

Abstract
Affinity maturation, the process in which somatic hypermutation and positive selection generate antibodies with increasing affinity for an antigen, is pivotal in acquired humoral immunity. We have studied the mechanism of affinity gain in a human B-cell lineage in which two main maturation pathways, diverging from a common ancestor, lead to three mature antibodies that neutralize a broad range of H1 influenza viruses. Previous work showed that increased affinity in the mature antibodies derives primarily from stabilization of the CDR H3 loop in the antigen-binding conformation. We have now used molecular dynamics simulations and existing crystal structures to identify potentially key maturation mutations, and we have characterized their effects on the CDR H3 loop and on antigen binding using further simulations and experimental affinity measurements, respectively. In the two maturation pathways, different contacts between light and heavy chains stabilize the CDR H3 loop. As few as two single-site mutations in each pathway can confer substantial loop stability, but none of them confers experimentally detectable stability on its own. Our results support models of the germinal center reaction in which two or more mutations can occur without concomitant selection and show how divergent pathways have yielded functionally equivalent antibodies. Proteins 2014; 83:771–780. © 2014 The Authors. Proteins: Structure, Function, and Bioinformatics Published by Wiley Periodicals, Inc.
Keywords: molecular dynamics, protein–protein interaction, kinetics measurements, protein evolution, acquired immunity
Introduction
Affinity maturation in the vertebrate immune system ensures production of antibodies with high affinity and specificity for an encountered antigen.1,2 In the course of this process, somatic mutations, primarily in the variable domains of the antibodies, accumulate in successive generations of B cells, which are selected in a germinal center based on effective engagement of antigen.3,4 A typical immune response consists of a population of antibodies that bind to various exposed sites (epitopes) on the antigen.5,6 Antibodies that bind an epitope conserved across multiple strains of a pathogen may be able to neutralize all such strains. Recently, such broadly neutralizing antibodies against HIV7–9 and influenza10–13 have been identified, raising the possibility of developing more effective vaccines against these rapidly evolving viruses.14–18
We have previously characterized a clonal lineage of broadly neutralizing human antibodies against H1 influenza viruses, in which the breadth of neutralization is a consequence of antibody mimicry of the viral host-cell receptor, sialic acid.19,20 Sequences of the mature antibodies were obtained by cloning paired, rearranged VH and VL genes derived from single B cells in a plasma sample obtained 7 days post vaccination;21 the H1 component in the vaccine (2007 TIV) was H1N1 A/Solomon Islands/3/2006. The lineage we have studied, referred to as the CH65 lineage, contains three mature antibodies in two branches, which diverge from an inferred last common intermediate, designated I-2. I-2 in turn descends from an inferred unmutated common ancestor (UCA) [Fig. 1(A)]. Two mature antibodies with very similar sequences, CH65 and CH66, constitute one branch; the third mature antibody, CH67, differs substantially in sequence and represents the other branch.20
Figure 1.
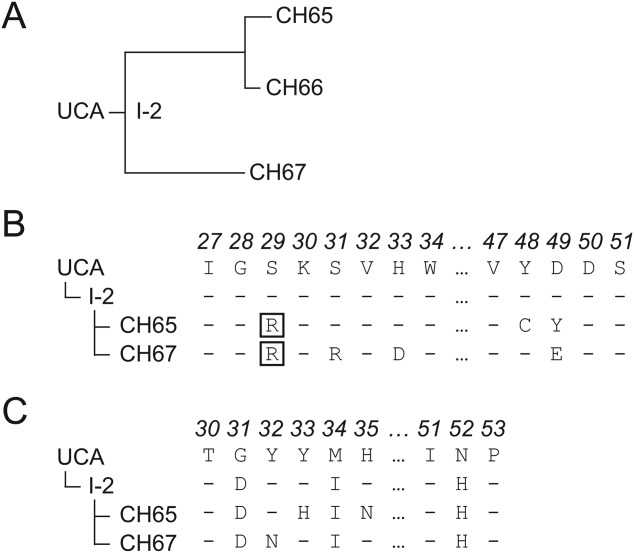
Maturation of the CH65 lineage. (A) Inferred phylogenetic tree of the antibodies in the CH65 lineage. (B) Sequences for a segment of the antibody light chain containing important mutations. The common mutation S29R is boxed. (C) Sequences for a segment of the antibody heavy chain, including the three mutations between UCA and I-2. The complete nucleotide and amino acid sequences are shown in Supporting Information Figure S4.
In comparing the two branches, we chose to study CH65 and CH67. This lineage binds the head domain of H1 influenza hemagglutinin (HA) by inserting the CDR H3 loop into the sialic acid binding pocket [Fig. 2(A)], as determined by X-ray crystallography and later reproduced by molecular dynamics (MD) simulations. MD simulations of binding between the UCA of the lineage and HA suggested that the UCA binds to HA in the same conformation as do the mature antibodies. Using X-ray crystallography, MD simulations, and binding measurements, we showed that the two branches in this lineage achieved affinity maturation by stabilizing the CDR H3 loop in the same antigen-binding conformation, despite largely diverged antibody sequences between the branches. The mechanism by which distinct mutational pathways have arrived at a common functional outcome is not evident from the positions and characteristics of the mutated amino acid residues.
Figure 2.
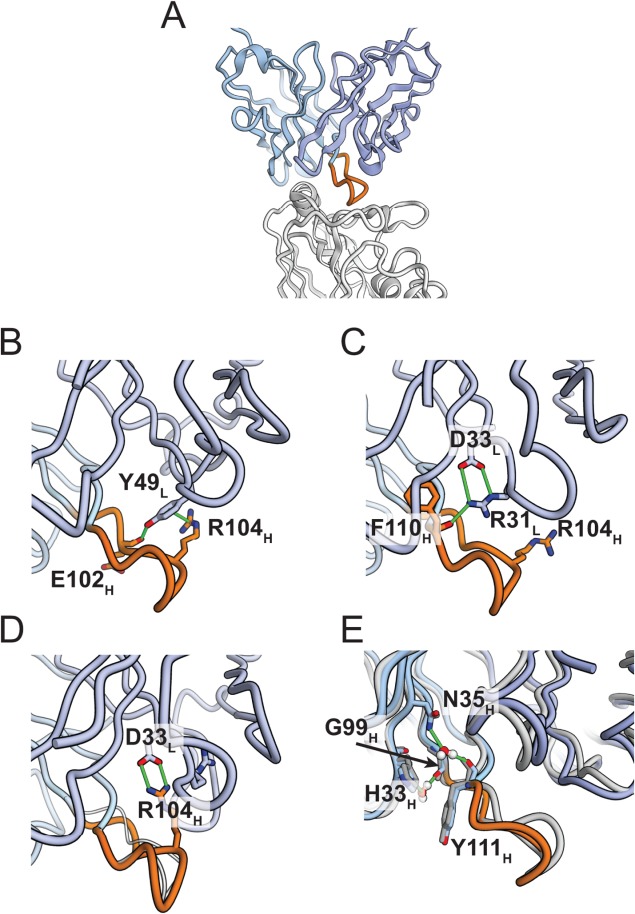
Distinct interactions in two main maturation pathways stabilizing the heavy-chain CDR H3 loop in the CH65 lineage. (A) X-ray crystal structure of CH65–HA complex (PDB ID: 3SM5). The heavy chain is colored in light blue, the light chain in dark blue, HA in light gray, and the CDR H3 loop in orange. The CDR H3 loop inserts into the receptor binding pocket of HA, mimicking the binding of sialic acid. (B) In the CH65 crystal structure, light-chain Y49 forms a cation–π interaction with R104 in the heavy-chain CDR H3, a finding observed with high probability in simulation. Y49 also forms a hydrogen bond with the backbone oxygen of E102H. The hydrogen bond and cation-π interaction are indicated by green lines. (C) In the CH67 crystal structure, R104 projects into the solvent. Hydrogen bonds from R31L to the carbonyl of F110H and from N32H to the carbonyl of A99H (not shown here) might contribute to stabilizing the CDR H3 loop; neither interaction could occur in CH65. (D) A structure from an MD simulation of CH67, in which light-chain D33 forms a salt bridge with R104 in the heavy-chain CDR3. In simulation, D33 forms alternating salt-bridge contacts with R31, as it does in the crystal structure, and with R104; the latter salt-bridge contributes to stabilization. (E) In one simulation of CH65HUCAL, two pairs of water-mediated hydrogen bonds were observed between H33H and G99H and between N35H and Y111H. The geometry of these interactions is compatible with the atomic coordinates in the CH65 crystal structure, although the water molecules are not included in the crystal structure. The simulated structure (shown in light and dark blue) is superimposed on the crystal structure (gray). Water molecules are shown with red (oxygen) and white (hydrogen) balls.
In the present study, we have connected specific mutations during affinity maturation with their effects on antigen binding by combining long-timescale MD simulations22 and binding measurements with mutated antibodies. We have identified for each branch two light-chain mutations that, when introduced into I-2, substantially stabilize the CDR H3 loop and increase affinity for antigen. The two observed maturation pathways have two different sets of mutations that share only one amino acid change in common, while maintaining the same binding conformation and epitope. Although a large number of mutations typically accumulate in broadly neutralizing antibodies over the course of affinity maturation, our results suggest that sets comprising a few key mutations can substantially enhance the affinity of suitable germline antibodies.
Methods
MD simulations
Simulations were carried out as previously described in Ref.20. All simulations used the AMBER99SB-ILDN force field23 for proteins and ions and the TIP3P model for water.24 Simulations were performed on a special-purpose machine, Anton,22 designed for MD simulations.
Conformational equilibrium constant estimation
To estimate fC (see text), a conformation of the apo antibody Fab was considered to be in the antigen-binding state if its CDR H3 loop was within 1 Å RMSD of any conformation sampled in a simulation of any antibody–HA complex that was included in Supporting Information Figure S2. The RMSD was calculated for the CDR H3 loop α carbons after alignment on the α carbons of the full antibody Fab.
Fab expression and purification
The heavy- and light-chain variable and constant domains of CH67, I-2, and the UCA Fabs were originally amplified as previously described.20 Fabs were produced by transient transfection of 293T cells using Lipofectamine 2000 (Invitrogen), following the manufacturer's suggested protocol. Supernatants were harvested and clarified from cellular debris by low-speed centrifugation 5 days after transfection. Fabs were purified using Ni-NTA agarose (Qiagen) or Co-NTA agarose (Clontech) followed by gel filtration chromatography on a Superdex 200 column (GE Healthcare). All mutants described in the text were created using the heavy- or light-chain DNA as a template and mutated at described positions using the QuikChange Mutagenesis (Agilent), following the manufacturer's suggested protocol. All constructs were confirmed by DNA sequencing.
Expression and purification of HA “head”
HA A/Solomon Islands/3/2006 was used as a template to clone the globular head of HA into a pFastBac vector modified for ligation-independent cloning (LIC), as previously described.20 Hi-5 cells were infected with recombinant baculovirus; the supernatant was harvested 72 h later and clarified by centrifugation. The HA head was purified by Ni-NTA agarose (Qiagen) followed by gel filtration chromatography on a Superdex 200 column (GE Healthcare). The C-terminal 6xHis tag was removed by treatment with PreScission protease (GE Healthcare) and repurified by orthogonal Ni-NTA agarose chromatography.
Interferometry and co-elution experiments
Interferometry experiments were performed with a Blitz instrument (forteBIO, Pall Corporation). In each of the experiments, the Fab was immobilized on a Ni-NTA biosensor, and cleaved HASI head, purified as described above, was added at various concentrations between 0.125 and 15 μM. All reactions were carried out at room temperature in buffer of 10 mM Tris-HCl, 150 mM sodium chloride at pH 8.0. Using the Advanced Kinetics program, a typical run included the following steps: baseline acquisition (≥30 s), loading of the Fab onto the NiNTA biosensor (≥90 s), additional baseline acquisition (≥30 s), association of HASI head (≥60 s), dissociation (≥60 s). The ka and kd parameters were determined and a KD obtained by applying a 1:1 binding isotherm after step correction at both the association and dissociation steps, using vendor-supplied software. All experiments were carried out with a minimum of four different concentrations of HASI head. Representative titration experiments are shown in Supporting Information Figure S5.
Size-exclusion chromatography (SEC) experiments were on Superdex 10/300 GL (GE Healthcare) equilibrated in 10 mM Tris-HCl, 150 mM sodium chloride at pH 8.0. HASI and Fab were incubated at room temperature for 15 min before loading. Column elution rate was 0.5 mL min−1 at 4 °C. Elution volume (Ve) was calculated using vendor-supplied software. Complexes and free Fab Ve values are shown in Supporting Information Table SI, and representative SEC traces of complexes, free Fabs, and HASI head are shown in Supporting Information Figure S6.
Results
Properties of the hybrid Fabs
Preconfiguration of the CDR H3 loop in the antigen-binding conformation is the principal consequence of affinity maturation in this lineage, and approximately half the total contact area between antibody and HA involves CDR H3. As an initial screen for mutations that contribute to CDR H3 loop stability, we constructed hybrid Fabs from the UCA and the mature CH65 and CH67 antibodies and used a simple co-elution assay to test for binding of the hybrids to a monomeric HA1 head from A/Solomon Islands/03/2006. Pairing either of the mature light chains with the UCA heavy chain (UCAHCH65L and UCAHCH67L) produced a hybrid that remained associated with HA head on size-exclusion chromatography; pairing either of the mature heavy chains with the UCA light chain gave detectable binding only for CH65HUCAL and not for CH67HUCAL (Supporting Information Table SI).
For a more detailed analysis, we performed long-timescale MD simulations of the free Fabs to observe the conformational states of the CDR H3 loop.25,26 The CDR H3 loop in CH67HUCAL stayed in the binding conformation only briefly and sampled other, diverse conformations for the remainder of the simulations (Supporting Information Figs. S2 and S3). CH65HUCAL stayed in the binding conformation throughout one simulation but departed from it in the other. In contrast, the CDR H3 loops in the chimeras with mature light chains (UCAHCH65L, UCAHCH67L, I-2HCH65L, and I-2HCH67L) remained in the binding conformation or revisited it more than once.
The kinetic and thermodynamic parameters of binding between the chimeras and HA (Table1) support the pattern of CDR H3 loop stabilization observed in MD simulations and the interaction with HA in the co-elution assay. The chimeras UCAHCH65L, UCAHCH67L, and CH65HUCAL have substantially higher affinities for HA than does the UCA, while CH67HUCAL has no detectable binding. The association rate of UCAHCH67L is lower than that of mature CH67 by a factor of only 2, and the association rate of UCAHCH65L is comparable to that of mature CH65, even though 13 (in CH67) and 12 (in CH65) mutations in the heavy chain distinguish the sequence of the chimera from that of the mature antibody. These results suggest that both branches of the CH65 lineage have accrued mutations within their respective light chains that contribute substantially to stabilizing the CDR H3 loop. In addition, the heavy chain of the CH65 branch, but not the CH67 branch, has also accrued mutations that independently stabilize the CDR H3 loop.
Table 1.
Experimentally Measured Kinetic Rate Constants ka and kd, Equilibrium Dissociation Constant of Fab–HA Binding KD, and Simulation-derived Equilibrium Constant KC Between Nonbinding and Binding Conformations for Various Fabs
| Heavy chain | Light chain | ka (103 M−1 s−1) | kd (s−1) | KD (10−6 M) | KC |
|---|---|---|---|---|---|
| UCA | UCA | N.B. | N.B. | >100 | 0.02 ± 0.01 |
| I-2 | UCA | N.B. | N.B. | >100 | 0.04 ± 0.02 |
| I-2 | UCAD49Y | N.B. | N.B. | >100 | 0.02 ± 0.01 |
| I-2 | UCAY48C | N.B. | N.B. | >100 | 0.10 ± 0.05 |
| I-2 | UCAD49Y,Y48C | 14 ± 7 | 0.11 ± 1E−2 | 8 ± 4 | 0.20 ± 0.07 |
| I-2 | UCAD49Y,Y48C,S29R | 12 ± 2 | 0.120 ± 5E−3 | 10 ± 2 | 0.60 ± 0.09 |
| I-2R104N | UCAD49Y,Y48C | 6 ± 2 | 0.20 ± 1E−2 | 31 ± 9 | 0.10 ± 0.04 |
| UCA | CH65 | 58 ± 8 | 0.067 ± 3E−3 | 1.2 ± 0.2 | 3 ± 2 |
| I-2 | CH65 | 31 ± 2 | 0.0320 ± 7E−4 | 1.02 ± 0.06 | 0.10 ± 0.04 |
| CH65 | UCA | 25 ± 5 | 0.082 ± 5E−3 | 3.3 ± 0.7 | 0.010 ± 0.004 |
| CH65 | CH65R29S | 26 ± 2 | 0.0320 ± 8E−4 | 1.21 ± 0.09 | 0.2 ± 0.2 |
| CH65 | CH65Y49D,C48Y | 29 ± 2 | 0.0280 ± 6E−4 | 0.97 ± 0.06 | 0.07 ± 0.03 |
| CH65 | CH65 | 42.9 ± 0.8 | 0.0130 ± 2E−4 | 0.30 ± 0.01 | 0.3 ± 0.1 |
| I-2 | UCAH33D | N.B. | N.B. | >100 | 0.010 ± 0.004 |
| I-2 | UCAS31R | N.B. | N.B. | >100 | 0.07 ± 0.03 |
| I-2 | UCAH33D,S31R | 14 ± 3 | 0.18 ± 1E−2 | 13 ± 3 | 0.3 ± 0.1 |
| I-2 | UCAH33D,S31R,S29R | N.B. | N.B. | >100 | 0.005 ± 0.003 |
| I-2R104N | UCAH33D,S31R | N.B. | N.B. | >100 | 0.010 ± 0.008 |
| UCA | CH67 | 70 ± 30 | 0.18 ± 2E−2 | 3 ± 1 | 0.2 ± 0.2 |
| I-2 | CH67 | 38.4 ± 0.7 | 0.0200 ± 2E−4 | 0.52 ± 0.01 | 0.05 ± 0.03 |
| CH67 | UCA | N.B. | N.B. | >100 | 0.05 ± 0.03 |
| CH67 | CH67R29S | 52 ± 1 | 0.0190 ± 3E−4 | 0.37 ± 0.01 | 0.20 ± 0.07 |
| CH67 | CH67D33H,R31S | 50 ± 10 | 0.067 ± 5E−3 | 1.5 ± 0.3 | 0.2 ± 0.06 |
| CH67 | CH67 | 150 ± 20 | 0.0400 ± 2E−4 | 0.26 ± 0.03 | 1.0 ± 0.8 |
“N.B.” indicates antibodies that bound HA at a strength below the detection limit (∼100 µM). The standard deviation is reported for each value.
Somatic light-chain mutations and the CDR H3 loop conformation
Analyses of the trajectories of long-timescale MD simulations and inspection of the high-resolution X-ray structures suggest specific, somatically mutated, light-chain residues that might contribute to configuring the CDR H3 loop.
In the CH65 light chain, two mutations appear to have direct effects on CDR H3: D49LY and Y48LC, both unique to the CH65-66 branch [Fig. 1(B)]. Y49L in mature CH65 has a π-cation interaction with R104H, and it forms a hydrogen bond with the backbone oxygen of E102H [Fig. 2(B)]. Both contacts are seen in the X-ray structure and frequently in our MD simulations. These interactions pull the CDR H3 loop toward the CDR L2 loop, holding the fomer in a geometry compatible with binding. In the UCA, a salt bridge, D95L-R104H, appears to stabilize the more ordered of the two non-binding conformers seen in the asymmetric unit of the UCA crystal structure. The other loop conformer in those crystals is a fully disordered one. At position 48, Y48L in the UCA would have a close contact with L101H at the N-terminal end of CDR H3, were the latter to adopt the binding conformation. Mutation of residue 48 to Cys relieves the steric incompatibility.
In the CH67 light chain, residues 49–58 (the C′C″ ridge of the light chain) have substantially higher thermal parameters, in the structures of both the CH67 Fab and its complex with the HA head, than they do in the UCA or in the CH65-HA complex. The same residues were also quite mobile in the MD simulations (Supporting Information Fig. S1). The disorder appears to arise from close contacts in CH67 between the side chain of Leu101H and those of L45L and Y48L, collisions that are alleviated in the UCA by the disordering of the CDR H3 loop and in CH65 by the Y48LC mutation. Displacement of residues 49–58 in CH67 correlates with mutations S31LR and H33LD, both unique to the CH67 branch [Fig. 1(B)]. In the CH67 crystal structures, D33L and R31L form a salt bridge, which the ordered conformation of residues 49–58 in the UCA and CH65 would exclude. In MD simulations of the CH67 Fab and of the CH67-HA complex, D33L alternately makes salt-bridge contacts with R31L and R104H. The former salt bridge [Fig. 2(C)] allows R31L to donate a hydrogen bond to the carbonyl of F110H, which probably contributes to CDR H3 conformational stability. The latter salt bridge likewise appears to anchor the CDR H3 loop in the geometry of the binding conformation [Fig. 2(D) and Supporting Information Movie S1]. A hydrogen bond from the side chain of somatically mutated N32H to the carbonyl of A99H may further stabilize the binding conformation of CDR H3.
Affinities of the mutated Fabs
We examined the influence of the light-chain mutations just described when they are introduced into the last common intermediate, I-2. (The I-2 light chain has no somatic mutations and hence is equivalent to the UCA light chain.) To test whether the above mutations indeed establish contacts that help stabilize the CDR H3 loop, we performed simulations of the Fabs of mutants I-2HUCALD49Y (the heavy chain of I-2 and the light chain of the UCA with mutation D49Y), I-2HUCALY48C, I-2HUCALD49Y,Y48C, I-2HUCALH33D, I-2HUCALS31R, I-2HUCALH33D,S31R, and I-2. From these simulations, we computed fC, the fraction of the apo antibody in the antigen-binding conformation (i.e., the equilibrium population of that state), and the corresponding conformational equilibrium constant, KC = fC/(1 − fC), for each mutant Fab (Table1). The single forward mutation Y48LC in I-2 produced only a modest increase in KC, and D49LY had a deleterious effect (Fig. 3 and Table1). Only when the two were both present did KC in the CH65 branch increase substantially. We observed a similar effect in the CH67 branch: only when both forward mutations, H33LD and S31LR, appeared together did KC increase significantly. Both double mutants, I-2HUCALD49Y,Y48C and I-2HUCALH33D,S31R, stabilized the CDR H3 loop in the antigen-binding conformation such that the corresponding KC values were similar to those of their respective mature antibodies, CH65 and CH67. The large KC values estimated from the simulations for the two double mutants predict that their affinity for HA will be substantially higher than that of I-2.
Figure 3.
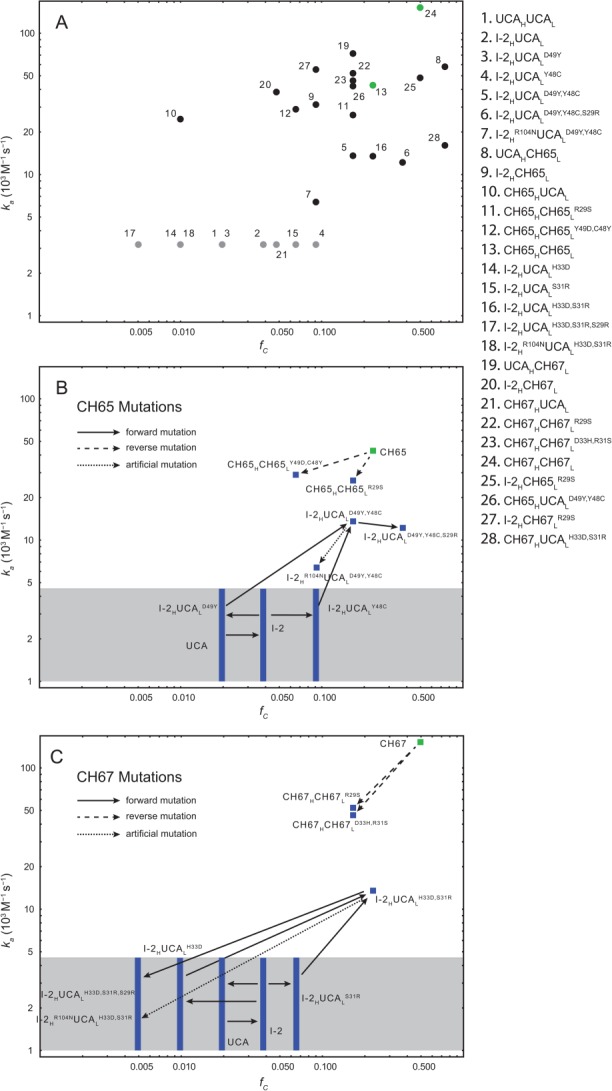
Log-log plot of association rate constants ka versus the fraction of antigen-binding conformation fC of various mutant Fabs. (A) All Fabs studied by MD simulations (Tables I and Supporting Information SI). The qualitative agreement between fC estimated from MD simulations and ka measured in kinetic experiments supports the finding that maturation in the CH65 lineage is driven by the stabilization of the CDR H3 loop in the binding conformation. The Pearson correlation coefficient between fC and ka, when both are available, is 0.26; perfect correlation is not to be expected, because factors other than CDR H3 loop stability also contribute to ka. Fabs whose affinities are below the interferometry sensitivity limit are represented by gray circles at an arbitrary vertical position, because their ka values are unknown. The mature antibodies are colored green. The hybrid Fab CH65HUCAL binds HA with intermediate affinity despite the low fC value estimated from MD simulations; its low fC value might be due to a lack of convergance of the MD simulations of finite length. (B) and (C) highlight the effects of mutations in CH65 (B) and CH67 (C) branches. The solid black arrows correspond to “forward” mutations (i.e., mutations that occurred in the maturation from the UCA to the mature antibody). The dashed arrows correspond to “reverse” mutations (i.e., mutations from the mature antibody to the UCA). The dotted black arrows correspond to the artificial mutation R104NH. Fabs whose affinities are below the interferometry sensitivity limit are shown in the gray box as bands at their respective fC values.
The measured affinities from biolayer interferometry (Table1) show that in the CH65 branch, neither D49LY nor Y48LC alone imparts measurable binding, but that the two together enhance the affinity of I-2 by >10-fold. The same is true for H33LD and S31LR in the CH67 branch. These results are consistent with our predictions from inspection of simulations and structures. In CH65, neither the relief of steric restrictions (Y48LC) nor the aromatic π-cation interaction (D49LY) is enough to bring affinity within the measurable range; in CH67, the S31LR and H33LD mutations are both necessary to generate their mutual salt bridge. When introduced into the UCA, neither pair of mutations conferred detectable affinity, suggesting that the three heavy-chain mutations that separate I-2 from the UCA [Fig. 1(C)] also contribute to the observed affinity gain. The crystal structures and the simulations do not, however, indicate a potential mechanism for this effect.
We also introduced the reverse double mutations into the background of a mature antibody. Simulating the reverse mutations in the corresponding mature antibodies yielded a decrease in KC commensurate with the increase in KC observed in the corresponding forward mutations. This decrease in KC led us to expect a decrease in the association rate and binding affinity. In the CH65 branch, the UCA light chain paired with the mature heavy chain (CH65HUCAL) bound HA head relatively strongly. The C48LY,Y49LD double mutation, introduced into the mature Fab, reduced affinity by a factor of ∼3 and association rate by ∼1.5, the latter being essentially equivalent to the association rate of CH65HUCAL. In the CH67 branch, the UCA light chain paired with the mature heavy chain (CH67HUCAL) had no detectable affinity in our assay. The D33LH,R31LS double mutation, introduced into the mature Fab, reduced the association rate by a factor of ∼3 and the affinity by more than fivefold, but binding was still readily detectable. The cumulative effects of other contributions evidently synergize with that of the R31L-D33L and D33L-R104H salt bridges in this branch of the lineage.
To test the role of R104H, which is unmutated during affinity maturation but is inferred from the MD simulations to contribute to stabilizing the CDR H3 loop in the double mutants I-2HUCALD49Y,Y48C and I-2HUCALH33D,S31R, we introduced into each of the double mutants a mutation, R104HN, that substitutes a neutral polar residue for arginine, creating the variants I-2HR104NUCALH33D,S31R and I-2HR104NUCALD49Y,Y48C. Introducing R104HN eliminated observable binding of I-2HUCALH33D,S31R with HA and somewhat reduced the affinity of I-2HUCALD49Y,Y48C (Table1). Correspondingly, in the MD simulations of the Fabs, R104HN reduced the stability of CDR H3 slightly in I-2HUCALD49Y,Y48C, but substantially in I-2HUCALH33D,S31R. Because R104H does not contact HA, the effect of the mutation is more likely to come from a change in the CDR H3 conformational equilibrium than from a change in the fixed-conformation affinity. The residual affinity and conformational stability of the mutant I-2HR104NUCALD49Y,Y48C could come from the hydrogen bond between Y49L and E102H.
Stabilization of the CDR H3 loop by heavy-chain mutations
Not all mutations that stabilize the CDR H3 loop occur in the light chain. The hybrid CH65HUCAL, which does not have a light-chain mutation, has relatively strong affinity for HA. Its CDR H3 loop may be stabilized by heavy-chain mutations Y33HH and H35HN, which are present in CH65 but not in CH67. In the simulation of CH65HUCAL in which the CDR H3 loop remains in the binding conformation, there are two water-mediated side chain–backbone hydrogen bonds: between H33H and G99H and between N35H and Y111H [Fig. 2(E)]. These interactions observed in the simulation are compatible with the atomic coordinates in the crystal structures and might help keep the CDR H3 loop in the binding conformation.
Mutations at the antigen–antibody interface
One plausible location for mutations with selective advantage in binding is the antigen–antibody interface. We tested whether any of the observed mutations at the HA–antibody contact could enhance affinity. We assayed the binding of HA1 head with various single-site mutant Fabs using the (semi-quantitative) co-elution assay and measured binding kinetics for a selected subset. None of the mutations that establish new polar interactions with HA—N26LD, S29LR, G31HD, N52HH, G63LD, and N68LT—yielded a detectable increase in affinity when introduced into either the UCA or I-2 (Supporting Information Tables SI and SII). This result is consistent with our previous conclusion that stabilization of the CDR H3 loop in the antigen binding conformation, rather than new or stronger noncovalent contacts at the molecular interface, is the principal contribution to affinity maturation in the CH65-67 lineage.
The S29LR mutation
A mutation of serine 29L to arginine occurred independently in the two branches of the lineage [Fig. 1(B)]. The codon usage is different in each of the two mutant antibodies: AGG in CH65 and CGT in CH67, both mutated from AGT in the UCA (Supporting Information Fig. S4). The mutations could have arisen at any point between I-2 and the mature antibodies. Introducing the reversion R29LS into CH65 lowers affinity for HA, with about equal contributions from retarding association and accelerating dissociation (Table1); the same mutation in a CH67 background has little net effect, reducing both the on and off rates by similar factors. The simulations likewise predict lower association rates (i.e., lower fC than for the fully mature antibodies) for both revertants. The forward mutation in a background of I-2HUCALY48C,D49Y (i.e., “early” in the CH65 branch) has very little apparent effect (although the simulation suggests some increase in rate of association), but it eliminates any detectable binding in a background of I-2HUCALS31R,H33D (i.e., “early” in the CH67 branch), consistent with a simulation of the same species, which has the lowest fC of any of the simulated variants.
Discussion
We draw two general conclusions from our long-timescale MD simulations, structures, and binding experiments with antibodies in the two branches of the CH65-67 lineage. First, both simulation and experiment suggest that in each of the two branches, a distinct pair of somatic mutations contributes disproportionately to the acquisition of higher stability in the CDR H3 loop, and either pair of mutations is sufficient to confer measurable HA binding on the common intermediate I-2. Second, the results of simulations and binding-kinetics measurements are broadly consistent with each other and with inferences from inspection of structures. Mutations that stabilize the CDR H3 loop in the MD simulations, with few exceptions, also increase the association rate in the binding-kinetics experiments (Fig. 3). This agreement directly supports our previous proposal20 that stabilization of the CDR loops in the binding conformation—described earlier in studies of affinity maturation with antibodies directed against small-molecule antigens27–29 and suggested for other protein antigens30—is the primary mechanism of affinity increase in the CH65 lineage.
The proposed mechanisms for preconfiguring the CDR H3 loop require eliminating interactions that disallow the binding-competent conformation as well as adding contributions that stabilize it. The good correlation of simulation and observed binding kinetics strengthens confidence in these proposals (Fig. 3). The very small proportion of the MD trajectory for the UCA during which we see the binding conformation and the interactions that contribute to alternative conformations support our inferences concerning likely unfavorable contributions to the unobserved, binding competent UCA and link the disorder observed in the crystal structure with residues somatically mutated in one or the other of the two branches.
The Y48LC, D49LY and H31LD, S33LR mutation pairs each removes steric interference with the binding-competent conformation of the CDR H3 loop; each also introduces apparent CDR H3 stabilizing contacts. Both mutation pairs gain interactions with R104H. Selection of one pair of mutations would therefore make it unlikely for the second pair to be selected, because the latter would no longer gain affinity through interactions with R104H. Neither of the individual mutations in these pairs has a substantial effect on its own. In the CH65 branch, Y48LC relieves steric interference with L101H, and D49LY adds a favorable interaction with R104H in a binding-competent conformation. In the CH67 branch, displacement of steric interference from the C′C″ loop of the light chain involves formation between the two somatically mutated residues of a salt bridge, and both D31L and R33L contribute new interactions to the binding-competent conformation of the CDR H3 loop. A common feature of the two pairs of mutations is that both of the component changes are needed to eliminate apparent contributions to the binding-incompetent CDR H3 conformation and that at least one of them also contributes to the binding-competent conformation. The inferred molecular mechanism for each of the branches thus supports correlated appearance of a somatically mutated pair.
How can a germinal center reaction select for a pair of somatic mutations, neither of which confers apparent fitness on its own? Recent work on cycling of B-cells between dark and light zones and on the timing of cell divisions suggests that more than one round of cell division (and hence of DNA replication and somatic mutation) can occur in the dark zone before a cell migrates back to the light zone, where antigen on follicular dendritic cells (FDCs) and follicular T helper (Tfh) cells promote selection for enhanced affinity.4,31 A selection regime that alternates several rounds of unselected proliferation with one or more rounds of selection will allow multiple mutations to accumulate and enhance the probability that the individual components of a double mutation will not be lost to negative selection before the two can appear together. Kinetic modeling showed some years ago that there are related, favorable properties of alternating periods of replication and selection.32,33
The reverse mutants CH65HCH65LC48Y,Y49D and CH67HCH67LD31H,R33S maintained sufficient CDR H3 stability and exhibited only modestly decreased affinity for HA (Fig. 3 and Table1), suggesting that the other mutations accumulated during affinity maturation (such as, possibly, the heavy-chain mutations Y33HH and H35HN in the CH65 branch) contributed additional rigidity to the CDR H3 loop. The overall dissociation constant of antibody–antigen binding can be written as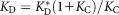 , where
, where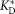 is the hypothetical dissociation constant for the antibody fixed in the antigen-binding conformation. In early antibodies such as I-2, KC ≪ 1, and thus
is the hypothetical dissociation constant for the antibody fixed in the antigen-binding conformation. In early antibodies such as I-2, KC ≪ 1, and thus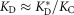 ; a change in KC will lead to an opposite change in KD by the same factor. In mature antibodies with larger values of KC, a change in KC will result in a smaller change in KD. The smaller effect of the reverse mutations on the affinity may be partially attributed to this asymmetry. In the limit of KC ≫ 1, KD is no longer sensitive to a moderate change in KC. Redundant stabilization may thus serve as a buffering mechanism: mutations that destabilize the binding conformation can be tolerated as long as the conformation has been stabilized adequately by prior mutations. Such buffering mechanisms may be a general characteristic of protein evolution.34
; a change in KC will lead to an opposite change in KD by the same factor. In mature antibodies with larger values of KC, a change in KC will result in a smaller change in KD. The smaller effect of the reverse mutations on the affinity may be partially attributed to this asymmetry. In the limit of KC ≫ 1, KD is no longer sensitive to a moderate change in KC. Redundant stabilization may thus serve as a buffering mechanism: mutations that destabilize the binding conformation can be tolerated as long as the conformation has been stabilized adequately by prior mutations. Such buffering mechanisms may be a general characteristic of protein evolution.34
During early stages of affinity maturation, parallel mutational pathways compete with each other for preferential proliferation of the corresponding B-cell progeny; the outcome of the competition will depend on the rate at which mutations enhance affinity for the stimulating immunogen. Analysis of the thermodynamic consequences of mutations in different contexts may complement sequence-based phylogenetic inference35,36 to reconstruct the mutational steps in affinity maturation. Correlating further studies of B-cell population dynamics during affinity maturation37,38 with the corresponding changes in antibody–antigen affinity will advance our understanding of this fundamental stage in the immune response.
Design of immunogens with high affinity for the UCA of a lineage known to have yielded a broadly neutralizing antibody is a potential strategy for inducing otherwise uncommon responses.17 A factor that may limit induction of some broadly neutralizing antibodies is the apparent requirement for complex affinity maturation pathways. Our study of the CH65 clonal lineage shows that these antibodies require relatively few mutations of the accumulated somatic mutations in the mature antibodies to achieve high-affinity HA binding and that more than one set of mutations has produced antibodies with similar properties. These observations may also have implications for the highly somatically mutated broadly neutralizing antibodies against HIV. If only a few “key” mutations are necessary to gain high affinity, targeting key intermediates of a clonal lineage with a designed immunogen may increase the likelihood of developing broad neutralization.
Acknowledgments
The authors thank Michael Eastwood for helpful discussions and a critical reading of the manuscript, Eric Kim and Ansgar Philippsen for assistance with some figures and movies, and Berkman Frank for editorial assistance. They thank members of the Harrison lab for helpful discussions and the Dana–Farber/Harvard Cancer Center DNA Resource Core. SCH is an investigator of the Howard Hughes Medical Institute. The authors note that SCH, AGS, BFH, HXL, MAM, and TBK have a patent on CH65 lineage antibodies.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Eisen HN, Siskind GW. Variations in affinities of antibodies during the immune response. Biochemistry. 1964;3:996–1008. doi: 10.1021/bi00895a027. [DOI] [PubMed] [Google Scholar]
- 2.McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc Natl Acad Sci USA. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 4.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 5.Caton AJ, Browniee GG. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype) Cell. 1982;31:417–427. doi: 10.1016/0092-8674(82)90135-0. [DOI] [PubMed] [Google Scholar]
- 6.de Alwis R, Smith SA, Olivarez NP, Messer WB, Huynh JP, Wahala WM, White LJ, Diamond MS, Baric RS, Crowe JE, Jr, de Silva AM. Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc Natl Acad Sci USA. 2012;109:7439–7444. doi: 10.1073/pnas.1200566109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, Yang Y, Zhu J, Nussenzweig MC, Sodroski J, Shapiro L, Nabel GJ, Mascola JR, Kwong PD. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, Pietzsch J, Fenyo D, Abadir A, Velinzon K, Hurley A, Myung S, Boulad F, Poignard P, Burton DR, Pereyra F, Ho DD, Walker BD, Seaman MS, Bjorkman PJ, Chait BT, Nussenzweig MC. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011;333:1633–1637. doi: 10.1126/science.1207227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, Santelli E, Stec B, Cadwell G, Ali M, Wan H, Murakami A, Yammanuru A, Han T, Cox NJ, Bankston LA, Donis RO, Liddington RC, Marasco WA. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, Vogels R, Brakenhoff JP, Kompier R, Koldijk MH, Cornelissen LA, Poon LL, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science. 2011;333:843–850. doi: 10.1126/science.1204839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez-Rodriguez BM, Agatic G, Bianchi S, Giacchetto-Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JP, Skehel JJ, Lanzavecchia A. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 13.Ekiert DC, Kashyap AK, Steel J, Rubrum A, Bhabha G, Khayat R, Lee JH, Dillon MA, O'Neil RE, Faynboym AM, Horowitz M, Horowitz L, Ward AB, Palese P, Webby R, Lerner RA, Bhatt RR, Wilson IA. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature. 2012;489:526–532. doi: 10.1038/nature11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei C-J, Boyington JC, McTamney PM, Kong WP, Pearce MB, Xu L, Andersen H, Rao S, Tumpey TM, Yang ZY, Nabel GJ. Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science. 2010;329:1060–1064. doi: 10.1126/science.1192517. [DOI] [PubMed] [Google Scholar]
- 15.Ofek G, Guenaga FJ, Schief WR, Skinner J, Baker D, Wyatt R, Kwong PD. Elicitation of structure-specific antibodies by epitope scaffolds. Proc Natl Acad Sci USA. 2010;107:17880–17887. doi: 10.1073/pnas.1004728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burton DR, Poignard P, Stanfield RL, Wilson IA. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science. 2012;337:183–186. doi: 10.1126/science.1225416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol. 2012;30:423–433. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jardine J, Julien JP, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang PS, MacPherson S, Jones M, Nieusma T, Mathison J, Baker D, Ward AB, Burton DR, Stamatatos L, Nemazee D, Wilson IA, Schief WR. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340:711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whittle JRR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, Kepler TB, Liao HX, Harrison SC. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc Natl Acad Sci USA. 2011;108:14216–14221. doi: 10.1073/pnas.1111497108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt AG, Xu H, Khan AR, O'Donnell T, Khurana S, King LR, Manischewitz J, Golding H, Suphaphiphat P, Carfi A, Settembre EC, Dormitzer PR, Kepler TB, Zhang R, Moody MA, Haynes BF, Liao HX, Shaw DE, Harrison SC. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc Natl Acad Sci USA. 2012;110:264–269. doi: 10.1073/pnas.1218256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moody MA, Zhang R, Walter EB, Woods CW, Ginsburg GS, McClain MT, Denny TN, Chen X, Munshaw S, Marshall DJ, Whitesides JF, Drinker MS, Amos JD, Gurley TC, Eudailey JA, Foulger A, DeRosa KR, Parks R, Meyerhoff RR, Yu JS, Kozink DM, Barefoot BE, Ramsburg EA, Khurana S, Golding H, Vandergrift NA, Alam SM, Tomaras GD, Kepler TB, Kelsoe G, Liao HX, Haynes BF. H3N2 influenza infection elicits more cross-reactive and less clonally expanded anti-hemagglutinin antibodies than influenza vaccination. PLoS One. 2011;6:e25797. doi: 10.1371/journal.pone.0025797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw DE, Dror RO, Salmon JK, Grossman JP, Mackenzie KM, Bank JA, Young C, Deneroff MM, Batson B, Bowers KJ, Chow E, Eastwood MP, Ierardi DJ, Klepeis JL, Kuskin JS, Larson RH, Lindorff-Larsen K, Maragakis P, Moraes MA, Piana S, Shan Y, Towles B. 2009. . Millisecond-scale molecular dynamics simulations on Anton. Proc Conf High Performance Computing, Networking, Storage Analysis (SC09). New York: Association for Computing Machinery;
- 23.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 25.Kuroda D, Shirai H, Jacobson MP, Nakamura H. Computer-aided antibody design. Protein Eng Des Sel. 2012;25:507–522. doi: 10.1093/protein/gzs024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kortkhonjia E, Brandman R, Zhou JZ, Voelz VA, Chorny I, Kabakoff B, Patapoff TW, Dill KA, Swartz TE. Probing antibody internal dynamics with fluorescence anisotropy and molecular dynamics simulations. MAbs. 2013;5:306–322. doi: 10.4161/mabs.23651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science. 1997;276:1665–1669. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- 28.Zimmermann J, Oakman EL, Thorpe IF, Shi X, Abbyad P, Brooks CL. Boxer SG, Romesberg FE. Antibody evolution constrains conformational heterogeneity by tailoring protein dynamics. Proc Natl Acad Sci USA. 2006;103:13722–13727. doi: 10.1073/pnas.0603282103. , III, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zimmermann J, Romesberg FE, Brooks CL, III, Thorpe IF. Molecular description of flexibility in an antibody combining site. J Phys Chem B. 2010;114:7359–7370. doi: 10.1021/jp906421v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Li H, Yang F, Smith-Gill SJ, Mariuzza RA. X-ray snapshots of the maturation of an antibody response to a protein antigen. Nat Struct Biol. 2003;10:482–488. doi: 10.1038/nsb930. [DOI] [PubMed] [Google Scholar]
- 31.Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature. 2014;509:637–640. doi: 10.1038/nature13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kepler TB, Perelson AS. Somatic hypermutation in B cells: an optimal control treatment. J Theor Biol. 1993;164:37–64. doi: 10.1006/jtbi.1993.1139. [DOI] [PubMed] [Google Scholar]
- 33.Kepler TB, Perelson AS. Cyclic re-entry of germinal center B cells and the efficiency of affinity maturation. Immunol Today. 1993;14:412–415. doi: 10.1016/0167-5699(93)90145-B. [DOI] [PubMed] [Google Scholar]
- 34.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein, evolution by conformational epistasis. Science. 2007;317:1544–1548. doi: 10.1126/science.1142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volpe JM, Cowell LG, Kepler TB. SoDA: implementation of a 3D alignment algorithm for inference of antigen receptor recombinations. Bioinformatics. 2006;22:438–444. doi: 10.1093/bioinformatics/btk004. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Zhou T, Zhu J, Zhang B, Georgiev I, Wang C, Chen X, Longo NS, Louder M, McKee K, O'Dell S, Perfetto S, Schmidt SD, Shi W, Wu L, Yang Y, Yang ZY, Yang Z, Zhang Z, Bonsignori M, Crump JA, Kapiga SH, Sam NE, Haynes BF, Simek M, Burton DR, Koff WC, Doria-Rose NA, Connors M. Mullikin JC, Nabel GJ, Roederer M, Shapiro L, Kwong PD, Mascola JR. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333:1593–1602. doi: 10.1126/science.1207532. ; NISC Comparative Sequencing Program, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berek C, Milstein C. Mutational drift and repertoire shift in the maturation of the immune response. Immunol Rev. 1987;96:23–41. doi: 10.1111/j.1600-065x.1987.tb00507.x. [DOI] [PubMed] [Google Scholar]
- 38.Shannon M, Mehr R. Reconciling repertoire shift with affinity maturation: the role of deleterious mutations. J Immunol. 1999;162:3950–3956. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.