

Abstract
The success of adoptive T cell gene transfer for treatment of cancer and HIV is predicated on generating a response that is both durable and safe. Here we report long term results from three clinical trials to evaluate gammaretroviral vector engineered T-cells for HIV. The vector encoded a chimeric antigen receptor (CAR) comprised of CD4 linked to the CD3-ζ signaling chain (CD4ζ). CAR T-cells were detected in 98% of samples tested for at least 11 years post-infusion at frequencies that exceed average T cell levels after most vaccine approaches. The CD4ζ transgene retained expression and function. There was no evidence of vector-induced immortalization of cells as integration site distributions showed no evidence of persistent clonal expansion or enrichment for integration sites near genes implicated in growth control or transformation. The CD4ζ T cells have stable levels of engraftment, with decay half-lives that exceed 16 years, in marked contrast to previous trials testing engineered T cells. These findings indicate that host immunosuppression prior to T cell transfer is not required in order to achieve long term persistence of gene-modified T cells. Further, our results emphasize the safety of T cells modified by retroviral gene transfer in clinical application, as measured in >500 patient years of follow up. Thus, previous safety issues with integrating viral vectors are hematopoietic stem cell or transgene intrinsic, and not a general feature of retroviral vectors. Engineered T cells are a promising form of synthetic biology for long term delivery of protein based therapeutics. These results provide a framework to guide the therapy of a wide spectrum of human diseases.
INTRODUCTION
Retroviral vectors have been associated with safety concerns in clinical applications (1). For example, when individuals with X-linked SCID (SCID-X1) were treated by gene transfer to restore the missing IL-2 receptor γ (IL2RG) gene to hematopoietic stem cells using gammaretroviral vectors, while 9 of 10 patients were successfully treated, 4 of the 9 developed T cell leukemia several years after gene therapy (2). Similarly, a lentiviral vector encoding β-globin flanked by insulator elements has been used to treat β-thalassemia successfully in one human subject, However, a clonal expansion was observed after integration in the HMGA2 locus, raising concerns regarding the long term safety of this approach (1). Thus, an issue is whether expansion of cells harboring vectors integrated near genes involved in growth control will inevitably result in clonal proliferation, or whether the safety concerns are cell type specific.
Adoptive transfer therapies are often further limited by a requirement for host lymphodepletion prior to T cell transfer (3). Until recently, persistence of gene modified T-cells in the absence of a strong selective advantage has been modest. However, in children with congenital immunodeficiency, persistence of gene corrected lymphocytes has been detected for at least 12 years (4). Similarly, in lymphopenic patients after hematopoietic stem cell transplantation, gene-marked EBV-specific cytotoxic T lymphocytes have been shown to persist for up to 9 years (5). The study reported here was undertaken as part of long-term follow-up for gene transfer studies using integrating vectors as mandated by the US Food and Drug Administration (http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/ucm072957.htm). T-cells expressing CD4ζ become activated upon binding HIV gp120 envelope protein on infected cells (6, 7). Between 1998 and 2005 three clinical studies evaluated the CD4ζ CAR expressed in autologous CD4+ and CD8+ T-cells in subjects with active viremia (8), or in T cell reconstituted patients with chronic HIV-1 infection (clinicaltrials.gov NCT01013415 and (9)). See Supplementary Material (SM) for protocol information, including other variables that were tested across the trials including dose and dose schedule, and the effect of IL-2 administration on cell persistence.
RESULTS
CD4ζ CAR T cells have T½ >16 years
To assess the durability of gene marking, total genomic DNA (gDNA) from peripheral blood mononuclear cell (PBMC) samples from 43 subjects collected between 1 and 11 years post-infusion were analyzed by quantitative PCR (qPCR) for CD4ζ. Remarkably, stable engraftment was observed in 212 of 221 subject samples (Fig. 1A). The majority of subjects had an average CD4ζ frequency in PBMC of 0.01 to 0.1%, with some exceeding 0.1%. (Fig. 1B–D). Linear mixed effects modeling (10) was used to measure their decay rates, indicating that their disappearance half-life (t1/2) was >16 years for the three trials (Table 1), suggesting CD4ζ modified T-cells may persist for decades. The extrapolation beyond year nine is subject to the unknown biology of whether or not decay remains linear after year 9. In contrast, persistence of gene modified T-cells has been modest in previous trials involving patients with cancer and HIV, with decay half lives of less than 30 days.
Figure 1. Persistence of CD4ζ modified CAR T-cells over 11 years post infusion.
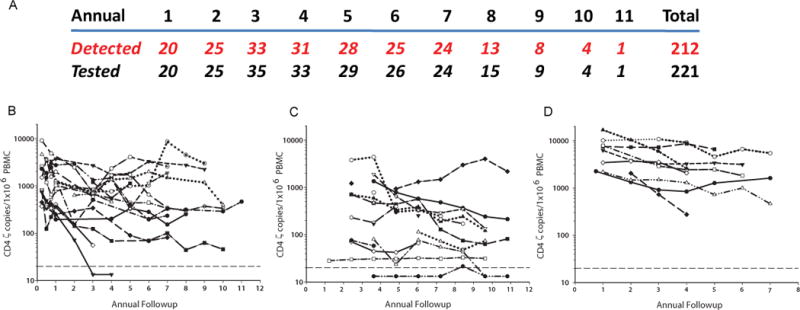
(A) Total samples tested at annual visits and the corresponding number of samples with detectable CD4-ζ. Persistence of CAR T cells for the 43 individual patients in the (B) Mitsuyasu (8), (C) Deeks (9) and (D) Aronson (clinicaltrials.gov NCT01013415) trials, at annual visits beginning at 1 year post infusion. The LOD for the assay is plotted as a dotted reference line.
Table 1.
Decay rates and half-lives of engrafted CD4-ζ cells in subjects on the 3 clinical protocols.
| Study | No. of subjects in followup | Avg no. of followup samples evaluated per patient | Decay rate (log10 copies/yr) | Half-Life (yrs post infusion) | Years to Reach LOD | ||||
|---|---|---|---|---|---|---|---|---|---|
| Estimate | 95% CI | P value | Estimate | 95% CI | Estimate | 95% CI | |||
| Deeks | 17 | 5.4 | −0.06 | −0.115, −0.006 | 0.034 | 24.5 | 13.3, 235.9 | 24.2 | 13.1, 233.4 |
| Mitsuyasu | 17 | 4.9 | −0.085 | −0.145, −0.025 | 0.005 | 16.5 | 10.5, 50.1 | 14.2 | 9.2, 42.5 |
| Aronson | 9 | 5 | −0.112 | −0.200, −0.023 | 0.013 | 17.5 | 10.2, 80.2 | 21.2 | 12.3, 98.3 |
| Combined | 43 | 5.3 | −0.074 | −0.107, −0.041 | 0.001 | ||||
Limit of quantitation is 26.6/1×106 or 1.42 copies on log10 scale; see figure 1 for data points. Note, although the estimated half-life and time to reach limit of detection (LOD) are listed as year post infusion, they were calculated using the estimated cell count beginning at year 1 (*or year 2 for Mitsuyasu study due to limitation in sample availability in year 1) rather than the actual initial infusion cell counts. In this analysis, there was on average 25 independent patients available (minimum: 10 patients) for estimating the rate of decline at each time point through year 9.
Decade long expression and function of CD4ζ CAR transgene
Gene silencing of integrating vectors is a potential limitation of retroviral gene therapy (11). To interrogate expression of CD4ζ, we isolated total RNA from 13 subjects 2–10 years post infusion, and measured transcriptional activity by RT-PCR for CD4ζ. All but two samples (the samples with lowest detectable engraftment) had measurable CD4ζ RNA (Fig. 2A) at levels which significantly correlated with DNA copies (Table S1). This indicates ongoing transcription of CD4ζ for at least 10 years after infusion of gene-modified T cells.
Figure 2. Transcriptional activity and CAR function in persisting cells.
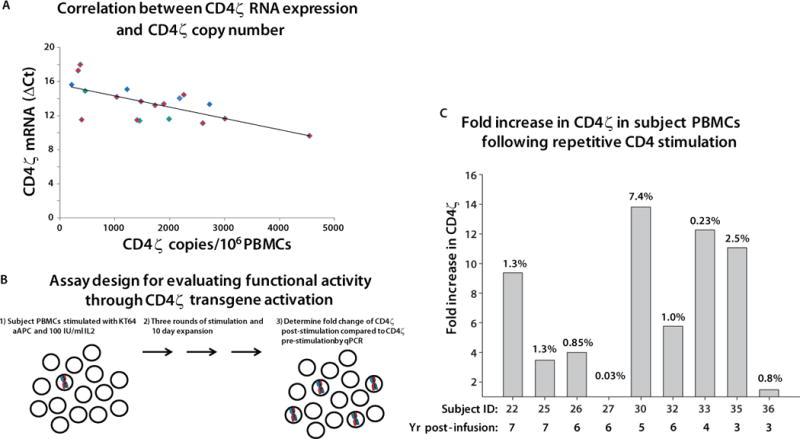
(A) The CD4ζ RNA level (y axis) is plotted versus the number of DNA CD4ζ DNA copies per million PBMC of each tested sample. Samples from the Deeks, Mitsuyasu and Aronson studies are plotted as red, blue and green symbols, respectively. CD4ζ RNA expression was calculated from the ΔCt values for RT-PCR of CD4ζ and GAPDH mRNA. GAPDH is expressed at a high level, so that greater expression of CD4ζ results in a smaller expression difference and so a smaller ΔCt. The values are significantly correlated by linear regression analysis (p=0.0018) testing whether rho=0 or not. No RT-controls were run in parallel and all plotted CD4ζ samples were negative, confirming the signal observed is due to RNA template. Two subjects did not have detectable CD4ζ RNA. (B) Design of the proliferation assay used to interrogate function of CD4-ζ CAR in T-cells. This assay was validated prior to employing as described in Fig. S1. Functionality is measured as the relative increase in the average copy number of CD4ζ cells following anti-CD4 antibody activation over percentage of CD4ζ before stimulation. (C) Fold-increase of CD4ζ expressing cells following three 10-day rounds of anti-CD4 mAb loaded irradiated K562 artificial antigen presenting cells expressing the high affinity Fc Receptor CD64 (KT64) and 100 IU of IL-2. CD4ζ copy numbers were evaluated from the gDNA of subject PBMCs before and after activation by qPCR analysis. The final percentage of CD4ζ in each culture is indicated by the number at the top of each bar. Each bar is designated at the bottom with the subject ID and year post-infusion of the sample.
We then evaluated whether the CD4ζ CAR was expressed and functional. CD4ζ positive cells could not be sorted for functional testing by standard methods such as cell surface staining and cytokine release due to (1) limiting numbers of cells in annual samples, (2) the inability to phenotypically distinguish native CD4 from CD4ζ, (3) CD4 expression by natural CD8 cells upon activation (12). Therefore, we developed an assay to enrich CD4ζ cells in response to ligation of CD4ζ using artificial antigen presenting cells (Fig. 2B; and Fig. S1A). We evaluated cryopreserved PBMC samples obtained 3–7 years after infusion. In seven of nine subjects, the ratio of the CD4ζ copies after stimulation showed a 3–13 fold increase in prevalence compared to before stimulation (Fig. 2C). Thus continued expression and function of CD4ζ may contribute to prolonged survival.
Genomic and epigenetic features of retroviral insertion sites
The durable persistence of the CD4ζ T-cells provided a unique opportunity to interrogate selection for preferred gammaretroviral integration sites over time in T cells, resulting from modulation of cellular growth control genes as a result of integration-mediated events such as distal effects from the LTR enhancer (2) or disruption of gene regulation (1). We isolated vector integration sites from 11 individuals with high level marking, and from whom samples were available in sufficient quantities, using LM-PCR and Mu-mediated recovery (13–15). A total of 202,435 sequence reads and 7,222 unique integration sites were determined from the infused cell product and post-infusion PBMC (Table S2).
We first examined the global integration site pattern relative to genomic features such as gene density, gene expression, and CpG islands. For comparison, we also analyzed integration sites from SCID-X1 gene-corrected subjects (16) and from MLV infected primary CD4+ T-cells infected in culture (17). Gammaretroviruses are known to integrate in gene dense, transcriptionally active regions near gene 5′ ends and CpG islands. Typical gammaretrovirus integration patterns were found in CD4ζ subjects (Fig. S2). An analysis over ChIP-Seq data sets querying 44 forms of histone post-translational modification and DNA binding proteins in T cells showed the expected associations with features enriched near gene 5′ ends. Comparison of global integration patterns in the CD4ζ subjects before and after infusion over genomic features and ChIP-Seq maps showed no notable differences.
Dynamics of modified cells can be tracked by detecting the prevalence of cell clones across multiple timepoints using the proportion of sequence reads corresponding to an integration site as a surrogate for cell abundance. By this approach, we failed to detect signs of persistent clonal expansion in CD4ζ subjects. Instead, integration sites were mostly unique at every time point (Table S3) and sites with elevated CD4ζ marking at one time point were absent at later timepoints (Fig. 3A). Another method for estimating clonal abundance is to count the number of independent recovery events that capture the same integration site (13). Most sites were recovered from only a single starting molecule (Table S3). Of the 15 sites recovered independently more than once (15 out of 682 sites), only two of the sites were in close proximity (<50kb) to cancer-related genes, a frequency indistinguishable from pre-infusion samples. We compared whether CD4ζ integration sites were enriched near genes marked by clusters of integration sites from SCID-X1 gene therapy in stem cells (16). These represent candidates for genes involved in clonal expansion or persistence. However, we did not find evidence for enrichment (Table S4). Because gammaretroviruses integrate near gene promoters, we also compared the frequency of integration near the 5′-ends of cancer-related genes in the pre-infusion gene-modified T cell product and post-infusion samples. We failed to detect significant enrichment over time (Fig. 3B).
Figure 3. Integration site analysis of CD4ζ modified CAR T cells.
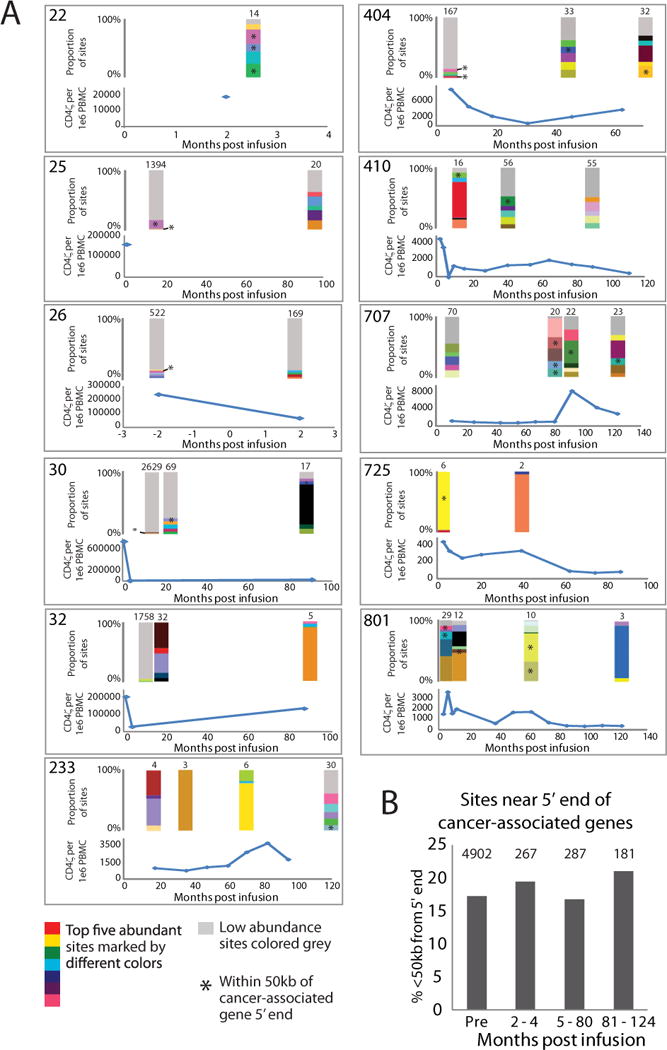
(A) Longitudinal abundance and dynamics of CD4ζ modified T-cells. The top-left corner of each panel shows the patient number. Q-PCR measurements of total CD4ζ copy number per million PBMCs are shown longitudinally for individual subjects (blue line). The x-axis shows months post infusion, the y axis shows Q-PCR vector copy number. Stacked bar graphs are shown directly above time points where integration sites were isolated and depict the relative abundance of integration sites based on the proportion of sequence reads detected using MseI and Tsp509I. The top five abundant sites are differentially colored with all other less abundant sites colored grey. The total number of unique sites detected at a given time point is shown above the corresponding bar graph. (B) Frequency of integration near cancer-associated gene 5′ ends. Integration sites were separated into four bins with one bin for preinfusion sites and three bins for post infusion sites (x-axis). The percent of sites found within 50kb from a gene 5′ end that were also within 50kb from a cancer-associated gene’s 5′ end are shown (y-axis). The number of sites <50kb from a gene’s 5′ end are shown at the top of each bin. No significant difference between the preinfusion and post infusion bins was found in pairwise comparisons using Fisher’s exact tests.
Retroviral gene transfer safety record in >500 patient years of observation
The stable level of engraftment with a functional transgene combined with the maturity of the clinical trials provides a unique opportunity to determine the safety and durability of gene transfer with integrating vectors. Clinical monitoring of the patients at yearly intervals has not detected any suspected or documented occurrences of hematologic disorders suggestive of retroviral genotoxicity. The clinical data set represents over 540 patient years without integration mediated toxicity, therefore, based on a Poisson distribution assumption, we are 95% confident that the true adverse event rate is less than 0.0068 per person-year, or equivalently, no more than one event in every ~147 years.
DISCUSSION
The safety of gene transfer with retroviral vectors has been difficult to establish due to a paucity of studies with persistently high levels of gene-modified cells. The persistence of gene modified T-cells has been modest in previous trials involving patients with cancer and HIV, with decay half lives of less than 30 days (18–27). Given the large number of subjects analyzed in this study and the extent of marking, it is likely that most targets in the human genome hosted vector integration, given that T cells with more than 2×1011 integration events were infused in the trials and that the human genome consists of approximately 3×109 base pairs. Thus the absence of adverse events and clonal expansion is unexpected given previous estimates at 10−6 to 10−8 adverse events per retrovirus insertion event (28). It is likely that mature human T cells are more resistant to insertional genotoxicity, consistent with the known resistance of mature mouse T-cells to transformation (29). In contrast, mature B cells do not appear to have this resistance to transformation (30). A potential mechanism for observations that mature T cells appear to offer “safe harbor” to integration events is that unlike B cells where homeostasis is regulated at the population level, the homeostasis of T cell mass is asserted at the clonal level by mechanisms involving intraclonal competition (31, 32). The essentially stable persistence of CD4ζ CAR T cells that we have observed is similar to the 14 year half life reported for vaccinia-specific human T cells (33). The notion of homeostatic regulation of CD4ζ T cells at the clonal level is consistent with the previous demonstration that CAR T cells continue to express their natural TCRs (6).
The mechanisms responsible for the high level persistence of CD4ζ CARs likely include the use of improved cell culture technology that promotes central memory cells (34), a non-immunogenic transgene, and signaling from the CD4ζ CAR moiety. The decay rates of CD4ζ modified T cells in the peripheral blood suggests persistence could last decades. One possible mechanism for long term persistence in this study could be repetitive CAR stimulation as a result of periodic encounters with HIV envelope (7), given that persistent replication of HIV-1 occurs in the presence of antiretroviral therapy (35). Alternatively, binding of the extracellular CD4 portion of the CD4ζ chimera with its low affinity ligand, MHC class II, might lead to signaling through the ζ chain to promote persistence (36). Both are supported findings that T cell persistence is enhanced by TCR signals to self ligands (37).
One limitation of the present study is that it is not yet possible to determine the survival rates of the CAR T cells in this study beyond 9 years, due to limited numbers of samples. The predicted decay rate is based on modeling that assumes continued linear decay rates that were observed during the initial 9 years. Another caveat is that the clinical safety that we have observed may be specific for T cells and/or CD4ζ; whether similar safety could be obtained in other cell types is unknown.
In addition to the safety features of CD4ζ CAR cells, an important clinical implication of our results is that the patients did not require conditioning with lymphoablative regimens in order to achieve stable engraftment that has been necessary with stem cell therapies and for T cell engraftment in cancer patients. The safety and long term persistence of engineered T cells provides a further rationale for cell based HIV eradication strategies (38). Finally, these findings provide a framework for the design of long term gene delivery strategies for genetic disorders and other benign conditions where chemotherapy is not feasible.
MATERIALS AND METHODS
Clinical Protocols
Each of the three CD4ζ trials had unique therapeutic protocols in anti-retroviral drug (ARV) treated HIV subjects. The Mitsuyasu study (8) was a phase II open label trial, infusing a single dose of 2–3×1010 CD4 and CD8 T cells per subject with detectable HIV loads divided into two groups: 1) those that received 6 million units of IL2 continuously infused over 4 days beginning 4 hours prior to T cell infusion or 2) those that received no IL-2. The Deeks study (9) was a phase II clinical trial treating ARV controlled HIV subjects (levels near limits of detection) and 1×1010 CD4/CD8 cells infused three times 2 weeks apart. Subjects were enrolled onto a cohort of 1) those receiving CD4ζ modified T-cells or 2) those receiving only unmodified T-cells. The Aronson study (clinicaltrials.gov NCT01013415) was a randomized, three cohort trial of subjects with ARV controlled HIV infections (levels below detection). Two cohorts received infusions of 8–9×109 CD4ζ modified CD4/8 T-cells with or without subcutaneous injections of 1.2 MIU/m2/day of IL-2 for 56 days. The third cohort group received IL-2 only. For each of these studies, subjects were intensely monitored and evaluated post-infusion in the first year for clinical responses, safety, and correlative effects. This report was initially undertaken as part of the FDA mandated long term follow-up for monitoring for delayed adverse events in patients receiving gene therapy using integrating vectors. Under this requirement, subjects participated in annual laboratory and physical exams up to 15 years post infusion. The collection of frozen PBMCs from the annual visits for each subject in each of the studies was compiled. The completeness of each subject’s annual visit follow up profile was primarily determined by the subjects compliance with protocol-specified study visits and to adequate specimen quality.
Determination of CD4ζ copy numbers
A qPCR assay was developed to detect the amplicon formed by the CD4 and zeta chimera. gDNA for qPCR was isolated from frozen pellets of 1×106 PBMCs obtained from the buffy coat of processed blood from subject annual visits using QIAamp DNA Micro Kit (Qiagene, 56304). qPCR was performed in 20µl volumes using the 384 well format on ABI HT7900. Validated CD4ζ and huGAPDH primer/probe sets were used to quantify respective copy numbers of subject samples from standard curves. Assay results were evaluated by ABI’s SDS2.3, Excel and Sigma Plot software. To determine the average CD4ζ copies/1×106 PBMCs, CD4ζ copy numbers were determined using four replicates of 250 ng gDNA, GAPDH copy numbers were determined using three replicates of 50 ng gDNA and then used to normalize the CD4ζ copies to 1E6 cells using the formula: 2× (CD4ζ copies/GAPDH copies) × 1 ×106. Taqman qPCR Assay performance criterion: Limit of Quantification (LOQ) is ~1 copy in 7600 cells, Limit of Detection (LOD) is ~1 copy in 38,000 cells, positive control within 80% of expected value, and R2 must be greater than 0.995.
Determination of CD4ζ decay slopes and half-life
A linear mixed effects model (10) with the time since infusion as an independent variable was used to estimate the rate of CD4ζ decay from year 1 to year 9 post-infusion. By using both random intercepts and random slopes, potential correlation among repeated measurements over time and between-subject variability in the initial values and as well as the rate of decay was modeled. For the combined data, the difference between studies in the year 1 cell counts was adjusted by including indicator variables of individual study in the model as covariates.
RT-PCR of CD4ζ subject samples
Cryopreserved subject PBMCs were thawed and allowed to recover overnight before total RNA was isolated using QIAgene RNAeasy Plus kit (74134). The RNA was reversed transcribed using mix hexamers and evaluated by taqman analysis for the detection of CD4ζ and the reference gene huGAPDH. A resulting positive Ct value for CD4ζ then subtracted away the Ct value of the GAPDH to determine deltaCt of CD4ζ relative to GAPDH. As a control for CD4ζ DNA contamination, a reverse transcription reaction without the reverse transcriptase was done for each sample. A negative signal was found in all control samples indicating samples had no contaminating DNA. The two samples with undetectable CD4ζ RNA described in figure 2A also had transgene copy numbers below 100, suggesting that 0.01% of CD4ζ modified T-cells in PBMCs may be the limit to detect CD4ζ RNA by this RT-Taqman procedure.
Integration site recovery and analysis
Purified genomic DNA was digested with MseI, Tsp509I, or for Mu-mediated recovery (13), with BanI. Samples with limiting DNA amounts were whole-genome amplified using the illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare) prior to digestion. PCR adapters were installed by T4 ligase or Mu Transposase. MseI and Tsp509I samples were then digested again using MscI to prevent recovery of vector sequence. Nested PCR was performed using conditions described previously (15) and primers specific to the CD4ζ vector LTR. Amplification products were purified using AmPure beads and sequenced using 454 pyrophosphate sequencing technology (39). Genomic sequences aligning within three base pairs of the LTR end and showing unique best alignments to the human genome by BLAT (hg18, version 36.1, >98% match score) were considered true integration sites. Comparisons to genomic features were carried out as described previously (40, 41) using Fisher’s exact tests, Chi square, or a combination of conditional logit, regression and Bayesian model averaging. An explanation of supplementary genomic heatmaps can be found in Ocwieja et al. 2011 supplementary text “Guide to Interpreting Genomic Heat Maps Summarizing Integration Site Distributions” (42). Identification of cancer-related genes was done using a collection of cancer-related gene lists from seven different sources (http://microb230.med.upenn.edu/protocols/cancergenes.html). Integration site datasets used and oligonucleotides used for analysis are listed in Tables S5 and S6. The CD4ζ integration sites were compared to integration sites recovered from other studies that used gammaretroviral vectors in T cells (17) and hematopoietic stem cells (16).
Supplementary Material
Supplementary Figure 1. Development and validation of the K562 activation assay for functional testing of CD4-ζ cells.
Supplementary Figure 2. Integration patterns for CD4-ζ are typical of gammaretroviral vectors.
Supplementary Table S1. CD4-ζ gene copy number and CD4-ζ transcriptional activity in persisting cells.
Supplementary Table S2. Integration sites recovered from CD4ζ subjects.
Supplementary Table S3. Integration sites recovered by >1 Mu Hop.
Supplementary Table S4. Integration sites near cluster genes.
Supplementary Table S5. Integration site datasets used in this study.
Supplementary Table S6. Oligonucleotides used in this study.
Acknowledgments
We are grateful for the study subject volunteers, research nurses and data managers who conducted the clinical study, staff of the Clinical Cell and Vaccine Production Facility, A. Chew, E. Veloso and A. Weintrob for regulatory support, H. Flaks and M. Dennis for clinical support, and Steven Sherwin and others formerly at Cell Genesys who provided samples.
Funding: Supported by NIH grant 1U19AI082628, the University of Pennsylvania Center for AIDS Research, and the Infectious Diseases Clinical Research Program, a Department of Defense program funded in part with federal funds from the National Institute of Allergy and Infectious Diseases under InterAgency Agreement Y1-AI-5072. T.B. is a Special Fellow of the Leukemia and Lymphoma Society. The contents and views expressed in this publication are the sole responsibility of the authors and do not necessarily reflect the views or policies of the Department of Defense, the Department of the Army, or the U.S. Government. Mention of trade names, commercial products, or organization does not imply endorsement by the U.S. Government.
Footnotes
Author Contributions: J.S. performed experiments and wrote first draft of manuscript; T.L.B. isolated integration sites and performed bioinformatic analysis, G.B-S. designed experiments; M.K, and J.L.R. suggested experimental approaches; W-T. H. conducted statistical analysis; G.P. conducted experiments; K.M.H. designed original clinical trials; A.N.V. conducted experiments; S.G.D, R.T.M., W.B.B., N.E.A. were responsible for patient care and clinical protocols; B.L.L manufactured cells and edited manuscript. J.S., TL.B, N.E.A, S.G.D, F.D.B. and C.H.J. edited the manuscript; C.H.J. designed the study. All authors discussed the results and interpretations.
Competing interests: The authors declare that they have no competing interests.
References and notes
- 1.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, Cavallesco R, Gillet-Legrand B, Caccavelli L, Sgarra R, Maouche-Chretien L, Bernaudin F, Girot R, Dorazio R, Mulder GJ, Polack A, Bank A, Soulier J, Larghero J, Kabbara N, Dalle B, Gourmel B, Socie G, Chretien S, Cartier N, Aubourg P, Fischer A, Cornetta K, Galacteros F, Beuzard Y, Gluckman E, Bushman F, Hacein-Bey-Abina S, Leboulch P. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, Macintyre E, Dal CL, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, Morecki S, Andolfi G, Tabucchi A, Carlucci F, Marinello E, Cattaneo F, Vai S, Servida P, Miniero R, Roncarolo MG, Bordignon C. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 4.Muul LM, Tuschong LM, Soenen SL, Jagadeesh GJ, Ramsey WJ, Long Z, Carter CS, Garabedian EK, Alleyne M, Brown M, Bernstein W, Schurman SH, Fleisher TA, Leitman SF, Dunbar CE, Blaese RM, Candotti F. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood. 2003;101:2563. doi: 10.1182/blood-2002-09-2800. [DOI] [PubMed] [Google Scholar]
- 5.Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, Bollard CM, Liu H, Wu MF, Rochester RJ, Amrolia PJ, Hurwitz JL, Brenner MK, Rooney CM. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts MR, Qin L, Zhang D, Smith DH, Tran AC, Dull TJ, Groopman JE, Capon DJ, Byrn RA, Finer MH. Targeting of human immunodeficiency virus-infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood. 1994;84:2878. [PubMed] [Google Scholar]
- 7.Yang OO, Tran AC, Kalams SA, Johnson RP, Roberts MR, Walker BD. Lysis of HIV-1-infected cells and inhibition of viral replication by universal receptor T cells. Proc Natl Acad Sci U S A. 1997;94:11478. doi: 10.1073/pnas.94.21.11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, Bakker A, Roberts MR, June CH, Jalali S, Lin AA, Pennathur-Das R, Hege KM. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785. [PubMed] [Google Scholar]
- 9.Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, Macken C, Richman DD, Kwok S, June CH, Lazar R, Broad DF, Jalali S, Hege KM. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination anti-retroviral therapy. Mol Ther. 2002;5:788. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 10.Fitzmaurice G, Laird N, Ware J. Applied longitudinal analysis. Wiley-Interscience; Hoboken, New Jersey: 2004. (Wiley series in probability and statistics). [Google Scholar]
- 11.Poleshko A, Palagin I, Zhang R, Boimel P, Castagna C, Adams PD, Skalka AM, Katz RA. Identification of cellular proteins that maintain retroviral epigenetic silencing: evidence for an antiviral response. Journal of Virology. 2008;82:2313. doi: 10.1128/JVI.01882-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitchen SG, LaForge S, Patel VP, Kitchen CM, Miceli MC, Zack JA. Activation of CD8 T cells induces expression of CD4, which functions as a chemotactic receptor. Blood. 2002;99:207. doi: 10.1182/blood.v99.1.207. [DOI] [PubMed] [Google Scholar]
- 13.Brady T, Roth SL, Malani N, Wang GP, Berry CC, Leboulch P, Hacein-Bey-Abina S, Cavazzana-Calvo M, Papapetrou EP, Sadelain M, Savilahti H, Bushman FD. A method to sequence and quantify DNA integration for monitoring outcome in gene therapy. Nucleic Acids Res. 2011;39:e72. doi: 10.1093/nar/gkr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ciuffi A, Ronen K, Brady T, Malani N, Wang G, Berry CC, Bushman FD. Methods for integration site distribution analyses in animal cell genomes. Methods. 2009;47:261. doi: 10.1016/j.ymeth.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17:1186. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang GP, Berry CC, Malani N, Leboulch P, Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M, Bushman FD. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood. 2010;115:4356. doi: 10.1182/blood-2009-12-257352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roth SL, Malani N, Bushman FD. Gammaretroviral integration into nucleosomal target DNA in vivo. J Virol. 2011;85:7393. doi: 10.1128/JVI.00635-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bordignon C, Notarangelo LD, Nobili N, Ferrari G, Casorati G, Panina P, Mazzolari E, Maggioni D, Rossi C, Servida P. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA-immunodeficient patients. Science. 1995;270:470. doi: 10.1126/science.270.5235.470. [DOI] [PubMed] [Google Scholar]
- 19.Morgan RA, Walker R, Carter CS, Natarajan V, Tavel JA, Bechtel C, Herpin B, Muul L, Zheng Z, Jagannatha S, Bunnell BA, Fellowes V, Metcalf JA, Stevens R, Baseler M, Leitman SF, Read EJ, Blaese RM, Lane HC. Preferential survival of CD4+ T lymphocytes engineered with anti-human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum Gene Ther. 2005;16:1065. doi: 10.1089/hum.2005.16.1065. [DOI] [PubMed] [Google Scholar]
- 20.Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, Lupton SD, Overell RW, Reynolds TC, Corey L, Greenberg PD. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med. 1996;2:216. doi: 10.1038/nm0296-216. [DOI] [PubMed] [Google Scholar]
- 21.Ranga U, Woffendin C, Verma S, Xu L, June CH, Bishop DK, Nabel GJ. Retroviral delivery of an antiviral gene in HIV-infected individuals. Proc Natl Acad Sci USA. 1998;95:1201. doi: 10.1073/pnas.95.3.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, Vanwaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA. Gene therapy with human and mouse T cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, Binder GK, Slepushkin V, Lemiale F, Mascola JR, Bushman FD, Dropulic B, June CH. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci USA. 2006;103:17372. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 25.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, Gopal AK, Pagel JM, Lindgren CG, Greenbergt PD, Riddell SR, Press OW. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, Yvon E, Weiss HL, Liu H, Rooney CM, Heslop HE, Brenner MK. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brentjens R, Rivière I, Park J, Davila M, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram HJ, Przbylowski M, Hollyman D, Usachencko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg DA, Jurcic JG, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baum C, Düllmann J, Li Z, Fehse B, Meyer J, Williams DA, von Kalle C. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood. 2003;101:2099. doi: 10.1182/blood-2002-07-2314. [DOI] [PubMed] [Google Scholar]
- 29.Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, Meyer J, Hartmann S, Hansmann ML, Fehse B, von Laer D. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- 30.Janz M, Dorken B, Mathas S. Reprogramming of B lymphoid cells in human lymphoma pathogenesis. Cell Cycle. 2006;5:1057. doi: 10.4161/cc.5.10.2737. [DOI] [PubMed] [Google Scholar]
- 31.Min B, Foucras G, Meier-Schellersheim M, Paul WE. Spontaneous proliferation, a response of naive CD4 T cells determined by the diversity of the memory cell repertoire. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3874. doi: 10.1073/pnas.0400606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science. 2006;312:114. doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- 33.Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. Cutting edge: long-term B cell memory in humans after smallpox vaccination. The Journal of Immunology. 2003;171:4969. doi: 10.4049/jimmunol.171.10.4969. [DOI] [PubMed] [Google Scholar]
- 34.Levine BL, Bernstein W, Craighead N, Lindsten T, Thompson CB, June CH. Effects of CD28 Costimulation on Long Term Proliferation of CD4+ T cells in the Absence of Exogenous Feeder Cells. J Immunol. 1997;159:5921. [PubMed] [Google Scholar]
- 35.Palmer S, Maldarelli F, Wiegand A, Bernstein B, Hanna GJ, Brun SC, Kempf DJ, Mellors JW, Coffin JM, King MS. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci USA. 2008;105:3879. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, Joachimiak A, Reinherz EL. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proceedings of the National Academy of Sciences. 2001;98:10799. doi: 10.1073/pnas.191124098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stefanova I, Dorfman JR, Germain RN. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature. 2002;420:429. doi: 10.1038/nature01146. [DOI] [PubMed] [Google Scholar]
- 38.Hütter G, Nowak D, Mossner M, Ganepola S, Müßig A, Allers K, Schneider T, Hofmann J, Kücherer C, Blau O. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. New England Journal of Medicine. 2009;360:692. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 39.Wang GP, Garrigue A, Ciuffi A, Ronen K, Leipzig J, Berry C, Lagresle-Peyrou C, Benjelloun F, Hacein-Bey-Abina S, Fischer A, Cavazzana-Calvo M, Bushman FD. DNA bar coding and pyrosequencing to analyze adverse events in therapeutic gene transfer. Nucleic Acids Res. 2008;36:e49. doi: 10.1093/nar/gkn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brady T, Lee YN, Ronen K, Malani N, Berry CC, Bieniasz PD, Bushman FD. Integration target site selection by a resurrected human endogenous retrovirus. Genes & Development. 2009;23:633. doi: 10.1101/gad.1762309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berry C, Hannenhalli S, Leipzig J, Bushman FD. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput Biol. 2006;2:e157. doi: 10.1371/journal.pcbi.0020157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ocwieja KE, Brady TL, Ronen K, Huegel A, Roth SL, Schaller T, James LC, Towers GJ, Young JAT, Chanda SK, Konig R, Malani N, Berry CC, Bushman FD. HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLOS Pathogens. 2011;7:e1001313. doi: 10.1371/journal.ppat.1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Development and validation of the K562 activation assay for functional testing of CD4-ζ cells.
Supplementary Figure 2. Integration patterns for CD4-ζ are typical of gammaretroviral vectors.
Supplementary Table S1. CD4-ζ gene copy number and CD4-ζ transcriptional activity in persisting cells.
Supplementary Table S2. Integration sites recovered from CD4ζ subjects.
Supplementary Table S3. Integration sites recovered by >1 Mu Hop.
Supplementary Table S4. Integration sites near cluster genes.
Supplementary Table S5. Integration site datasets used in this study.
Supplementary Table S6. Oligonucleotides used in this study.