shading of individual bars.
Antibody administration
Mice were given intraperitoneal injections of 250 μg of anti-TNF-α monoclonal antibody (mAb; clone MP6-XT3) 1 day before infection with C. difficile or injections of 250 μg of anti-Gr-1 mAb (clone RB6-8C5) 1 day before and 1 day after infection. Mouse serum (Sigma) injections were administered to control mice.
Histology
Colonic tissue was collected from the midpoint of the colon and either flash-frozen in liquid nitrogen for subsequent DNA extraction or stored in RNAlater (Life Technologies, Grand Island, NY) for RNA analysis. Whole colons were excised from representative mice and prepared for histological analysis as described previously.8,10 Cassettes were processed, paraffin embedded, sectioned and used to prepare haematoxylin & eosin stained slides (McClinchey Histology Lab Inc., Stockbridge, MI).
Representative images were acquired on an Olympus BX40 light microscope (Olympus Corporation, Center Valley, PA) using a QImaging MicroPublisher RTV 5·0 5 megapixel camera (QImaging Corporation, Surrey, BC, Canada) at a total magnification of × 400. Images were acquired using QCapture Suite PLUS (QImaging Corporation) version 3.1.3.10. Image and panel assembly were performed in Adobe Photoshop CS5, version 12.0 (Adobe, San Jose CA). Image processing was restricted to global adjustments of brightness, contrast and image size.
Histopathological examination
Histological sections were coded, randomized and scored in a blinded manner. The slides were first scored categorically on a 0–5 scale for epithelium damage and for inflammation, using defined criteria. Epithelium damage was scored as follows: 0, intact epithelium; 1, minimal, scattered goblet cell loss with no significant epithelium destruction and no histologically defined loss of surface integrity; 2, widespread moderate goblet cell loss with no significant epithelium destruction and no histologically defined loss of surface integrity; 3, moderate to extensive widespread goblet cell loss with scattered epithelium destruction and histologically defined loss of surface integrity; 4, extensive epithelium and goblet cell destruction with histologically defined loss of surface integrity; 5, severe epithelium destruction and goblet cell destruction with widespread histologically apparent loss of surface integrity. Inflammation was scored as follows: 0, no inflammation; 1, minimal multifocal leucocytic infiltrates; 2, moderate multifocal leucocytic infiltrates and low level oedema (greater submucosal involvement); 3, significant multifocal leucocytic infiltrates, oedema, submucosal involvement; 4, extensive multifocal leucocytic infiltrates, oedema, extensive submucosal involvement; 5, severe multifocal leucocytic infiltrates, extensive oedema and submucosal involvement with luminal involvement and/or abscess formation. The slides were then assigned an overall score using a rank-order scoring system. The total of both categorical scores (epithelium damage and inflammation) were used to rank all the slides in the study in order of increasing severity of histopathological changes (1 = least, 16 = most). This method offers significant advantages over straight categorical scoring systems for comparing histological changes between groups and has been previously reported for scoring C. difficile-induced intestinal pathology.10,30,31
RNA isolation and expression analysis
RNA was isolated and purified from colonic tissue as described previously.9 RNA quality was assessed using an Agilent bioanalyzer (Agilent Technologies, Santa Clara, CA), and the concentration of RNA was assessed using a Nanodrop instrument (Thermo Fisher Scientific, Waltham, MA). Complementary DNA was generated using the RT2 First Strand kit (Qiagen, Valencia, CA), and gene expression was then determined using RT2 PCR cards (Qiagen). To account for any variation between RT2 PCR cards, cross-card normalization was performed as described previously.9,32 The mean Ct values of two internal control genes was subtracted from the Ct of the gene in question to generate ΔCt (dCt) values.33 The 2−ddCt method was used to calculate fold change gene expression for all comparisons.34 All reactions were run on a LightCycler 480 (Roche, Indianapolis, IN).
DNA isolation, amplicon library preparation, 454 pyrosequencing and microbiome community analysis
All procedures and analyses were performed as previously described.9 Briefly, DNA was isolated from rinsed colonic tissue and V3-V5 16S ribosomal RNA gene amplicon libraries were generated. They were then sequenced on a Roche 454 GS Junior Titanium platform according to the manufacturer's specifications. Bacterial 16S rRNA gene sequences were processed using the microbial ecology software suite mothur35 to generate operational taxonomic units (OTUs) at a 3% level of difference (approximating species-level differences). These data were then imported into the software package R and analysed using the R add on-package vegan.36 Rank abundance plots were generated by selecting for the OTUs that contributed to > 0·5% of the population. The content of each treated tissue was ordered according to the average rank order of its untreated counterpart. Taxonomic classification of an OTU was assigned within mothur by identifying the consensus sequence of the OTU and assigning taxonomy using a Bayesian classifier trained on an Ribosomal Database Project training set (classify.otu).
Quantification of C. difficile colonization
Mucosal C. difficile colonization was determined using a species-specific quantitative PCR of DNA isolated from colonic tissue. The C. difficile-specific qPCR was performed as described previously.8,37 All reactions were carried out in a total volume of 10 μl. Each reaction contained 2 μl of template primers, 6·25 pmol forward and reverse tcdB primers, and 1 pmol tcdB probe. The cycling conditions and probe and primer sequences are identical to those used previously.8 Raw Ct values were normalized to signal from a single-copy host internal control gene to generate dCt values.8,34 The dCt values were then converted to ‘C. difficile genomes/g tissue’ using a standard curve generated with known amounts of vegetative C. difficile and colonic tissue.
Colonic leucocyte isolation
Colonic leucocytes were isolated as described previously2,9,38 with modification. Colonic tissue was minced with serrated scissors and incubated in 20 ml of Hanks’ balanced salt solution supplemented with 2·5% fetal bovine serum, 5 mm EDTA and 1 mm dithiothreitol for 20 min at 37°. After washing, tissue was incubated in 20 ml Hanks’ balanced salt solution supplemented with 2·5% fetal bovine serum, 400 U/ml collagenase type 3 (Worthington Biochemical, Freehold NJ), and 0·5 mg/ml DNase I (Roche) for 60 min at 37°. Samples were then resuspended in 20% Percoll (Sigma) in PBS, and centrifuged at 900 g for 30 min at room temperature. The resulting single-cell suspensions were stained for analysis of cell surface marker expression via flow cytometry.
Flow cytometry and staining
Flow cytometric staining was performed as described previously.9 Briefly, cells were blocked with unlabelled FcRIII/II, and subsequently stained with fluorescently labelled antibodies for 30 min. Cells were then washed and finally suspended in stabilizing fixative (BD Biosciences, San Jose, CA). All data were collected on a three-laser Canto II using FACSDiva software (BD Biosciences). All subsequent analysis was performed using FlowJo (Treestar, Ashland, OR). The following antibodies were used for flow cytometric analysis of intestinal leucocytes. CD11c (clone HL3), CD45 (clone 30-F11), CD11b (clone M1/70), Gr-1 (clone RB6-8C5), and Ly6C (clone AL-21) as well as FcRIII/II (clone 2·4G2). All antibodies were purchased from BD Biosciences and Biolegend (San Diego, CA).
Statistical analysis
Unpaired two-tailed t-tests were used to identify statistically significant differences in gene expression between untreated and ceftriaxone-treated mice. For all other analyses, statistically significant changes were identified using a one-way analysis of variance with Tukey's post hoc test for multiple comparisons. For all quantitative PCR data (colonic gene expression and C. difficile colonization), statistical analysis was performed on normalized dCt values. Significance was set at P ≤ 0·05 in all analyses.
Results
Effect of ceftriaxone treatment on the colonic microbiota
To assess the effect of ceftriaxone treatment on the diversity and membership of the colonic microbiota, animals were given ceftriaxone (0·5 g/l) in their drinking water for 4 days and analysed 4 days later (Fig.1a). 454-pyrosequencing analysis of 16S amplicon libraries generated from colonic tissue from untreated and ceftriaxone-treated mice allowed for characterization and comparisons of the colonic mucosa-associated microbiota between groups.39 This analysis revealed that 4 days of ceftriaxone treatment was sufficient to significantly decrease the diversity of the mucosa-associated microbiota, compared with untreated mice, at 4 days post-antibiotic (Fig.1b). While untreated animals possessed a complex microbiota comprised primarily of members of the phyla Bacteroidetes and Firmicutes (Fig.1c), ceftriaxone treatment was associated with a marked shift in the membership of the community towards a composition dominated by a single Firmicute; Enterococcaceae (Fig.1d). These experiments demonstrated that ceftriaxone treatment was sufficient to significantly decrease the diversity and markedly alter the membership of the colonic microbiota even 4 days after the cessation of antibiotic treatment.
Effect of ceftriaxone treatment on colonic gene expression
To investigate the effect of ceftriaxone treatment and the associated loss of microbial diversity on immune regulation the colonic mucosa, quantitative RT-PCR was used to examine colonic cytokine expression following ceftriaxone treatment. Ceftriaxone treatment was associated with significantly increased expression of the chemokines Ccl5 and Cxcl9, and significantly decreased expression of Ccl11, Ccl3 and Cxcl2 (Fig.2a,b). Additionally, Il2, Il10, Il5 and Il3 expression was all significantly increased following ceftriaxone treatment, while expression of Il23a, Il17f, Il1b, Il17a, Tgfb and Slpi was decreased (Fig.2c–e). Examination of histopathological sections from the colons of ceftriaxone-treated mice revealed no evidence of cellular infiltration or inflammation in these animals (see Supporting information, Figure S1). Taken together, these data demonstrate that ceftriaxone treatment alone induces significant changes in the cytokine expression pattern in the colonic mucosa but does not result in overt inflammatory histopathological changes.
Figure 2.
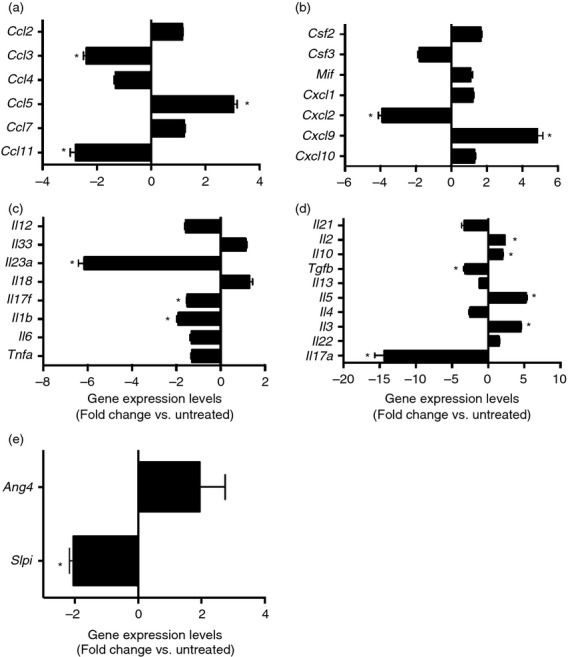
(a–e) Effect of ceftriaxone treatment on colonic inflammatory gene expression (day 4). Mice were treated as outlined in Figure1. Host gene expression was measured by quantitative PCR as described in the methods; n ≥ 9 per group. Data are shown as mean ± SEM fold change gene expression of ceftriaxone-treated animals compared with untreated mice. *P < 0·05 as compared with untreated animals.
Clostridium difficile infection following ceftriaxone treatment
To determine if ceftriaxone treatment was sufficient to permit colonization and infection by C. difficile, ceftriaxone-treated mice were challenged with vegetative cells (105 colony-forming units) from C. difficile strain VPI 10463 2 days after the cessation of ceftriaxone treatment and were followed for an additional 2 days (Fig.1a). Significant colonization by C. difficile was detected in the colonic mucosa of infected mice via quantitative PCR by 2 days post challenge (Fig.3a). Colonization did not cause a shift in the membership (Fig.1c–e) or diversity (Fig.1b) of the colonic microbiota beyond that attributable to ceftriaxone treatment alone. Clostridium difficile colonization of ceftriaxone-treated mice resulted in an influx of inflammatory cells into the colon and marked epithelial damage, indicative of active infection (Fig.4). Taken together, these data demonstrate that ceftriaxone treatment renders mice susceptible to C. difficile colonization with the concomitant development of colitis.
Figure 3.
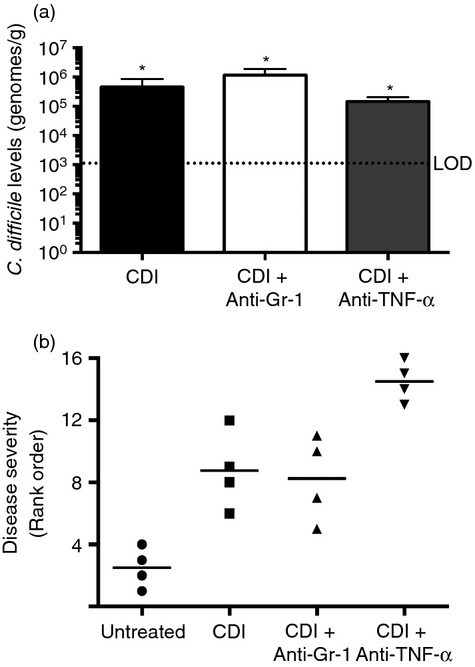
(a) Mucosal Clostridium difficile colonization as determined by species-specific quantitative PCR (Day 4). n ≥ 9 mice per group. Data are shown as mean ± SEM. CDI = C. difficile infected. LOD = Limit of Detection. *P < 0·05 as compared with untreated animals. Mice were treated as outlined in Fig.1 and the methods. (b) Rank order analysis of colonic histopathological sections from Untreated, C. difficile-infected, C. difficile-infected and anti-Gr-1-treated, C. difficile infected and anti-tumour necrosis factor-α (TNFα) treated animals (day 4). Slides were ordered on the basis of disease severity, with 16 having the most severe histopathology. n = 4 mice per group.
Figure 4.
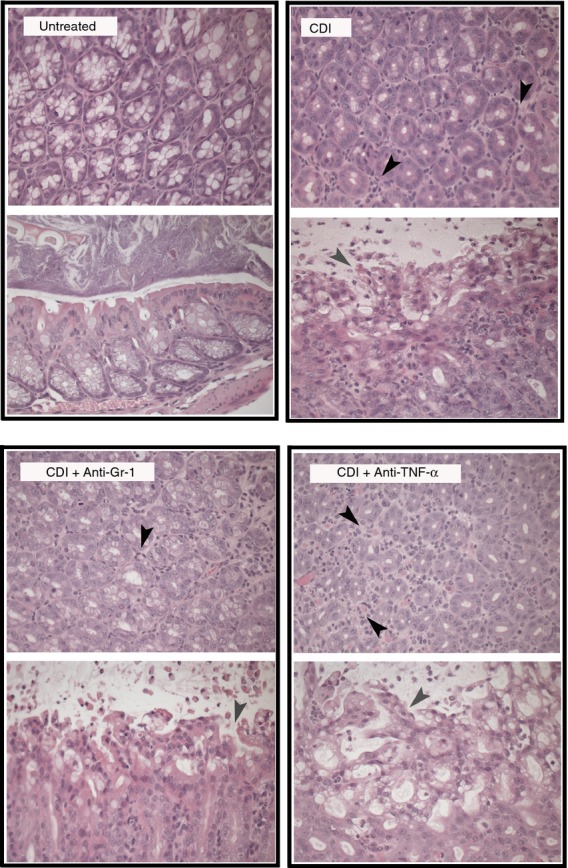
Photomicrographs of representative haematoxylin & eosin (H&E) -stained colonic sections from Untreated, Clostridium difficile-infected, C. difficile-infected and anti-Gr-1-treated, C. difficile-infected and anti- tumour necrosis factor-α (TNF-α)-treated animals (day 4). Cross-sections of colonic crypts (upper images) and longitudinal sections of the epithelial–luminal interface (lower images) are shown for each treatment. Black arrowheads indicate infiltrating inflammatory cells, while grey arrowheads indicate areas of epithelial damage. All animals were treated as outlined in Fig.1. Total magnification for all images is 400 ×. CDI = C. difficile infected.
Host responses to C. difficile colitis
In order to characterize the mucosal response to C. difficile colitis following ceftriaxone treatment, colonic gene expression was examined. Infection with C. difficile was associated with increased expression of Inos, Slpi, Il1b, Il6, Il17f, Ifng, Il17a, Il22, Il2, Il33 and Tnfa (Fig. 7 and 8c). Expression of Il18, Il23, Il12, Il3, Il4, Il5, Il13, Il10, Tgfb and Ang4 was not induced in response to C. difficile infection (Fig. 7 and 8c). Consistent with the robust cellular recruitment observed in histological sections from C. difficile-infected mice (Fig.4), there was increased expression of the CC chemokines Ccl2, Ccl3, Ccl4, Ccl5 and Ccl7, but not Ccl11 or Ccl24 (Fig. 6a,b) and the expression of the CXC chemokines Cxcl1 and Cxcl2, as well as the neutrophil stabilization factors Csf2 and Csf3 were also increased (Fig. 6c,d). Hence, these data indicate that the mucosal response to C. difficile infection is characterized by increased expression of CC and CXC chemokines and inflammatory cytokines including Il1b, Il6, Il17f and Tnfa.
Figure 7.
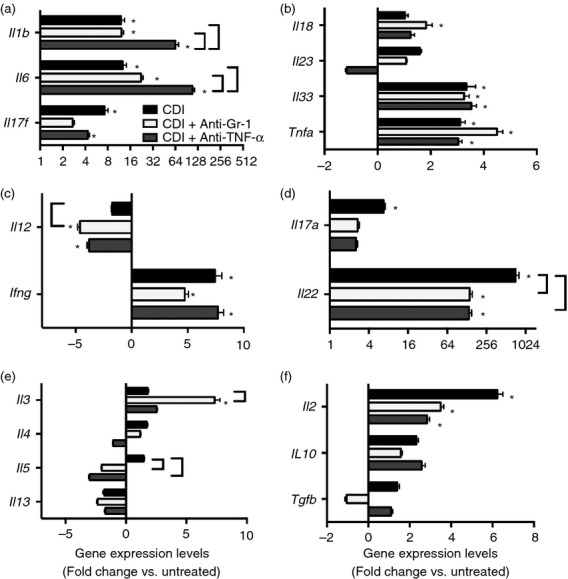
(a–g) Effect of anti-Gr-1 and anti-tumour necrosis factor-α (TNF-α ) treatment on colonic inflammatory cytokine expression during C. difficile colitis. Host gene expression was measured via quantitative PCR as described in the methods; n ≥ 7 per group. Data are shown as mean ± SEM fold change gene expression of C. difficile infected (black bars), C. difficile infected and anti-Gr-1 treated (white bars), and C. difficile infected and anti-TNF-α treated (grey bars) animals as compared with untreated mice. CDI = C. difficile infected *P < 0·05 compared with untreated animals. Brackets indicate P < 0·05 for the differences between the indicated groups.
Figure 8.
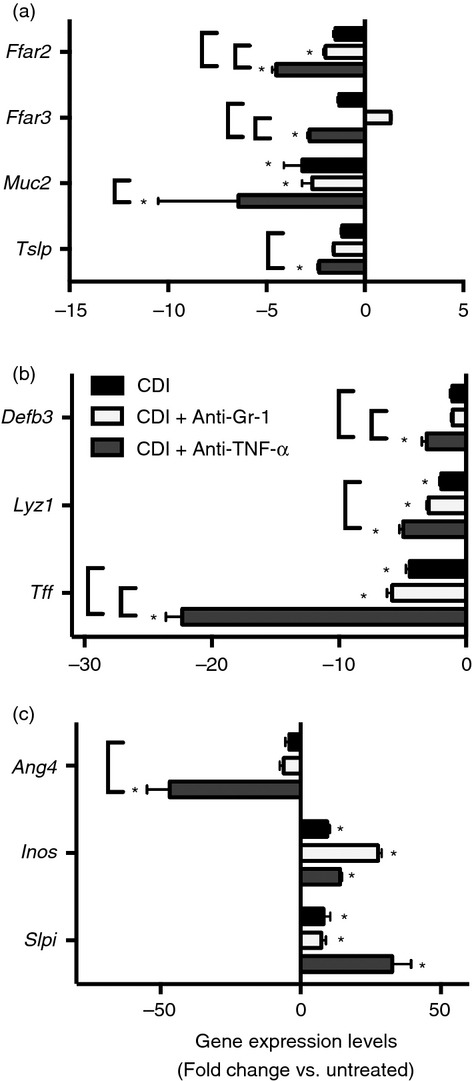
(a–c) Effect of anti-Gr-1 and anti-tumour necrosis factor-α (TNF-α) treatment on colonic gene expression during Clostridium difficile colitis. Host gene expression was measured via quantitative PCR as described in the Materials and methods; n ≥ 7 per group. Data are shown as mean ± SEM fold change gene expression of C. difficile-infected (black bars), C. difficile-infected and anti-Gr-1-treated (white bars), and C. difficile-infected and anti-TNF-α-treated (grey bars) animals as compared to untreated mice. CDI = C. difficile infected *P < 0·05 as compared to untreated animals. Brackets indicate P < 0·05 for the differences between the indicated groups.
Figure 6.
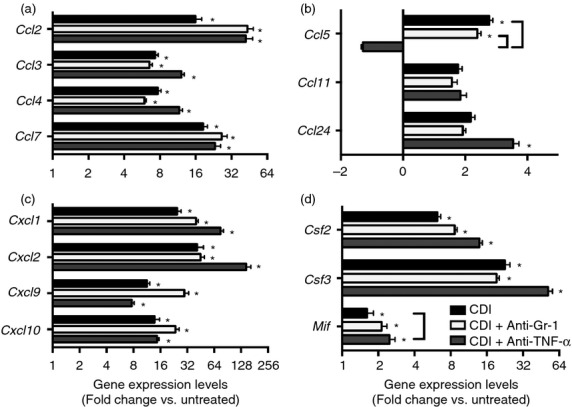
(a–d) Effect of anti-Gr-1 and anti-tumour necrosis factor-α (TNF-α) treatment on colonic inflammatory chemokine expression during C. difficile colitis (day 4). Colonic gene expression was measured via quantitative PCR as outlined in the Materials and methods. n ≥ 8 per group. Data are shown as mean ± SEM fold change gene expression of C. difficile infected (black bars), C. difficile-infected and anti-Gr-1-treated (white bars), and C. difficile infected and anti-TNFα treated (gray bars) animals as compared with untreated mice. *P < 0·05 compared with untreated animals. Brackets indicate P < 0·05 for the differences between the indicated groups.
Flow cytometry was used to further delineate the leucocyte populations recruited to the colon following C. difficile infection. Analysis of the side scatter (SSC) and forward scatter (FSC) parameters of CD45+ leucocytes revealed a drastic influx of FSCMid SSCHigh leucocytes in response to C. difficile infection (Fig.5a). Nearly all of the recruited FSCMid SSCHigh cells were CD11bHigh CD11cLow inflammatory myeloid cells (Fig.5b). Within the CD11bHigh CD11cLow population, there were two populations, the largest of which were Ly6CMid Gr-1High neutrophils, and a smaller population of Ly6CHigh Gr-1Mid monocytes (Fig.5c). Hence, these data demonstrate marked recruitment of two distinct Gr-1+ leucocyte populations, Ly6CMid Gr-1High neutrophils and Ly6CHigh Gr-1Mid monocytes, to the colon in response to C. difficile colitis.
Figure 5.
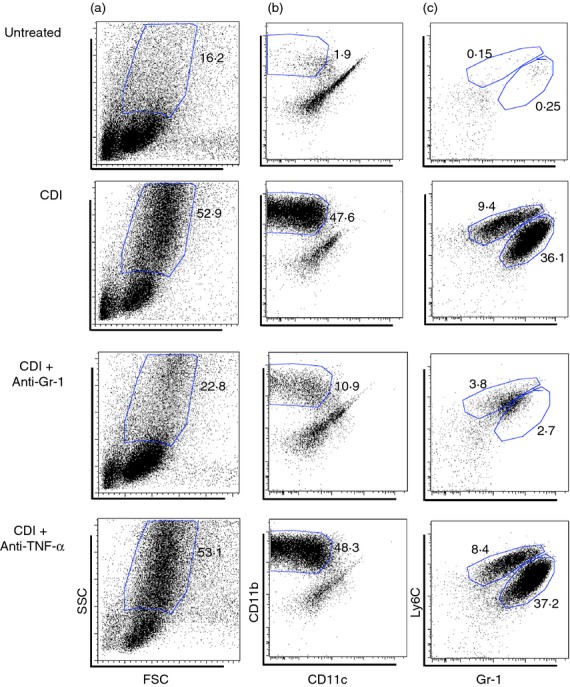
Flow cytometric analysis of colonic leucocytes from Untreated, Clostridium difficile-infected, C. difficile-infected and anti-Gr-1-treated, C. difficile infected and anti-tumour necrosis factor (TNF-α ) -treated animals (day 4). (a) Analysis of forward-scatter (FSC) and side-scatter (SSC) properties of total colonic CD45+ leucocytes. (b) Analysis of CD11b and CD11c expression profiles of SSCHigh FSCLow CD45+ leucocytes as defined in panel (a). (c) Analysis of Ly6C and Gr-1 expression profiles of the CD11bHigh CD11cLow population defined in panel (b). CDI = C. difficile infected. The number in bold type indicates the percentage of total CD45+ leucocytes contained within the indicated gate.
The role of Gr-1+cells during C. difficile colitis
To determine the role of Gr-1+ cells in supporting the mucosal inflammatory response to C. difficile infection, animals were treated with an anti-Gr-1 mAb (clone RB6-8C5) 1 day before and 1 day after C. difficile infection (Fig.1a). There was a marked reduction in the frequency of CD11bHigh CD11cLow inflammatory myeloid cells, as well as a reduction in both the Ly6CMid Gr-1High neutrophil and Ly6CHigh Gr-1Mid monocyte populations following anti-Gr-1 treatment (Fig.5b,c). However, anti-Gr-1 treatment had no effect on C. difficile levels in the colonic mucosa (Fig.3a).
Compared with C. difficile infection alone, Il5, Il12 and Il22 expression was significantly reduced, and Il3 expression was significantly increased, in C. difficile-infected anti-Gr-1-treated mice (Fig. 7c–e). However, expression of Il1b, Il6, Csf2, Csf3, Tnfa and Il33 was unchanged following anti-Gr-1 treatment (Fig.6 and 7). Expression of Cxcl1, Cxcl2, Ccl2 and Ccl3 was also not significantly affected by anti-Gr-1 treatment (Fig.6a,c). Anti-Gr-1 treatment did not protect against the colonic histopathology that developed during the normal course of infection (Fig.3b and 4). Taken together, these data indicate that the expression of CC and CXC chemokines, the expression of inflammatory cytokines including Tnfa, Il1b and Il6, and the development of severe colonic histopathology during C. difficile colitis are all independent of the presence of Gr-1+ cells within the colon.
The role of TNF-α during C. difficile colitis
To examine the role of TNF-α in promoting inflammatory myeloid cell recruitment and epithelial damage in response to C. difficile infection, mice were treated with a TNF-α neutralizing mAb (clone MP6-XT3) 1 day before infection (Fig.1a). Clostridium difficile colonization within the colonic mucosa was equivalent between anti-TNF-α-treated and C. difficile-infected animals (Fig.3a). There was near identical recruitment of CD11bHigh CD11cLow inflammatory myeloid cells following anti-TNF-α treatment compared with that seen in response to C. difficile infection alone (Fig.5b). Ly6CMid Gr-1High neutrophil and Ly6CHigh Gr-1Mid monocyte frequencies were also unchanged following anti-TNF-α treatment (Fig.5c). Consistent with this observation, Cxcl1, Cxcl2, Ccl2 and Ccl3 expression was not reduced in these mice (Fig.6a,c). While the expression of Il33, Ifng and Il17f was not significantly reduced by anti-TNF-α treatment, expression of Mif, Il1b and Il6 was all significantly increased following anti-TNF-α treatment (Fig.6d and 7). Furthermore, expression of Ccl5, Il5 and Il22 was all significantly lower in anti-TNF-α-treated mice (Fig.6b and 7d,e).
Epithelial damage was prominent in anti-TNF-α-treated mice (Fig.4) and anti-TNF-α treatment was associated with more severe colonic histopathology (Fig.3b). Anti-TNF-α treatment was also associated with a significant decrease in the expression levels of the colonic goblet cell gene Ang4, the free fatty acid receptors Ffar2 and Ffar3, epithelium-derived effector and signal molecules encoded by Tslp, Tff, Defb3 and Lyz1, in addition to a trend toward higher expression levels of Slpi compared with C. difficile infection alone (Fig.8). Additionally, there was a trend towards decreased expression of the goblet cell-specific gene Muc2 after anti-TNF-α treatment (Fig.8a). Taken together, these data indicate that anti-TNF-α treatment was associated with enhanced colonic epithelial dysfunction/destruction and can augment the epithelial inflammatory milieu during C. difficile infection.
Discussion
Together, these data demonstrate that ceftriaxone-treated mice are a robust model for investigating the innate acute inflammatory response to C. difficile infection. In addition to altering the composition of the colonic microbiome, ceftriaxone treatment alone was also capable of modulating colonic cytokine and chemokine expression 4 days after the cessation of antibiotic treatment. While ceftriaxone treatment was associated with significantly altered expression of several genes induced during C. difficile colitis, the induction of the inflammatory cytokines and chemokines associated with C. difficile infection was significantly greater than that seen with ceftriaxone treatment alone (see Supporting information, Figure S2). Additionally, ceftriaxone treatment alone did not produce any histopathological changes in the colonic mucosa (see Supporting information, Figure S1). Hence, while ceftriaxone treatment is capable of modulating colonic gene expression, we propose it is not a confounding factor in evaluating the host inflammatory response in this model of infectious colitis.
Anti-TNF-α treatment was associated with an augmented epithelial inflammatory response to C. difficile infection. Previous studies have reported a role for TNF-α signalling in promoting myeloid cell recruitment during mucosal inflammation.23,24 TNF-α can enhance the expression of CCL3 during chemically induced pulmonary inflammation,24 and interference with TNF-α signalling reduces neutrophil recruitment in response to acute allergic airway inflammation.23 TNF-α can contribute to inflammatory cytokine expression and tissue damage during mucosal inflammation.25,40 Directly applicable to gastrointestinal inflammation, during 2,4,6-trinitrobenzenesulphonic acid colitis, TNF-α signalling promotes both IL-18 and TNF-α expression as well as the development of intestinal histopathology.25 Anti-TNF-α treatment had no effect on Cxcl1, Cxcl2, Ccl2 and Ccl3 expression during C. difficile colitis, and we observed no reduction in neutrophil and monocyte recruitment under these conditions. However, colonic expression of Il1b and Il6 was increased following anti-TNF-α treatment. Hence, despite its relatively low level of expression, TNF-α is involved in the inflammatory response during C. difficile colitis but the induction of CC and CXC chemokines and subsequent myeloid cell recruitment during infection are largely TNF-α-independent.
While TNF-α is generally held to be a pro-inflammatory mediator that can participate in indirect epithelial damage, there is precedent for an epithelium cytoprotective role for this cytokine, as our data suggest may occur in this infection model.41–43 TNF-α signalling can protect against severe intestinal histopathology during Citrobacter rodentium infection.41 Furthermore, cellular infiltration and intestinal epithelial damage during DSS colitis are enhanced in the absence of TNF-α.42,43 Following anti-TNF-α treatment we observed decreased expression of the free fatty acid receptors Ffar2 and Ffar3, as well as Defb3, Lyz1 and Tslp, consistent with the severe colonic epithelial damage present in these animals. Anti-TNF-α treatment was also associated with significantly decreased expression of Ang4 and a trend towards decreased expression of Muc2, two genes whose expression is specific to goblet cells within the gastrointestinal tract,44–46 suggesting reduced goblet cell function. Additionally, colonic histopathology was most severe following anti-TNF-α treatment. Hence, our data are consistent with these other models of colitis where TNF-α plays a role in protecting the colonic epithelium from damage during inflammation, and suggests an epithelial-protective role for TNF-α during acute, severe C. difficile infection.
One model that could explain the association between anti-TNF-α treatment and increased inflammatory responses and epithelial damage is that TNF-α signalling may restrain host inflammatory responses by promoting the clearance of translocated commensal bacteria. Previous studies have demonstrated the dissemination of commensal bacterial to distal sites during C. difficile infection,1,2 and have also suggested a role for these translocated bacteria in promoting innate inflammatory responses, including the production of IL-1β.1 TNF-α signalling can activate macrophages and enhance their bactericidal capacity,47 and mucosal macrophages represent a large resident population within the colonic lamina propria with the capacity to eliminate invading bacteria.48 Hence, in the absence of TNF-α signalling, translocated commensal bacteria may be less efficiently cleared by macrophages within the colonic lamina propria, leading to increased activation of inflammatory pathways and enhanced colonic tissue damage.
Despite the robust recruitment of Ly6CMid Gr-1High neutrophils and Ly6CHigh Gr-1Mid monocytes in response to C. difficile infection, we observed no reduction in colonic expression of CC and CXC chemokines or proinflammatory cytokines including Il1b, Il6, Il33 and Tnfa following anti-Gr-1 treatment. Neutrophil recruitment is commonly associated with the development of inflammation at mucosal sites23,49,50 and neutrophils are a well-documented source of inflammatory cytokines, including IL-6, as well as the neutrophil chemokine IL-8.51–53 While several studies have investigated the role of neutrophils in reducing host mortality and preventing the translocation of bacteria during C. difficile infection,2,3 to our knowledge the role of neutrophils as a cellular source of chemokine and cytokine expression has not been extensively investigated. Despite effective depletion of both neutrophils and monocytes following our anti-Gr-1 treatment, colonic CC and CXC chemokine expression as well as Il1b, Il6, Il33 and Tnfa expression was unchanged. Consistent with the significant reduction in Il22 expression, a cytokine with a vital role in promoting wound healing and preventing epithelial damage at mucosal sites,54–56 we observed no protection from intestinal epithelial damage in anti-Gr-1-treated mice. Taken together, our data strongly suggest that the recruited Gr-1+ populations, including neutrophils, are not responsible for the robust inflammatory cytokine expression or epithelial damage observed during C. difficile infection.
One potential explanation for our observation that depletion of Gr-1+ cell populations had minimal effect on colonic cytokine and chemokine expression is that Gr-1+ cells, including neutrophils, are not a major cellular source of these inflammatory mediators during the host response to C. difficile colitis. A recent study has reported decreased intestinal IL-1β and CXCL1 production following neutrophil depletion.1 However, that study used a less severe model of disease with 100% survival of infected animals at 5 days-post infection,1 while the model used in the current study, similar to other models based on the third-generation cephalosporin cefoperazone,8,10 resulted in high morbidity by 2 days post infection. Hence, in the context of such an overwhelming infection, alternative pathways and sources of inflammatory cytokine expression may respond and effectively supersede the cytokine production by neutrophils observed in less severe models. Additionally, Hasegawa et al.1 used a combination of seven antibiotics to permit C. difficile infection, compared with the single antibiotic, ceftriaxone, used in the current study. These two antibiotic treatments will likely result in different colonic microbial communities, whose members may differentially stimulate host cell subsets and result in the activation of distinct inflammatory pathways in response to the same challenge.
Acknowledgments
We thank the staff of the University of Michigan Flow Cytometry Core for their assistance in performing the flow cytometry experiments. We also thank Sue Foltin for her help with the pyrosequencing experiments, and Charles Frank for his assistance with the mouse experiments. We thank Dr Merritt Gillilland III for critical review of this manuscript. This work was supported by National Institutes of Health grants U19 AI090871 (GBH and VBY) and P30 DK034933 (GBH and VBY). Support was also provided by the Host-Microbiome Initiative (HMI) of the University of Michigan Medical School (GBH and VBY).
Glossary
Abbreviations:
- GM-CSF
Granulocyte-Macrophage Colony Stimulating Factor
- TNF-α
Tumor Necrosis Factor Alpha
- IL
Interleukin
- IFNγ
Interferon Gamma
- CCL
Chemokine (C-C Motif) Ligand
- CXCL
Chemokine (C-X-C Motif) Ligand
- CSF
Colony Stimulating Factor
- MIF
Macrophage Migration Inhibitory Factor
- iNOS
Inducible Nitric Oxide Synthase (NOS2)
- RegIIIγ
Regenerating Islet-Derived 3 Gamma
- SLPI
Secretory leukocyte peptidase inhibitor
- Tgfβ1
Transforming Growth Factor beta 1
- Ang4
Angiogenin 4
- TCCFA
Taurocholate Cycloserine Cefoxitin Fructose Agar
- TNBS
2,4,6-trinitrobenzenesulfonic acid
- BHIS
Brain Heart Infusion Supplemented with 0.1% Cysteine
- SSC
Side Scatter
- FSC
Forward Scatter
Author contributions
AJM and GBH conceived, designed and interpreted the experiments; VBY, NRF and RAM contributed to their design and interpretation. AJM, JRE, NRM, KEH and RAM performed the experiments. AJM, RM, JRE, KEH and GBH analysed the data. AJM and GBH wrote the manuscript, and all other authors provided comments and advice on the manuscript.
Disclosures
Vincent B. Young is on the advisory board of ViroPharma in relation to the development of a non-toxigenic C. difficile strain for the management of C. difficile infection. All other authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Photomicrographs of representative Hematoxylin and Eosin (H&E) stained colonic sections from both uninfected (untreated) and ceftriaxone treated mice (day 4).
Figure S2. (a–e) Comparison of ceftriaxone treatment alone and Clostridium difficile infection following ceftriaxone treatment on colonic inflammatory gene expression (day 4).
References
- Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G, Inohara N. Protective role of commensals against Clostridium difficile infection via an IL-1β-mediated positive-feedback loop. J Immunol. 2012;189:3085–91. doi: 10.4049/jimmunol.1200821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G, Inohara N. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol. 2011;186:4872–80. doi: 10.4049/jimmunol.1003761. [DOI] [PubMed] [Google Scholar]
- Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun. 2012;80:2989–96. doi: 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarchum I, Liu M, Lipuma L, Pamer EG. Toll-like receptor 5 stimulation protects mice from acute Clostridium difficile colitis. Infect Immun. 2011;79:1498–503. doi: 10.1128/IAI.01196-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A, Lynch M, Smith SM, et al. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog. 2011;7:e1002076. doi: 10.1371/journal.ppat.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Jarchum I, Equinda M, et al. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun. 2012;80:62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP. A mouse model of Clostridium difficile-associated disease. Gastroenterology. 2008;135:1984–92. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile Infection. Gut Microbes. 2011;2:145–58. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadighi Akha AA, Theriot CM, Erb Downward JR, et al. Acute infection of mice with Clostridium difficile leads to eIF2α phosphorylation and pro-survival signalling as part of the mucosal inflammatory response. Immunology. 2013;140:111–22. doi: 10.1111/imm.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot CM, Koumpouras CC, Carlson PE, Bergin II, Aronoff DM, Young VB. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes. 2011;2:326–34. doi: 10.4161/gmic.19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wang H, Zhang Y, Chen K, Davis B, Feng H. Mouse relapse model of Clostridium difficile infection. Infect Immun. 2011;79:2856–64. doi: 10.1128/IAI.01336-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai M, Henke MO, Rubin BK. Macrolide antibiotics as immunomodulatory medications: proposed mechanisms of action. Pharmacol Ther. 2008;117:393–405. doi: 10.1016/j.pharmthera.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Vickers IE, Smikle MF. The immunomodulatory effect of antibiotics on the secretion of tumour necrosis factor α by peripheral blood mononuclear cells in response to Stenotrophomonas maltophilia stimulation. West Indian Med J. 2006;55:138–41. doi: 10.1590/s0043-31442006000300002. [DOI] [PubMed] [Google Scholar]
- Tauber SC, Nau R. Immunomodulatory properties of antibiotics. Curr Mol Pharmacol. 2008;1:68–79. [PubMed] [Google Scholar]
- Shalit I, Halperin D, Haite D, Levitov A, Romano J, Osherov N, Fabian I. Anti-inflammatory effects of moxifloxacin on IL-8, IL-1β and TNF-α secretion and NFκB and MAP-kinase activation in human monocytes stimulated with Aspergillus fumigatus. J Antimicrob Chemother. 2006;57:230–5. doi: 10.1093/jac/dki441. [DOI] [PubMed] [Google Scholar]
- Lopez-Boado YS, Rubin BK. Macrolides as immunomodulatory medications for the therapy of chronic lung diseases. Curr Opin Pharmacol. 2008;8:286–91. doi: 10.1016/j.coph.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Cigana C, Assael BM, Melotti P. Azithromycin selectively reduces tumor necrosis factor α levels in cystic fibrosis airway epithelial cells. Antimicrob Agents Chemother. 2007;51:975–81. doi: 10.1128/AAC.01142-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigana C, Nicolis E, Pasetto M, Assael BM, Melotti P. Anti-inflammatory effects of azithromycin in cystic fibrosis airway epithelial cells. Biochem Biophys Res Commun. 2006;350:977–82. doi: 10.1016/j.bbrc.2006.09.132. [DOI] [PubMed] [Google Scholar]
- Yamasawa H, Oshikawa K, Ohno S, Sugiyama Y. Macrolides inhibit epithelial cell-mediated neutrophil survival by modulating granulocyte macrophage colony-stimulating factor release. Am J Respir Cell Mol Biol. 2004;30:569–75. doi: 10.1165/rcmb.2003-0105OC. [DOI] [PubMed] [Google Scholar]
- Bijie H, Kulpradist S, Manalaysay M, Soebandrio A. In vitro activity, pharmacokinetics, clinical efficacy, safety and pharmacoeconomics of ceftriaxone compared with third and fourth generation cephalosporins: review. J Chemother. 2005;17:3–24. [PubMed] [Google Scholar]
- Nahata MC, Barson WJ. Ceftriaxone: a third-generation cephalosporin. Drug Intell Clin Pharm. 1985;19:900–6. doi: 10.1177/106002808501901203. [DOI] [PubMed] [Google Scholar]
- Harrison CJ, Bratcher D. Cephalosporins: a review. Pediatr Rev. 2008;29:264–7. doi: 10.1542/pir.29-8-264. ; quiz 73. [DOI] [PubMed] [Google Scholar]
- Lukacs NW, Strieter RM, Chensue SW, Widmer M, Kunkel SL. TNF-α mediates recruitment of neutrophils and eosinophils during airway inflammation. J Immunol. 1995;154:5411–7. [PubMed] [Google Scholar]
- Smith RE, Strieter RM, Phan SH, Lukacs N, Kunkel SL. TNF and IL-6 mediate MIP-1α expression in bleomycin-induced lung injury. J Leukoc Biol. 1998;64:528–36. [PubMed] [Google Scholar]
- Shen C, de Hertogh G, Bullens DM, Van Assche G, Geboes K, Rutgeerts P, Ceuppens JL. Remission-inducing effect of anti-TNF monoclonal antibody in TNBS colitis: mechanisms beyond neutralization? Inflamm Bowel Dis. 2007;13:308–16. doi: 10.1002/ibd.20005. [DOI] [PubMed] [Google Scholar]
- Sun X, He X, Tzipori S, Gerhard R, Feng H. Essential role of the glucosyltransferase activity in Clostridium difficile toxin-induced secretion of TNFα by macrophages. Microb Pathog. 2009;46:298–305. doi: 10.1016/j.micpath.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan CE, Sukhumavasi W, Bierly AL, Denkers EY. Understanding the multiple functions of Gr-1+ cell subpopulations during microbial infection. Immunol Res. 2008;40:35–48. doi: 10.1007/s12026-007-0061-8. [DOI] [PubMed] [Google Scholar]
- Nakano H, Yanagita M, Gunn MD. CD11c+ B220+ Gr-1-)cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med. 2001;194:1171–8. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, Sibley LD. Gr1+ inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–17. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland T. A survey of discriminant methods used in toxicological histopathology. Toxicol Pathol. 2001;29:269–73. doi: 10.1080/019262301317052567. [DOI] [PubMed] [Google Scholar]
- Holland T. The comparative power of the discriminant methods used in toxicological pathology. Toxicol Pathol. 2005;33:490–4. doi: 10.1080/01926230590965382. [DOI] [PubMed] [Google Scholar]
- Sadighi Akha AA, Harper JM, Salmon AB, Schroeder BA, Tyra HM, Rutkowski DT, Miller RA. Heightened induction of proapoptotic signals in response to endoplasmic reticulum stress in primary fibroblasts from a mouse model of longevity. J Biol Chem. 2011;286:30344–51. doi: 10.1074/jbc.M111.220541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Kindt, et al. pp. 17–3. vegan: Community Ecology Package. R Package version 1,1.17-3 Edn. 2010. 2010.
- Luna RA, Boyanton BL, Jr, Mehta S, Courtney EM, Webb CR, Revell PA, Versalovic J. Rapid stool-based diagnosis of Clostridium difficile infection by real-time PCR in a children's hospital. J Clin Microbiol. 2011;49:851–7. doi: 10.1128/JCM.01983-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc. 2007;2:2307–11. doi: 10.1038/nprot.2007.315. [DOI] [PubMed] [Google Scholar]
- Erb-Downward JR, Thompson DL, Han MK, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20:87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves NS, Ghaem-Maghami M, Monteleone G, Frankel G, Dougan G, Lewis DJ, Simmons CP, MacDonald TT. Critical role for tumor necrosis factor α in controlling the number of lumenal pathogenic bacteria and immunopathology in infectious colitis. Infect Immun. 2001;69:6651–9. doi: 10.1128/IAI.69.11.6651-6659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y, Takagi T, Handa O, et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-α-deficient mice. J Gastroenterol Hepatol. 2003;18:560–9. doi: 10.1046/j.1440-1746.2003.03034.x. [DOI] [PubMed] [Google Scholar]
- Noti M, Corazza N, Mueller C, Berger B, Brunner T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med. 2010;207:1057–66. doi: 10.1084/jem.20090849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau SK, Weiss LM, Chu PG. Differential expression of MUC1, MUC2, and MUC5AC in carcinomas of various sites: an immunohistochemical study. Am J Clin Pathol. 2004;122:61–9. doi: 10.1309/9R66-73QE-C06D-86Y4. [DOI] [PubMed] [Google Scholar]
- Audie JP, Janin A, Porchet N, Copin MC, Gosselin B, Aubert JP. Expression of human mucin genes in respiratory, digestive, and reproductive tracts ascertained by in situ hybridization. J Histochem Cytochem. 1993;41:1479–85. doi: 10.1177/41.10.8245407. [DOI] [PubMed] [Google Scholar]
- Forman RA, deSchoolmeester ML, Hurst RJ, Wright SH, Pemberton AD, Else KJ. The goblet cell is the cellular source of the anti-microbial angiogenin 4 in the large intestine post Trichuris muris infection. PLoS One. 2012;7:e42248. doi: 10.1371/journal.pone.0042248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt AM, Mowat AM. Mucosal macrophages and the regulation of immune responses in the intestine. Immunol Lett. 2008;119:22–31. doi: 10.1016/j.imlet.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Farooq SM, Stillie R, Svensson M, Svanborg C, Strieter RM, Stadnyk AW. Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2009;329:123–9. doi: 10.1124/jpet.108.145862. [DOI] [PubMed] [Google Scholar]
- Godinez I, Raffatellu M, Chu H, et al. Interleukin-23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect Immun. 2009;77:387–98. doi: 10.1128/IAI.00933-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The neutrophil as a cellular source of chemokines. Immunol Rev. 2000;177:195–203. doi: 10.1034/j.1600-065x.2000.17706.x. [DOI] [PubMed] [Google Scholar]
- Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 2010;49:1618–31. doi: 10.1093/rheumatology/keq045. [DOI] [PubMed] [Google Scholar]
- Takahashi GW, Andrews DF, 3rd, Lilly MB, Singer JW, Alderson MR. Effect of granulocyte-macrophage colony-stimulating factor and interleukin-3 on interleukin-8 production by human neutrophils and monocytes. Blood. 1993;81:357–64. [PubMed] [Google Scholar]
- Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–72. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–9. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Ogawa A, Mizoguchi E, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–44. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Photomicrographs of representative Hematoxylin and Eosin (H&E) stained colonic sections from both uninfected (untreated) and ceftriaxone treated mice (day 4).
Figure S2. (a–e) Comparison of ceftriaxone treatment alone and Clostridium difficile infection following ceftriaxone treatment on colonic inflammatory gene expression (day 4).