

Abstract
Although runt-related transcription factor 2 (RUNX2) is known to be an essential key transcription factor for osteoblast differentiation and bone formation, RUNX2 also plays a pivotal role in the regulation of p53-dependent DNA damage response. In the present study, we report that, in addition to p53, RUNX2 downregulates pro-apoptotic TAp73 during DNA damage-dependent cell death. Upon adriamycin (ADR) exposure, human osteosarcoma-derived U2OS cells underwent cell death in association with an upregulation of TAp73 and various p53/TAp73-target gene products together with RUNX2. Small interfering RNA-mediated silencing of p73 resulted in a marked reduction in ADR-induced p53/TAp73-target gene expression, suggesting that TAp73 is responsible for the ADR-dependent DNA damage response. Immunoprecipitation and transient transfection experiments demonstrated that RUNX2 forms a complex with TAp73 and impairs its transcriptional activity. Notably, knockdown of RUNX2 stimulated ADR-induced cell death accompanied by a massive induction of TAp73 expression, indicating that RUNX2 downregulates TAp73 expression. Consistent with this notion, the overexpression of RUNX2 suppressed ADR-dependent cell death, which was associated with a remarkable downregulation of TAp73 and p53/TAp73-target gene expression. Collectively, our present findings strongly suggest that RUNX2 attenuates the transcriptional activity and ADR-mediated induction of TAp73, and may provide novel insights into understanding the molecular basis behind the development and/or maintenance of chemoresistance. Thus, we propose that the silencing of RUNX2 might be an attractive strategy for improving the chemosensitivity of malignant cancers.
Structured digital abstract
p73andRUNX2colocalizefluorescence microscopy(View interaction)
RUNX2physically interactswithp73byanti bait coip(1,2)
Keywords: cell death, DNA damage, p53, RUNX2, TAp73
Introduction
The appropriate DNA damage response that monitors and maintains genomic integrity is considered to be a critical barrier against tumorigenesis. Tumor suppressor p53 plays a pivotal role in the regulation of the DNA damage response. p53 is divided into three chracteristic functional domains, including the NH2-terminal transactivation domain (TA), the central sequence-specific DNA-binding domain (DB) and the COOH-terminal oligomerization domain (OD). As expected from its structural property, p53 acts as a nuclear transcription factor and has the ability to transactivate numerous target genes implicated in the induction of cell cycle arrest (p21WAF1 and 14-3-3σ), DNA repair (GADD45), cellular senescence (p21WAF1) and/or apoptotic cell death (BAX,PUMA and NOXA) [1–3]. Among these cellular processes, the promotion of apoptotic cell death has a prominent role in tumor suppression by eliminating cells with seriously damaged DNA. Upon cellular insult, such as DNA damage, oncogene activation and hypoxia, p53 is quickly activated by sequential post-translational modifications, including phosphorylation (Ser-15, Ser-20 and Ser-46) and acetylation (Ace-373 and Ace-382), thereby exerting its pro-apoptotic function via the subsequent transactivation of its target genes [1–3].
Unfortunately, p53 is mutated and loses its tumor-suppressive function in approximately half of human cancerous cells [4–6]. Over 90% of p53 mutations are detectable within the genomic region encoding its central sequence-specific DNA-binding domain, suggesting that the sequence-specific transactivation activity of p53 is tightly linked to its pro-apoptotic function. By contrast to a short-lived wild-type p53, mutant p53 shows a prolonged half-life [3]. Additionally, mutant p53 aquires an oncogenic potential and also displays a dominant-negative behavior against wild-type p53 [7]. Moreover, cancerous cells expressing p53 mutant sometimes exhibit chemoresistant phenotypes [7,8]. Of note, mutant p53 transactivates myriad of genes involved in many different aspects of tumorigenesis such as MYC,EGFR and MDR1 [8]. p73 is a member of p53 tumor suppressor family and acts as a negative regulator of tumorigenesis by inducing cell cycle arrest and/or apoptotic cell death via its ability to transactivate overlapping set of p53-target genes [9,10]. Similar to p53, p73 has been shown to accumulate in response to certain DNA damaging agents such as cisplatin and adriamycin (ADR) and to play a crucial role in the regulation of DNA damage response [11]. It has been reported that DNA damage-mediated transcriptional activation of p73 is modulated by E2F-1 [12–14]. Importantly, p73 produces two distinct gene products, including a transactivation domain-containing isoform (TAp73) and a transactivation-deficient NH2-terminally truncated isoform (ΔNp73). ΔNp73 arises from the alternative promoter usage [15]. Although p73 is rarely mutated in human cancerous cells [16], TAp73-deficient mice showed an increased susceptibility to spontaneous and carcinogen-induced tumors with a peculiar promotion of lung adenocarcinomas, implying that TAp73 possesses a tumor-suppressive capability [17]. It is worth noting that TAp73 is required for p53-dependent apoptotic cell death, whereas TAp73 alone has the ability to induce apototic cell death in the absence of wild-type p53 [18]. By contrast, ΔNp73 lacking the NH2-terminal transactivation domain has an oncogenic function and acts as a dominant-negative inhibitor toward TAp73, as well as wild-type p53 [15,19]. Thus, the intracellular balance between the amounts of anti-apoptotic ΔNp73 and pro-apoptotic p53/TAp73 might be a critical determinant of cell fate. The mammalian runt-related transcription factor (RUNX) family is composed of three members, including RUNX1, RUNX2 and RUNX3. RUNX family members contain an evolutionarily conserved 128-amino acids runt domain, which is required for specific DNA-binding (ACC(A/G)CA) and heterodimerization with its cofactor CBFβ (core-binding factor β) [20,21]. CBFβ does not bind directly to DNA but enhances RUNX proteins binding to DNA [22]. Among the RUNX family members, RUNX2 was shown to play a key role in the regulation of proper osteoblast differentiation and bone formation [23,24]. By contrast to RUNX2, RUNX1 and RUNX3 are known to act as essential regulators of hematopoietic and gastric development, respectively [25–28]. Indeed, RUNX2-deficient mice completely failed to form mineralized bone [23,24] and mice overexpressing RUNX2 displayed osteopenia with multiple fractures [29]. Consistent with these observations, RUNX2 transactivates various target genes involved in the promotion of osteogenic differentiation, such as type 1 collagen,osteopontin and osteocalcin [30,31].
Alternatively, there is growing evidence to suggest that RUNX2 has a pro-oncogenic function in a wide variety of cells. For example, overexpression of RUNX2 in transgenic mice was found to predispose mice to lymphoma [32]. Intriguingly, Browne et al. [33] reported that RUNX2 induces the expression of pro-survival Bcl-2 and thus contributes to the development of malignant phenotypes in prostate cancer cells. Furthermore, current evidence indicates that RUNX2 is a critical pathological factor in human metastatic breast, pancreatic, thyroid, prostate and colon cancer cells [34–37]. Thus, it is likely that RUNX2 is closely involved in malignant cancer cell proliferation, chemoresistance, migration and invasion. To overcome the malignant phenotypes of human cancers, a precise understanding of the molecular mechanism(s) with respect to how aggressive cancers develop and maintain resistance to chemotherapeutic agents is required. As noted above, chemoresistance of cancerous cells is often attributed to an impaired functioning of p53 as a result of genetic mutation and/or sequestration by the other proteins. Recently, we described that RUNX2 attenuates p53-dependent apoptotic cell death in response to DNA damage [38], indicating that RUNX2 contributes to chemoresistance by inhibiting p53.
In the present study, we report that RUNX2 has the ability to suppress transcriptional activity as well as ADR-mediated induction of TAp73, thereby inhibiting the proper DNA damage response of cancerous cells.
Results
Simultaneous induction of TAp73 and RUNX2 during ADR-mediated cell death
To determine whether a functional interaction exists between pro-apoptotic TAp73 and pro-oncogenic RUNX2 during the DNA damage response, we investigated the expression level of TAp73 and RUNX2 in response to ADR. Accordingly, human osteosarcoma-derived p53-proficient U2OS cells were exposed to increasing doses of ADR. Consistent with our recent observations [38], U2OS cells underwent cell death after ADR treatment (Fig.1). Under our experimental conditions, non-apoptotic cell death was barely detectable, as examined by annexin V and propidium iodide (PI) double-staining experiments. Whole cell lysates and total RNA were then prepared and analyzed by immunoblotting and RT-PCR, respectively. As shown in Fig.2A, ADR-mediated accumulation of γH2AX and cleaved poly(ADP-ribose) polymerase (PARP) was detectable, indicating that DNA damage-induced cell death takes place in response to ADR. As expected, ADR treatment led to an induction of TAp73, as well as RUNX2, in association with a remarkable upregulation of p53/TAp73-target gene products, such as p21WAF1 and BAX. Additionally, RT-PCR analysis revealed that ADR-dependent induction of TAp73 and RUNX2 is regulated at the transcription level (Fig.2B).
Figure 1.
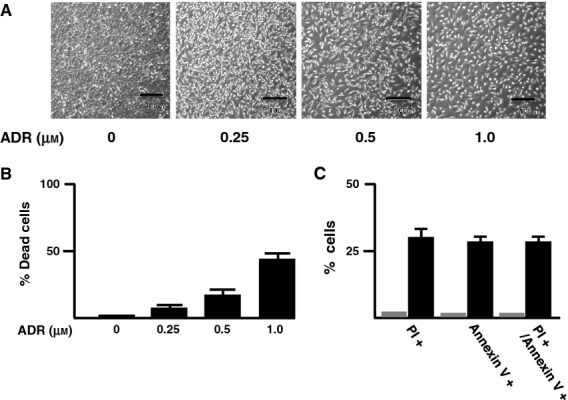
U2OS cells undergo cell death in response to ADR. (A) Phase-contrast micrograph. U2OS cells were treated with the indicated concentrations of ADR or left untreated. Twenty-four hours after treatment, phase-contrast micrographs were taken. Scale bar = 100 μm. (B) Trypan-blue exclusion assay. U2OS cells were treated as described in (A). Twenty-four hours after treatment, cells were trypsinized and stained with 0.4% of trypan blue. Cells restricting trypan-blue entry were considered viable. (C) Annexin V and PI double staining. U2OS cells were treated with 0.5 μm of ADR (filled boxes) or left untreated (gray boxes). Twenty-four hours after treatment, cells were subjected to annexin V and PI double staining.
Figure 2.

ADR-mediated induction of TAp73 and RUNX2 in U2OS cells. U2OS cells were exposed to the indicated concentrations of ADR. Twenty-four hours after treatment, whole cell lysates and total RNA were prepared and subjected to (A) immunoblotting and (B) RT-PCR, respectively. Actin and GAPDH were used as a loading and an internal control, respectively.
Because p53 was also induced and phosphorylated at Ser-15 in response to ADR, it is likely that U2OS cells undergo cell death in a p53 and/or a TAp73-dependent manner. To confirm whether TAp73 could play a central role in the regulation of ADR-mediated cell death, U2OS cells were transfected with control small interfering RNA (siRNA) or with siRNA targeting p73. Transfected cells were then treated with or without ADR. Twenty-four hours after treatment, whole cell lysates and total RNA were extracted and analyzed for the expression of p53/TAp73-target genes. As seen in Fig.3A, knockdown of p73 suppressed an ADR-mediated induction of p21WAF1 and BAX, whereas ADR-dependent upregulation of RUNX2 was basically unaffected. RT-PCR analysis demonstrated that depletion of p73 impairs ADR-dependent transcriptional upregulation of various p53/TAp73-target genes, including p21WAF1, 14-3-3σ, BAX,PUMA and NOXA, indicating that, similar to p53, TAp73 is required for ADR-mediated DNA damage response (Fig.3B).
Figure 3.
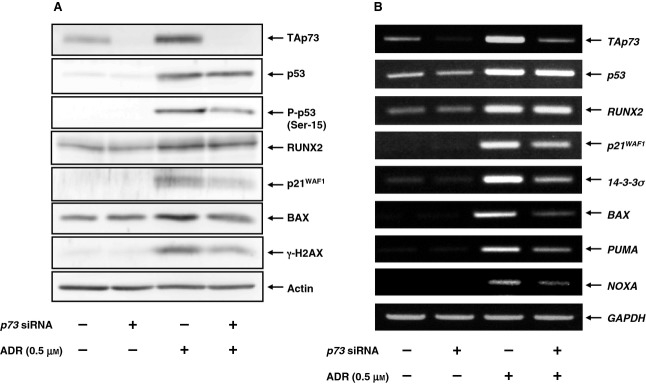
Knockdown of p73 attenuates ADR-mediated induction of p53/TAp73-target gene expression. U2OS cells were transfected with control siRNA or with siRNA against p73. Twenty-four hours after transfection, cells were treated with 0.5 μm of ADR or left untreated. Twenty-four hours after treatment, whole cell lysates and total RNA were extracted and processed for (A) immunoblotting and (B) RT-PCR, respectively.
Complex formation between TAp73 and RUNX2 in response to ADR
The above observations showing that TAp73 and RUNX2 are simultaneously induced after ADR treatment prompted us to examine whether TAp73 could form a complex with RUNX2. Accordingly, U2OS cells were stimulated with ADR or left untreated and then subjected to indirect immunofluorescence staining followed by subsequent confocal microscopy analysis. As shown in Fig.4A, TAp73 and RUNX2 were induced to accumulate and co-localize in cell nucleus in response to ADR, suggesting that TAp73 might interact with RUNX2 in the cell nucleus. To further confirm this possibility, we performed co-immunoprecipitation experiments using whole cell lysates prepared from ADR-treated U2OS cells. As shown in Fig.4B, the anti-p73 immunoprecipitates contained the endogenous RUNX2. Similaly, the reciprocal experiments demonstrated that TAp73 is co-precipitated with the endogenous RUNX2. Intriguingly, we failed to detect RUNX2/TAp73 complex in the absence of ADR (data not shown). Thus, our present results imply that TAp73 forms a complex with RUNX2 in cell nucleus in response to DNA damage.
Figure 4.
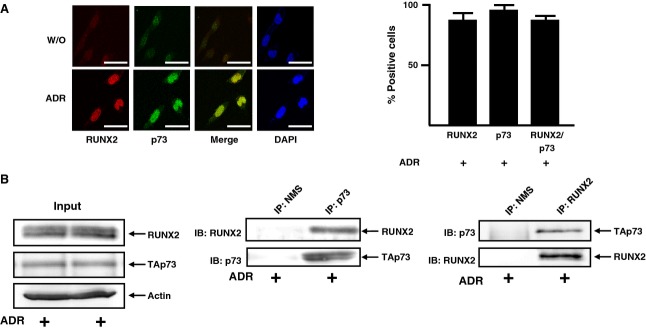
Complex formation between TAp73 and RUNX2 in response to ADR. (A) Nuclear co-localization of TAp73 with RUNX2 after ADR exposure. U2OS cells were exposed to 0.5 μm of ADR (lower) or left untreated (upper). Twenty-four hours after treatment, cells were simultaneously incubated with monoclonal anti-RUNX2 (red) and polyclonal anti-p73 (green) antibodies. Cell nuclei were stained with DAPI (blue). Merged images (yellow) indicated the nuclear co-localization of TAp73 with RUNX2. Scale bar = 25 μm (magnification, × 630) (left). Percentages of RUNX2-, p73- or RUNX2/p73-positive cells were indicated (right). (B) Co-immunoprecipitation. An equal amount of whole cell lysates prepared from ADR-treated U2OS cells was immunoprecipitated with normal mouse serum (NMS) or with monoclonal anti-p73 antibody. The immunoprecipitates were analyzed by immunoblotting with the indicated antibodies (middle). The reciprocal experiments (right) and 1 : 20 inputs (left) are also shown.
RUNX2 inhibits the transcriptional activity of TAp73
We next investigated a functional consequence of the complex formation between TAp73 and RUNX2. Accordingly, U2OS cells were transfected with or without increasing amounts of RUNX2 expression plasmid. Forty-eight hours post-transfection, whole cell lysates and total RNA were isolated and analyzed by immunoblotting and RT-PCR, respectively. As described previously [38], RUNX2 had an undetectable effect on the expression level of p53 (Fig.5A). Overexpression of RUNX2 caused a marked decrease in the expression level of p53/TAp73-target genes, and similar results were also obtained in p53-deficient human lung carcinoma H1299 cells (data not shown). Of note, overexpression of RUNX2 in U2OS cells resulted in an accelerated cell proliferation in a dose- and time-dependent fashion (Fig.5B,C), raising the possibility that RUNX2-mediated downregulation of p53/TAp73-target gene products might contribute to the promotion of cell proliferation. Because U2OS and H1299 cells expressed wild-type TAp73, it is likely that, in addition to p53, RUNX2 might also inhibit the transcriptional activity of TAp73, thereby enhancing their unregulated cell proliferation.
Figure 5.
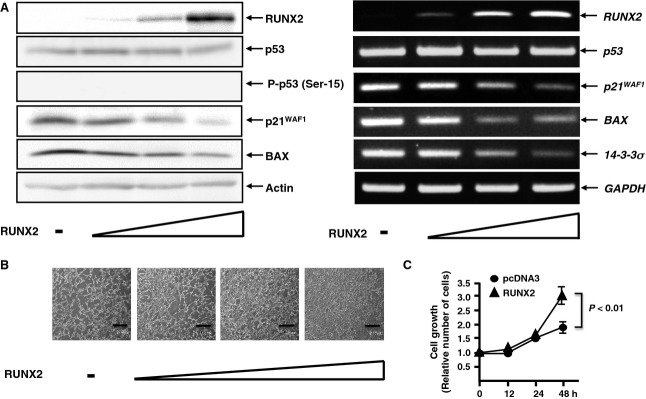
RUNX2 represses the expression of p53/p73-target genes. (A) U2OS cells were transfected with or without the increasing amounts of the expression plasmid for RUNX2 (0.5, 1.0 or 2.0 μg). Forty-eight hours after transfection, whole cell lysates and total RNA were isolated and analyzed by immunoblotting (left) and RT-PCR (right), respectively. (B, C) RUNX2 promotes cell proliferation. U2OS cells were transfected as described in (A). Forty-eight hours after transfection, phase-contrast micrographs were taken. Scale bar = 100 μm (B). U2OS cells were transfected with 2.0 μg of the expression plasmid encoding RUNX2. At the indicated time points, the number of viable cells was measured (C).
To adequately address this issue, H1299 cells were transfected with the indicated combinations of the expression plasmids. Forty-eight hours post-transfection, whole cell lysates and total RNA were prepared and processed for immunoblotting and RT-PCR, respectively. As clearly shown in Fig.6, overexpression of TAp73α stimulated the expression of p53/TAp73-target genes, such as BAX,PUMA and NOXA, whereas co-expression of TAp73α with RUNX2 markedly reduced their TAp73α-dependent transcriptional activation. Consistent with these results, luciferase reporter assays demonstrated that RUNX2 remarkably decreases TAp73α-mediated enhancement of luciferase activities driven by p53/TAp73-responsible human BAX and NOXA promoters in a dose-dependent manner (data not shown). These observations strongly suggest that RUNX2 inhibits the transcriptional activity of TAp73.
Figure 6.
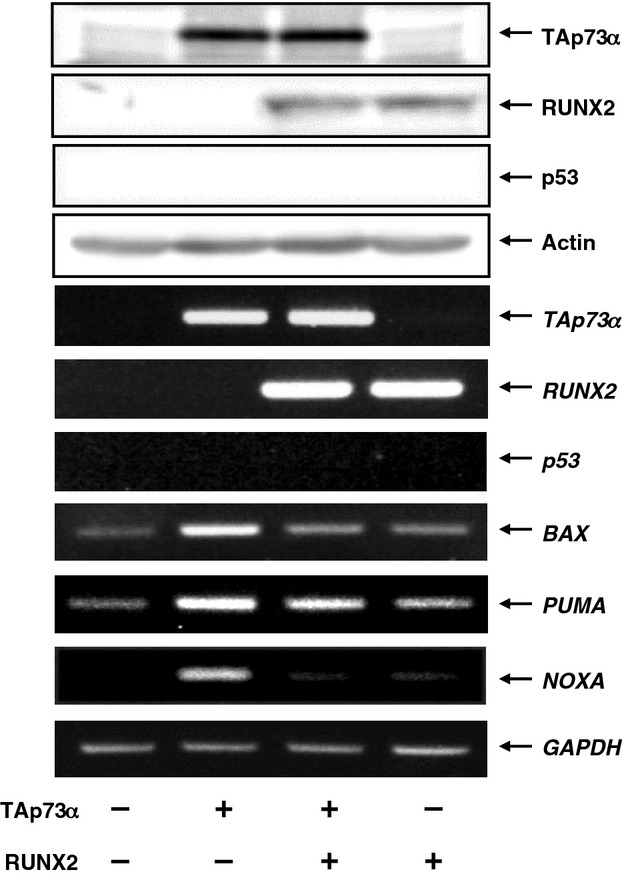
RUNX2 inhibits the transcriptional activity of TAp73. H1299 cells were transfected with the indicated combinations of the expression plasmids. Forty-eight hours after transfection, whole cell lysates and total RNA were prepared and subjected to immunoblotting (upper) and RT-PCR (lower), respectively.
Knockdown of RUNX2 enhances ADR-mediated cell death in association with the transcriptional induction of TAp73
Recently, we reported that depletion of RUNX2 in U2OS cells augments p53-dependent cell death after ADR exposure [38]. These observations led us to examine a possible effect of RUNX2 knockdown on TAp73 expression. Accordingly, U2OS cells were transfected with control siRNA or with siRNA targeting RUNX2, followed by incubation with or without ADR for 24 h. In accordance with our recent observations [38], RUNX2 was successfully knocked down under our experimental conditions (Fig.7A) and RUNX2-depleted cells underwent cell death in response to ADR (42%) much more efficiently than ADR-treated control cells (24%), as examined by DNA ladder formation (Fig.7B) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Fig.8). To check whether silencing of RUNX2 could affect the expression level of TAp73 after ADR treatment, total RNA was prepared and analyzed by RT-PCR. As clearly shown in Fig.7C, ADR-mediated induction of p53/TAp73-target genes, such as p21WAF1, BAX and PUMA, was further enhanced in RUNX2-knockdown cells. Of note, knockdown of RUNX2 had a negligible effect on p53 in cells exposed to ADR, whereas ADR-mediated upregulation of TAp73 was further enhanced upon the silencing of RUNX2 expression. Additionally, depletion of RUNX2 resulted in a slight increase in the steady-state expression level of TAp73. These observations suggest that RUNX2 has an ability to suppress ADR-dependent induction of TAp73, and TAp73 contributes at least in part to the higher sensitivity to ADR of RUNX2-depleted cells.
Figure 7.

siRNA-mediated knockdown of RUNX2 enhances ADR-dependent cell death and expression of TAp73. (A) Knockdown of RUNX2. U2OS cells were transfected with control siRNA or with siRNA against RUNX2. Twenty-four hours after transfection, cells were treated with 0.5 μm of ADR or left untreated. After 24 h of incubation, cells were stained with anti-RUNX2 antibody (green). Cell nuclei were stained with DAPI (blue). Scale bar = 25 μm. (B) Genomic DNA fragmentation. U2OS cells were transfected as described in (A). Twenty-four hours post-transfection, cells were exposed to 0.5 μm of ADR or left untreated. Twenty-four hours after treatment, attached and floating cells were collected, and their genomic DNA was prepared and analyzed by 1% agarose gel electrophoresis. (C) RT-PCR analysis of the endogenous TAp73. U2OS cells were transfected as described in (A). Transfected cells were treated with 0.5 μm of ADR or left untreated. Twenty-four hours after treatment, total RNA was prepared and subjected to RT-PCR analysis.
Figure 8.

Knockdown of RUNX2 in U2OS cells enhances the sensitivity to ADR. U2OS cells were transiently transfected with control siRNA or with siRNA targeting RUNX2. Twenty-four hours after transfection, cells were treated with 0.5 μm of ADR or left untreated. Twenty-four hours after treatment, cells were subjected to TUNEL assay, and number of TUNEL-positive cells (red) was scored. Cell nuclei were stained with DAPI. Scale bar = 50 μm (magnification, × 100).
RUNX2 attenuates ADR-mediated cell death in association with the downregulation of TAp73
To further confirm whether RUNX2 could downregulate the expression of TAp73 during ADR-mediated cell death, U2OS cells were transfected with the empty plasmid or with RUNX2 expression plasmid. Twenty-four hours post-transfection, cells were incubated with or without 0.5 μm of ADR. As expected, overexpression of RUNX2 suppressed ADR-mediated cell death (Fig.9), whereas ADR-mediated cell death was further stimulated in the presence of exogenous p53. Similar results were also obtained in a trypan-blue exclusion assay (data not shown). Under these experimental conditions, whole cell lysates and total RNA were prepared and analyzed for the expression level of TAp73 by immunoblotting and RT-PCR, respectively. As shown in Fig.10A, ADR-mediated proteolytic cleavage of PARP, caspase-9 and induction of p53/TAp73-target gene products were notably suppressed by exogenous RUNX2. Similar results were also obtained in RT-PCR analysis (Fig.10B). Intriguingly, ADR treatment led to a remarkable induction of TAp73, whereas ADR-mediated upregulation of TAp73 was impaired in cells expressing exogenous RUNX2. As expected, overexpression of RUNX2 also abrogated ADR-dependent transcriptional induction of TAp73 and various p53/TAp73-target genes in H1299 cells (Fig.11).
Figure 9.

RUNX2 prohibits ADR-mediated cell death. U2OS cells were transfected with the empty plasmid, the expression plasmid for RUNX2 (upper panels) or with the expression plasmid for p53 (lower panels), followed by additional incubation with or without 0.5 μm of ADR. Twenty-four hours after treatment, attached and floating cells were collected and analyzed by fluorescence-activated cell sorting.
Figure 10.

RUNX2 suppresses ADR-mediated induction of TAp73. U2OS cells were transfected as described in Fig.9. Twenty-four hours after transfection, cells were treated with 0.5 μm of ADR or left untreated. Twenty-four hours after treatment, whole cell lysates and total RNA were prepared and analyzed by (A) immunoblotting and (B) RT-PCR, respectively.
Figure 11.
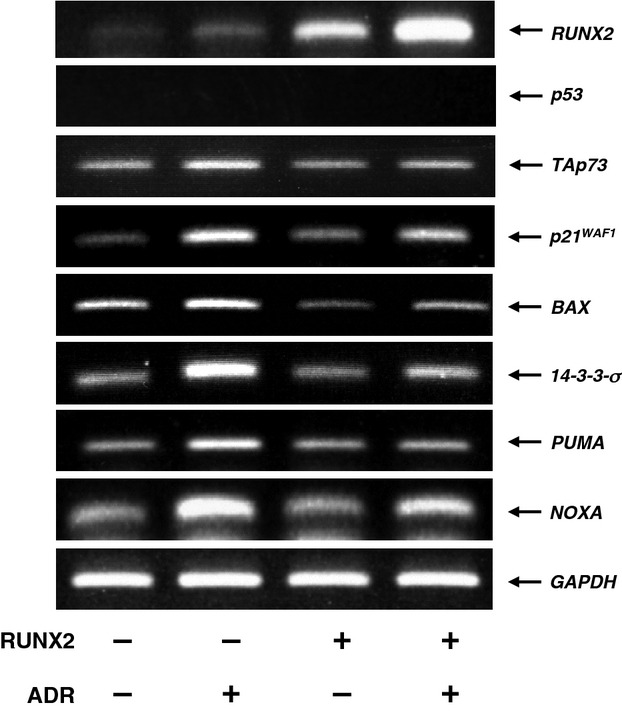
RUNX2 inhibits ADR-mediated upregulation of TAp73 in H1299 cells. H1299 cells were transiently transfected with the empty plasmid or with the expression plasmid for RUNX2. Twenty-four hours after transfection, cells were treated with 0.5 μm of ADR or left untreated. Twenty-four hours after treatment, total RNA was prepared and subjected to RT-PCR.
Taken together, our present findings strongly suggest that RUNX2 helps the resistance of cancerous cells to DNA damage by downregulation of TAp73.
Discussion
In the present study, we report that the transcriptional activity and ADR-mediated induction of pro-apoptotic TAp73 are significantly impaired by RUNX2. Because RUNX2 prohibits both wild-type p53 and TAp73, it is likely that the inhibition of RUNX2 might be a promising anti-cancer therapy for chemoresistant malignant cancers.
It is well established that, similar to p53, TAp73 is activated through DNA damage-mediated post-translational modifications such as phosphorylation and acetylation. For example, TAp73 was tyrosine phosphorylated at Tyr-99 by nonreceptor tyrosine kinase c-Abl in response to DNA damage, and c-Abl-mediated phosphorylation of TAp73 enhanced its stability as well as its transcriptional activity [39–41]. Alternatively, Costanzo et al. [42] demonstrated that DNA damage-induced acetylation of TAp73 at Lys-321, Lys-327 and Lys-331 stimulates its pro-apoptotic activity. According to our immunoprecipitation experiments, we successfully detected RUNX2/TAp73 complex in cells exposed to ADR (Fig.4). By contrast, we failed to detect RUNX2/TAp73 complex in untreated cells, which might the result of the low expression level of RUNX2 and TAp73 or the absence of post-translationally modified active TAp73. Because RUNX2 significantly reduced the transcriptional activity of TAp73, it is likely that RUNX2 could preferentially associate with the transcriptionally active form of TAp73. Although plausible, this possiblity remains to be tested experimentally.
According to the results of the present study, overexpression of RUNX2 suppressed the ADR-dependent induction of TAp73 and siRNA-mediated knockdown of RUNX2 further enhanced ADR-dependent upregulation of TAp73, indicating that RUNX2 might repress TAp73 expression at the transcritional level. In support of this notion, luciferase reporter assays demonstrated that luciferase activity driven by human TAp73 promoter (-2713/+20) is remarkably reduced in response to increasing amounts of exogenous RUNX2 (data not shown). Intriguingly, we identified several putative RUNX2-recognition sites (ACC(A/G)CA) within this promoter region (data not shown). RUNX/CBFβ complex is known to activate or repress the transcription of key regulators of growth, survival and differentiation pathways [43]. Recently, McGee-Lawrence et al. [44] reported that RUNX2 represses the transcription of its target genes by recruiting histone deacetylases (HDACs) to their promoters. We have also found that RUNX2 interacts with HDAC6, thereby inhibiting the transcription of p53/TAp73-target genes in response to DNA damage [38]. It is possible that RUNX2 downregulates the transcription of TAp73 by recruiting HDACs to the putative RUNX2-binding site located within TAp73 promoter region; however, additional experiments are required to adequately address this issue.
Alternatively, it has been demonstrated that transcriptional repressor ZEB1 which has anti-apoptotic potential, inhibits the transcription of TAp73 [45,46]. Aghdassi et al. [47] reported that ZEB1-mediated recruitment of HDAC1 and HDAC2 onto the E-cadherin promoter results in its downregulation. Under our experimental conditions, the expression level of ZEB1 was markedly increased in response to exogenous RUNX2, and siRNA-mediated knockdown of RUNX2 resulted in a massive downregulation of ZEB1 in U2OS cells (data not shown), raising the possibility that the expression of ZEB1 is regulated under the control of RUNX2. Based on these observations, it is conceivable that RUNX2-mediated transcriptional activation of ZEB1 causes a marked repression of TAp73 or that RUNX2 facilitates the efficient recruitment of the repressor complex containing ZEB1 onto the putative RUNX2-binding site within 5′-upstream region of TAp73 and/or ZEB1-binding site located in the first intron of TAp73, thereby attenuating TAp73 transcription.
As described previously [18], TAp73 was required for p53-dependent cell death, whereas TAp73 was capable of promoting cell death in a p53-independent manner. In support of these observations, the overexpression of TAp73 alone in p53-deficient pancreatic cancer cells triggered cell death [48]. In addition, Willis et al. [49] demonstrated that TAp73 exerts its growth-suppressive activity in cancerous cells expressing mutant p53. Previously, Chen et al. [50] found that p53 transactivates TAp73 through a functional p53-responsible element located within its 5′-upstream region. Under our experimental conditions, overexpression of FLAG-p53 stimulated transcription of TAp73 in U2OS cells (data not shown). Given that TAp73 acts as an essential cofactor for p53 [18], RUNX2 knockdown-mediated enhancement of pro-apoptotic activity of p53 might be at least in part attiributed to p53-dependent induction of TAp73. It was worth noting that siRNA-mediated knockdown of p73 strongly suppresses ADR-dependent upregulation of p53/TAp73-target gene expression even in the presence of active p53 (Fig.3), implying that TAp73 plays a critical role during ADR-mediated cell death. Because TAp73 is rarely mutated in a variety of human cancers [16], it is likely that silencing of RUNX2 enhances chemosensitivity of malignant cancers expressing functionally impaired-p53 through up-regulation of TAp73.
In summary, we provided evidence showing that RUNX2 downregulates TAp73 in response to DNA damage. Therefore, we propose that silencing of RUNX2 might be a novel strategy for improving chemosensitivity of malignant cancers.
Experimental procedures
Cells and transient transfection
p53-proficient human osteosarcoma-derived U2OS and p53-deficient human lung carcinoma-derived H1299 cells were grown in Dulbecco's modified Eagle's medium supplemented with heat-inactivated 10% FBS (Invitrogen, Carlsbad, CA, USA) and penicillin-streptomycin at 37 °C in 5% CO2. For transient transfection, cells were transiently transfected with the indicated combinations of the expression plasmids by using Lipofectamine 2000 reagent in accordance with the manufacturer's instructions (Invitrogen). The expression plasmids used in the present study were: pcDNA3 empty plasmid, pcDNA3-TAp73α and pcDNA3-RUNX2.
Trypan-blue exclusion assay
The number of dead cells was determined by the trypan-blue exclusion assay. Cells were exposed to the indicated concentrations of ADR. Twenty-four hours after the treatment, cells were trypsinized and stained with trypan-blue (0.4%) at room temperature for 10 min. Cells restricting trypan-blue entry were considered viable.
Annexin V and PI double staining
U2OS cells were treated with the indicated concentration of ADR or left untreated. Twenty-four hours after treatment, cells were washed in PBS and incubated with annexin V-FITC solution containing PI (50 μg·mL−1) at room temperature for 5 min in accordance with the manufacturer's instructions (PromoKine, Heiderberg, Germany). After the incubation, cells were washed in PBS, fixed in 2% formaldehyde at room temperature for 30 min and then observed under a confocal microscope (Leica, Milton Keynes, UK).
RNA extraction and RT-PCR
Total RNA was prepared from the indicated cells using RNeasy mini kit (Qiagen, Hilden, Germany) in accordance with the manufacturer's instructions. The integrity of total RNA was verified by visualization of rRNAs after electrophoresis in agarose gel. One microgram of total RNA for each sample was reverse-transcribed using SuperScript II reverse transcriptase (Invitrogen) in the presence of random primers. The resultant cDNA was subjected to PCR-based amplification using the specific primer sets for TAp73, 5′-TCTGGAACCAGACAGCACCT-3′ (sense) and 5′-GTGCTGGACTGCTGGAAAGT-3′ (antisense); p53, 5′-CTGCCCTCAACAAGATGTTTTG-3′ (sense) and 5′-CTATCTGAGCAGCGCTCATGG-3′ (antisense); RUNX2, 5′-TCTGGCCTTCCACTCTCAGT-3′ (sense) and 5′-GACTGGCGGGGTGTAAGTAA-3′ (antisense); p21WAF1, 5′-ATGAAATTCACCCCCTTTCC-3′ (sense) and 5′-CCCTAGGCTGTGCTCACTTC-3′ (antisense); 14-3-3σ, 5′-GAGCGAAACCTGCTCTCAGT (sense) and 5′-CTCCTTGATGAGGTGGCTG-3′ (antisense); BAX, 5′-TTTGCTTCAGGGTTTCATCC-3′ (sense) and 5′-CAGTTGAAGTTGCCGTCAGA-3′ (antisense); PUMA, 5′-GCCCAGACTGTGAATCCTGT-3′ (sense) and 5′-TCCTCCCTCTTCCGAGATTT-3′ (antisense); NOXA, 5′-CTGGAAGTCGAGTGTGCTACT-3′ (sense) and 5′-TCAGGTTCCTGAGCAGAAGAG-3′ (antisense); GAPDH, 5′-ACCTGACCTGCCGTCTAGAA-3′ (sense) and 5′-TCCACCACCCTGTTGCTGTA-3′ (antisense). The PCR products were electrophoresed on 1.5% agarose gels and their amounts were evaluated by staining with ethidium bromide. GAPDH was used as an internal control.
Immunofluorescence
Cells were exposed to 0.5 μm of ADR or left untreated. Twenty-four hours after treatment, cells were washed twice in ice-cold PBS, fixed in 3.7% folmaldehyde in PBS at room temperature for 30 min, permeabilized with 0.1% Triton X-100 in PBS at room temperature for 5 min and then blocked with 3% BSA in PBS at room temperature for 1 h. After blocking, cells were washed in ice-cold PBS and simultaneously incubated with mouse monoclonal anti-RUNX2 (8G5; Medical & Biological Laboratories, Nagoya, Japan) and rabbit polyclonal anti-p73 (Bethyl Laboratories, Montgomery, TX, USA) antibodies at room temperature for 1 h followed by the incubation with rhodamine-conjugated goat anti-mouse IgG and FITC-conjugated goat anti-rabbit IgG (Invitrogen) at room temperature for 1 h. After the extensive washing, cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Peterborough, UK). Fluorescent images were captured using a confocal microscope.
Immunoblot analaysis
Cells were washed twice in ice-cold PBS and immediately lysed in Laemmli buffer as described previously [38]. Equal amounts of cell lysates were resolved by 10% SDS/PAGE and electro-transferred onto poly(vinylidene difluoride) membrane. Immunoblot analyses were performed as described previously [19] using the primary antibodies: mouse monoclonal anti-p53 (DO-1; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-p73 (Ab-4; NeoMarkers, Fremont, CA, USA), mouse monoclonal anti-γH2AX (2F3; BioLegend, San Diego, CA, USA), rabbit polyclonal anti-p21WAF1 (Santa Cruz Biotechnology), rabbit polyclonal anti-phospho-p53 at Ser-15 (Cell Signaling Technology, Beverly, MA, USA), rabbit polyclonal anti-RUNX2 (Cell Signaling Technology), rabbit polyclonal anti-E2F-1 (Cell Signaling Technology), rabbit polyclonal anti-PARP (Cell Signaling Technology), rabbit polyclonal anti-caspase-9 (Cell Signaling Technology) and rabbit polyclonal anti-Actin (20–33; Sigma, St Louis, MO, USA) antibodies. Immunoreactive bands were visualized by an enhanced chemiluminescence system (ECL; GE Healthcare, Little Chalfont, UK) in accordance with the manufacturer's instructions.
Immunoprecipitation
For immunoprecipitation, cells were exposed to 0.5 μm of ADR for 24 h and then lysed in lysis buffer containing 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 2.7 mm KCl, 1% Triton X-100 and protease inhibitor mixture (Sigma). Equal amounts of cell lysates (500 μg of protein) were first precleared with 30 μL of protein G-Sepharose beads for 1 h at 4 °C and then incubated with monoclonal anti-p73 antibody overnight at 4 °C followed by the incubation with 30 μL of protein G-Sepharose beads for 2 h at 4 °C. The beads were extensively washed with lysis buffer and the immunoprecipitates were eluted by boiling in SDS-sample buffer. The bound proteins were separated by 10% SDS/PAGE.
siRNA interference
Cells were transfected with control siRNA or with siRNA directed against RUNX2 (10 nm) using Lipofectamine 2000 transfection reagent in accordance with the manufacturer's recommendations. Twenty-four hours after transfection, cells were treated with 0.5 μm of ADR or left untreated. Twenty-four hours after treatment, total RNA was prepared and analyzed for the expression level of RUNX2 by RT-PCR.
Luciferase reporter assay
Cells were seeded at a density of 5 x 104 cells per 12-well plate and allowed to attach overnight. Cells were then transfected with the constant amount of TAp73α expression plasmid (25 ng), 100 ng of luciferase reporter plasmid carrying p53/p73-responsible human BAX or NOXA promoter and 10 ng of renilla luciferase expression plasmid, together with or without increasing amounts of the expression plasmid for RUNX2. Equal DNA concentration in each experiment (500 ng per well) was maintained by adding the appropriate amounts of the empty plasmid to the DNA mixture. Forty-eight hours after transfection, cell lysates were prepared and their luciferase activities were measured in accordance with the protocol for the dual-luciferase system (Promega, Madison, WI, USA). The resultant firefly luciferase values were normalized by those of renilla luciferase. The experiments were performed in parallel and repeated at least three times.
TUNEL assay
TUNEL assay was performed by using an In Situ Cell Detection Kit (Roche Applied Science, Mannheim, Germany) in accordance with the manufacturer's protocol. In brief, cells were washed twice in PBS, fixed in freshly prepared 4% paraformaldehyde for 1 h at room temperature, permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min at room temperature, and then incubated with the enzyme solution in a humidified atmosphere for 1 h at 37 °C in the dark. After the reaction, cells were washed in PBS three times and fluorescent images were taken using a confocal microscope.
Statistical analysis
Statistical significance was determined by two-sided paired Student's t-test. P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (grant number 23501278).
Glossary
- ADR
adriamycin
- DAPI
4′,6-diamidino-2-phenylindole
- HDAC
histone deacetylase
- HRP
horseradish peroxidase
- PARP
poly(ADP-ribose) polymerase
- PI
propidium iodide
- RUNX2
runt-related transcription factor 2
- siRNA
small interfering RNA
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Author contributions
TO and HN planned the experiments. TO, HS, MN, KH and HY performed the experiments. TO analyzed the data. MS and KF contributed reagents or other essential materials. TO wrote the paper.
References
- 1.Prives C. Hall PA. The p53 pathway. J Pathol. 1999;187:112–126. doi: 10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.0.CO;2-3. &. [DOI] [PubMed] [Google Scholar]
- 2.Sionov RV. Haupt Y. The cellular response to p53: the decision between life and death. Oncogene. 1999;18:6145–6157. doi: 10.1038/sj.onc.1203130. &. [DOI] [PubMed] [Google Scholar]
- 3.Vousden KH. Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. &. [DOI] [PubMed] [Google Scholar]
- 4.Vogelstein B. Kinzler KW. p53 function and dysfunction. Cell. 1992;70:523–526. doi: 10.1016/0092-8674(92)90421-8. &. [DOI] [PubMed] [Google Scholar]
- 5.Harris CC. p53: at the crossroads of molecular carcinogenesis and risk assessment. Science. 1993;262:1980–1981. doi: 10.1126/science.8266092. [DOI] [PubMed] [Google Scholar]
- 6.Vogelstein B, Lane D. Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. &. [DOI] [PubMed] [Google Scholar]
- 7.Strano S, Dell'Orso S, Mongiovi AM, Monti O, Lapi E, Di Agostino S, Fontemaggi G. Blandino G. Mutant p53 proteins: between loss and gain of function. Head Neck. 2007;29:488–496. doi: 10.1002/hed.20531. &. [DOI] [PubMed] [Google Scholar]
- 8.Strano S, Dell'Orso S, Di Agostino S, Fontemaggi G, Sacchi A. Blandino G. Mutant p53: an oncogenic transcription factor. Oncogene. 2007;26:2212–2219. doi: 10.1038/sj.onc.1210296. &. [DOI] [PubMed] [Google Scholar]
- 9.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 10.Jost CA, Marin MC. Kaelin WG., Jr p73 is a simian p53-related protein that can induce apoptosis. Nature. 1997;389:191–194. doi: 10.1038/38298. &. [DOI] [PubMed] [Google Scholar]
- 11.Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC. Kaelin WG., Jr Chemosensitivity linked to p73 function. Cancer Cell. 2003;3:403–410. doi: 10.1016/s1535-6108(03)00078-3. &. [DOI] [PubMed] [Google Scholar]
- 12.Lissy NA, Davis PK, Irwin M, Kaelin WG., Jr Dowdy SF. A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature. 2000;407:642–645. doi: 10.1038/35036608. &. [DOI] [PubMed] [Google Scholar]
- 13.Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, Flores ER, Tsai KY, Jacks T, Vousden KH, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407:645–648. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 14.Stiewe T. Pützer BM. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet. 2000;26:464–469. doi: 10.1038/82617. &. [DOI] [PubMed] [Google Scholar]
- 15.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR. Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–306. doi: 10.1126/science.289.5477.304. &. [DOI] [PubMed] [Google Scholar]
- 16.Ikawa S, Nakagawara A. Ikawa Y. p53 family genes: structural comparison, expression and mutation. Cell Death Differ. 1999;6:1154–1161. doi: 10.1038/sj.cdd.4400631. &. [DOI] [PubMed] [Google Scholar]
- 17.Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F. Jacks T. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–373. doi: 10.1016/j.ccr.2005.02.019. &. [DOI] [PubMed] [Google Scholar]
- 18.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F. Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564. doi: 10.1038/416560a. &. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa T, Takahashi M, Ozaki T, Watanabe Ki K, Todo S, Mizuguchi H, Hayakawa T. Nakagawara A. Autoinhibitory regulation of p73 by Delta Np73 to modulate cell survival and death through a p73-specific target element within the Delta Np73 promoter. Mol Cell Biol. 2002;22:2575–2585. doi: 10.1128/MCB.22.8.2575-2585.2002. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blyth K, Cameron ER. Neil JC. The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer. 2005;5:376–387. doi: 10.1038/nrc1607. &. [DOI] [PubMed] [Google Scholar]
- 21.Ito Y. RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Adv Cancer Res. 2008;99:33–76. doi: 10.1016/S0065-230X(07)99002-8. [DOI] [PubMed] [Google Scholar]
- 22.Anglin I. Passaniti A. Runx protein signaling in human cancers. Cancer Treat Res. 2004;119:189–215. doi: 10.1007/1-4020-7847-1_10. &. [DOI] [PubMed] [Google Scholar]
- 23.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 24.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi A, Satake M, Yamaguchi-Iwai Y, Bae SC, Lu J, Maruyama M, Zhang YW, Oka H, Arai N, Arai K, et al. Positive and negative regulation of granulocyte-macrophage colony-stimulating factor promoter activity by AML1-related transcription factor, PEBP2. Blood. 1995;86:607–616. [PubMed] [Google Scholar]
- 26.Okuda T, van Deursen J, Hiebert SW, Grosveld G. Downing JR. AML1 the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. &. [DOI] [PubMed] [Google Scholar]
- 27.Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, Ito Y. Littman DR. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. &. [DOI] [PubMed] [Google Scholar]
- 28.Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 29.Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Himeno M, Narai S, Yamaguchi A. Komori T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol. 2001;155:157–166. doi: 10.1083/jcb.200105052. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welch RD, Jones AL, Bucholz RW, Reinert CM, Tjia JS, Pierce WA, Wozney JM. Li XJ. Effect of recombinant human bone morphogenetic protein-2 on fracture healing in a goat tibial fracture model. J Bone Miner Res. 1998;13:1483–1490. doi: 10.1359/jbmr.1998.13.9.1483. &. [DOI] [PubMed] [Google Scholar]
- 31.Karsenty G. The genetic transformation of bone biology. Genes Dev. 1999;13:3037–3051. doi: 10.1101/gad.13.23.3037. [DOI] [PubMed] [Google Scholar]
- 32.Vaillant F, Blyth K, Terry A, Bell M, Cameron ER, Neil J. Stewart M. A full-length Cbfa1 gene product perturbs T-cell development and promotes lymphomagenesis in synergy with myc. Oncogene. 1999;18:7124–7134. doi: 10.1038/sj.onc.1203202. &. [DOI] [PubMed] [Google Scholar]
- 33.Browne G, Nesbitt H, Ming L, Stein GS, Lian JB, McKeown SR. Worthington J. Bicalutamide-induced hypoxia potentiates RUNX2-mediated Bcl-2 expression resulting in apoptosis resistance. Br J Cancer. 2012;107:1714–1721. doi: 10.1038/bjc.2012.455. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kayed H, Jiang X, Keleg S, Jesnowski R, Giese T, Berger MR, Esposito I, Löhr M, Friess H. Kleeff J. Regulation and functional role of the Runt-related transcription factor-2 in pancreatic cancer. Br J Cancer. 2007;97:1106–1115. doi: 10.1038/sj.bjc.6603984. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Endo T, Ohta K. Kobayashi T. Expression and function of Cbf a-1/Runx2 in thyroid papillary carcinoma cells. J Clin Endocrinol Metab. 2008;93:2409–2412. doi: 10.1210/jc.2007-2805. &. [DOI] [PubMed] [Google Scholar]
- 36.Onodera Y, Miki Y, Suzuki T, Takagi K, Akahira J, Sakyu T, Watanabe M, Inoue S, Ishida T, Ohuchi N, et al. Runx2 in human breast carcinoma: its potential roles in cancer progression. Cancer Sci. 2010;101:2670–2675. doi: 10.1111/j.1349-7006.2010.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sase T, Suzuki T, Miura K, Shiiba K, Sato I, Nakamura Y, Takagi K, Onodera Y, Miki Y, Watanabe M, et al. Runt-related transcription factor 2 in human colon carcinoma: a potent prognostic factor associated with estrogen receptor. Int J Cancer. 2012;131:2284–2293. doi: 10.1002/ijc.27525. [DOI] [PubMed] [Google Scholar]
- 38.Ozaki T, Wu D, Sugimoto H, Nagase H. Nakagawara A. Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis. 2013;4:e610. doi: 10.1038/cddis.2013.127. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr, Levrero M. Wang JY. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. &. [DOI] [PubMed] [Google Scholar]
- 40.Agami R, Blandino G, Oren M. Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399:809–813. doi: 10.1038/21697. &. [DOI] [PubMed] [Google Scholar]
- 41.Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, Lu H, Kharbanda S, Weichselbaum R. Kufe D. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399:814–817. doi: 10.1038/21704. &. [DOI] [PubMed] [Google Scholar]
- 42.Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, Fontemaggi G, Fanciulli M, Schiltz L, Blandino G, et al. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol Cell. 2002;9:175–186. doi: 10.1016/s1097-2765(02)00431-8. [DOI] [PubMed] [Google Scholar]
- 43.Wang CQ, Jacob B, Nah GS. Osato M. Runx family genes, niche, and stem cell quiescence. Blood Cells Mol Dis. 2010;44:275–286. doi: 10.1016/j.bcmd.2010.01.006. &. [DOI] [PubMed] [Google Scholar]
- 44.McGee-Lawrence ME, Li X, Bledsoe KL, Wu H, Hawse JR, Subramaniam M, Razidlo DF, Stensgard BA, Stein GS, van Wijnen AJ, et al. Runx2 protein represses Axin2 expression in osteoblasts and is required for craniosynostosis in Axin2-deficient mice. J Biol Chem. 2013;288:5291–5302. doi: 10.1074/jbc.M112.414995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fontemaggi G, Gurtner A, Strano S, Higashi Y, Sacchi A, Piaggio G. Blandino G. The transcriptional repressor ZEB regulates p73 expression at the crossroad between proliferation and differentiation. Mol Cell Biol. 2001;21:8461–8470. doi: 10.1128/MCB.21.24.8461-8470.2001. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bui T, Sequeira J, Wen TC, Sola A, Higashi Y, Kondoh H. Genetta T. ZEB1 links p63 and p73 in a novel neuronal survival pathway rapidly induced in response to cortical ischemia. PLoS One. 2009;4:e4373. doi: 10.1371/journal.pone.0004373. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, Friess H, Büchler M, Evert M, Lerch MM, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61:439–448. doi: 10.1136/gutjnl-2011-300060. [DOI] [PubMed] [Google Scholar]
- 48.Rödicker F. Pützer BM. p73 is effective in p53-null pancreatic cancer cells resistant to wild-type TP53 gene replacement. Cancer Res. 2003;63:2737–2741. &. [PubMed] [Google Scholar]
- 49.Willis AC, Pipes T, Zhu J. Chen X. p73 can suppress the proliferation of cells that express mutant p53. Oncogene. 2003;22:5481–5495. doi: 10.1038/sj.onc.1206505. &. [DOI] [PubMed] [Google Scholar]
- 50.Chen X, Zheng Y, Zhu J, Jiang J. Wang J. p73 is transcriptionally regulated by DNA damage, p53, and p73. Oncogene. 2001;20:769–774. doi: 10.1038/sj.onc.1204149. &. [DOI] [PubMed] [Google Scholar]