

Abstract
Psychological stress is implicated in the etiology of many common chronic diseases and mental health disorders. Recent research suggests that inflammation may be a key biological mediator linking stress and health. Nevertheless, the neurocognitive pathways underlying stress-related increases in inflammatory activity are largely unknown. The present study thus examined associations between neural and inflammatory responses to an acute laboratory-based social stressor. Healthy female participants (n = 31) were exposed to a brief episode of stress while they underwent an fMRI scan. Blood samples were taken before and after the stressor, and plasma was assayed for markers of inflammatory activity. Exposure to the stressor was associated with significant increases in feelings of social evaluation and rejection, and with increases in levels of inflammation. Analyses linking the neural and inflammatory data revealed that heightened neural activity in the amygdala in response to the stressor was associated with greater increases in inflammation. Functional connectivity analyses indicated that individuals who showed stronger coupling between the amygdala and the dorsomedial prefrontal cortex (DMPFC) also showed a heightened inflammatory response to the stressor. Interestingly, activity in a different set of neural regions was related to increases in feelings of social rejection. These data show that greater amygdala activity in response to a stressor, as well as tighter coupling between the amygdala and the DMPFC, are associated with greater increases in inflammatory activity. Results from this study begin to identify neural mechanisms that might link stress with increased risk for inflammation-related disorders such as cardiovascular disease and depression.
Keywords: Amygdala, Medial prefrontal cortex, Inflammation, IL-6, Stress, Social stress, Social rejection, fMRI, Neuroimaging
1. Introduction
Psychological stress is implicated in the onset and progression of many common and costly chronic diseases, including cardiovascular disease, chronic pain conditions, and major depressive disorder (Cohen et al., 2007; Kendler et al., 1999; Steptoe and Kivimäki, 2012). An emerging body of evidence suggests that inflammation may be a key biological mechanism by which stress affects health (Baker et al., 2012; Miller et al., 2009; Slavich et al., 2010). Indeed, psychological stressors can induce increases in inflammation (Slavich and Irwin, 2014; Kiecolt-Glaser et al., 2003; Rohleder, 2014; Steptoe et al., 2007), which might contribute to the development of disease (Capuron and Miller, 2004; Choy and Panayi, 2001; DellaGiola and Hannestad, 2010; Raison and Miller, 2013; The Emerging Risk Factors Collaboration, 2010). Despite this growing literature linking stress, inflammation, and poor health, little is known about the neurocognitive mechanisms that underlie stress-induced changes in inflammatory activity.
Given our limited knowledge of the neural mechanisms linking stress and inflammation, the aim of the present study was to examine neural and inflammatory responses to a social stressor. We hypothesized that greater activity in neural regions known to activate during threatening experiences, including the amygdala, the dorsal anterior cingulate cortex (dACC), and the periaqueductal gray (PAG), would be associated with increases in inflammation. This hypothesis was based in part on animal research suggesting a critical role for these threat-related brain regions in translating stress into inflammatory-related conditions. For example, lesions to the amygdala or the anterior cingulate prevent stress from exacerbating inflammatory-induced gastric pathology, while electrical stimulation of these regions leads to heightened inflammatory-related symptoms (Henke, 1982). Furthermore, human neuroimaging research suggests that these regions are often activated during tasks that involve processing social threats (e.g., threatening facial expressions, social rejection; (Eisenberger, 2012; Kross et al., 2011; Whalen et al., 2001), and social stressors are among the most potent psychological activators of inflammatory responses (Dickerson et al., 2009; Murphy et al., 2013; Sheridan et al., 2000). Finally, the dACC and amygdala have dense anatomical projections to regions that play a role in inflammatory responding (e.g., hypothalamus, brainstem; Eisenberger and Cole, 2012; Irwin and Cole, 2011), thus providing further evidence that they may play a role in stress-induced inflammation.
In addition to testing whether activation of threat-related neural regions may be related to inflammatory responses to stress, we also examined if functional connectivity between these regions and pre-frontal cortical structures may be related to stress-induced changes in inflammation. In animal work, stimulation of a region analogous to the human dorsomedial prefrontal cortex (DMPFC) has been shown to amplify transient amygdala responses to threat (Burgos-Robles et al., 2009), providing evidence of an “aversive amplification circuit” involving DMPFC–amygdala coupling (Robinson et al., 2012). Furthermore, a growing body of human research suggests that there is increased functional connectivity between DMPFC and threat-related limbic structures during negative emotional states and among individuals with mood disorders (Etkin et al., 2011). This raises the intriguing possibility that greater functional connectivity between the DMPFC and amygdala during stress may also be related to heightened inflammatory responses.
To investigate the relationships between threat-related neural activity, as well as functional connectivity, and inflammatory responses to stress, healthy young women (N = 31) were scanned using fMRI while they were exposed to an acute episode of social stress. Blood samples taken before and after the stressor were assayed for levels of the inflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α). Both of these inflammatory cytokines are activated in response to stress (Rohleder, 2014; Steptoe et al., 2007), and are associated with chronic disease and depression (Choy and Panayi, 2001; Howren et al., 2009). We hypothesized that greater activity in neural regions often associated with processing threat (i.e., the amygdala) would be associated with greater inflammatory responses to the stressor. We also explored the possibility that stronger functional connectivity between threat-related neural regions (i.e., the amygdala) and cortical regions implicated in sustaining threat responses (i.e., DMPFC) would be associated with heightened inflammatory activity. We focused this investigation on women, given that females are at heightened risk for developing inflammatory-related diseases (e.g., depression, rheumatoid arthritis; Nolen-Hoeksema, 2001; Tengstrand et al., 2004), are more sensitive to the negative effects of social stress (Stroud et al., 2011, 2002), and may be more likely to show an exaggerated inflammatory response to a stressor (Prather et al., 2009; Rohleder et al., 2001; Steptoe et al., 2002).
2. Methods and materials
2.1. Participants
Participants were 31 healthy young-adult females (M age = 19 years; Range = 18–22 years). The sample self-identified as 32% Asian/Asian American, 23% Hispanic/Latina, 22% Mixed/Other, 13% African American, and 10% White. All participants provided written informed consent, and procedures were approved by the UCLA Institutional Review Board. Participants were paid $135 for participating.
2.2. Procedure
Interested participants responded to an advertisement for a study on “how the brain and body respond to first impressions.” Prospective participants were screened via telephone, and excluded from further participation if they endorsed any of the following exclusionary criteria: acute cold or flu symptoms during the fMRI session, current or prior chronic physical illness, current or lifetime history of Axis-I psychiatric disorder, allergies or autoimmune diseases; major sleep disturbance in the past six weeks; tobacco use; current prescription medication use, including hormonal birth control; excessive caffeine use (i.e., >8 caffeinated beverages per day), Body Mass Index over 30, left-handed, claustrophobic, or metal in the body.
Participants who met all inclusionary criteria were then invited to the lab where we confirmed their psychiatric status using the Structured Clinical Interview for DSM-IV Axis I Disorders (First et al., 1995). Next, participants completed a video recorded “impressions interview” that lasted approximately ten minutes, in which they responded to questions such as “What would you most like to change about yourself?” and “What are you most proud of?” Participants were told that in the next session for the study, they would meet another participant, and the experimenters would choose one person to form an impression of the other based on the video of the interview. Meanwhile, the other person would be scanned while they saw the impression being formed of them.
The fMRI session occurred within 2 days of the interview session. Upon arrival at the scanner, participants met a female confederate, whom they believed was also participating in the study. After a brief introduction, participant and confederate were taken to separate testing rooms where a nurse inserted an indwelling catheter into the participant’s left (non-dominant) forearm, through which blood samples were taken. Following at least 45 min of acclimation time, a first baseline blood sample was taken (approximately 55 min before the stressor).
Following the blood collection, participant and confederate were reunited and told that the experimenters had decided that the confederate was going to watch the participant’s video and form an impression of her, while the participant would undergo the fMRI scan and view the confederate’s impressions. After being familiarized with the impression formation task (see below), a second baseline blood sample was drawn (approximately 35 min prior to the stressor). Next, the confederate was seated in front of a computer screen in the scanner control room, while the participant was set up in the scanner. Following structural scans, the confederate supposedly evaluated the participant’s interview, and the participant received feedback about how she was supposedly coming across. Participants also viewed the confederate’s feedback about a nature video (not included in the present study). After the scan, the participant returned to the testing room to complete questionnaires; additional blood samples were collected 30, 60, and 90 min after the termination of the stressor. After the final sample, participants were probed regarding any suspicion about the cover story, and were fully debriefed. No participants indicated that they thought the feedback was fake or that the confederate was a member of our research team.
2.3. fMRI social stress task
We induced social stress using procedures similar to those in a prior study (Eisenberger et al., 2011). Briefly, during the scan, participants viewed a video of a mouse cursor moving around a screen that displayed 24 “adjective buttons”, which they believed was a live interface of the confederate’s impressions of their interview. Feedback adjectives were divided into one-third positive (e.g., “intelligent”), one-third neutral (e.g., “practical”), and one-third negative words (e.g., “annoying”). The cursor selected (by depressing) a new adjective button every 11–12 s (see Fig. 1). Over the course of the scan, participants received positive, neutral, and negative feedback (15 “trials” of each valence), and every time an adjective was selected, participants responded to the question “How do you feel?” using a button box with 4 buttons (1 = really bad, 4 = really good; reverse-coded for analyses). The feedback task was preceded and followed by a fixation crosshair (10 s each), which formed the implicit baseline.
Fig. 1.
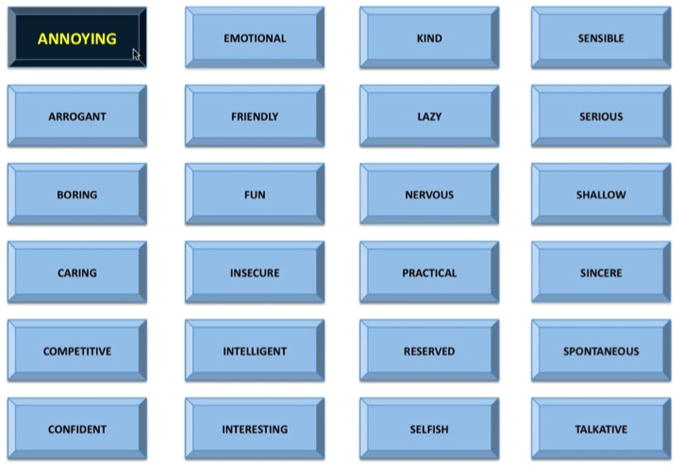
Social stress task used in the fMRI scanner. Participants viewed a grid of adjective “buttons”, and every 11–12 s, one of the “buttons” was depressed by a mouse cursor that was supposedly controlled by an evaluator (actually a pre-made video). Pictured is an example of a negative word (i.e., annoying) being selected.
2.4. Self-reports of social evaluation and social rejection
Participants were asked five questions before and after the scan, which served as measures of social evaluation and social rejection. Participants indicated the extent to which they felt “evaluated” and “judged” by the confederate, on a seven-point scale (1 = not at all, 7 = very much), which were combined to form a measure of feelings of evaluation (α = .84). Participants also indicated the extent to which they agreed with the following statements (1 = not at all, 7 = very much): “I feel like the other participant likes me; I feel like the participant has a positive impression of my interview; I feel the other participant accepts me.” Responses to these three items were reverse coded (i.e., so higher number indicate greater feelings of rejection) and combined to form a measure of social rejection perception (α = .88). Changes in self-reported feelings of social evaluation and rejection were marginally significantly correlated (r = .317, p = .082).
2.5. Inflammatory responses
Inflammatory responses were assessed at two baseline (BL) time points prior to the stressor and three time points after the stressor. Blood was drawn into EDTA Vacutainer tubes, held on ice until the completion of all blood draws, then centrifuged for collection of plasma and frozen at −80 °C until assays were performed. Concentrations of IL-6 and TNF-α were measured in duplicate using high sensitivity enzyme-linked immunosorbent assays (ELISAs; R&D Systems, Minneapolis, MN) according to the manufacturer’s protocols; all samples from a single participant were assayed on the same plate. The lower limit of detection for these assays is 0.2 pg/mL and 0.5 pg/mL for IL-6 and TNF-α, respectively.1 Within- and between-assay coefficients of variation were <9% for both IL-6 and TNF-α ELISAs. All cytokine data were positively skewed, so raw values were log transformed to normalize the distribution prior to statistical testing. Though log transformed values were used in statistical analyses, raw mean and median values are reported in text for ease of interpretation. Analyses linking the neural and inflammatory data focus exclusively on cytokines that showed a significant change in response to the task.
2.6. fMRI image acquisition
Imaging data were acquired using a Siemens Trio 3.0 Tesla MRI scanner at the UCLA Staglin Center for Cognitive Neuroscience. First, we acquired a T1-weighted MPRAGE anatomical image for functional image registration and normalization (slice thickness = 1 mm, 176 slices, TR = 2300 ms, TE = 2.98 ms, flip angle = 9 degrees, matrix = 256 × 256, FOV = 256 mm). Then, we acquired 288 functional T2-weighted EPI volumes, during the stress task (slice thickness = 3 mm, gap = 1 mm, TR = 2000 ms, TE = 25 ms, flip angle = 90 degrees, matrix = 64 × 64, FOV = 200 mm.
2.7. Data analysis
Neuroimaging data were pre-processed and analyzed using Statistical Parametric Mapping (SPM8; Wellcome Department of Cognitive Neurology, London, UK). Pre-processing included image realignment to correct for head motion, normalization into Montreal Neurologic Institute space (resampled at 3 × 3 × 3 mm), and spatial smoothing using an 8 mm Gaussian kernel, full width at half maximum, to increase signal-to-noise ratio. All imaging coordinates are reported in Montreal Neurological Institute (MNI) format.
Following pre-processing, a general linear model was constructed for each participant. The selection of each feedback word (lasting 3 s) and the subsequent 8–9 s (until the next word was selected) were modeled as a block, and were convolved with a canonical hemodynamic response function. Our regressor-of-interest coded for the type of feedback presented (positive, neutral, negative), and we included the six motion parameters as covariates. For each model, the time series was high-pass filtered using a 128 Hz function, and serial autocorrelation was modeled as an AR(1) process. For the current study, we focused on neural activity during the negative feedback trials compared to the neutral feedback trials. Following estimation, we computed linear contrasts for each participant that compared BOLD signal during the negative feedback trials to BOLD signal during neutral feedback. Contrast images for each participant were then entered into random effect analyses at the group level for statistical inference.
Functional connectivity analyses were conducted using a generalized psychophysiological interaction analysis (gPPI; McLaren et al., 2012), with left and right amygdala anatomical regions-of-interest (ROIs) as seeds. At the individual subject level, we extracted a deconvolved time course averaged across voxels in each amygdala ROI. This time course was then included in a generalized PPI model, together with a PPI regressor for each of the variables of interest (i.e., negative feedback, neutral feedback), as well as motion parameters. The resulting PPI connectivity estimates were then taken to the group level, where we conducted an independent samples t-test, in which we grouped participants into “high responder” and “low responder” groups (based on a median split of inflammatory responses), and examined differences in PPI connectivity between the groups. This analysis allowed us to determine which neural regions were correlated with the time course of activity in the amygdala, during negative feedback > neutral feedback, for those who showed a higher inflammatory responses to the social stressor compared to those who showed a smaller change in inflammation.
A statistical threshold of p < .005, 40 voxels, which corresponds to a false-discovery rate of .05, as determined by Monte Carlo simulations conducted in the AFNI program 3dClustSim (parameters: individual voxel p-value = 0.005; 10,000 simulations; FWHM 8 mm in each direction x, y, and z; whole-brain mask including 44,428 resampled voxels), was used for all main-effect analyses and regressions with self-report data. For analyses involving the inflammatory data, we used a more liberal threshold of p < .005, 10 voxels. Given that this is the first study to examine neural and inflammatory responses to a social stressor, and the difficulty of comparing neural activity at one point in time with peripheral biological responses collected hours later, using a more liberal threshold allowed us to increase sensitivity for detecting any relations between neural activity and inflammatory responses.
3. Results
3.1. Manipulation check
To ensure that participants felt worse in response to receiving negative feedback compared to positive or neutral feedback, we examined their ratings of how they felt after each adjective was selected. As expected, there was a significant effect of feedback valence on these ratings, F(2,60) = 240.42, p < .001, such that participants felt significantly worse in response to receiving negative feedback (M = 3.27, SD = .51) compared to neutral feedback (M = 1.97, SD = .37, t(30) = 15.15, p < .001) or positive feedback (M = 1.40, SD = .41, t(30) = 16.95, p < .001). Participants also felt worse after receiving neutral feedback, compared to positive feedback, t(30) = 9.71, p < .001.
3.2. Psychological responses to the stressor
Next, we examined if the stressor led to changes in feelings of evaluation and perceptions of social rejection. Participants reported feeling significantly more evaluated following the stressor (pre-stress M = 2.87, SD = 1.85; post-stress M = 4.97, SD = 1.41 t(30) = −8.14, p<.001). Participants also reported feeling significantly more socially rejected following the stressor (pre-stress M = 2.62, SD = .80; post-stress M = 3.52, SD = .95 t(30) = −4.07, p < .001. Together, these data suggest that the “impressions task” was successful in creating an experience of social stress that increased feelings of evaluation and rejection.
3.3. Inflammatory responses to the stressor
Next, we examined if the stressor was associated with increases in levels of pro-inflammatory cytokines. We found a significant increase in IL-6 over time, F(4,116) = 48.55, p < .001, but no significant change in TNF-α, F(4,120) = 0.70, p = .59. Follow-up pairwise-comparisons of the IL-6 data indicated no difference between the two baseline measures (M BL1 = 1.23 pg/mL, SD = 1.01; M BL2 = 1.22 pg/mL, SD = .96; p = .87); thus, these two measures were combined to form an average baseline used in the remainder of the analyses. Additional pairwise-comparisons revealed significant increases in IL-6 for each post-stress time point compared to the average baseline (T30: M=1.80pg/mL, SD = 1.23; t(30) = −6.17, p<.001; T60: M = 2.92pg/mL, SD = 1.56; t(29) =−6.27, p<.001; T90: M = 3.64pg/mL, SD = 3.10; t(30) =−8.25, p<.001). For the remainder of our analyses, we focus on the change in IL-6 from the 90 min time point compared to the combined baseline, as IL-6 levels were at their highest at this time point.2
We also explored if changes in IL-6 in response to the stress task were correlated with the psychological responses reported above. There were no significant correlations between IL-6 responses and in-scanner ratings of the negative feedback or the neutral feedback, changes in feelings of evaluation, or perceptions of social rejection (all ps > .77).
3.4. Neural responses to the stressor
Turning to the fMRI data, we first examined the neural regions that were active when participants received negative, compared to neutral, feedback (regardless of inflammatory response). Results from this contrast revealed significant clusters of activation in DMPFC and MPFC (extending into pregenual anterior cingulate cortex [pACC] and dACC), bilateral ventrolateral prefrontal cortex (VLPFC), bilateral temporal parietal junction (TPJ), bilateral posterior superior temporal sulcus (pSTS), bilateral temporal poles, occipital lobe, and cerebellum (for a full list of activations, see Supplementary Table 1 and Supplementary Fig. 1). Thus, when receiving negative feedback (compared to neutral feedback), participants showed greater activity in regions commonly activated during tasks that involve (a) thinking about other people (DMPFC, MPFC/pACC, TPJ, pSTS, temporal poles), (b) processing threat or distress (dACC, AI), and (c) regulating emotion (VLPFC).
3.5. Linking fMRI and psychological responses to stress
To explore neural activity during the stressor that was associated with changes in perceptions of evaluation and feelings of rejection, we regressed participants’ change in these measures into the contrast of negative feedback > neutral feedback. There were no significant correlations between neural activity during negative (vs. neutral) feedback and change in feelings of evaluation. However, greater increases in feelings of social rejection in response to the stressor were associated with heightened activity in the medial prefrontal cortex (MPFC), the bilateral hippocampus, and the posterior cingulate cortex (PCC) in response to negative feedback (for a complete list of regions, see Supplementary Table 2 and Supplementary Fig. 2).
3.6. Linking fMRI and inflammatory responses to stress
The main goal of the present study was to examine the neural regions that were associated with inflammatory responses to stress. To accomplish this goal, we regressed participants’ change in IL-6 from baseline to T90 into the contrast of negative feedback > neutral feedback. Results of this whole-brain regression analysis revealed significant, positive correlations between activity in the left amygdala (−21, −4, 11) and IL-6 responses (see Fig. 2 and Table 1). Thus, participants who showed greater activity in a key threat-related neural region during negative evaluations also showed greater IL-6 responses to the stressor. Activity in the subgenual anterior cingulate cortex (−3,14, −8), and the left middle temporal gyrus (−48, −73, 25) was also correlated with IL-6 responses. No neural activity was negatively correlated with IL-6 responses.
Fig. 2.
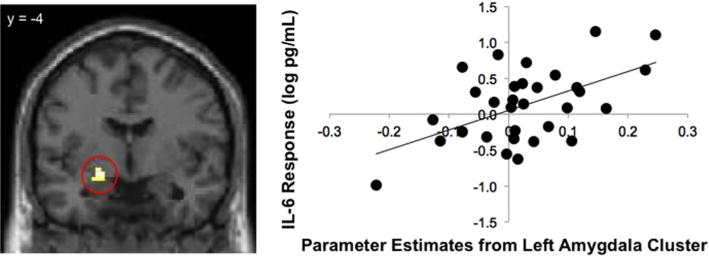
. Relations between neural activity in the left amygdala (from the contrast of negative > neutral social feedback) and inflammatory responses to the social stressor as measured by log-transformed IL-6 increases from baseline to T90 (in pg/mL). The left side depicts the cluster within left amygdala that was positively correlated with IL-6 responses from a whole-brain regression analysis, and the right side shows a scatter plot of parameter estimates from the left amygdala cluster and IL-6 responses.
Table 1.
Regions that were significantly positively correlated with IL-6 response (T90-baseline) during the contrast of negative social feedback > neutral social feedback.
| Anatomical region | BA | x | y | z | t | r | k |
|---|---|---|---|---|---|---|---|
| Amygdala | n/a | −21 | −4 | −11 | 3.46 | .54 | 10 |
| subACC | 25 | −3 | 14 | −8 | 3.36 | .53 | 10 |
| Middle temporal gyrus | 39 | −48 | −73 | 25 | 3.61 | .56 | 16 |
Notes. All activations thresholded at p < .005, 10 voxels. BA refers to putative Brodmann’s Area; x, y, and z refer to MNI coordinates in the left–right, anterior– posterior, and inferior–superior dimensions, respectively; t refers to the t score at those coordinates (local maxima); r refers to the Pearson correlation coefficient relating neural activity in that cluster and IL-6 response; k refers to the number of voxels in each cluster.
3.7. Functional connectivity
In our last set of analyses, we explored the functional connectivity of the amygdala with other brain regions during negative feedback to examine if individuals who showed greater functional coupling of the amygdala and DMPFC would show a higher IL-6 response to the stressor. A PPI analysis revealed that there was greater functional connectivity between the left amygdala and the DMPFC/temporal poles (but only when the cluster extent threshold was lowered to 9 voxels) for high IL-6 responders (i.e., those above the median IL-6 change of 1.4 pg/mL) compared to low IL-6 responders (i.e., those below the median IL-6 change). There was also stronger functional coupling between the right amygdala and the DMPFC for high IL-6 responders (compared to low responders; see Fig. 3 and Table 2 for a complete list of regions).
Fig. 3.
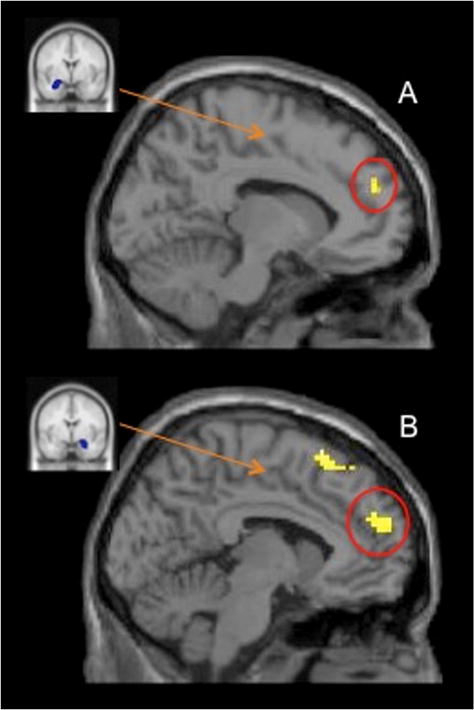
Panel A depicts the anatomical ROI of the left amygdala that was used as a seed region in the PPI analysis (left), and the region in DMPFC that was more strongly correlated with left amygdala activity for high IL-6 responders (compared to low IL-6 responders) during negative (vs. neutral) feedback (right). Panel B depicts the anatomical ROI of the right amygdala that was used as a seed region in the PPI analysis (left), and the region in DMPFC that was more strongly correlated with right amygdala activity for high IL-6 responders (compared to low IL-6 responders) during negative (vs. neutral) feedback (right).
Table 2.
Regions that were positively correlated with the amygdala seeds in the psychophysiological interaction analysis.
| Anatomical region | BA | x | y | z | t | k |
|---|---|---|---|---|---|---|
| Left amygdala seed | ||||||
| High IL-6 responders > low IL-6 responders | ||||||
| DMPFC | 9 | −9 | 50 | 25 | 3.62 | 9 |
| Temporal pole | 38 | 36 | 14 | −20 | 5.13 | 106 |
| 38 | −36 | 17 | −23 | 3.87 | 23 | |
| IFG | 45 | −54 | 20 | 16 | 3.69 | 14 |
| Low IL-6 responders > high IL-6 responders | ||||||
| VLPFC | 11 | −21 | 41 | −17 | 4.75 | 11 |
| Hippocampus | n/a | 18 | −22 | −11 | 3.45 | 10 |
| Occipital | −30 | −82 | −8 | 4.04 | 26 | |
| −33 | −73 | 22 | 3.41 | 12 | ||
| Right amygdala seed | ||||||
| High IL-6 responders > low IL-6 responders | ||||||
| DMPFC | 9 | −15 | 50 | 31 | 4.03 | 79 |
| 8/9 | 9 | 44 | 46 | 3.54 | 57 | |
| SMA | 6 | −6 | 20 | 64 | 3.47 | 28 |
| IFG | 47 | −33 | 26 | 14 | 3.40 | 14 |
| 47/11 | −36 | 35 | −5 | 3.69 | 61 | |
| 45 | −54 | 20 | 16 | 4.00 | 57 | |
| n/a | 45 | 26 | −11 | 3.43 | 16 | |
| Superior temporal gyrus | 38 | −51 | −1 | −11 | 3.92 | 12 |
| Middle temporal gyrus | n/a | −45 | −25 | −8 | 3.67 | 10 |
| 39 | −51 | −76 | 22 | 4.37 | 30 | |
| Low IL-6 responders > High IL-6 responders | ||||||
| No sig. activity |
Notes. All activations thresholded at p < .005, 10 voxels. BA refers to putative Broadmann’s Area; x, y, and z refer to MNI coordinates in the left–right, anterior–posterior, and inferior–superior dimensions, respectively; t refers to the t score at those coordinates (local maxima); k refers to the number of voxels in each cluster. The following abbreviations are used for specific brain regions: DMPFC = dorsomedial prefrontal cortex; IFG = inferior frontal gyrus; VLPFC = ventrolateral prefrontal cortex; SMA = supplementary motor area.
4. Discussion
Inflammation is hypothesized to be a key biological mediator of the relationship between psychological stress and the onset and course of chronic disease and psychiatric illness. However, the neurocognitive systems engaged during stress that lead to increases in inflammation are largely unknown. To address this issue, the present study investigated how neural activity during a social stressor is linked with stressor-evoked inflammatory activity. Results demonstrated that greater neural activity in the left amygdala in response to negative social feedback was related to greater stressor-evoked changes in IL-6. In addition, functional connectivity analyses revealed that individuals who showed more tightly coupled activity of the amygdala and the DMPFC during negative feedback showed heightened inflammatory responses to the stressor. Taken together, these results suggest that greater activity in a key threat-related neural region (i.e., amygdala), and stronger coupling between brain regions involved in sustaining or amplifying threat responses (i.e., DMPFC and amygdala), are associated with heightened inflammatory responses to stress.
Results from the present study are the first to show that amygdala activity is associated with greater inflammatory responses to a stressor. These findings diverge from those of one prior investigation, which showed that the activation of different regions of the “threat network” (i.e., dACC, anterior insula) were associated with increases in inflammation (Slavich et al., 2010). However, a number of methodological differences between the present study and the prior investigation may account for these distinct relationships, including the use of different stress tasks during fMRI scanning (social evaluation vs. social rejection), different ways of measuring inflammation (plasma vs. oral fluids), and different experimental designs (one session vs. multiple sessions with different tasks). Taken together, these two studies demonstrate an important role for threat-related neural regions in linking stress and inflammation (Muscatell and Eisenberger, 2012), though the particular brain regions within this network that are related to inflammatory activity in any given study may vary as a function of a variety of factors.
The current findings linking amygdala activity and inflammation complement and extend prior research that has linked amygdala responses with other measures of stress-related physiological activation (Muscatell and Eisenberger, 2012). Further demonstrating the relevance of this brain region for physical and mental health, amygdala hyperactivity has also been linked with preclinical atherosclerosis (Gianaros et al., 2009) and major depression (Drevets, 2000). Results from the present study converge with prior research to suggest a possible mechanism whereby amygdala activity during stress is associated with heighted inflammation, which, over time, may lead to compromised physical and mental health. It is also interesting to note that, in the present study, we did not observe significant amygdala activity during negative feedback for the entire sample; rather, only those individuals who showed increased amygdala activity during the stressor also showed greater inflammatory responses. This pattern of activity highlights the possibility that heightened amygdala activity in response to a stressor may be an important “risk factor” for inflammatory-related diseases.
In terms of how amygdala activity increases inflammation, one possibility is that the amygdala may activate the sympathetic nervous system, which can then drive increases in inflammation. In support of this possibility, research has shown that the amygdala has strong, efferent projections to brainstem regions such as the locus coeruleus and pons that are known to play a role in the generation of sympathetic responses to threat and stress (LeDoux et al., 1988). Furthermore, sympathetic activation has been shown to lead to increases in inflammation (Bierhaus et al., 2003; DeRijk et al., 1994; Kop et al., 2008; van Gool et al., 1990), while pharmacologically blocking sympathetic activation attenuates the inflammatory response to stress (Bierhaus et al., 2003). Thus, amygdala activity during stress may lead to a cascade of physiological responses, starting with sympathetic activation and ultimately resulting in greater inflammation. Future research should investigate this issue by simultaneously measuring neural, sympathetic, and inflammatory responses to a stressor, and testing if sympathetic activation mediates the relation between amygdala activity and inflammatory responses.
Results from connectivity analyses demonstrating tighter coupling between the amygdala and the DMPFC for high IL-6 responders are consistent with data from animal and human studies of fear and anxiety, which suggest that the DMPFC may provide top-down influence on the amygdala to create an “aversive amplification” circuit during conditions of threat or stress (Robinson et al., 2012). For example, electrical stimulation of the prelimbic cortex, the rodent analog of human DMPFC/dACC (Milad et al., 2009, 2007), has been shown to increase activity in the amygdala and subsequent behavioral indicators of fear (Sierra-Mercado et al., 2011; Vidal-Gonzalez et al., 2006). Other work from animals suggests that prelimbic cortex is responsible for sustaining transient amygdala responses to threat (Burgos-Robles et al., 2009), providing further evidence that it is the co-activation of DMPFC and amygdala during threat that drives increases in behavioral and physiological stress responses. Finally, accumulating evidence in human neuroimaging studies suggests that DMPFC–amygdala connectivity is observed in many negative emotional states (Etkin et al., 2011), and that co-activation of these regions during threat is associated with increases in anxiety, negative affect and greater attention to threatening cues (Robinson et al., 2012; Ochsner and Gross, 2005). Results from the current study suggest, for the first time, that functional connectivity between the amygdala and the DMPFC may also drive inflammatory responses to stress, providing additional evidence for the importance of these regions in orchestrating physiological responses to stress.
In addition to exploring the neural systems associated with inflammatory responses, the current study examined how neural activity during negative social feedback is linked with stressinduced changes in feelings of social evaluation and rejection. Interestingly, we found that activity in a different set of neural regions from those related to inflammation – namely, the MPFC, PCC and hippocampus, were associated with changes in self-reported feelings of social rejection following the stressor. These results suggest the possibility that different neural systems may underlie psychological and physiological responses to a stressor, with activity in basic threat-related neural regions linked with physiological changes, and neural systems involved in thinking about the self (i.e., MPFC, PCC, hippocampus) relating to psychological changes. An interesting avenue for future research will be to explore if these different neural systems are related to the development of distinct patterns of psychiatric symptoms, with the “psychological circuit” more strongly associated with the cognitive and affective symptoms of depression, and the “physiological circuit” more closely associated with the somatic or vegetative symptoms of depression (Inagaki et al., 2013).
The present study represents an important step in elucidating the neurocognitive systems that are related to inflammatory responses to stress (Slavich et al., 2010). However, the study is not without limitations. First, all participants were healthy, young adult females, which limits the generalizability of the findings. However, given that women are at heightened risk for developing some inflammatory-related diseases (e.g., depression, rheumatoid arthritis; (Nolen-Hoeksema, 2001; Tengstrand et al., 2004) and are more sensitive to the negative effects of social stress (Stroud et al., 2011, 2002), the present findings are relevant for a number of critical public health issues. Second, we did not include a nonstress control group, and we thus cannot be certain that the observed increases in IL-6 are due solely to exposure to the stressor, nor can we conduct a formal test of mediation to examine the neural mechanisms linking stress and inflammation. Along similar lines, all participants in the present study were exposed to negative, neutral, and positive social feedback; thus, we could not determine whether one type of feedback was most strongly associated with increases in inflammation. Finally, a fairly lenient statistical threshold was used for analyses examining correlations between neural activity and inflammatory responses; replication with larger samples will be necessary to determine the strength of the effects observed in the current investigation.
In conclusion, the present study provides novel evidence for the role of the amygdala in inflammatory responses to stress. Across correlational and connectivity analyses, results demonstrated that activity in the amygdala and coupling between the amygdala and the DMPFC during negative social feedback was related to increases in levels of IL-6. These findings represent an exciting first step in understanding the neurocognitive processes that are engaged during stress, and that may translate features of the external social environment into immunological changes that affect health.
Supplementary Material
Acknowledgments
This work was supported by a National Alliance for Research on Schizophrenia and Depression (NARSAD) Young Investigator Award (NIE), a UCLA Cousins Center for Psychoneuroimmunology Seed Grant (NIE), a UCLA Clinical & Translational Science Institute (CTSI) Seed Grant (NIE), the NIH/National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant Number UL1TR000124, the UCLA Older Americans Independence Center Inflammatory Biology Core (funded by NIA/NIH Grant Number AG028748), a Canadian Institutes of Health Research Post-Doctoral Research Fellowship (KD), a National Science Foundation Graduate Research Fellowship (KAM), and NIH Pre-Doctoral Institutional Training Grant T32 MH015750 (KAM).
The authors wish to thank the Staglin IMHRO Center for Cognitive Neuroscience at UCLA, the UCLA Clinical and Translational Research Center, and the Cousins Center for Psychoneuroimmunology Inflammatory Biology Core Laboratory for supporting this research. We also acknowledge the significant contributions of Brittany Branscomb, Stephanie Chan, Christie Fung, Joyce Gorman, Ashley Guzman, Adrienne Healey, Evelyn Ho, Patil Kodchian, Becky Phan, and Shizue Reid in their roles as confederates and research assistants; Sten Witzel for serving as CTRC protocol manager; Christian Perez and Tuff Witarama for performing inflammatory assays; Bob Spunt and Jared Torre for help with fMRI data analysis, and members of the UCLA Social & Affective Neuroscience Laboratory for comments on previous drafts.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bbi.2014.06.201.
Footnotes
Two TNF-α samples (one at the BL1 time point, one at the 60 min time point) were below the detectable limit of the assay, and were thus set to half of the lower limit of detection (.25), a commonly-used approach for dealing with such values (LaFleur et al., 2011). Furthermore, we were unable to obtain a blood sample for one participant at the 60-min time point due to difficulty with the indwelling catheter.
To ensure that the observed increases in IL-6 were not simply due to anxiety associated with being in the neuroimaging environment, we ran a separate sample of 10 participants (5 females) through the identical experimental procedure outside the MRI scanner (though we did not include a 90 min post-stress sample, as in the present study). Results from this pilot study also showed a significant increase in IL-6 (from baseline to 60 min post-evaluation) in response to the social evaluation F(3,27) = 9.70, p < .001, which was of similar magnitude to the increase we observed in the present study (partial eta squared for pilot study = .52; partial-eta squared for current study, only including time-points that match pilot study = .54). These data suggest that the increases in IL-6 observed in the present study were not simply due to being in the neuroimaging environment.
Conflict of interest
The authors report no biomedical financial interests or potential conflicts of interest.
References
- Baker DG, Nievergelt CM, O’Connor DT. Biomarkers of PTSD: neuropeptides and immune signaling. Neuropharmocology. 2012;62:663–673. doi: 10.1016/j.neuropharm.2011.02.027. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, Ferstl R, von Eynatten M, Wendt T, Rudofsky G, Joswig M, Morcos M, Schwaninger M, McEwen B, Kirschbaum C, Nawroth PP. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci USA. 2003;100:1920–1925. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ. Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J Neurosci. 2009;29:8474–8482. doi: 10.1523/JNEUROSCI.0378-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-a. Biol Psychiatry. 2004;56:819–824. doi: 10.1016/j.biopsych.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Choy EHS, Panayi GS. Mechanisms of disease: cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. JAMA. 2007;298:1685–1687. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- DellaGiola N, Hannestad J. A critical review of human endotoxin administration as an experimental paradigm of depression. Neurosci Biobehav Rev. 2010;34:130–143. doi: 10.1016/j.neubiorev.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRijk RH, Boelen A, Tilders FJH, Berkenbosch F. Induction of plasma interleukin-6 by circulating adrenaline in the rat. Psychoneuroendocrinology. 1994;19:155–163. doi: 10.1016/0306-4530(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Dickerson SS, Gable SL, Iriwn MR, Aziz N, Kemeny ME. Social-evaluative threat and pro-inflammatory cytokine regulation: an experimental laboratory investigation. Psychol Sci. 2009;20:1237–1244. doi: 10.1111/j.1467-9280.2009.02437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC. Functional anatomical abnormalities in limbic and pre-frontal cortical structures in major depression. Prog Brain Res. 2000;126:413–431. doi: 10.1016/S0079-6123(00)26027-5. [DOI] [PubMed] [Google Scholar]
- Eisenberger NI. The pain of social disconnection: examining the shared neural underpinnings of physical and social pain. Nat Rev Neurosci. 2012;13:421–434. doi: 10.1038/nrn3231. [DOI] [PubMed] [Google Scholar]
- Eisenberger NI, Cole SW. Social neuroscience and health: neurophysiological mechanisms linking social ties with physical health. Nat Neurosci. 2012;15:669–674. doi: 10.1038/nn.3086. [DOI] [PubMed] [Google Scholar]
- Eisenberger NI, Inagaki TK, Muscatell KA, Haltom KEB, Leary MR. The neural sociometer: brain mechanisms underlying state self-esteem. J Cogn Neurosci. 2011;23:3448–3455. doi: 10.1162/jocn_a_00027. [DOI] [PubMed] [Google Scholar]
- Etkin A, Egner T, Kalisch R. Emotional processing in anterior cingulate cortex and medial prefrontal cortex. Trends Cogn Sci. 2011;15:85–93. doi: 10.1016/j.tics.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First MB, Gibbon M, Spitzer RL, Williams JBW. User’s Guide for the Structured Clinical Interview for DSM-IV Axis I disorders (SCID-I, Version 2.0, Final Version) New York State Psychiatric Institute; New York (NY): 1995. [Google Scholar]
- Gianaros PJ, Hariri AR, Sheu LK, Muldoon MF, Sutton-Tyrrell K, Manuck SB. Preclinical atherosclerosis covaries with individual differences in reactivity and functional connectivity of the amygdala. Biol Psychiatry. 2009;65:943–950. doi: 10.1016/j.biopsych.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke PG. The telencephalic limbic system and experimental gastric pathology: a review. Neurosci Biobehav Rev. 1982;6:381–390. doi: 10.1016/0149-7634(82)90047-1. [DOI] [PubMed] [Google Scholar]
- Howren MB, Lamkin DL, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71:171–186. doi: 10.1097/PSY.0b013e3181907c1b. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Akechi T, Okuyama T, Sugawara Y, Kinoshita H, Shima Y, Terao K, Mitsunaga S, Ochiai A, Uchitomi Y. Associations of interleukin-6 with vegetative but not affective depressive symptoms in terminally ill cancer patients. Support Care Cancer. 2013;21:2097–2106. doi: 10.1007/s00520-013-1767-x. [DOI] [PubMed] [Google Scholar]
- Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol. 2011;11:625–632. doi: 10.1038/nri3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, Preacher KJ, MacCallum RC, Atkinson C, Malarkey WB, Glaser R. Chronic stress and age-related increases in pro-inflammatory cytokine IL-6. Proc Natl Acad Sci USA. 2003;100:9090–9095. doi: 10.1073/pnas.1531903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kop WJ, Weissman NJ, Zhu J, Bonsall RW, Doyle M, Stretch MR, Glaes SB, Krantz DS, Gottdiener JS, Tracy RP. Effects of acute mental stress and exercise on inflammatory markers in patients with coronary artery disease and healthy controls. Am J Cardiol. 2008;101:767–773. doi: 10.1016/j.amjcard.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Kross E, Berman MG, Mischel W, Smith EE, Wager TD. Social rejection shares somatosensory representations with physical pain. Proc Natl Acad Sci USA. 2011;108:6270–6275. doi: 10.1073/pnas.1102693108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFleur B, Lee W, Billhiemer D, Lockhart C, Liu J, Merchant N. Statistical methods of assays with limits of detection: serum bile acid as a differentiator between patients with normal colons, adenomas, and colorectal cancer. J Carcinog. 2011;10:1–19. doi: 10.4103/1477-3163.79681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren DG, Ries ML, Xu G, Johnson SC. A generalized form of context-dependent psychophysiological interactions (gPPI): a comparison to standard approaches. NeuroImage. 2012;61:1277–1286. doi: 10.1016/j.neuroimage.2012.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ, Pitman RK, Orr SP, Fischl B, Rauch SL. A role for the human dorsal anterior cingulate cortex in fear expression. Biol Psychiatry. 2007;62:1191–1194. doi: 10.1016/j.biopsych.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66:1075–1082. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Cole SW. Health psychology: developing biologically plausible models linking the social world and physical health. Annu Rev Psychol. 2009;60:501–524. doi: 10.1146/annurev.psych.60.110707.163551. [DOI] [PubMed] [Google Scholar]
- Murphy MLM, Slavich GM, Rohleder N, Miller GE. Targeted rejection triggers differential pro- and anti-inflammatory gene expression in adolescents as a function of social status. Clin Psychol Sci. 2013;1:30–40. doi: 10.1177/2167702612455743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscatell KA, Eisenberger NI. A social neuroscience perspective on stress and health. Soc Personal Psychol Compass. 2012;6:890–904. doi: 10.1111/j.1751-9004.2012.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen-Hoeksema S. Gender differences in depression. Curr Dir Psychol Sci. 2001;10:173–176. [Google Scholar]
- Ochsner KN, Gross JJ. The cognitive control of emotion. Trends Cogn Sci. 2005;9:242–249. doi: 10.1016/j.tics.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Prather AA, Carroll JE, Fury JM, McDade KK, Ross D, Marsland AL. Gender differences in stimulated cytokine production following acute psychological stress. Brain Behav Immun. 2009;23:622–628. doi: 10.1016/j.bbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Miller AH. The evolutionary significance of depression in Pathogen Host Defense (PATHOS-D) Mol Psychiatry. 2013;18:15–37. doi: 10.1038/mp.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson OJ, Charney DR, Overstreet C, Vytal K, Grillon C. The adaptive threat bias in anxiety: amygdala–dorsomedial prefrontal cortex coupling and aversive amplification. NeuroImage. 2012;60:523–529. doi: 10.1016/j.neuroimage.2011.11.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohleder N. Stimulation of systemic low-grade inflammation by psychosocial stress. Psychosom Med. 2014;76:181–189. doi: 10.1097/PSY.0000000000000049. [DOI] [PubMed] [Google Scholar]
- Rohleder N, Schommer NC, Hellhammer DH, Engel R, Kirschbaum C. Sex differences in glucocorticoid sensitivity of proinflammatory cytokine production after psychosocial stress. Psychosom Med. 2001;63:966–972. doi: 10.1097/00006842-200111000-00016. [DOI] [PubMed] [Google Scholar]
- Sheridan JF, Stark JL, Avitsur R, Padgett DA. Social disruption, immunity, and susceptibility to viral infection: role of glucocorticoid insensitivity and NGF. Ann N Y Acad Sci. 2000;917:894–905. doi: 10.1111/j.1749-6632.2000.tb05455.x. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull. 2014;140:774–815. doi: 10.1037/a0035302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavich GM, Way BM, Eisenberger NI, Taylor SE. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc Natl Acad Sci USA. 2010;107:14817–14822. doi: 10.1073/pnas.1009164107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavich GM, O’Donovan A, Epel ES, Kemeny ME. Black sheep get the blues: a psychobiological model of social rejection and depression. Neurosci Biobehav Rev. 2010;35:39–45. doi: 10.1016/j.neubiorev.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steptoe A, Kivimäki M. Stress and cardiovascular disease. Nat Rev Cardiol. 2012;9:360–370. doi: 10.1038/nrcardio.2012.45. [DOI] [PubMed] [Google Scholar]
- Steptoe A, Owen N, Kunz-Ebrecht S, Mohamed-Ali V. Inflammatory cytokines, socioeconomic status, and acute stress responsivity. Brain Behav Immun. 2002;16:774–784. doi: 10.1016/s0889-1591(02)00030-2. [DOI] [PubMed] [Google Scholar]
- Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun. 2007;21:901–912. doi: 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Stroud LR, Salovey P, Epel ES. Sex difference in stress responses: social rejection versus achievement stress. Biol Psychiatry. 2002;52:318–327. doi: 10.1016/s0006-3223(02)01333-1. [DOI] [PubMed] [Google Scholar]
- Stroud CB, Davila J, Hammen C, Vrshek-Schallhorn S. Severe and nonsevere events in first onset versus recurrences of depression: evidence for stress sensitization. J Abnorm Psychol. 2011;120:142–154. doi: 10.1037/a0021659. [DOI] [PubMed] [Google Scholar]
- Tengstrand B, Ahlmén M, Hafström I. The influence of sex on rheumatoid arthritis: a prospective study of onset and outcome after 2 years. J Rheumatol. 2004;31:214–222. [PubMed] [Google Scholar]
- The Emerging Risk Factors Collaboration. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gool J, van Vugt H, Helle M, Aarden LA. The relation among stress, adrenalin, interleukin-6 and acute phase proteins in the rat. Clin Immunol Immunopathol. 1990;57:200–210. doi: 10.1016/0090-1229(90)90034-n. [DOI] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem. 2006;13:728–733. doi: 10.1101/lm.306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen PJ, Shin LM, McInerney SC, Fischer H, Wright CI, Rauch SL. A functional MRI study of human amygdala responses to facial expression of fear versus anger. Emotion. 2001;1:70–83. doi: 10.1037/1528-3542.1.1.70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.