

Abstract
Tuberculosis (TB) lesions are extremely complex and dynamic. Here, we review the multiple types and fates of pulmonary lesions that form following infection by Mycobacterium tuberculosis and the impact of this spatial and temporal heterogeneity on the bacteria they harbor. The diverse immunopathology of granulomas and cavities generates a plethora of microenvironments to which M. tuberculosis bacilli must adapt. This in turn affects the replication, metabolism, and relative density of bacterial subpopulations, and consequently their respective susceptibility to chemotherapy. We outline recent developments that support a paradigm shift in our understanding of lesion progression. The simple model according to which lesions within a single individual react similarly to the systemic immune response no longer prevails. Host-pathogen interactions within lesions are a dynamic process, driven by subtle and local differences in signaling pathways, resulting in diverging trajectories of lesions within a single host. The spectrum of TB lesions is a continuum with a large overlap in the lesion types found in latently infected and active TB patients. We hope this overview will guide TB researchers in the design, choice of read-outs, and interpretation of future studies in the search for predictive biomarkers and novel therapies.
Keywords: tuberculosis, immunopathology, lesion dynamics, biomarkers, animal models
Introduction
Tuberculosis lesions in humans are very complex with a wide range of pathological, microbiological, and immunological features that often evolve over a period of many months, sometimes even years, prior to the emergence of symptoms and diagnosis of disease. Most exposed individuals never develop any signs of disease, although they show immunologic evidence of infection and are considered ‘latent’. Among those that develop active disease, we typically distinguish between ‘primary’ disease that occurs rapidly after exposure in a small percentage of patients (typically who have an underlying condition of impaired immunity) and the more common ‘post-primary’ disease that in many individuals occurs years or decades later, again often triggered by altered immune status. Primary disease is characterized by hematologic spread and a characteristic miliary pattern of disease, while postprimary disease includes a wide spectrum of pathologies. This spectrum is observed between individual patients with disease. For instance some patients develop large air-filled cavities (typically only a minority), while others develop only solitary nodules in the lung parenchyma or have infection limited to the lymphatic system or other extra-pulmonary sites. Importantly though, a wide spectrum of pathology is observed even within a single individual patient at a given moment in time. In this review, we focus on current thinking about the spectrum of lesions seen in pulmonary tuberculosis, with an eye on how the delicate interplay between the immune system and the bacterium has consequences for the host that are determined at the very local level of the individual lesion.
A brief history of perspectives on the pathology of active TB lesions
In the late nineteenth century, tuberculosis was rampant in Europe, infecting between 70 and 90% of the urban population (1). It is no surprise, therefore, that the most comprehensive early studies on the nature of various types of tuberculous lesions in man were first published in French, during the 1940s by Georges Canetti (reviewed in (2)). Canetti based his observations on the results of 1500 autopsies of TB patients performed during a 7-year period. He carefully documented three different features from a large variety of tuberculous lesions from humans: (i) the histopathologic characteristics of the lesion; (ii) the number of acid-fast bacilli observed using Ziehl staining by microscopic examination; and (iii) the number of culturable bacilli on egg medium. His work documented in extensive detail three very different types of lesions in man, which could be further divided into various subtypes. His first category included lesions characterized by marked inflammation including vasodilatation, edema, fibrinous exudate, and an influx of lymphocytes. Canetti's second class of lesions was that characterized by a cellular influx of monocytes transforming into giant cells which are characteristic of a classical granuloma or tubercle. And his final category consisted of caseous necrotic lesions, which were characterized by necrosis and homogenization. These necrotic lesions can remain solid with very few bacilli, and evolve to ultimate sclerosis. Solid caseous necrosis could also evolve by softening of the caseum accompanied by an enormous bacillary load, which he associated with the onset of clinical tuberculosis. Canetti also argued that although transitional lesion types exist, they evolve independently and never convert from one type into another.
A second significant body of work that influences our understanding of lesion morphology and formation comes from the careful rabbit studies of Arthur Dannenberg and Max Lurie (3). By comparing outbred rabbits (which are intrinsically resistant to infection with Mycobacterium tuberculosis) and inbred rabbits that showed enhanced susceptibility to infection, they proposed that tuberculosis disease could be classified into five stages: (i) the ingestion of bacilli by pulmonary alveolar macrophages; (ii) bacterial growth in non-activated macrophages recruited from blood circulation; (iii) the arrest of bacterial growth by delayed-type hypersensitivity (DTH) causing the initiation of formation of solid caseous lesions; (iv) the onset of cell-mediated immunity that is thought to stabilize caseous lesions; and (v) liquefaction of the caseous center, with extracellular bacterial growth and cavity formation. Dannenberg emphasized that the stages are not distinct but blend into each other, and stages 3, 4, and 5 can all be present in the same lung and even in different parts of the same lesion. Thus, they argue, lesion development is not a sequential linear process, and tuberculous lesions may change over time from one type to another so that the balance of disease as a whole may fluctuate between periods of exacerbation and remission. In light of current data to be discussed in this review, especially from PET and CT analysis in rabbits, macaques, and TB patients (4–7), even this slightly more nuanced interpretation may be an oversimplification of the complex, multi-trajectory process of lesion evolution. Fig. 1 recapitulates the major types of pulmonary lesions seen in human TB, active, and latent.
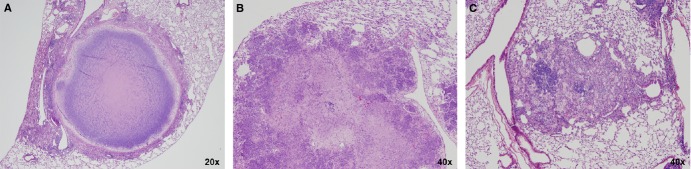
Figure 1.

(A). Early non-necrotizing granuloma; the lymphocyte rim (L) appears bluish owing to the prominent nuclei and small cytoplasm; the center of the granuloma contains a majority of epitheloid histiocytes (H) or macrophages, which are large cells with pale abundant cytoplasm. (B). Necrotizing granuloma: the central necrosis appears as pink amorphous material (N), surrounded by a layer of cellular infiltrates as well as a fibrotic rim (F) which clearly defines the granuloma from the surrounding lung alveoli. (C). Masson's trichrome stain of a necrotizing granuloma showing the collagenous areas in blue. The thick fibrotic layer (F) is composed of fibroblasts, collagen and new vessels, and scattered chronic inflammatory cells. Note the prominent vascularization of the fibrotic layer. (D). Calcified granuloma with sclerotic rim (S) associated with absence of mononuclear infiltrates and no visible infectious activity; Ca: area of calcification/mineralization. (E). Tuberculous pneumonia: confluent aggregates of epitheloid histiocytes mixed with Langhans’ and multinuclear giant cells (arrows). The more bluish areas are lymphoid aggregates. These are the same cells that make up a granuloma, but in the absence of a fibrotic wall they have spread to fill alveolar space. (F). Wall of cavitating tuberculous granuloma demonstrating a trilaminar structure with inner necrosis (N), histiocyte aggregates (H) and peripheral fibrosis (F). The fibrotic wall contains mononuclear infiltrates which indicates a still active and progressing tuberculous lesion. The more friable caseous material has been removed during tissue processing.
The comparative pathology of ‘latent infections’
Latent tuberculosis infection (LTBI) refers to clinically asymptomatic individuals who demonstrate immunological sensitization traditionally defined by a DTH response using the tuberculin skin test [more modern interferon-γ release assays from whole blood stimulated with TB-specific antigens are available but not as widely used in TB-endemic areas (8)]. Examining lesions from LTBI cases has been historically attractive as a means to study an apparently effective immune response to challenge with TB. Our knowledge of the histopathology and location/number of bacilli in human LTBI is based on autopsies of patients who died of other causes with incidental abnormalities from whom lung biopsies were retrieved and analyzed. In the lungs of these individuals, lesions were generally less cellular than those found in active disease, with minimal inflammation, prominent sclerosis, fibrous encapsulation, and often calcification. In the early TB literature, they were described as ‘scars’ or ‘arrested lesions’ that remain after a primary granuloma heals (2,3). The size and rate of healing were proposed as potential predictors of the likelihood of reactivation.
The paradigm of a simple binary distribution between active disease and latent infection has dramatically evolved in recent years spurred on by insights from non-human primates where infection and outcome could be more carefully controlled (9–11). In cynomolgous macaques infected with low dose M. tuberculosis, about half of all animals develop active disease and half convert their skin test but show no other signs of disease. This latent disease in non-human primates can be ‘reactivated’ by administration of α-TNF antibodies, making this entirely analogous to LTBI in humans (12,13). Pathologically and microbiologically, both active and latently infected monkeys present a range of disease and lesion types that are quantitatively distinct but not necessarily exclusive. The implication of these studies are that the current definition of LTBI includes a range of individuals, from those who have completely cleared the infection (and thus are at no future risk of ever developing active disease) to those who are incubating actively replicating bacteria in the absence of clinical symptoms (who are at higher risk of developing disease). In addition, low-level disease in the absence of symptoms may be associated with prolonged periods of time when individuals are potentially at some risk of transmitting disease. This spectrum model is causing a radical shift in our thinking about treatment rationales for LTBI. If there was a way to identify individuals at highest risk for the development and transmission of disease, and target these TB patients for treatment, then instead of aiming to treat the one-third of all humans with LTBI, we would be faced with a much smaller patient number. Important strides have been made recently in identifying blood transcriptional signatures that appear promising for distinguishing TB from other diseases and potentially LTBI from active disease (9,14,15). Ongoing studies to establish the spectrum of LTBI pulmonary lesion types by (18F)-2-fluorodeoxyglucose (FDG) positron emission tomography (PET) and computed tomography (CT), and relate these findings to immunological responses by transcriptional profiling, are nearly complete, and promise to solidify this thinking.
Lesions are diverse and dynamic in humans and animal models
The dynamic nature of human lesions
Because TB is a chronic disease with a long gestation period between infection and presentation of symptoms, there has been a strong tendency to think of the disease pathology to be slowly evolving and relatively stable. Recently, we have had the opportunity to perform serial FDG-PET/CT imaging of TB patients who were infected with extensively drug-resistant TB and randomized into a 2-month delayed start phase 2 trial of linezolid (16). Five of the subjects in this study were allocated to receive FDG-PET/CT scans at the time of entry and 2 months later, the day before they started taking linezolid. These scans were therefore performed in untreated TB patients to explore how stable these radiologic response markers were over time (the patients were purposely untreated because they had to be stably failing to respond to treatment for 6 months before enrollment and were not allowed any change in treatment regimen for that 2 months period). In terms of lung involvement, all five subjects were scored as fairly stable over that 2-month period (less than 10% change in total volume of abnormal lung density or in total FDG uptake). In contrast, at the level of individual lesions there were dramatic fluctuations in the size and FDG avidity, generally taken as a surrogate of local inflammatory activity since it labels primarily neutrophils, macrophages, and dendritic cells (17) (Fig. 2). Thus, the simple model according to which most lesions within a single individual behave similarly in response to the systemic immune response no longer prevails.
Figure 2.
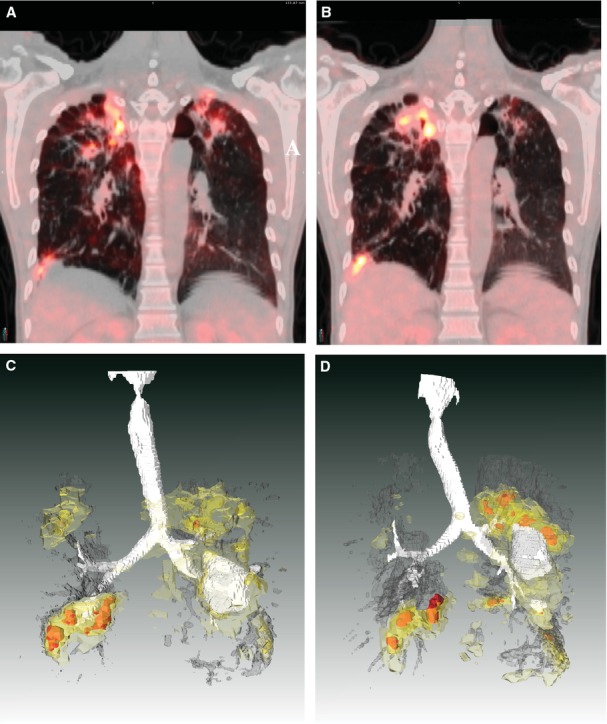
TB lesions evolve independently in humans. This patient had non-responsive extensively drug-resistant TB and was enrolled into the delayed arm of a randomized clinical trial of linezolid. The subject had failed to respond to any chemotherapy and was hospitalized for 2 months while awaiting study drug. Upon admission, his baseline (18F)-2-fluorodeoxyglucose positron emission tomography (FDG-PET) computed tomography (CT) scan showed extensive bilateral disease. A coronal section of this subjects’ scan is shown in (A), projected at a maximum SUV (Standardized Uptake Value, a method to standardize FDG uptake accounting for body weight and decay time) of 10. The same coronal slice at the same SUV is shown from a repeated scan after 2 months (immediately prior to starting linezolid therapy) in (B). Apical lesions in the left lung (right side of PET/CT image) are resolving while apical lesions in the right lung have progressed. (C, D) The same scan data are presented from the full 3D volume of the chest cavity. White represents air, either in the trachea and airways or inside of cavitary lesions. Dark gray represents areas of radiodensity between −100 and 200 Hounsfeld Units associated with TB lesions in both lungs. FDG uptake is represented by light yellow (SUV 2-3) darker yellow (SUV 3-5) and red (SUV >5). These are posterior views, reversed from above for clarity. Each lesion complex appears to progress or regress independently of the others. The complex in the left superior lobe (on the left in these drawings) appears to resolve while the lesions in the right upper and lower lobes appear to progress significantly. The lesion in the left inferior lobe appears relatively stable with little change.
Spectrum and divergent trajectories of lesions in animal models
Despite their heterogeneity, many lesions found in the lungs of humans infected with TB display necrosis in the center surrounded by a peripheral rim of fibrosis (Fig. 1). In addition, the presence and extent of cavitary disease have often been found to be correlates of poor clinical outcome, development of resistance, and relapse (18–20). To obtain results that can be leveraged to design clinical trials, animal models should aim to replicate the major pathophysiological conditions existing in human pulmonary tuberculosis lesions. Important characteristics include (i) the development of caseous necrosis surrounded by a collagen rim, which can affect drug penetration as well as the nutrient supply available to bacilli; (ii) hypoxia, which can influence the metabolic state of M. tuberculosis bacilli; (iii) intracellular as well as extracellular populations of bacilli, which can impact drug efficacy; and (iv) liquefaction and cavity formation. This review does not aim to provide an exhaustive summary of the pathology characteristics of all animal species used to model TB, but rather focuses on a few models to illustrate the lesion dynamics of TB infection/disease. The most prominent or unique pathological features of major animal models are summarized in Table 1.
Table 1.
Lesion features across animal models
| Mycobacterial strain(s) | Lesion types |
Prominent features | References | ||||
|---|---|---|---|---|---|---|---|
| Cellular | Caseating | Calcified | Cavitating | ||||
| Zebrafish | M. marinum | Yes | Yes | No | No | Cellular granulomas which undergo progressive necrosis and pigment deposition, with large numbers of bacteria found within necrotic (but non-fibrotic) granuloma-like structures | (133,134) |
| Standard mouse strains | Various M. tuberculosis | Yes | No | No | No | Inflammatory or cellular lesions composed predominantly of admixed epithelioid (occasionally foamy) macrophages with large numbers of lymphocytes and few neutrophils present, containing bacteria which are primarily located intracellularly in macrophages. These lesions lack both necrosis and fibrosis with no distinct delineation from adjacent lung tissue | (135) |
| C3HeB/FeJ mouse | M. tuberculosis H37Rv, Erdman | Yes | Yes | No | Occasional | Heterogeneous pathology with the signature lesion consisting of caseous necrotic granulomas composed of a neutrophil-dominated central core that degenerates over time into an amorphous, acellular caseum (characterized by very high bacterial loads) surrounded by a band of intact neutrophils and a distinct rim of foamy macrophages at the peripheral margin. The core region is encapsulated by a collagen rim deposited by fibroblasts intermixed with epithelioid and activated macrophages, and few lymphocytes Exudative lesions: acute inflammatory response predominated by granulocytes, which fill alveolar spaces in a loose exudate containing variable amounts of fibrin and may harbor numerous bacteria |
(22,23,26,136) |
| Wistar Rat | M. tuberculosis H37Rv | Yes | Occasional | No | No | (137) | |
| Guinea Pig | Various M. tuberculosis | Yes | Yes | Yes | No | Granulomas with solid caseous centers, resulting from the complete necrosis of immune cells. All nuclei and cellular contours have disappeared, forming the so called cheese-like core or ‘caseum’. Solid caseum contains relatively low bacterial numbers, and is generally delineated by layers of lymphocytes, activated macrophages surrounded by a collagen rim | (76,138) |
| New Zealand White Rabbit | HN878 | Yes | Yes | No | Yes |
Solid caseous granulomas as described above, as well as liquefied necrotic lesions in which the caseous mass becomes fissured and fragmented into small particles with a soft and semi-liquid consistency (resembling thick sour milk), often containing high bacillary numbers which are extracellular. Liquefaction is caused by proteolysis and an active DTH (delayed-type hypersensitivity) response Cavities: the remnants of liquefied lesions after releasing their content into an airway. They are delineated by a fibrous capsule, with generally high bacterial numbers (both extracellular and intracellular) seen in the inner liquefied area of the cavitary wall, which also appear in expelled sputum |
(4,33,36) |
| Goat | M. bovis,M. Caprae | Yes | Yes | Yes | Yes | Solid and liquefied granulomas, calcified lesions and cavities | (139,140) |
| Minipig | M. tuberculosis H37Rv | Yes | Yes | No | No | (141) | |
| Marmoset | M. tuberculosis CDC1551 | Yes | Yes | No | Yes | Combination of all lesion types described above, in a small non-human primate (0.3–0.4 kg), thus covering the major lesion types seen in human TB | (5) |
| Marmoset | M. tuberculosis K04, M. africanum N0091 | Yes | Yes | No | No | Infection with these alternate strains generates a spectrum of disease progression, with various extents of cavitation and extrapulmonary disease | (5) |
| Macaque | M. tuberculosis Erdman | Yes | Yes | Yes | Yes | Large spectrum of lesion types, including those described above, as well as fibro-calcific granulomas: lesions with a central area of mineralization surrounded by prominent peripheral fibrosis, which sometimes appear as part of the lesion healing process following chemotherapy. A small proportion of animals also develop TB pneumonia: a form of the disease in which the pulmonary tissue is effaced by dense and poorly organized sheets of inflammation, composed of mixed macrophages including Langhans’ and multinucleated giant cells which host large numbers of bacilli, lymphocytes and granulocytes | (30,38,130,142) |
| New Zealand White Rabbit (Latent TB) | CDC1551 | Yes | Yes | No | No | Microbiological control of the infection and establishment of latent-like infection with undetectable bacteria that can be reactivated upon immune suppression | (34,35) |
| Macaque (Latent TB) | M. tuberculosis Erdman | Yes | Yes | Yes | No | Low dose bronchial instillation with M. tuberculosis Erdman – the same strain that causes active disease – results in a subset of animals developing latent infection as seen in humans. Pathologically and microbiologically, latently infected monkeys present a range of manifestations and lesion types – such as spontaneous mineralization of caseous granulomas typical of human LTBI – which has contributed to the discovery of the ‘latent spectrum’ concept | (10,13,29,143) |
The standard laboratory mouse models have provided critical information in TB drug development, but have not been successfully employed to determine prospectively the duration of combination treatments required to achieve sterile cure. The drawback of these models, is their lack of advanced lung lesion types, as the progression of disease rarely reaches stages of extensive necrosis and calcification in the lungs. Whereas, the most widely used laboratory mouse strains such as BALB/c and C57BL/6 show little evidence of necrosis, a mouse strain with a recessive allele, the C3HeB/FeJ mouse, does form necrotic lesions after infection with M. tuberculosis. This model was initially adapted and described for tuberculosis by Igor Kramnik and colleagues (21) and is thus referred to by most as the ‘Kramnik mouse model’. Although C3HeB/FeJ mice had no overt immunodeficiency, the mice were highly susceptible to M. tuberculosis infection. Moreover, these mice developed heterogeneous pulmonary granulomas that more closely resembled human lesions, thus overcoming a significant limitation of conventional mouse models of TB. Following either intravenous infection with a dose of 105 CFU or aerosol infection with 50–75 CFU per mouse, the lung bacterial load reaches high numbers concomitant with severe and diverse lung pathology (21). Necrotic microfoci are observed 3–4 weeks following infection, progressing to highly organized encapsulated lesions at later stages of disease, when the pulmonary bacterial load can reach 108 CFU or higher (21,22). In these C3HeB/FeJ mice, lesion necrosis occurs in lungs only, whereas infection is better controlled in other organs such as spleen and liver (21). This mouse model has successfully lent itself to advanced imaging techniques such as FDG-PET/CT enabling longitudinal studies of disease progression, response to drug treatment and relapse in real time (7,23). As discussed in a subsequent section, the distinct and diverse pathology of C3HeB/FeJ has important implications in terms of response to therapy (22,24–26), highlighting the critical role of the pulmonary pathology in drug efficacy. Neutrophils are the dominant cell type in C3HeB/FeJ mice, and therefore various recent studies have been reporting on the major role of neutrophils in regulation of inflammation of disease using this mouse model (27,28). The benefits of the C3HeB/FeJ mouse model are its small size and cost-effectiveness, and the small amount of test compound required in early drug discovery. C3HeB/FeJ mice only occasionally develop cavitary disease after infection with M. tuberculosis H37Rv or Erdman, and therefore this mouse strain is less amenable to transmission studies.
The non-human primate (NHP) model of TB provides an ideal tool that captures most disease pathologies seen in human TB. Among all laboratory animal models, cynomolgus macaques appear to deliver the most natural and closest reproduction of the full spectrum of human TB (12). Low dose infection via bronchoscopic instillation generates a range of manifestations, from complete resolution to fulminant active TB, as is thought to be the case in humans. The central caseous material of most granulomas undergoes mineralization over time, with fewer epithelioid macrophages and giant cells as well as reduced lymphocytic infiltration in the cellular rim (29). These characteristics together with the progressive appearance of fibrous connective tissue result in a narrow fibrocalcific interface (Table 1). Overall, these properties are consistent with containment of infection. As seen in human TB, longitudinal PET-CT imaging and bacterial load evaluation in the cynomolgous macaques revealed that host response (measured as standard 18F-glucose uptake volume ratio) and bacterial growth within individual lesions are independent and dynamic during infection, within the same host. Fibrocalcific lesions represented sites of bacterial control, whereas caseous sites were associated with poor control, overall in agreement with early microscopy studies in human tissues (2). While these features all strongly resemble the spectrum of human disease, they also highlight one major issue with the model in that this full spectrum of outcomes results in relatively large groups of animals required to make meaningful comparisons, particularly when using end points measured only at necropsy. Studies using FDG-PET/CT enable the tracking of lesional variability in both the host response and bacterial growth (30), as well as progressing and regressing lesions within the same animal. In that sense, they significantly reduce the group size requirements and increase the ‘translatability’ of the effects to human clinical trials, but this remains an expensive and challenging model (17).
Other NHP models of TB have been explored in some detail. The common marmoset is a highly susceptible animal species that develops significant pathology within 6–8 weeks of infection. While similar in some sense to human primary disease, the pathology includes necrotic lesions and cavities more typically associated with postprimary human disease. The course of the disease and the sites of infection depend upon the TB strain clade used to infect the marmosets, consistent with the observed epidemiological associations of these same strain clades in human populations. The small size of this species makes it highly amenable to drug studies for which larger primates require prohibitive amounts of test substance. However, the marmoset has intrinsic limitations for immunological studies since only small amounts of blood and cells are usually obtainable (5). Rhesus macaques have also been widely used in vaccine studies, since these animals are, like marmosets, highly susceptible to TB and do not contain the infection (31,32).
The rabbit model of TB disease presents several aspects of the human pathology, such as fibrotic granulomas with caseous necrotic foci that harbor small persisting mycobacterial populations that have adapted to changing conditions of oxygen tension, pH, and nutrient availability. Aerosol inoculation of outbred New Zealand White (NZW) rabbits can generate a spectrum of disease states and progression, mostly dependent on the infecting M. tuberculosis strain (34,35,33,36). The various manifestations encompass a wide range of disease states from active chronic cavitary disease to fully controlled bacterial growth resulting in sub-symptomatic latent infection. Such dynamics of disease progression offers the opportunity to study the impact of lesion heterogeneity on immunological correlates of vaccine response, as well as pharmacokinetics and efficacy of anti-TB drugs at the lesion level (37). Similar to the NHP, individual rabbit lesions followed longitudinally by PET-CT had very different fates, ranging from complete resolution to significant progression (4).
What determines the fate of individual lesions
The factors of the host immune response that determine the differential outcome of the disease following a M. tuberculosis infection and the development of the heterogeneity in pulmonary lung lesions within one patient are not fully understood. The local immune environment rather than the systemic immune response appears to be the determining factor in lesion dynamics. A recent report on the NHP model using sophisticated molecular tracking of M. tuberculosis strains showed that the fate of individual lesions varies substantially within one host. In addition, this study suggests that lesion heterogeneity is in part due to differential host-mediated killing of the residing pathogen at the lesion level after the onset of adaptive immunity (38). This bacterial killing can occur to the point of lesional sterilization, although in the case of TB pneumonia for instance, no evidence of bacterial reduction was seen. Accordingly, the production of T-cell cytokines by individual granulomas is highly heterogeneous within a single animal. The authors made the important conclusion that individual lesions follow diverse, at times overlapping, trajectories, which ultimately determine the clinical outcome of infection.
The influence of host-specific factors on lesion fate
Although the mechanisms driving divergent lesion progression are far from being elucidated, several pieces of the puzzle have started to emerge. At the host level, a fine balance between pro- and anti-inflammatory eicosanoids appears to play a major role in disease exacerbation and the development of necrotic foci (39,40). Eicosanoids are lipid mediators derived from arachidonic acid, mainly prostaglandins, leukotrienes, lipoxins, and resolvins. To provide optimal protection, i.e. limit bacterial growth while minimizing tissue damage and necrosis, the right balance between prostaglandins (41,42), lipoxins, and leukotrienes (42) is required. Key players in this regulatory cascade are IL-1β, leukotriene A4 hydrolase (LTA4H), and TNFs (43), and these are functionally linked via eicosanoids (40,44). While these networks have been studied systemically by linking concentrations of lipid mediators in serum/ELF to genetic polymorphisms or genetic knockouts, it is reasonable to speculate that similar variations at the lesion level influence the fate of individual granulomas. Host-pathogen interactions within lesions are a dynamic process, most likely influenced by subtle spatial and temporal factors, resulting in diverging trajectories of lesion progression. Tilting the balance toward induction of the prostaglandin versus lipoxin branch of the eicosanoid pathway could result in two lesions taking dramatically different path within the same lung. Many studies have focused on the role of lipid mediators in necrotic and apoptotic paths of infected macrophages, and shown that eicosanoids regulate the death modality of M. tuberculosis-infected macrophages (41). The results suggest that early events taking place within an aggregate of immune cells could seal the fate of a developing granuloma. Local differences in environmental and microbiological conditions could drive neighboring granulomas into either excessive necrosis or adequate control of microbial growth (45).
In C3HeB/FeJ mice, subtle differences in the local immunological cascades may contribute to lesion type and fate. Several of the key immune features associated with lesion type are observed upon low dose aerosol M. tuberculosis infection. The polymorphic lung lesions found in C3HeB/FeJ mice could be categorized into three types based on cellular composition and subsequent differential immunological control (Fig. 3). Type I lesions are the fibrous encapsulated pulmonary lesions with central liquefactive necrosis, which show a layer of foamy macrophages around a core composed of neutrophilic debris surrounded by a collagen rim with interstitial functional macrophages. Type II lesions are less organized with a massive recruitment of neutrophils, resulting in large areas of cellular necrosis throughout the lung parenchyma, and Type III lesions develop as a result of the accumulation of lymphocytes, epithelioid and foamy macrophages, as well as small pockets of neutrophils. Importantly, these lesion types in C3HeB/FeJ mice do change and progress over time but do not appear to interconvert once established. This was clearly demonstrated in C3HeB/FeJ mice infected with a less virulent strain of M. tuberculosis, which initially produced only Type III lesions and never exhibited caseous necrosis, even 20 weeks after aerosol infection (A. Lenaerts, unpublished results). By comparing all three lesion types, the bacterial burden was found to be inversely proportional to the number of lymphocytes present in these pulmonary lesions. As a result, these three lesion types represent vastly different levels of host immunological control within the lung during infection. Type III lesions contained abundant lymphocytes and controlled bacterial replication more efficiently, and progressed at a rate slower than the other two lesion types (Table 1). These observations are consistent with the initiation of a strong adaptive immune response capable of controlling bacterial replication and limiting host immunopathology. It is the failing of the adaptive immune response that is ultimately responsible for the characteristic liquefactive necrotic lesions observed in the C3HeB/FeJ mice.
Figure 3.
Heterogeneity in lesion types observed in C3HeB/FeJ mice infected with M. tuberculosis Erdman. Type I lesions (A) in C3HeB/FeJ mice are defined as the caseous necrotic lesions composed of a neutrophil-dominated central core region that degenerates over time into an amorphous, acellular caseum surrounded by a band of intact neutrophils and a distinct rim of foamy macrophages at the peripheral margin. The foamy macrophages contain numerous intracellular bacilli, while large numbers of extracellular bacilli are dispersed within the acellular caseum. The core region is encapsulated by a collagen rim deposited by fibroblasts intermixed with epithelioid and activated macrophages, and few lymphocytes. Type II lesions (B) observed in C3HeB/FeJ mice closely resemble PMN alveolitis occasionally observed in human tuberculosis patients, as described by Canetti (2). These lesions are predominantly composed of necrotizing neutrophils, but lack the fibrotic encapsulation seen in the Type I lesions. Type II lesions present as fulminant granulocytic pneumonia, containing high bacterial numbers extracellular and intracellular within in neutrophils, and very few if any detectable lymphocytes, and negatively impact mouse survival. Type III lesions (C) in C3HeB/FeJ mice are similar to lesions observed in BALB/c mice following aerosol infection. These inflammatory lesions are composed predominantly of epithelioid and foamy macrophages with large numbers of lymphocytes present, only containing few bacteria which are primarily located within macrophages.
The influence of M. tuberculosis strain-specific factors on lesion fate
A recent report using the rabbit model elegantly showed that in a matter of hours after infection with M. tuberculosis, differences in host gene expression were seen that determined the long-term outcome of M. tuberculosis infection in rabbit lungs (46). The investigators used two M. tuberculosis strains with varying virulence to demonstrate the effects of the early inflammation-associated innate immune response on the progression of pathology in individual lesions. The M. tuberculosis strain HN878, a ‘hyper-virulent strain’ (in standard models) of the Beijing family, shows rapid disease progression leading to cavitary disease in rabbits as well as in Guinea pigs (47). CDC1551 is a hyper-immunogenic strain from the Euro-American lineage that is more efficiently controlled in rabbit lungs, leading to containment of the pathogen and latent infection. The most prominent differences were the early recruitment of neutrophils in the lungs of HN878 infected animals, compared to animals infected with CDC1551. Increased recruitment of polymorphonuclear (PMN) cells is expected to exacerbate the local inflammatory response, as addressed below. Other differences seen after infection by the most virulent M. tuberculosis strain, both at 3 hours and 4 weeks post infection, were components of the inflammatory response and STAT-1 activation, recruitment and activation of macrophages, and PMN cell stimulation. Overall, HN878 infection was shown to be associated with exacerbated lung inflammation, followed by a slow and sub-optimal activation of both host innate and adaptive immune responses, resulting in progressive granulomatous response with large necrotic and liquefied lesions (48). This highlights that variations in bacterial products have an overall influence on immunologic response and suggests that, in studying the local immune response, both strain and host genetic backgrounds likely play a role even at very early stages.
The critical role of neutrophils in determining the fate of TB lesions
Increasing attention has been devoted to the role of neutrophils in pulmonary tuberculosis. While neutrophils may play a prominent role in preventing infection or in augmenting the initiation of protective immunity early after infection (49), growing evidence supports a distinctly negative association between a strong PMN response and clinical outcome once the infection has been established (50,51). High numbers of neutrophils present in bronchoalveolar lavage or peripheral blood of pulmonary TB patients were shown to correlate with cavitary disease and longer duration of sputum smear positivity, poor prognosis, and higher mortality (51,50,52). More recently, Eum et al. (53) reported that numerous infected neutrophils were present in the sputum of patients with active disease, and that many of the bacilli in these patients appeared to be actively replicating within the airways and in association with neutrophils. Another study identified a transcriptional signature comprised of neutrophil-driven interferon inducible genes obtained from peripheral blood that correlated with the severity of disease, further underscoring a role for PMN in TB pathogenesis (14,15). The ability of neutrophils to interact with macrophages, facilitate TB replication, and induce significant pathology has stimulated thinking about their potential importance in the context of understanding lesional dynamics (54).
Animal models of TB infection such as the rabbit, NHP, and specific mouse strains with human-like pathology can reproduce pulmonary neutrophilia, typically under conditions of high bacterial load, using virulent strains of TB or decreased host immunity. In IFN-γ knockout mice (GKO) and C3HeB/FeJ mice, the prolonged neutrophil response appears to represent a compensatory reaction by the host in response to a failure to initiate a protective Th1 immune response. In C3HeB/FeJ mice, Type I and Type II lesions are both lymphopenic and neutrophil-dominated following aerosol infection (A. Lenaerts, unpublished data). These neutrophils are highly permissive for bacterial replication with little evidence of bactericidal activity, consistent with a recent report demonstrating RD1-dependent M. tuberculosis subversion of neutrophil-mediated oxidative killing (55). Although the notion of distinct neutrophil subsets is controversial (56), evidence exists to suggest that neutrophils exhibit significant phenotypic plasticity in their effector functions in response to diverse stimuli (57). We should therefore be cautious in ascribing to all neutrophils what may only be characteristic of one particular subset. It is plausible that both protective and disease-promoting, phenotypically distinct, subpopulations of neutrophils may exist in response to TB infection, and that the balance of these responses may determine the heterogeneity of pulmonary lesion types. Strategies that specifically suppress neutrophil recruitment and persistence within the lung, particularly with regard to the IFN-γ/IL-17 axis of immunity (58), may augment conventional TB therapeutic regimens by denying the pathogen a preferred replicative niche, and by limiting neutrophil-mediated pulmonary immunopathology.
The impact of lesion heterogeneity on bacterial populations
Heterogeneity of bacterial location and numbers in lesions
M. tuberculosis is both extracellular and intracellular in the tissues of infected individuals, including within seemingly uninvolved lung tissue which hosts a complex spectrum of inflammation and repair. Canetti provided an early comprehensive report on bacillary location and numbers in the polymorphic lesion types found in human disease (2). He pointed out that the bacterial load is moderate where macrophages exist, numerous where PMNs predominate, and practically nil where only fibrin is observed. Sites of fibrous alveolitis and fully mineralized lesions were described as devoid of bacteria (2). Canetti highlighted the technical difficulties of using chromogenic stains and therefore studied more than 100 oil immersion microscopy fields per lesion to generate data. Despite major technological progress since those early days, the relative ratio and clinical relevance of intracellular and extracellular mycobacterial populations remains the matter of long-standing debates, as well as the cell types in which they reside (60,61). Because M. tuberculosis has the unique ability to survive within and disseminate from the phagolysosome of activated macrophages, this particular population has been the focus of intensive research efforts, particularly in the context of therapeutics development. The characterization of bacilli found in sputum and bronchoalveolar lavage fluid of TB patients, however, revealed a larger proportion of bacteria within neutrophils than macrophages, with smaller numbers inside epithelial cells (53). It has also been argued that M. tuberculosis may in fact spend most of its human lung ‘life-cycle’ extracellularly, with the intracellular phase being little more than a brief interlude during which the bacterium must replicate to increase its chances of transmission and physiologically adapt prior to switching to an extracellular phase (60). Extracellular bacilli residing within the caseum of necrotic foci could be particularly important as they constitute the reservoir from which large bacterial numbers emerge when a closed nodule encounters an airway and develops into an aerated cavity (Fig. 1). At the luminal surface of cavities, bacilli are either extracellular or intracellular (within cell types such as neutrophils and macrophages). The relatively low bacterial numbers and technical challenges associated with their visualization likely contribute to our inability to resolve this controversy. Current acid-fast stains used to visualize M. tuberculosis in tissue sections have a number of drawbacks, as M. tuberculosis bacilli are known to lose their ‘acid-fastness’ in vivo and in vitro under certain conditions (61–64). The acid-fastness of mycobacteria in mice reduces significantly over time (65). Likewise, staining with auramine-rhodamine revealed a gradual decrease in bacterial ‘stainability’ within the necrotic lesions of C3HeB/FeJ mice, while these bacilli could still be visualized by Ziehl-Neelsen (22), demonstrating that bacterial phenotypes and physiology evolve over time. This was also confirmed by combining various detection techniques, whereby bacterial phenotypes could be differentiated in a seemingly single microenvironment such as necrotic lesions (66). The development of fluorescent stains has facilitated the detection of tubercle bacilli owing to the higher signal-to-noise ratio (67). The recent evaluation of a novel fluorescent acid-fast staining approach has shown that the nucleic acid-binding dye SYBR Gold stains nearly all bacilli, regardless of their metabolic state and cell wall (68). Its most unique characteristic is its exceptionally high signal-to-noise ratio due to a >1000-fold enhanced fluorescence after binding to DNA/RNA, thereby overcoming issues of background fluorescence. SYBR Gold staining enabled visualization of intracellular and extracellular bacteria in both type I and type II lesions of C3HeB/FeJ mice, whereas very few bacteria could be visualized in type III lesions (A. Lenaerts, unpublished results). These relatively low numbers combined with poor acid-fastness make it difficult to visualize bacteria either pre- or posttherapy. SYBR Gold thus promises to be a valuable tool to detect sparsely distributed bacilli in animal and human tissues, to establish the relative importance and relevance of intracellular and extracellular populations, and to locate the bacilli most recalcitrant to therapy.
In animal models that develop necrotic caseous lesions, such as NHPs and rabbits, as well as in human TB, the number of bacilli per lesion is generally much lower than in C3HeB/FeJ mice, except for liquefactive and cavitary lesions. Furthermore, not all lesions contain viable bacteria, even in animals with active disease. In rabbits with chronic TB disease and mature lesions, 20–30% of the granulomas harbored no detectable CFU at 15–20 weeks post infection, while most granulomas with culturable bacteria contained anywhere between 10 and 104 CFU, with higher numbers found in necrotic than in cellular lesions, as expected (V. Dartois, unpublished data). In macaques with chronic active disease, individual animals presented with 0 to 75% sterile lesions, while 20–100% of the lesions were sterile in latently infected monkeys, showing that lesion sterilization is not solely a characteristic of latent disease. Interestingly, the number of CFU present in the lesions of latent and active macaques largely overlapped, ranging between 10 and 105 bacteria (38). Although latent TB is loosely equated to bacterial containment in some inactive form, it is now clear that a wide range of bacillary burdens could exist in apparently homogeneous asymptomatic populations. These highly variable lesion-centric numbers, even within a single animal, corroborate the diverging immunopathologic trajectories of individual lesions and marked lesion-to-lesion variability in host-mediated killing. Measuring cumulative bacterial burden using quantitative PCR at the lesion level is possible owing to the long residence time of M. tuberculosis DNA after the bacilli have been killed (39,70). Thus, CFU numbers can be related to local killing capacity and in turn correlated with lesion-specific histopathology. This emerging methodology promises to be a valuable tool to study host-pathogen cross-talks in TB pathology, bacterial killing, and lesion progression.
Environmental conditions in necrotic lesions
The lesion heterogeneity seen in human lungs generates a plethora of microenvironmental conditions at the various locations where the bacilli reside. Hypoxia, nutrient limitation and shift, pH and oxidative stress are thought to be present in caseating pulmonary lesions and in the phagolysosome of infected macrophages. Each of these conditions induces metabolic and physiologic adaptations, affecting anti-TB chemotherapy and most likely contributing to its long duration and requirement for multi-drug treatment.
Hypoxia has been postulated as one of the environmental conditions driving mycobacteria in vitro into a ‘persister’ state (70–72), thereby potentially affecting their responsiveness to drugs. It is a key distinguishing feature of caseous granulomas in both active disease and latent infection (73,74). Numerous studies have demonstrated that the standard laboratory mouse fails to develop significant necrosis and thus hypoxia in lung lesions (73,75). Therefore, animal models showing a broader range of pathological features have been proposed to assess drug activity against persisting bacteria found in hypoxic lesions. Hypoxia has been demonstrated in regions with cellular necrosis as well as in solid and liquefactive caseous lesions (Table 1) in humans, rabbits, non-human primates, Guinea pigs, and C3HeB/FeJ mice (22,74,76,77). The studies all used pimonidazole, a 2-nitroimidazole able to identify regions of hypoxia (defined as <4 μM O2 saturation or O2 tension lower than 10 mm Hg) in animal organs after injection. When interpreting these results, one should keep in mind the relatively ‘fluid’ definition of hypoxic/anaerobic conditions, from 10% oxygen tension to induce the dormancy regulon in M. tuberculosis (78), to 1 and 0.06% in the microaerophilic non-replicating phase I and II of Wayne's in vitro model (79), and the <0.05% oxygen tension required for bioreductive activation of pimonidazole (80). Many micro-compartments inside lesions could exhibit relatively low oxygen tension, while not being positively stained by pimonidazole. New probes that could detect and measure a range of oxygen tensions will likely reveal a spectrum of oxygenation with local consequences on bacterial metabolism and drug efficacy.
Current belief holds that the caseum of necrotic lesions might be acidic. This assumption is mostly based on the fact that early necrosis results in the release of phagolysosome content which is itself acidic. While this is a reasonable hypothesis, it is also likely that the drop in pH is a transient event. Recently, pH measurements of excised and homogenized type I necrotic lesions from chronically infected C3HeB/FeJ mice, using pH indicator strips, revealed a slightly basic range of 7.4–8.0 (A. Lenaerts, unpublished results). This suggests that the caseum of mature lesions may be slightly basic rather than acidic. This crude assessment was similar to an earlier report published in 1934 (81). However, chemostat culture of hypoxemic M. tuberculosis revealed that the bacillus actively secretes succinic acid to maintain proton-motive force across its membranes which may cause localized acidic regions near bacilli within caseum (82). Analytical and intracellular pH probes are required to accurately measure pH in various lesion types and inside M. tuberculosis bacilli residing in these lesions. These early results, however, indicate that a range of pH should be considered for in vitro screens of chemical libraries: a ‘low’ pH range to reproduce the conditions encountered by intracellular bacteria in the phagolysosome of macrophages or local areas of caseum, and a slightly basic medium to reflect the conditions seen in necrotic lesions. The slightly basic pH of necrotic lesions could be one of the factors contributing to the modest PZA efficacy observed in the C3HeB/FeJ compared to BALB/c mice, where the bacilli are primarily intracellular in activated macrophages (22).
Caseum is rich in lipids such as cholesterol, cholesterol esters, and triglycerides (83), but its exact molecular composition has not been fully elucidated. This important question is currently the focus of unbiased lipidomics, proteomics, and metabolomics projects using sensitive analytical tools such as NMR, MALDI, and mass spectrometry. In vitro and ex vivo expression profiling studies have shown that M. tuberculosis adapts to a lipid-rich environment by inducing cholesterol and triglyceride utilizing enzymes (84–87). While this finding suggests that the pathogen adapts to a fat-based diet as a matter of necessity, there is also evidence that M. tuberculosis contributes to this nutrient shift by inducing the formation of lipid bodies in infected macrophages. The contents of these lipid bodies are either released into the caseum upon macrophage necrosis and could thus be used by extracellular bacilli, or metabolized by intracellular bacteria and imported to form intracytoplasmic lipid inclusions (88). Concomitant to this nutrient shift, M. tuberculosis is thought to acquire a ‘dormant’ or persisting phenotype (89,90).
Inside the phagolysosome of macrophages, the pathogen is exposed to oxidative and nitrosative stress, owing to the production of reactive species. M. tuberculosis has the unique ability to survive and counteract this hostile environment, because it is equipped to resist those insults, subvert the host pathways responsible for the production of these reactive species and interfere with phagolysosome biogenesis and integrity (91,92). Again, these properties require metabolic and physiological adaptations that are selectively acquired in specific niches, affecting the replication status and relative susceptibility to antimicrobial agents.
Whereas it is relatively straightforward to analyze bacterial populations to define a physiological or metabolic state in vitro, only limited information can be obtained from the analysis of in vivo samples derived from tissues. It has been challenging to perform metabolomic, proteomic, or biochemical studies with the few organisms obtained from infected tissues. We thus have a limited understanding of how expression data generated in vitro translate at the lesion level. Transcriptome analyses of M. tuberculosis in cultured macrophages, in mouse infection models, and in human lung lesions (86,93–96) have shown that (i) the glyoxylate shunt appears to be required during intracellular growth confirming that M. tuberculosis survives by scavenging host lipids (97–99); (ii) gluconeogenesis is required for growth in vivo (100); and (iii) there is a shift to anaerobic respiration during latent infection as well as in the persister population that emerges during treatment of active TB (101–103). There are no published studies addressing the in vivo transcriptomics profile of the persisting bacterial population during drug treatment. Highly sensitive genetic tools such as multiplex RT-PCR for M. tuberculosis-specific genes (70) could address this question in sputum samples, as well as in human or animal lung lesions.
Irreversible lesions – the importance of cavities
In humans, caseous necrosis can sometimes lead to liquefaction and cavity formation (Fig. 1). Cavitation is the process whereby the liquefied caseum discharges through an eroded airway or pleural space and empties the cavity. It is one of the hallmarks of TB and presents some unique features. Studies of resected lungs from patients having surgery for drug-resistant TB revealed that the luminal surface of cavities are nearly completely devoid of CD4+ and CD8+ T cells, preventing direct contact of these cells with infected macrophages at the cavity surface and providing a highly permissive environment for bacterial growth (104). Cavities represent a process of extravasation of the formerly parenchymal contents of a lesion into the airways and thus reflect a process in which the disease inverts from inside the body to outside the body. Solid caseum originates from the degradation of viable tissue and recruited inflammatory cells, and liquefied caseum forms by hydrolysis of solid caseum after it is formed. Although the process is not entirely known, it has been shown that hydrolytic enzymes (105,106), lack of vascularity causing a decrease in enzyme inhibitors, and the DTH response (107) play key roles in liquefaction and cavity formation. Cavity formation is irreversible pathologically, even with effective chemotherapy cavities typically collapse but do not resolve into healthy lung. In some circumstances in NHP and humans, the initiation of chemotherapy appears to coincide with cavitation of some lesions, an area that requires significantly more study as this residual pathology may contribute to permanent loss of pulmonary function.
Cavitary disease in TB patients has been correlated with difficulty to treat successfully, higher relapse rates, transmission, and increase in the development of drug resistance. Cavitary disease is not observed in mouse models and only infrequently in Guinea pigs, which develop solid caseous necrosis containing low bacterial numbers which then continue to mineralize. Although cavitary lesions occur naturally in C3HeB/FeJ mice following aerosol infection, it is a relatively infrequent event. A current focus in at least two laboratories is to develop strategies to make cavitary lesions more reproducible in C3HeB/FeJ mice. Only the rabbit, marmoset, and macaque have consistently shown cavitary disease, and their presence/development can be tracked or even predicted by PET/CT imaging (5,108,109). They are thus important tools for transmission studies and evaluation of drugs and drug combinations to shorten chemotherapy.
The impact of lesion heterogeneity on drug distribution
The observation that lesion progression, structure, and microenvironments affect the physiology of bacteria that reside in them has been recognized and studied for quite some time. What has emerged more recently is the concept that these lesion-specific properties also affect drug penetration and drug access. As described in the previous section, pulmonary lesions are highly heterogeneous both in space and time, including within a single host. They evolve from fully cellular granulomas to necrotic and/or fibrotic lesions which can in turn form open cavities in direct contact with an airway. Primary lesions that successfully contain the pathogen often develop as calcified granulomatous lesions, which are thought to be relatively stable and less dynamic than other lesion types (110,111). How do these various structures affect drug distribution?
As the necrotic granuloma ages and expands, the ratio between the surrounding leukocyte coat and the necrotic core becomes smaller, suggesting that necrosis expands at the expense of the surrounding cell layers, rather than by proportional growth due to increased leukocyte recruitment from the circulation (110). Staining of endomucin or CD31 (markers used primarily to demonstrate the presence of endothelial cells in histological tissue sections) in human TB lesions revealed denser vascularization of uninvolved lung in patients with latent than active TB. The fibrotic walls of cavities and latently infected granulomas contained high vessel densities (61), suggesting that drugs have adequate access to the outer layers of mature granulomas and cavities (Fig. 1C). In M. avium-infected mouse granulomas, quantitative comparison of the number of microvessels close to the necrotic core with that measured at the periphery of granulomas confirmed a severe reduction in vascularization in areas directly subtending the necrotic center, with caseous foci being totally devoid of functional vessels. At the peripheral fringes of the granulomas, the vasculature appeared abundant and compressed by the adjacent granuloma (112). Thus, vascularization is extremely heterogeneous across lesion types and lesion compartments, leading to impaired blood flow and consequently drug supply.
Inside the cellular layers of granulomas, expression of active transporters and efflux pumps on the surface of leukocytes vary according to cell type, activation/infection status, and other microenvironmental factors. Active uptake and efflux of antibiotics by macrophages has been quantified in vitro, most extensively for broad spectrum antibiotics such as the fluoroquinolones (113), but also for selected anti-TB agents (114,115). In general, drugs that are known to accumulate into macrophages in vitro have shown favorable distribution into the leukocyte coat of necrotic granulomas ((116), V. Dartois, unpublished data). A fully quantitative translation of such in vitro findings at the lesion level, during a typical dosing interval, will be critical to interpret efficacy results as a function of pharmacokinetics at the site of action.
The fibrotic rims that encapsulate cavities are among the most densely vascularized areas and thus constitute less of an obstacle to pharmacotherapy than once assumed based on the long-standing observation that cavities are most difficult to treat and have a negative prognostic impact. The most problematic niche is more likely the caseous center of necrotic lesions and cavities, where all vascular architecture has been destroyed, leading to failed immunity and lack of drug supply from blood. To reach the center of necrotic granulomas where quiescent extracellular bacilli can be found in relatively large numbers (3,104), drugs thus have to diffuse from the cellular rim across the entire necrotic center without the assistance of active or facilitated transport mechanisms. Factors that drive diffusion of a drug across a concentration gradient are the physicochemical properties of the small molecule, the viscosity and pH of the medium (caseum in this case), and the interactions between the drug and the medium such as binding of the drug to macromolecules. MALDI mass spectrometry imaging (117), an emerging imaging modality that allows the visualization of unlabeled drugs in tissue sections, is ideal to generate 2D ion maps of TB drugs in infected lungs, showing that several drugs and drug classes diffuse poorly through caseous material, and exhibit radically different patterns of heterogeneous distribution. An important consequence of uneven drug distribution is the creation of spatial and temporal windows of monotherapy, potentially driving the emergence of drug resistance in selected niches. A complete understanding of the impact of multidimensional lesion heterogeneity on bacterial populations and drug pharmacokinetics would provide an invaluable opportunity to rationally design new drug regimens that close the door to resistance development and accelerate cure.
Although this is only a hypothesis at this point, we propose that host-directed therapies (HDTs) that reduce the extent of necrosis and caseation might provide a secondary and desirable pharmacokinetic effect. Many currently pursued HDTs have the potential to curb granuloma progression, by either suppressing or reverting necrosis. For example, manipulation of the eicosanoid pathway could shift the balance away from lipoxin-induced necrosis through inhibition of 5-lipoxygenase by Zileuton (40). Other candidates that could interfere with the necrosis of lipid-laden macrophages or lesion progression in general are statins (118), PDE4 inhibitors (119), vitamin D (120), and anti-inflammatory agents that curb atherosclerotic plaque progression (121–123). Since, caseous foci are the niches where many antibiotics struggle to penetrate, reducing or preventing the development of necrosis could significantly improve anti-TB drug efficacy by reducing or removing a problematic compartment.
The impact of heterogeneity on response to therapy at the lesion level
Two important clinical implications emerge from lesion heterogeneity, in terms of the relative effectiveness of drug therapy within different lesions and lesion compartments. Lesion characteristics differentially affect (i) the pharmacokinetics of TB drugs, i.e. their ability to cross all barriers between the blood compartment and the intrabacterial molecular target; and (ii) the relative susceptibility of the bacilli contained within these lesions due to metabolic realignments that may induce phenotypic drug tolerance. These combined factors result in marked interlesional and intralesional variability in drug-mediated killing. The concept of bacterial subpopulations that are differentially susceptible to specific anti-TB agents is not new. Based on numerous clinical trials conducted by the BMRC with very large patient cohorts (reviewed in (124)), Mitchison (125) proposed a model of selective killing/inhibition of specific subpopulations by individual drugs that make up the first line regimen. This ‘special population theory’ accounts for spatial and temporal fluctuations in the microenvironment, resulting in differential susceptibility to antimicrobial agents. Mitchison postulated that actively growing organisms, mostly extracellular bacilli in areas of caseation, are primarily killed by isoniazid. Semi-dormant organisms inhibited by an acid environment, either in areas of acute inflammation or in phagolysosomes, are mostly killed by pyrazinamide; slow or non-replicating bacteria with spurts of metabolism are the primary targets of rifampicin.
Mitchison's model (125) has formed the basis of in-depth studies on the impact of lesional microenvironment on local drug response. The markedly different pathology seen in C3HeB/FeJ and BALB/c mice offers a unique opportunity to study the impact of lesion heterogeneity on drug efficacy. Several anti-TB agents have demonstrated inferior efficacy in C3HeB/FeJ mice, compared to BALB/c mice, where structured and mature lesions do not form (22). Furthermore, treatment of C3HeB/FeJ mice with single agents often times elicits a heterogeneous response, whereby two clusters of mice generally emerge: one in which a clear response is observed and another in which either poor or no efficacy is seen. For example, treatment with rifamycins was efficacious in a subset of mice, resulting in large standard deviations in each treatment group (E. Nuermberger, A. Lenaerts, unpublished results). Upon further analysis, the heterogeneous treatment response appeared to be directly linked to the diverse histopathology observed in the lungs of individual animals (Fig. 3). In-depth studies are underway to correlate lesion-specific pathology, drug exposure, and treatment response for multiple TB drugs. Powerful tools such as PET and CT scan analysis in conjunction with histopathology will be instrumental to connect these read-outs. Thus, lesion heterogeneity has practical implications regarding the use of C3HeB/FeJ mice in drug candidate evaluation. Important recommendations for optimal use of the mouse model include (i) collection of all five lung lobes for bacterial enumeration to mitigate variability and increase accuracy; (ii) use of larger mouse numbers per treatment group to increase statistical power; (iii) allowing sufficient time for the caseous necrotic pathology to develop; and (iv) histological verification of the pulmonary pathology at treatment start.
The most striking efficacy discrepancy between C3HeB/FeJ and BALB/c mouse strains was seen with pyrazinamide (PZA) and clofazimine (CFZ). For both drugs, only moderate bacterial killing was detected in the lungs of C3HeB/FeJ mice, while they were highly effective in BALB/c mice (22,24). In C3HeB/FeJ mice, CFZ was active only when treatment was initiated prior to the formation of necrotic granulomas, whereas its activity barely significant when administered after the formation of caseous necrotic lesions. In contrast, CFZ remained effective throughout infection in spleens, which never show necrosis, similar to BALB/c mice. These results point to the pathological progression of disease as the predominant factor contributing to the attenuated efficacy of CFZ in C3HeB/FeJ lungs. Recent tissue distribution studies indicate that poor penetration of CFZ in the caseous foci of necrotic lesions likely contributes to the limited activity of CFZ in C3HeB/FeJ mice (V. Dartois, unpublished data). The decreased potency of CFZ under hypoxic conditions – specifically present in necrotic lesions – is another likely contributing factor (22). Indeed, CFZ activity was found to strongly correlate with the level of aeration of an in vitro M. tuberculosis culture, in agreement with the proposed mechanism of action of CFZ through the generation of reactive oxygen species only under aerobic conditions (126). Thus, lesion heterogeneity in C3HeB/FeJ mice revealed both pharmacokinetic and pharmacodynamic liabilities of CFZ. Additional follow-up studies have been planned to support future clinical use of CFZ: long-term treatment to assess its sterilizing potential, testing in combination regimens, and confirmation in other animal models.
Intracellular bacteria create an additional hurdle on the path of TB drugs from the blood compartment to their bacterial targets: to be active, a therapeutic agent must permeate the host cells in which the pathogen resides. In addition, the microenvironmental conditions encountered within the phagolysosome may significantly impair drug action. Because different drugs, drug classes, and even their metabolites can exhibit differential activity and penetration properties in phagocytic cells, this adds to the heterogeneity of response to therapy. Predicting accumulation and activity of therapeutic agents inside phagocytic cells in vitro has been challenging, because the results can differ significantly between cell lines, and the predictive value of specific cell lines has not been established. Sutezolid, a new oxazolidinone currently in Phase II clinical development, has a unique profile in this regard. Its major metabolite, which reaches concentrations in plasma several times those of the parent, drives the killing of extracellular bacteria, while the parent compound sutezolid is mostly active against intracellular bacilli, as determined in an ex vivo whole blood bactericidal assay (127). Although the contribution of accumulation versus intrinsic activity inside host cells has not yet been established, this is the first report of a drug candidate and its metabolite acting on different mycobacterial subpopulations. Mathematical modeling of ex vivo pharmacological data has been used to recommend doses and dosing regimens in upcoming clinical trials (128).
Metronidazole, an agent that requires hypoxic to anaerobic conditions for its activation, is another drug whose efficacy is influenced by the local environment. Metronidazole was not active in BALB/c mice (129), whose lesions lack sufficient hypoxia (73,75). However, the drug also failed to demonstrate activity in C3HeB/FeJ mice, where hypoxia does develop within necrotic lesions, as visualized by piminazole staining (22). Activity was demonstrated in rabbits against M. bovis (74) and in macaques (130). Metronidazole evaluation in Guinea pigs failed due to toxicity (131). A possible explanation for the lack of activity in C3HeB/FeJ mice is the small granuloma size resulting in residual oxygen within the necrotic lesions, compared to the larger lesion size in other animal models. High resolution measurements of oxygen tension would be critical to interpret these efficacy results. While no clinical data are available to confirm the activity of metronidazole in TB patients (132), this case study highlights the importance of using the appropriate animal model for evaluating a compound, especially when a specific condition or environment is required for drug activity. These conditions might not always be known, particularly in early drug development. Therefore, we argue that results obtained in a standard mouse strain must be confirmed or challenged in a second species with more advanced and heterogeneous lesion types, before proceeding with preclinical drug combination and clinical trials.
In rabbits with chronic active disease, lesions were tracked by PET-CT imaging following treatment with first line anti-TB drugs. Lesion volume, density, and maximum standard uptake value (coined ‘PET avidity’) showed a diversity of response at the lesion level, within single animals (4). In macaques with active TB, the number of CFU per granuloma measured at the end of treatment with first- and second-line drugs was highly variable, from no detectable bacteria to >104 CFU per lesion (17,30). Similarly, the percentage of sterile lesions posttreatment varied between animals, from 40 to 100% (17). Lesions with the highest bacterial burden left at the end of therapy were caseous lesions and areas of TB pneumonia. In a marmoset trial comparing the efficacy of the current 1st line drug combination (isoniazid, rifampicin, pyrazinamide and ethambutol, or HRZE) with one of the first regimens used in the 1950s (isoniazid and streptomycin, or HS), cavities were much more recalcitrant to treatment with HS than closed nodules (C.E. Barry, unpublished results).
These findings highlight some of the limitations of current efficacy read-outs such as bacterial burdens in whole lungs or bacterial counts in sputum. If we are to dramatically shorten TB therapy, we need to ‘understand the system’, i.e. determine how specific lesion types and the bacteria they host respond to individual drugs and why. Only this type of analytical approach can deliver a significant shortening of TB treatment by combining drugs that complement each other in their ability to reach and kill all bacterial subpopulations.
Outlook
Since the 1950s, when Canetti described the various manifestations of tuberculous pulmonary lesions, we have acquired a much broader, though still incomplete, picture of the impact of this diversity on bacteriological responses at the lesion level. Spatial and temporal lesion dynamics are the result of local fluctuations in immune cascades and microenvironments, which in turn impact the cellular and microbiological contents of granulomas and cavities. Knowledge and awareness of these diverging trajectories are critical to optimally design and interpret preclinical experiments and clinical research. It is now clear that lesion-centric read-outs are required to study biomarkers of disease progression, responses to new drug candidates and regimens, and immunological correlates of vaccine efficacy.
Efforts invested in trying to identify systemic predictors of disease outcome, such as progression from latent infection to active disease, have met limited success. The marked inter-lesion variability reviewed here suggests that we may need to revisit our approaches and develop invasive and non-invasive methods that enable probing individual lesions rather than seeking a systemic response in the blood. PET-CT imaging has recently emerged as promising methodology. In particular, the development of novel PET tracers to monitor the immune and microbiological dynamics has the potential to broaden the scope of PET imaging as a biomarker of drug response at the lesion level.
Differential therapeutic responses of individual granulomas and cavities observed in animal models where single lesions can be isolated have highlighted the need to quantify bacterial burden in single lesions rather than whole lungs. Likewise, sputum counts and sputum conversion only reflect the status of lesions that have open access to an airway, i.e. mostly cavities. Here again, alongside classical techniques, non-invasive imaging methodologies targeting mycobacteria are ideally suited to longitudinally track bacterial kill kinetics within lesions. Such approaches can address important questions, which remain unanswered so far and are critical in interpreting clinical trial results: which lesion types are most recalcitrant to therapy and why? Are these the source of disease relapse?
Despite rapid progress over the last decade, vaccine development still awaits a major breakthrough, mostly due to the lack of predictive biomarkers that would enable (i) patient stratification; (ii) selection of latently infected individuals at higher risk of reactivation, thus reducing the number of study participants, cost and duration of vaccine trials; and (iii) assessment of vaccine efficacy. Awareness of lesion heterogeneity within a single individual may provide new directions and set the stage for the discovery of predictive biomarkers and the selection of better surrogate end points.
Acknowledgments
The authors are funded by the TB Drug Accelerator program of the Bill and Melinda Gates Foundation, as well as NIH/NIAID IDIQ Contract, Task Order HHSN272201000009I/01 (Anne Lenaerts), the Intramural Research Program of the NIH, NIAID (Clifton Barry) and NIH grants 1R01AI111967-01 and 1R01AI106398-01 (Véronique Dartois). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does it mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The authors have no conflicts of interest to declare.
References
- 1.Barnes DS. The Making of a Social Disease – Tuberculosis in Nineteenth-Century France. Berkeley, Los Angeles, London: University of California Press; 1995. [Google Scholar]
- 2.Canetti G. The Tubercle Bacillus. New York: Springer Publishing COmpany; 1955. [Google Scholar]
- 3.Dannenberg AM., Jr . Pathogenesis of Human Pulmonary Tuberculosis. Washington, DC: ASM Press; 2006. [Google Scholar]
- 4.Via LE, et al. Infection dynamics and response to chemotherapy in a Rabbit model of tuberculosis using (18F)2-fluoro-deoxy-D-glucose positron emission tomography and computed tomography. Antimicrob Agents Chemother. 2012;56:4391–4402. doi: 10.1128/AAC.00531-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Via LE, et al. Differential virulence and disease progression following Mycobacterium tuberculosis complex infection of the common marmoset (Callithrix jacchus) Infect Immun. 2013;81:2909–2919. doi: 10.1128/IAI.00632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagci U, et al. A computational pipeline for quantification of pulmonary infections in small animal models using serial PET-CT imaging. EJNMMI Res. 2013;3:55. doi: 10.1186/2191-219X-3-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murawski AM, et al. Imaging the evolution of reactivation pulmonary tuberculosis in mice using 18F-FDG PET. J Nucl Med. 2014 doi: 10.2967/jnumed.114.144634. doi: 10.2967/jnumed.114.144634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esmail H, et al. The ongoing challenge of latent tuberculosis. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130437. doi: 10.1098/rstb.2013.0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawn SD, Wood R, Wilkinson RJ. Changing concepts of “latent tuberculosis infection” in patients living with HIV infection. Clin Dev Immunol. 2011;2011 doi: 10.1155/2011/980594. doi: 10.1155/2011/980594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barry CER, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young DB, Gideon HP, Wilkinson RJ. Eliminating latent tuberculosis. Trends Microbiol. 2009;17:183–188. doi: 10.1016/j.tim.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Capuano SV, 3rd, et al. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect Immun. 2003;71:5831–5844. doi: 10.1128/IAI.71.10.5831-5844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin PL, et al. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect Immun. 2009;77:4631–4642. doi: 10.1128/IAI.00592-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berry MP, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bloom CI, et al. Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PLoS ONE. 2013;8:e70630. doi: 10.1371/journal.pone.0070630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee M, et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N Engl J Med. 2012;367:1508–1518. doi: 10.1056/NEJMoa1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coleman MT, et al. PET/CT monitoring demonstrates a therapeutic response to oxazolidinones in Mycobacterium tuberculosis infected macaques and humans. Sci Transl Med. 2014;6:265ra167. doi: 10.1126/scitranslmed.3009500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aber VR, Nunn AJ. Short term chemotherapy of tuberculosis. Factors affecting relapse following short term chemotherapy. Bull Int Union Tuberc. 1978;53:276–280. [PubMed] [Google Scholar]
- 19.Chang KC, et al. A nested case-control study on treatment-related risk factors for early relapse of tuberculosis. Am J Respir Crit Care Med. 2004;170:1124–1130. doi: 10.1164/rccm.200407-905OC. [DOI] [PubMed] [Google Scholar]
- 20.Benator D, et al. Rifapentine and isoniazid once a week versus rifampicin and isoniazid twice a week for treatment of drug-susceptible pulmonary tuberculosis in HIV-negative patients: a randomised clinical trial. Lancet. 2002;360:528–534. doi: 10.1016/s0140-6736(02)09742-8. [DOI] [PubMed] [Google Scholar]
- 21.Kramnik I, et al. Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2000;97:8560–8565. doi: 10.1073/pnas.150227197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Driver ER, et al. Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56:3181–3195. doi: 10.1128/AAC.00217-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis SL, et al. Noninvasive pulmonary (18F)-2-fluoro-deoxy-D-glucose positron emission tomography correlates with bactericidal activity of tuberculosis drug treatment. Antimicrob Agents Chemother. 2009;53:4879–4884. doi: 10.1128/AAC.00789-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irwin SM, et al. Limited activity of clofazimine as a single drug in a mouse model of tuberculosis exhibiting caseous necrotic granulomas. Antimicrob Agents Chemother. 2014;58:4026–4034. doi: 10.1128/AAC.02565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skerry C, et al. Adjunctive TNF inhibition with standard treatment enhances bacterial clearance in a murine model of necrotic TB granulomas. PLoS ONE. 2012;7:e39680. doi: 10.1371/journal.pone.0039680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vilaplana C, et al. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J Infect Dis. 2013;208:199–202. doi: 10.1093/infdis/jit152. [DOI] [PubMed] [Google Scholar]
- 27.Marzo E, et al. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis. 2014;94:55–64. doi: 10.1016/j.tube.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Obregon-Henao A, et al. Gr1(int)CD11b+ myeloid-derived suppressor cells in Mycobacterium tuberculosis infection. PLoS ONE. 2013;8:e80669. doi: 10.1371/journal.pone.0080669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin PL, et al. Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum. 2010;62:340–350. doi: 10.1002/art.27271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin PL, et al. Radiologic responses in cynomolgous macaques for assessing tuberculosis chemotherapy regimens. Antimicrob Agents Chemother. 2013;57:4237–4244. doi: 10.1128/AAC.00277-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharpe SA, et al. Establishment of an aerosol challenge model of tuberculosis in rhesus macaques and an evaluation of endpoints for vaccine testing. Clin Vaccine Immunol. 2010;17:1170–1182. doi: 10.1128/CVI.00079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darrah PA, et al. Aerosol vaccination with AERAS-402 elicits robust cellular immune responses in the lungs of rhesus macaques but fails to protect against high-dose Mycobacterium tuberculosis challenge. J Immunol. 2014;193:1799–1811. doi: 10.4049/jimmunol.1400676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manabe YC, et al. Different strains of Mycobacterium tuberculosis cause various spectrums of disease in the rabbit model of tuberculosis. Infect Immun. 2003;71:6004–6011. doi: 10.1128/IAI.71.10.6004-6011.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manabe YC, et al. The aerosol rabbit model of TB latency, reactivation and immune reconstitution inflammatory syndrome. Tuberculosis. 2008;88:187–196. doi: 10.1016/j.tube.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subbian S, et al. Spontaneous latency in a rabbit model of pulmonary tuberculosis. Am J Pathol. 2012;181:1711–1724. doi: 10.1016/j.ajpath.2012.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subbian S, et al. Chronic pulmonary cavitary tuberculosis in rabbits: a failed host immune response. Open Biol. 2011;1:110016. doi: 10.1098/rsob.110016. doi: 10.1098/rsob.110016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kjellsson MC, et al. Pharmacokinetic evaluation of the penetration of antituberculosis agents in rabbit pulmonary lesions. Antimicrob Agents Chemother. 2012;56:446–457. doi: 10.1128/AAC.05208-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin PL, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med. 2014;20:75–79. doi: 10.1038/nm.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tobin DM, Ramakrishnan L. TB: the Yin and Yang of lipid mediators. Curr Opin Pharmacol. 2013;13:641–645. doi: 10.1016/j.coph.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayer-Barber KD, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511:99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8:668–674. doi: 10.1038/nrmicro2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tobin DM, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140:717–730. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tobin DM, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148:434–446. doi: 10.1016/j.cell.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lalvani A, Behr MA, Sridhar S. Innate immunity to TB: a druggable balancing act. Cell. 2012;148:389–391. doi: 10.1016/j.cell.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 45.Mattila JT, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol. 2013;191:773–784. doi: 10.4049/jimmunol.1300113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Subbian S, et al. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun Signal. 2013;11:60. doi: 10.1186/1478-811X-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palanisamy GS, et al. Clinical strains of Mycobacterium tuberculosis display a wide range of virulence in guinea pigs. Tuberculosis. 2009;89:203–209. doi: 10.1016/j.tube.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 48.Reed MB, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–87. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 49.Martineau AR, et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. 2007;117:1988–1994. doi: 10.1172/JCI31097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barnes PF, et al. Predictors of short-term prognosis in patients with pulmonary tuberculosis. J Infect Dis. 1988;158:366–371. doi: 10.1093/infdis/158.2.366. [DOI] [PubMed] [Google Scholar]
- 51.Lowe DM, et al. Neutrophilia independently predicts death in tuberculosis. Eur Respir J. 2013;42:1752–1757. doi: 10.1183/09031936.00140913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Condos R, et al. Local immune responses correlate with presentation and outcome in tuberculosis. Am J Respir Crit Care Med. 1998;157(3 Pt 1):729–735. doi: 10.1164/ajrccm.157.3.9705044. [DOI] [PubMed] [Google Scholar]
- 53.Eum SY, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010;137:122–128. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowe DM, et al. Neutrophils in tuberculosis: friend or foe? Trends Immunol. 2012;33:14–25. doi: 10.1016/j.it.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 55.Corleis B, et al. Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell Microbiol. 2012;14:1109–1121. doi: 10.1111/j.1462-5822.2012.01783.x. [DOI] [PubMed] [Google Scholar]
- 56.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 57.Tsuda Y, et al. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity. 2004;21:215–226. doi: 10.1016/j.immuni.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 58.Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012;12:581–591. doi: 10.1038/nri3259. [DOI] [PubMed] [Google Scholar]
- 60.Orme IM. A new unifying theory of the pathogenesis of tuberculosis. Tuberculosis. 2014;94:8–14. doi: 10.1016/j.tube.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ulrichs T, et al. Modified immunohistological staining allows detection of Ziehl-Neelsen-negative Mycobacterium tuberculosis organisms and their precise localization in human tissue. J Pathol. 2005;205:633–640. doi: 10.1002/path.1728. [DOI] [PubMed] [Google Scholar]
- 62.Deb C, et al. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS ONE. 2009;4:e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhatt A, et al. Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc Natl Acad Sci USA. 2007;104:5157–5162. doi: 10.1073/pnas.0608654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan Y, et al. The effect of oxygenated mycolic acid composition on cell wall function and macrophage growth in Mycobacterium tuberculosis. Mol Microbiol. 1998;29:1449–1458. doi: 10.1046/j.1365-2958.1998.01026.x. [DOI] [PubMed] [Google Scholar]
- 65.Seiler P, et al. Cell-wall alterations as an attribute of Mycobacterium tuberculosis in latent infection. J Infect Dis. 2003;188:1326–1331. doi: 10.1086/378563. [DOI] [PubMed] [Google Scholar]
- 66.Ryan GJ, et al. Multiple M. tuberculosis phenotypes in mouse and guinea pig lung tissue revealed by a dual-staining approach. PLoS ONE. 2010;5:e11108. doi: 10.1371/journal.pone.0011108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steingart KR, et al. Fluorescence versus conventional sputum smear microscopy for tuberculosis: a systematic review. Lancet Infect Dis. 2006;6:570–581. doi: 10.1016/S1473-3099(06)70578-3. [DOI] [PubMed] [Google Scholar]
- 68.Ryan GJ, Shapiro HM, Lenaerts AJ. Improving acid-fast fluorescent staining for the detection of mycobacteria using a new nucleic acid staining approach. Tuberculosis. 2014;94:511–518. doi: 10.1016/j.tube.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 69.Munoz-Elias EJ, et al. Replication dynamics of Mycobacterium tuberculosis in chronically infected mice. Infect Immun. 2005;73:546–551. doi: 10.1128/IAI.73.1.546-551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Galagan JE, et al. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature. 2013;499:178–183. doi: 10.1038/nature12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wayne LG, Sohaskey CD. Nonreplicating persistence of mycobacterium tuberculosis. Annu Rev Microbiol. 2001;55:139–163. doi: 10.1146/annurev.micro.55.1.139. [DOI] [PubMed] [Google Scholar]
- 72.Boon C, Dick T. How Mycobacterium tuberculosis goes to sleep: the dormancy survival regulator DosR a decade later. Future Microbiol. 2012;7:513–518. doi: 10.2217/fmb.12.14. [DOI] [PubMed] [Google Scholar]
- 73.Tsai MC, et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol. 2006;8:218–232. doi: 10.1111/j.1462-5822.2005.00612.x. [DOI] [PubMed] [Google Scholar]
- 74.Via LE, et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and non-human primates. Infect Immun. 2008;76:2333–2340. doi: 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aly S, et al. Oxygen status of lung granulomas in Mycobacterium tuberculosis-infected mice. J Pathol. 2006;210:298–305. doi: 10.1002/path.2055. [DOI] [PubMed] [Google Scholar]
- 76.Lenaerts AJ, et al. Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother. 2007;51:3338–3345. doi: 10.1128/AAC.00276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harper J, et al. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis. 2012;205:595–602. doi: 10.1093/infdis/jir786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Voskuil MI, et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. 2003;198:705–713. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996;64:2062–2069. doi: 10.1128/iai.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raleigh JA, et al. Hypoxia and vascular endothelial growth factor expression in human squamous cell carcinomas using pimonidazole as a hypoxia marker. Cancer Res. 1998;58:3765–3768. [PubMed] [Google Scholar]
- 81.Koller F, Leuthardt F. Nekrose und autolyse beitrag zur kenntnis der dystrophischen verkalkung. Klin Wochenschr. 1934;43:1527–1529. [Google Scholar]
- 82.Watanabe S, et al. Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog. 2011;7:e1002287. doi: 10.1371/journal.ppat.1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim MJ, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. 2010;2:258–274. doi: 10.1002/emmm.201000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deb C, et al. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J Biol Chem. 2006;281:3866–3875. doi: 10.1074/jbc.M505556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raju B, et al. Gene expression profiles of bronchoalveolar cells in pulmonary TB. Tuberculosis. 2008;88:39–51. doi: 10.1016/j.tube.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dhouib R, et al. Watching intracellular lipolysis in mycobacteria using time lapse fluorescence microscopy. Biochim Biophys Acta. 2011;1811:234–241. doi: 10.1016/j.bbalip.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 88.Daniel J, et al. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011;7:e1002093. doi: 10.1371/journal.ppat.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garton NJ, et al. Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med. 2008;5:e75. doi: 10.1371/journal.pmed.0050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Caire-Brandli I, et al. Reversible lipid accumulation and associated division arrest of Mycobacterium avium in lipoprotein-induced foamy macrophages may resemble key events during latency and reactivation of tuberculosis. Infect Immun. 2014;82:476–490. doi: 10.1128/IAI.01196-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Simeone R, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 2012;8:e1002507. doi: 10.1371/journal.ppat.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vergne I, et al. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2005;102:4033–4038. doi: 10.1073/pnas.0409716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cappelli G, et al. Profiling of Mycobacterium tuberculosis gene expression during human macrophage infection: upregulation of the alternative sigma factor G, a group of transcriptional regulators, and proteins with unknown function. Res Microbiol. 2006;157:445–455. doi: 10.1016/j.resmic.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 94.Schnappinger D, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tailleux L, et al. Probing host pathogen cross-talk by transcriptional profiling of both Mycobacterium tuberculosis and infected human dendritic cells and macrophages. PLoS ONE. 2008;3:e1403. doi: 10.1371/journal.pone.0001403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Talaat AM, et al. The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc Natl Acad Sci USA. 2004;101:4602–4607. doi: 10.1073/pnas.0306023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bishai W. Lipid lunch for persistent pathogen. Nature. 2000;406:683–685. doi: 10.1038/35021159. [DOI] [PubMed] [Google Scholar]
- 98.McKinney JD, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 99.Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Marrero J, et al. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci USA. 2010;107:9819–9824. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fritz C, et al. Dependence of Mycobacterium bovis BCG on anaerobic nitrate reductase for persistence is tissue specific. Infect Immun. 2002;70:286–291. doi: 10.1128/IAI.70.1.286-291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hutter B, Dick T. Molecular genetic characterisation of whiB3, a mycobacterial homologue of a Streptomyces sporulation factor. Res Microbiol. 1999;150:295–301. doi: 10.1016/s0923-2508(99)80055-2. [DOI] [PubMed] [Google Scholar]
- 103.Weber I, et al. Anaerobic nitrate reductase (narGHJI) activity of Mycobacterium bovis BCG in vitro and its contribution to virulence in immunodeficient mice. Mol Microbiol. 2000;35:1017–1025. doi: 10.1046/j.1365-2958.2000.01794.x. [DOI] [PubMed] [Google Scholar]
- 104.Kaplan G, et al. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect Immun. 2003;71:7099–7108. doi: 10.1128/IAI.71.12.7099-7108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weiss C, Tabachnick J, Cohen HP. Mechanism of softening of tubercles. III. Hydrolysis of protein and nucleic acid during anaerobic autolysis of normal and tuberculous lung tissue in vitro. AMA Arch Pathol. 1954;57:179–193. [PubMed] [Google Scholar]
- 106.Rojas-Espinosa O, et al. The role of cathepsin D in the pathogenesis of tuberculosis. A histochemical study employing unlabeled antibodies and the peroxidase-antiperoxidase complex. Am J Pathol. 1974;74:1–17. [PMC free article] [PubMed] [Google Scholar]
- 107.Yamamura Y, Ogawa Y, Maeda H. Prevention of tuberculous cavity formation by desensitization with tuberculin-active peptide. Am Rev Respir Dis. 1974;109:594–601. doi: 10.1164/arrd.1974.109.6.594. [DOI] [PubMed] [Google Scholar]
- 108.Luna B, et al. In vivo prediction of tuberculosis cavity formation in rabbits. J Infect Dis. 2014;211:481–485. doi: 10.1093/infdis/jiu449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Langermans JA, et al. Divergent effect of bacillus Calmette-Guerin (BCG) vaccination on Mycobacterium tuberculosis infection in highly related macaque species: implications for primate models in tuberculosis vaccine research. Proc Natl Acad Sci USA. 2001;98:11497–11502. doi: 10.1073/pnas.201404898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ulrichs T, Kaufmann SH. New insights into the function of granulomas in human tuberculosis. J Pathol. 2006;208:261–269. doi: 10.1002/path.1906. [DOI] [PubMed] [Google Scholar]
- 111.Leong FJ. Pathology of tuberculosis in the human lung. In: Leong FJ, Dartois V, Dick T, editors. A Color Atlas of Comparative Pathology of Pulmonary Tuberculosis. CRC Press; 2011. pp. 53–81. [Google Scholar]
- 112.Aly S, et al. Interferon-gamma-dependent mechanisms of mycobacteria-induced pulmonary immunopathology: the role of angiostasis and CXCR3-targeted chemokines for granuloma necrosis. J Pathol. 2007;212:295–305. doi: 10.1002/path.2185. [DOI] [PubMed] [Google Scholar]
- 113.Van de Velde S, et al. Contrasting effects of human THP-1 cell differentiation on levofloxacin and moxifloxacin intracellular accumulation and activity against Staphylococcus aureus and Listeria monocytogenes. J Antimicrob Chemother. 2008;62:518–521. doi: 10.1093/jac/dkn232. [DOI] [PubMed] [Google Scholar]
- 114.Rey-Jurado E, et al. Activity and interactions of levofloxacin, linezolid, ethambutol and amikacin in three-drug combinations against Mycobacterium tuberculosis isolates in a human macrophage model. Int J Antimicrob Agents. 2013;42:524–530. doi: 10.1016/j.ijantimicag.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 115.Baik J, et al. Multiscale distribution and bioaccumulation analysis of clofazimine reveals a massive immune system-mediated xenobiotic sequestration response. Antimicrob Agents Chemother. 2013;57:1218–1230. doi: 10.1128/AAC.01731-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Prideaux B, et al. High-sensitivity MALDI-MRM-MS imaging of moxifloxacin distribution in tuberculosis-infected rabbit lungs and granulomatous lesions. Anal Chem. 2011;83:2112–2118. doi: 10.1021/ac1029049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Prideaux B, Stoeckli M. Mass spectrometry imaging for drug distribution studies. J Prot. 2012;75:4999–5013. doi: 10.1016/j.jprot.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 118.Skerry C, et al. Simvastatin increases the in vivo activity of the first-line tuberculosis regimen. J Antimicrob Chemother. 2014;69:2453–2457. doi: 10.1093/jac/dku166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Subbian S, et al. Phosphodiesterase-4 inhibition combined with isoniazid treatment of rabbits with pulmonary tuberculosis reduces macrophage activation and lung pathology. Am J Pathol. 2011;179:289–301. doi: 10.1016/j.ajpath.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Salamon H, et al. Cutting edge: vitamin D regulates lipid metabolism in Mycobacterium tuberculosis infection. J Immunol. 2014;193:30–34. doi: 10.4049/jimmunol.1400736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Corson MA. Darapladib: an emerging therapy for atherosclerosis. Ther Adv Cardiovasc Dis. 2010;4:241–248. doi: 10.1177/1753944710375820. [DOI] [PubMed] [Google Scholar]
- 122.Serruys PW, et al. Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–1182. doi: 10.1161/CIRCULATIONAHA.108.771899. [DOI] [PubMed] [Google Scholar]
- 123.Ridker PM, Luscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J. 2014;35:1782–1791. doi: 10.1093/eurheartj/ehu203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946-1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999;3(10 Suppl 2):S231–S279. [PubMed] [Google Scholar]
- 125.Mitchison DA. The action of antituberculosis drugs in short-course chemotherapy. Tubercle. 1985;66:219–225. doi: 10.1016/0041-3879(85)90040-6. [DOI] [PubMed] [Google Scholar]
- 126.Yano T, et al. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: a pathway for the generation of bactericidal levels of reactive oxygen species. J Biol Chem. 2011;286:10276–10287. doi: 10.1074/jbc.M110.200501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wallis RS, et al. Whole blood bactericidal activity during treatment of pulmonary tuberculosis. J Infect Dis. 2003;187:270–278. doi: 10.1086/346053. [DOI] [PubMed] [Google Scholar]
- 128.Zhu T, et al. Population pharmacokinetic/pharmacodynamic analysis of the bactericidal activities of sutezolid (PNU-100480) and its major metabolite against intracellular mycobacterium tuberculosis in ex vivo whole-blood cultures of patients with pulmonary tuberculosis. Antimicrob Agents Chemother. 2014;58:3306–3311. doi: 10.1128/AAC.01920-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dhillon J, et al. Metronidazole has no antibacterial effect in Cornell model murine tuberculosis. Int J Tuberc Lung Dis. 1998;2:736–742. [PubMed] [Google Scholar]
- 130.Lin PL, et al. Metronidazole prevents reactivation of latent Mycobacterium tuberculosis infection in macaques. Proc Natl Acad Sci USA. 2012;109:14188–14193. doi: 10.1073/pnas.1121497109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hoff DR, et al. Metronidazole lacks antibacterial activity in guinea pigs infected with Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2008;52:4137–4140. doi: 10.1128/AAC.00196-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Carroll MW, et al. Efficacy and safety of metronidazole for pulmonary multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2013;57:3903–3909. doi: 10.1128/AAC.00753-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ramakrishnan L. Looking within the zebrafish to understand the tuberculous granuloma. Adv Exp Med Biol. 2013;783:251–266. doi: 10.1007/978-1-4614-6111-1_13. [DOI] [PubMed] [Google Scholar]
- 134.Swaim LE, et al. Mycobacterium marinum infection of adult zebrafish causes caseating granulomatous tuberculosis and is moderated by adaptive immunity. Infect Immun. 2006;74:6108–6117. doi: 10.1128/IAI.00887-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Basaraba RJ. Experimental tuberculosis: the role of comparative pathology in the discovery of improved tuberculosis treatment strategies. Tuberculosis. 2008;88(Suppl 1):S35–S47. doi: 10.1016/S1472-9792(08)70035-0. [DOI] [PubMed] [Google Scholar]
- 136.Rosenthal IM, et al. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob Agents Chemother. 2012;56:4331–4340. doi: 10.1128/AAC.00912-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Singhal A, et al. BCG induces protection against Mycobacterium tuberculosis infection in the Wistar rat model. PLoS ONE. 2011;6:e28082. doi: 10.1371/journal.pone.0028082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Palanisamy GS, et al. Disseminated disease severity as a measure of virulence of Mycobacterium tuberculosis in the guinea pig model. Tuberculosis. 2008;88:295–306. doi: 10.1016/j.tube.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gonzalez-Juarrero M, et al. Experimental aerosol Mycobacterium bovis model of infection in goats. Tuberculosis. 2013;93:558–564. doi: 10.1016/j.tube.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 140.Sanchez J, et al. Microscopical and immunological features of tuberculoid granulomata and cavitary pulmonary tuberculosis in naturally infected goats. J Comp Pathol. 2011;145:107–117. doi: 10.1016/j.jcpa.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 141.Gil O, et al. Granuloma encapsulation is a key factor for containing tuberculosis infection in minipigs. PLoS ONE. 2010;5:e10030. doi: 10.1371/journal.pone.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kaushal D, et al. The non-human primate model of tuberculosis. J Med Primatol. 2012;41:191–201. doi: 10.1111/j.1600-0684.2012.00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lin PL, et al. Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun. 2006;74:3790–3803. doi: 10.1128/IAI.00064-06. [DOI] [PMC free article] [PubMed] [Google Scholar]