

Abstract
Circuit computation requires precision in the timing, extent, and synchrony of principal cell (PC) firing that is largely enforced by parvalbumin-expressing, fast-spiking interneurons (PVFSIs). To reliably coordinate network activity PVFSIs exhibit specialized synaptic and membrane properties that promote efficient afferent recruitment such as expression of high-conductance, rapidly gating, GluA4-containing AMPA receptors (AMPARs). We found that PVFSIs upregulate GluA4 during the second postnatal week coincident with increases in the AMPAR clustering proteins NPTX2 and NPTXR. Moreover, GluA4 is dramatically reduced in NPTX2−/−/NPTXR−/− mice with consequent reductions in PVFSI AMPAR function. Early postnatal NPTX2−/−/NPTXR−/− mice exhibit delayed circuit maturation with a prolonged critical period permissive for giant depolarizing potentials. Juvenile NPTX2−/−/NPTXR−/− mice display reduced feedforward inhibition yielding a circuit deficient in rhythmogenesis and prone to epileptiform discharges. Our findings demonstrate an essential role for NPTXs in controlling network dynamics highlighting potential therapeutic targets for disorders with inhibition/excitation imbalances such as schizophrenia.
Introduction
Excitatory afferent recruitment of perisomatic inhibition by PVFSIs dictates synaptic integration properties of downstream excitatory PCs (Pouille and Scanziani, 2001; Gabernet et al., 2005). Such feedforward inhibition provides temporal constraints upon excitation-spike coupling that allow for coordination of firing rates in PC assemblies (Bartos et al., 2007; Klausberger and Somogyi, 2008). Indeed depression of AMPA and/or NMDA receptors (AMPAR/NMDAR) selectively within PVFSIs disrupts their recruitment yielding deficits in PC entrainment that may underlie cognitive deficits associated with psychiatric disorders such as schizophrenia, and predispose circuits to inhibition/excitation (I/E) imbalances that promote epilepsy or Alzheimer’s disease (Fuchs et al., 2007; Belforte et al., 2010; Korotkova et al., 2010; Caputi et al., 2012; Maheshwari et al., 2013). However, relative to PCs, little is known regarding establishment and regulation of excitatory synapses onto PVFSIs. Recently, we found that the immediate early gene product neuronal pentraxin 2 (NPTX2; or NARP) regulates synaptic drive selectively onto PVFSIs by promoting activity-dependent accumulation of GluA4-containing AMPARs (Chang et al., 2010). This regulation by NPTX2 critically dictates PVFSI recruitment to maintain circuit I/E balance following perturbations to network activity. The importance of such homeostatic control of PVFSI synaptic integration is highlighted by findings that NPTX2−/− mice display increased sensitivity to kindling-induced epilepsy and fail to exhibit ocular dominance plasticity (Chang et al., 2010; Gu et al., 2013).
Studies revealing a role for NPTX2 in controlling excitatory drive on hippocampal PVFSIs were complicated by the requirement to pretreat mice with stimuli that induce NPTX2 (Chang et al., 2010). In prior studies, this was accomplished by administering maximal electroconvulsive seizure (MECS) 12 to 18 hrs prior to analysis. We sought to avoid the need for MECS since it will induce other factors that could also contribute to changes in excitability. NPTX2 belongs to the neuronal pentraxin family that also includes the neuronal pentraxin 1 (NPTX1) and neuronal pentraxin receptor/3 (NPTXR/3). All three NPTXs are expressed in adult brain, and form disulfide linked mixed assemblies that bind AMPARs (Xu et al., 2003; Sia et al., 2007). Only NPTX2 is induced by activity (Tsui, et al., 1996; Xu et al., 2003). NPTXR is unique in possessing a transmembrane domain and can function to anchor NPTX complexes to plasma membranes. Based on their known co-functionality, we examined NPTX2−/−/NPTXR−/− mice and found they exhibit basal deficits in PVFSI AMPAR function. Accordingly, NPTX2−/−/NPTXR−/− mice provide a unique opportunity to assess the impact of NPTXs on synapse development, as well as cellular and circuit function, without a requirement for perturbed activity. We report that combined loss of NPTX2 and NPTXR markedly reduces GluA4 expression leading to reduced PVFSI AMPAR function and feedforward inhibition. The resulting I/E imbalance in NPTX2−/−/NPTXR−/− mice disrupts hippocampal rhythmogenesis, promotes epileptic activity, and impairs hippocampal-dependent working memory.
Results and Discussion
In the mature hippocampus GluA4-containing AMPARs play an important role at excitatory synapses on PVFSIs due to their fast kinetics and high conductance which promote efficient synaptic recruitment (Geiger et al., 1995; Geiger et al., 1997; Fuchs et al., 2007). This specialized role is highlighted by the limited expression of GluA4 within the mature hippocampus being excluded from PCs and segregated to a sparse population of large cells concentrated around stratum pyramidale of the CA1-CA3 regions and at the hilar-granule cell layer border in the dentate gyrus consistent with PVFSI localization (Fig. 1A). However, early developmental progression of GluA4 in PVFSIs remains largely unknown due to late PV onset (Seto-Ohshima et al., 1990). To address this, we employed Nkx2-1-cre:RCE GFP or Nkx2-1-cre:tdTOM RFP mice that report the majority of medial ganglionic eminence (MGE) derived interneurons, including PVFSIs (Tricoire et al., 2010). Immunohistochemical (IHC) analyses revealed the emergence of GluA4+/GFP+ (or RFP+, together considered XFP+) cells at postnatal day 5 (P5), then increasing roughly tenfold by P14, and leveling off through P40 (Fig. 1B–C). This contrasts with findings that GluA4 in hippocampal homogenates is high at P1 then decreases becoming undetectable by P18 (Zhu et al., 2000), highlighting the importance of cell-type specific evaluation for minority cell populations within a circuit. However, our findings are consistent with progressive upregulation of GluA4 mRNA within putative PVFSIs over the first two postnatal weeks (Okaty et al., 2009). Thus, GluA4 can be grouped together with a number of hallmark proteins expressed by mature PVFSIs such as PV itself, Kv3 channels, synaptotagmin 2, Nav1.1 sodium channels, and 2-pore domain potassium leak channels (TWIK1 and TASK1/3) that are coordinately upregulated during the first few postnatal weeks and function in a concerted fashion to differentiate immature interneurons fated to become PVFSIs into fast perisomatic targeting inhibitory devices (Du et al., 1996; Doischer et al., 2008; Okaty et al., 2009; Goldberg et al. 2011).
Figure 1. Developmental profile of hippocampal GluA4 expression.
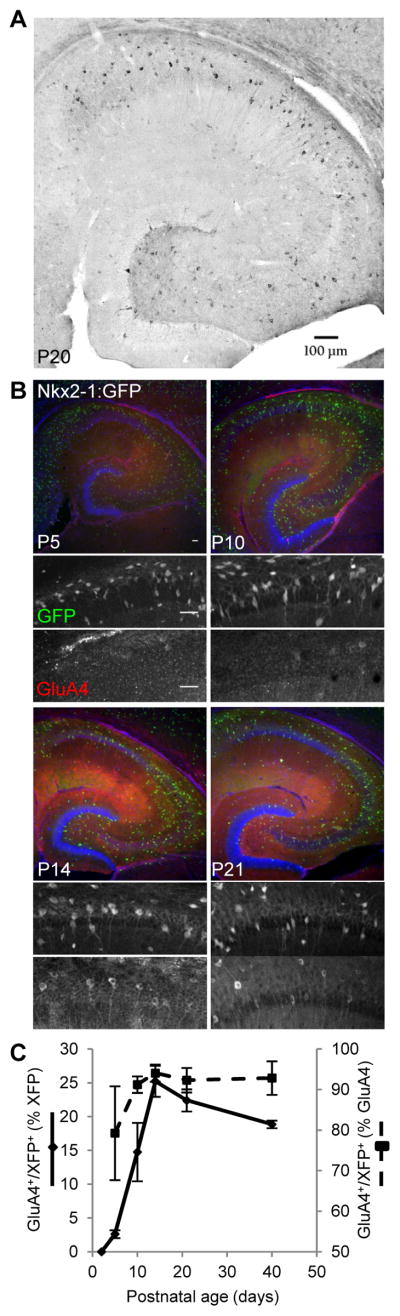
(A) Representative image illustrating the mature immunolabeling pattern of GluA4 in wild type hippocampus (bar, 100 μm). (B Low magnification (10X) and digitally zoomed panels reveal) expression of GFP and GluA4 in representative sections from Nkx2-1-cre:RCE mice at the indicated ages (bars, 100/50 μm for low/high mag. images respectively). (C) Group data summarizing the developmental expression of GluA4/XFP double positive cells as percentages of the XFP+ population (solid line, left axis) and GluA4+ population (dashed line, right axis). Mice/sections counted: 2/6, P2; 3/9, P5; 4/10, P10, P14, P21 and P40.
The developmental increase in GluA4 temporally coincides with upregulation of the AMPAR interacting partners NPTX2 and NPTXR (Cho et al., 2008). Based on their known co-functionality we examined the necessity of these NPTXs for normal PVFSI function by probing GluA4 expression in P21-30 mice lacking NPTX2 and NPTXR (NPTX2−/−/NPTXR−/−). Surprisingly, specific somatic GluA4 signal was absent throughout the hippocampus of NPTX2−/−/NPTXR−/− mice (Fig. 2A–C). This loss was particularly striking for PV+ cells, which comprised the vast majority of GluA4+ cells in wild type tissue (Fig. 2B–C). To confirm the loss of GluA4 throughout NPTX2−/−/NPTXR−/− PVFSIs we also probed hippocampal cultures. Double labeling for PV and GluA4 confirmed a profound reduction in GluA4 signal throughout the dendritic arbors of PV-expressing cells cultured from NPTX2−/−/NPTXR−/− mice (Fig. 2D–E). Even when GluA4+ cells were encountered in NPTX2−/−/NPTXR−/− cultures, dendritic expression was comparatively weak (supplemental Fig. 1A), suggesting that synaptic GluA4 is not spared at the expense of somatic extrasynaptic GluA4. Indeed subcellular evaluation of GluA4 at asymmetric synapses onto PV immunopositive processes in hippocampal sections confirmed that PVFSIs in NPTX2−/−/NPTXR−/− mice are deficient in synaptic GluA4 content (Fig. 2F–G). Importantly, GluA4 expression persisted in PV+ cells cultured from either NPTX2−/− or NPTXR−/− mice and the total number of PV+ cells expressing GluA4 was similar to wild types (Fig. 2E and supplemental Fig 1A). These observations are consistent with our prior findings of continued GluA4 expression in NPTX2−/− PVFSIs (Chang et al., 2010) and further indicate that NPTX2 and NPTXR compensate for each other to some degree in regulating PVFSI GluA4. Indeed presynaptically derived NPTX2 and NPTXR have both individually been implicated in AMPAR clustering (Sia et al., 2007; Chang et al., 2010). As loss of NPTX2 or NPTXR alone does not lead to compensatory increases of other pentraxins (Bjartmar et al., 2006), residual basal NPTX2 and NPTXR appear capable of driving significant GluA4 expression. Consistent with our IHC findings biochemical analyses of hippocampal synaptosomal and synaptic plasma membrane preparations (SPMs) further confirmed a dramatic reduction in GluA4 levels in NPTX2−/−/NPTXR−/− mice relative to wild type and NPTX2−/− or NPTXR−/− mice (Fig. 2H–J). Moreover, loss of GluA4 in NPTX2−/−/NPTXR−/− mice was evident in cerebellum (supplemental Fig. 1B–C), suggesting a conserved role for pentraxins in regulating GluA4 expression throughout the nervous system.
Figure 2. Loss of GluA4 in NPTX2−/−/NPTXR−/− mice.

(A) GluA4 expression in representative sections from P21 WT and NPTX2−/−/NPTXR−/− mice (top panels; 10X, bar 200μm) with zoomed images of CA1 and DG (bottom panels; bar 50μm). (B) Representative images illustrating GluA4 and PV expression in CA1 of P21WT and NPTX2−/−/NPTXR−/− mice (20X, bar 50μm). (C) Summary plots of GluA4+/PV+ double positive cells as percentages of the GluA4+ population (left axis) and the density of GluA4+ cells (right axis) in WT and NPTX2−/−/NPTXR−/− mice. Mice/sections counted: 5/12, WT; 6/14, NPTX2−/−/NPTXR−/−. (D) Representative images of PV (green), GluA4 (red), and DAPI (blue) staining in dissociated hippocampal cultures from wild type and NPTX2−/−/NPTXR−/− mice. Low magnification images (upper, bar 150 μm) show merged fluorescence signals and digitally zoomed panels (lower panels, bar, 40 μm) from boxed dendritic regions above highlight GluA4 and PV signals in isolation. (E) Histogram summarizing the percentage of PV+ cells that express GluA4+ in wild type, NPTX2−/−, NPTXR−/−, and NPTX2−/−/NPTXR−/− mice. A total of 127 PV+ cells from 19 culture dishes of wild type pups from three different litters, 208 PV+ cells from 19 culture dishes of NPTX2−/−/NPTXR−/− mice from 3 different litters, 548 PV+ cells from 12 dishes of NPTX2−/− pups from 3 litters, and 310 PV+ cells from 12 culture dishes from 3 NPTXR−/− litters were examined, with each litter representing a single n. For each litter paired wild type and knockout cultures were produced, processed, and analyzed in parallel. (F) Sample electron micrographs from hippocampal sections of wild type and NPTX2−/−/NPTXR−/− mice illustrating asymmetric synapses (arrowheads) in PV immunopositive processes (15 nm gold, *) probed for GluA4 (5nm gold, arrows in WT) (bars 100 nm). (G) Histogram summarizing the density of GluA4 immunogold label observed at asymmetric synapses onto PV immunopositive dendrites in wild type and NPTX2−/−/NPTXR−/− mice. Insets show the percentage of GluA4 labeled versus unlabeled asymmetric synapses in PV immunopositive processes of wild type (top) and NPTX2−/−/NPTXR−/− (bottom) mice. A total of 152 assymetric synapses from 2 wild type mice and a total of 82 assymetric synapses from 3 NPTX2−/−/NPTXR−/− mice were examined. (H) Representative sample western blots for GluA4, along with β-actin, from hippocampal synaptosomal preparations obtained from wild type, NPTX2−/−, NPTXR−/−, and NPTX2−/−/NPTXR−/− mice. (I) Plotted is a quantitative summary of GluA4 levels in hippocampal synaptosomal preparations from wild type, NPTX2−/−, NPTXR−/−, and NPTX2−/−/NPTXR−/− mice. The amount of GluA4 is expressed relative to β-actin and a total of 18 mice from 14 independent litters for each genotype were paired and analyzed in parallel. (J) At left is a representative sample immunoblot of discrete biochemical fractions from wild type hippocampus probed for GluA4 illustrating preferential partitioning of GluA4 into the SPM fraction (S2= post-nuclear supernatant, P2= pellet 2, S3= supernatant 3, I= input for sucrose gradient, LM, light membranes; MM, microsomal membranes; SPM, synaptic plasma membranes; Mito, mitochondria). At right is a representative sample immunoblot of wild type and NPTX2−/−/NPTXR−/− SPMs run in parallel and probed for GluA4 (upper blot) and actin (lower blot) illustrating the loss of GluA4 signal in NPTX2−/−/NPTXR−/− hippocampal synapses (SPM GluA4/actin = 5.8±1.3 and 0.57±0.08 for WT and NPTX2−/−/NPTXR−/− respectively; p=0.02, n=3 mice per genotype). Values plotted throughout are mean±s.e.m.; * p<0.05, Student’s t-test or Mann-Whitney U Test as appropriate. See also Figure S1.
Maturation and circuit integration of PVFSIs has been linked to the formation of perineuronal nets (PNNs) that accumulate preferentially around these cells and control critical period plasticity during network development (Pizzorusso et al., 2002; Gogolla et al., 2009). Interestingly, PNN formation surrounding PVFSIs proceeds with a developmental profile that directly parallels GluA4 expression within PVFSIs (Fig. 3A–B). Moreover, PNNs are implicated in NPTX2-mediated GluA4 clustering in mature PVFSIs as a homeostatic response to elevated circuit activity (Chang et al., 2010). Thus, it is conceivable that failure of NPTX2−/−/NPTXR−/− PVFSIs to upregulate GluA4 relates to deficient PNN formation. However, we found no difference in PNNs labeled with Wisteria floribunda agglutinin (WFA) surrounding PV+ cells of wild type and NPTX2−/−/NPTXR−/− hippocampal sections, revealing that NPTX2 and NPTXR are not instructive for PNNs (Fig. 3C–D). In addition to PNNs the AMPAR accessory protein stargazing/TARPγ2 is enriched in PVFSIs and has been implicated in regulating PVFSI AMPAR content (Tomita et al., 2003; Maheshwari et al., 2013; Tao et al., 2013). Consistent with these reports immunoprecipitation of GluA4 from hippocampal synaptic plasma membranes co-precipitates stargazin in wild type mice (Fig. 3E) raising the possibility that the loss of GluA4 in NPTX2−/−/NPTXR−/− mice occurs secondarily to loss of stargazin. Indeed in stargazin mutant mice GluA4 is reportedly reduced at parallel fiber-interneuron synapses of the cerebellum (Shevtsova and Leitch, 2012) and selectively lost in nucleus reticularis of the thalamus (RTN) (Barad et al., 2012). However, biochemical analyses revealed continued expression of stargazin in the hippocampus of NPTX2−/−/NPTXR−/− mice with preferential partitioning into the SPM fraction similar to wild type mice (Fig. 3F). Moreover, an antibody that recognizes stargazin (and TARPγ8) continues to label NPTX2−/−/NPTXR−/− PV+ cells (Fig. 3G) and GluA4 remains present in stargazin−/− mice (supplemental Fig. 1D). The continued expression of GluA4 in PVFSIs of stargazin−/− mice conflicts with findings at corticothalamic inputs to RTN and may reflect compensation in hippocampal PVFSIs by TARPγ8 which also co-precipitated with hippocampal GluA4 (data not shown). Nonetheless, our findings clearly indicate that loss of GluA4 in NPTX2−/−/NPTXR−/− mice is not related to changes in stargazin. We also probed whether NPTX2−/−/NPTXR−/− mice exhibit altered expression of ErbB4, a receptor tyrosine kinase implicated in PVFSI AMPAR expression and localization (Fazzari et al., 2010; Ting et al., 2011). While ErbB4 was found to interact with GluA4 we did not detect any changes in ErbB4 levels of NPTX2−/−/NPTXR−/− mice (supplemental Fig. 1E–G). Despite the profound reduction of GluA4 protein in NPTX2−/−/NPTXR−/− mice, in situ hybridization and quantitative PCR revealed similar expression of GluA4 mRNA in wild type and NPTX2−/−/NPTXR−/− mice (Fig. 3H–K). Thus, NPTX2−/−/NPTXR−/− PVFSIs are not deficient in GluA4 transcription, suggesting a role in regulating GluA4 translation or protein stability.
Figure 3. Normal PNN, stargazin, and GluA4 mRNA levels in NPTX2−/−/NPTXR−/− mice.

(A) Representative images of PNN staining with WFA in Nkx2-1-cre:RCE hippocampal sections obtained at the developmental time points indicated (bar 100μm). (B) Group data plot illustrating the time course for PNN formation around GFP+ cells in Nkx2-1-cre:RCE mice. Mice/sections counted: 2/6, P2; 3/3, P5; 2/6, P10; 3/5 P14; 2/6 P21; and 3/5 P40. (C,D) Representative images (C) and group data (D) illustrating the co-localization of PNNs and PV+ cells in hippocampal sections obtained from P21-30 wild type and NPTX2−/−/NPTXR−/− mice (bar 35μm). Mice/sections counted: 5/5, WT; 2/5, NPTX2−/−/NPTXR−/−. (E) Representative immunoblots illustrating that pull-down of GluA4 from hippocampal synaptic plasma membranes co-precipitates stargazin (γ2) as well as GluA4 itself (IP lanes). Also shown are the input (IN) and flow through (FT) materials probed with the same antibodies (comparing FT/IN signals revealed that 55±9% of available SPM stargazin co-precipitated with GluA4, n=4 mice). (F) Representative western blots of the indicated cell fractions from wild type and NPTX2−/−/NPTXR−/− hippocampi probed with an antibody that recognizes stargazin (γ2) and TARPγ8 (γ8; upper blot) or GluA4 (lower blot). SPM stargazin/actin = 0.38±0.06 and 0.34±0.06 for wild type and NPTX2−/−/NPTXR−/− mice respectively, p=0.66, n=3 mice per genotype. (G) Representative images of PV+ cells from wild type and NPTX2−/−/NPTXR−/− mice cultures confirming stargazin/γ8 expression in PV+ cells in both genotypes (scale bar 50 μm). (H) Representative single trial qPCR amplification plots for GluA4 mRNA (solid black), PV (grey), and GADPH (hashed black) from wild type and NPTX2−/−/NPTXR−/− hippocampi. Relative fluorescence intensities (deltaRn) are plotted against PCR cycle numbers on a logarithmic scale. (I) Group data summarizing the abundance of GluA4 mRNA (relative to GADPH) in hippocampi from wild type and NPTX2−/−/NPTXR−/− mice. 3 independent experiments using RNA from 3 independent litters of each genotype were performed. (J,K) Representative sample images (J) and group data summary (K) of wild type and NPTX2−/−/NPTXR−/− hippocampal sections probed for PV and GluA4 mRNA by fluorescent in situ hybridization (bar 50μm). Cells/sections/mice counted: 165/4/2, WT; 196/6/3, NPTX2−/−/NPTXR−/−. See also Figure S1.
To determine if the loss of GluA4 in NPTX2−/−/NPTXR−/− PVFSIs translates to a functional synaptic deficit we examined excitatory inputs to PVFSIs. NPTX2−/−/NPTXR−/− mice exhibited a severe reduction in AMPA/NMDA ratios at both granule cell- and medial perforant path-inputs to PVFSIs (Fig. 4A–B). This was not accompanied by changes in paired-pulse ratio (PPR) or AMPAR rectification, suggesting that presynaptic function is unaltered and that residual AMPARs remain calcium-permeable (Fig. 4A–B), likely comprised of GluA1 homomers (Geiger et al., 1997). As pentraxins do not interact with NMDARs the altered AMPA/NMDA ratio in NPTX2−/−/NPTXR−/− mice likely reflects reduced AMPAR-mediated transmission rather than enhanced NMDAR function. Indeed AMPAR-mediated spontaneous excitatory postsynaptic currents (sEPSCs) exhibited slower decay kinetics and were reduced in amplitude and frequency in NPTX2−/−/NPTXR−/− PVFSIs versus wild type controls (Fig. 4C–D). Interestingly, deficits in PVFSI AMPAR function were not observed in either NPTX2−/− or NPTXR−/− single knockouts (Fig. 4B,D) despite modest decreases in total hippocampal GluA4 probed biochemically (Fig. 2I). This lack of synaptic phenotype in single knockouts is consistent with our prior findings (Chang et al., 2010) and the continued expression of GluA4 in most PVFSIs of NPTX2−/− or NPTXR−/− mice (Fig. 2E).
Figure 4. Impaired PVFSI AMPAR function and feedforward inhibition in NPTX2−/−/NPTXR−/− mice.

(A) Representative PVFSIs recorded in wild type and NPTX2−/−/NPTXR−/− mice with corresponding AMPA and NMDA EPSC traces evoked by granule cell stimulation (bars, 100μm). (B) Summary for AMPA/NMDA ratios in PVFSIs at granule cell (MF, n=16cells/8mice, 4cells/4mice, 8cells/5mice, 15cells/7mice for WT, NPTX2−/−, NPTXR−/−, NPTX2−/−/NPTXR−/− respectively; *p=0.00007 vs. WT, Mann-Whitney U test) and medial perforant path (PP, n=15 cells/6mice, 6 cells/3mice, 7cells/3mice for WT, NPTXR−/− and NPTX2−/−/NPTXR−/− respectively; *p=0.02 vs. WT Mann-Whitney U test) inputs for indicated genotypes (PP inputs not determined (ND) for NPTX2−/− mice). Also plotted are PPRs (5Hz) of MF-PVFSI EPSCs. Inset, I–V relation for AMPAR-mediated transmission, and sample traces, at PP inputs to NPTX2−/−/NPTXR−/− PVFSIs (n=5cells/3mice). (C) Representative sEPSC recordings (left) and ensemble average sEPSC (right) recorded in WT and NPTX2−/−/NPTXR−/− PVFSIs. (D) Summary of PVFSI sEPSC properties for the indicated genotypes (n=39cells/16mice, 23cells/8mice, 14cells/7mice, 20cells/9mice for WT, NPTX2−/−, NPTXR−/−, NPTX2−/−/NPTXR−/− respectively; *p=0.002, 0.03, 0.01, and 0.01 for Amplitude, Taudecay, Frequency and Frequency, respectively vs. WT) and cumulative probability plots comparing WT and NPTX2−/−/NPTXR−/− PVFSIs (insets). (E) Schematic (above, left) and sample traces (below, left) illustrating methodology for recording SC-CA1 feedforward inhibition, along with representative sample recordings in wild type and NPTX2−/−/NPTXR−/− mice (right). (F) Group data summary plot of I/E ratios observed using train stimulation in wild type and NPTX2−/−/NPTXR−/− mice (n=10cells/3mice and 9cells/3mice for wild type and NPTX2−/−/NPTXR−/− respectively; p=0.04, 0.005, 0.02, 0.008 for P1,P3,P4,P5 respectively, Mann-Whitney U test). Inset shows SC-CA1 excitatory field potential recording (fEPSP) input-output relations, with traces from representative recordings, for wild type and NPTX2−/−/NPTXR−/− mice. (G) Image of a representative NPTX2−/−/NPTXR−/− dentate PVFSI (bar, 100μm) and associated voltage responses (lower inset) to hyperpolarizing current injection (-200pA) as well as depolarizing current injections peri-threshold (250pA) and twice peri-threshold (500pA) for action potential firing (inset bars, 250ms/20mV). (H) Histogram summarizing resting membrane potentials (Vrest), input resistances (Rin), action potential thresholds (Vthresh), firing frequencies at twice threshold current injection (f @2Xthresh), and maximal firing frequencies (fmax) measured in wild type (n=16 cells/7 mice) and NPTX2−/−/NPTXR−/− (n=9 cells/4 mice) PVFSIs. (I) Continuous traces (upper) from representative CA1 PC recordings in wild type (left) and NPTX2−/−/NPTXR−/− (right) mice illustrating the effects of oxytocin receptor agonist ((Thre4, Gly7)-oxytocin, TGOT) treatment to evoke release from PVFSIs on sIPSCs. Traces below show the indicated regions on an expanded time scale. (J) Summary time course plot of the effects of TGOT on sIPSC amplitudes in wild type (n=6 cells from 3 mice) and NPTX2−/−/NPTXR−/− (n=7 cells from 3 mice) CA1 PCs. sIPSC amplitudes measured during 30s epochs during TGOT treatment were normalized to the average amplitude measured prior to TGOT application. Also plotted is the effect of TGOT in wild type CA1 PCs treated with Omega-Agatoxin (AgTX, 500 nM; n = 4 cells from 2 mice) to prevent release from PVFSIs confirming that TGOT selectively drives sIPSC output from PVFSIs (Hefft and Jonas, 2005; Owen et al., 2013). Values plotted throughout are average±s.e.m. Recordings throughout were made in slices from mice aged P15-P30. See also Figure S2.
Reduced AMPAR function in PVFSIs is expected to disrupt efficient synaptic recruitment of these cells, and thus, compromise feedforward perisomatic inhibition promoting a circuit I/E imbalance in downstream PCs (Pouille and Scanziani, 2001; Gabernet et al., 2005). To probe whether reduced AMPAR function in NPTX2−/−/NPTXR−/− PVFSIs alters I/E balance we examined disynaptic inhibition in the Schaffer collateral-CA1 pyramidal cell (SC-CA1 PC) circuit. Compared to wild types, NPTX2−/−/NPTXR−/− mice exhibited reduced feedforward inhibition when normalized to monosynaptic excitation in SC-CA1 PC recordings (Fig. 4E–F). As SC-CA1 PC excitation was not different between wild type and NPTX2−/−/NPTXR−/− slices (inset Fig. 4F) the altered I/E ratio reflects a reduction in recruited inhibition rather than increased excitatory drive at SC-CA1 synapses. Importantly, total density of PVFSIs was comparable between wild type and NPTX2−/−/NPTXR−/− mice indicating that similar numbers of PVFSIs are available for recruitment in the two genotypes (PV+ cell density of NPTX2−/−/NPTXR−/− was 91±7% of that observed in wild types, eg. Figs. 2B, 3C). Indeed the developmental trajectory of hippocampal PV expression was similar in wild type and NPTX2−/−/NPTXR−/− mice (supplemental Fig. 2A–B). Moreover, basic membrane properties of NPTX2−/−/NPTXR−/− PVFSIs were comparable to wild types (Fig. 4G–H). Finally, PVFSI GABA release probed with the oxytocin receptor agonist TGOT, which selectively drives PVFSI firing (Owen et al., 2013), was similar in wild type and NPTX2−/−/NPTXR−/− slices (Fig. 4I–J). Thus, reduced disynaptic inhibition in NPTX2−/−/NPTXR−/− mice likely reflects diminished PVFSI afferent driven recruitment due to impaired AMPAR function within these cells rather than deficits in PVFSI numbers, impaired spike generation, or transmitter release. Interestingly, the reduced PVFSI AMPAR function and consequent disruption of feedforward inhibition in NPTX2−/−/NPTXR−/− are similar in magnitude to observations in GluA4−/− mice (Fuchs et al., 2007). This is consistent with the almost complete absence of GluA4 in NPTX2−/−/NPTXR−/− mice and reinforces the critical role of NPTXs in regulating GluA4 expression to control PVFSI circuit integration.
Failure of circuits to effectively recruit PVFSIs and the ensuing I/E imbalance has the capacity to disrupt coherent population activity since PVFSIs critically entrain PC assemblies (Bartos et al., 2007; Klausberger and Somogyi, 2008). Thus, we next probed physiological and pathological hippocampal network activity in NPTX2−/−/NPTXR−/− mice. We first examined the immature hippocampus beginning on postnatal day 6 (P6) when circuit activity is dominated by giant depolarizing potentials (GDPs). GDPs occur spontaneously in immature networks to drive correlated population activity and are paced by depolarizing GABAergic inhibition prior to the onset of mature chloride gradients within PCs (Ben-Ari et al., 2007). In recordings from CA3 PCs we reliably observed GDPs in wild type and NPTX2−/−/NPTXR−/− mice between P6-8 in every PC examined (Fig. 5A–D). Then between P9-P13 the occurrence of GDPs decreased in both genotypes, but with distinct temporal dynamics (Fig. 5C). In wild types less than 50% of CA3 PCs exhibited GDPs at P11 and no GDPs were detected beyond this age. In contrast, greater than 50% of the PCs examined in NPTX2−/−/NPTXR−/− mice still exhibited GDPs at P12, yielding a significant increase in the overall frequency of GDPs across all recordings at P11-P12 (Fig. 5D). This altered time course of GDPs within the NPTX2−/−/NPTXR−/− hippocampus is reminiscent of disrupted critical period plasticity and synaptic refinement in the developing visual system of pentraxin deficient mice (Bjartmar et al., 2006; Koch and Ullian, 2010; Gu et al., 2013) suggesting a general role for synaptic integration of PVFSIs in coordinating circuit maturation.
Figure 5. Prolonged critical period for GDP expression in NPTX2−/−/NPTXR−/− mice.

(A,B) Representative sample traces illustrating GDPs recorded in slices from wild type (A) and NPTX2−/−/NPTXR−/− mice with individual GDPs illustrated below on an expanded time scale. (C–F) Histograms summarizing the developmental profiles (ages indicated along x-axis) for the percentage of CA3 PCs exhibiting GDPs (C), GDP frequency averaged across recorded cells for each stage of development (D), GDP half widths observed across development (E) and the average total charge transferred through GDPs at each stage of development (F) in wild type and NPTX2−/−/NPTXR−/− slices. For wild type recordings the total number of cells and mice recorded at each time point (P6-7 to P13) were: 15/3;7/2;12/2;10/2;12/2;11/3;6/1. For NPTX2−/−/NPTXR−/−mice: 19/3;18/3;14/2;24/3;13/2;14/2;8/1. * in D p=0.015, Mann-Whitney U test. Note, as GDPs were not observed at P12 and 13 in wild type mice we did not perform any statistical comparisons with GDP properties of NPTX2−/−/NPTXR−/− mice at these stages. (G–H) Summary plots illustrating the effects of cannabinoid receptor antagonism with WIN 55,212-2 mesylate (WIN, 5μM) and NMDAR antagonism with DL-APV (APV, 100μM) on GDP frequencies in wild type and NPTX2−/−/NPTXR−/− mice. Plotted are GDP frequencies observed in each recording before (Ctl) and after WIN or APV treatment. In G **, p=0.007 and 9.7×10−4 for WT and NPTX2−/−/NPTXR−/− respectively (Wilcoxon test). In H *, p=0.026 and 0.015 for wild type and NPTX2−/−/NPTXR−/− respectively (paired t-test).
Despite the prolonged developmental window to observe GDPs in NPTX2−/−/NPTXR−/− mice other GDP features, including frequency at younger ages, duration, and charge transfer were comparable to wild type mice indicating similar synaptic dynamics underlying GDP emergence and pacing in both genotypes (Fig. 5D–F). Indeed GDP frequency in both wild type and NPTX2−/−/NPTXR−/− mice was sensitive to modulation by cannabinoid and NMDA receptor tone as previously reported (Fig. 5G–H) (Ben-Ari et al., 1989; Bernard et al., 2005). Thus, rather than directly participating in the cellular and synaptic mechanisms that drive GDPs, PVFSIs likely function to terminate the permissive window for GDP generation perhaps by triggering the establishment of hyperpolarizing chloride gradients following initiation of perisomatic innervation (Ben-Ari et al., 2007). This model is consistent with pacing of GDPs by dendritic inhibition rather than perisomatic inhibition (Ben-Ari et al., 2007), a feature highlighted by the strong regulation by cannabinoid receptors which likely operate at dendrite targeting cholecystokinin-expressing interneurons (Morozov and Freund, 2003; Lee et al., 2010).
Cessation of the critical period for GDP generation in the maturing hippocampus signals a transition from spontaneously internal circuit generated network activity to mature oscillatory patterns driven by behavior such as sensory processing, spatial navigation, learning, and memory retrieval. Unlike GDPs, pacing of mature network rhythms critically relies upon PVFSI-mediated perisomatic inhibition to entrain PC assemblies, particularly in the gamma frequency range (Bartos et al., 2007; Klausberger and Somogyi, 2008). To determine whether changes in I/E balance observed in NPTX2−/−/NPTXR−/− mice alters circuit rhythmogenesis in juvenile mice we examined carbachol (CCh) induced gamma oscillations (Mann et al., 2005). Field recordings in the CA3 PC layer of hippocampal slices revealed that gamma power was markedly impaired in NPTX2−/−/NPTXR−/− slices compared to interleaved wild type slices (Fig. 6A–F). Consistent with our PVFSI synaptic analyses, gamma power in NPTX2−/− and NPTXR−/− single knockout mice was comparable to wild types (Fig. 6C–D). While average peak gamma frequency was similar between the four genotypes (Fig. 6E), both NPTXR−/− and NPTX2−/−/NPTXR−/− mice had greater variability in gamma frequency, indicating less stable oscillations (Fig. 6F). The minor phenotype in the absence of just NPTXR−/− may reflect a small deficit in excitatory drive onto PVFSIs relating to reduced sEPSC frequency in this mouse (Fig. 4D).
Figure 6. Impaired gamma oscillations in NPTX2−/−/NPTXR−/− mice both in vitro and in vivo.

(A,B) In vitro gamma oscillations detected in field recordings (top and middle) from CA3 in slices from wild type (A) or NPTX2−/−/NPTXR−/− (B) mice after bath application of 25μM carbachol (CCh), with corresponding Wavelet transform (bottom). (C) Power density spectra from representative recordings of CCh-evoked gamma oscillations in wild type (black), NPTX2−/− (red), NPTXR−/− (blue) and NPTX2−/−/NPTXR−/− (green) mice. Inset shows corresponding sample traces. (D) NPTX2−/−/NPTXR−/− mice had significant gamma oscillation deficits (Log10 peak gamma power, WT (n=21) vs NPTX2−/− (n=8) vs NPTXR−/− (n=15) vs NPTX2−/−/NPTXR−/− (n=24); −2.89 ± 0.11 vs −3.17 ± 0.14 vs −2.92 ± 0.16 vs −3.43 ± 0.12; p=0.0074, one-way ANOVA with post hoc Bonferroni multiple comparisons); inset shows cumulative frequency distributions of the peak gamma power for WT and NPTX2−/−/NPTXR−/− mice. (E) No significant differences in peak gamma frequency were observed (WT vs NPTX2−/− vs NPTXR−/− vs NPTX2−/−/NPTXR−/−: 33.7 ± 0.75 Hz vs 34.1 ± 1.00 Hz vs 31.7 ± 1.23 Hz vs 33.5 ± 1.07 Hz; F=0.898, p=0.447, one-way ANOVA). (F) NPTXR−/− and NPTX2−/−/NPTXR−/− mice displayed significantly greater variability in gamma frequency (deviation from mean frequency, WT vs NPTX2−/− vs NPTXR−/− vs NPTX2−/−/NPTXR−/−: 1.80 ± 0.43 Hz vs 2.34 ± 0.47 Hz vs 4.04 ± 0.59 Hz vs 4.51 ± 0.65 Hz; H=16.64, p=0.0008, Kruskal-Wallis test with post hoc Dunn’s multiple comparisons test). (G, H) Representative in vivo recordings from CA1 stratum pyramidale of wild type (G) or NPTX2−/−/NPTXR−/− (H) mice, showing raw traces (top), the same traces band-pass filtered between 25 and 90Hz to show the gamma band (middle) and band-pass filtered between 5 and 10Hz to show the theta band (bottom). Traces at right with expanded time scale are from boxed regions of the longer time compressed traces. (I) Normalized power spectra, averaged from all mice in wild type (black, n=6) or NPTX2−/−/NPTXR−/− (green n=6) groups. (J) NPTX2−/−/NPTXR−/− mice displayed a significant reduction in peak gamma power (z-score, WT vs NPTX2−/−/NPTXR−/− : 2.13 ± 0.27 vs 1.34 ± 0.20; t=2.410; p=0.0367; Student’s t test) and (K) peak gamma frequency WT vs NPTX2−/−/NPTXR−/− : 38.0 ± 0.86 vs 33.2 ± 1.03; t=3.608; p=0.0048; Student’s t test). (L) NPTX2−/−/NPTXR−/− mice did not display significant differences in peak theta power (z-score, WT vs NPTX2−/−/NPTXR−/− : 4.38 ± 1.01 vs 5.37 ± 0.71; t=0.7938; p=0.4457, Student’s t test) but (M) did display a small but significant decrease in peak theta frequency (WT vs NPTX2−/−/NPTXR−/− : 7.50 ± 0.18 vs 6.75 ± 0.28; t=2.236; p=0.0493; Student’s t test). *, p <0.05 **, p < 0.01; ***, p < 0.001 vs wild type. In vitro recordings were performed in slices from mice aged P15-21 and in vivo recordings were performed in P32-34 mice.
To determine whether naturally occurring in vivo oscillations are disrupted in NPTX2−/−/NPTXR−/− mice we performed extracellular recordings from hippocampal CA1 in awake, head-fixed mice. Analyses of the local field potential recorded in stratum pyramidale revealed clear episodes of theta and gamma activity during active states in aged-matched wild type and NPTX2−/−/NPTXR−/− mice indicating that these rhythms remain intact in the mutants (eg. Fig. 6G–H). However, examination of the averaged power spectra across all recordings revealed deficient power in the gamma frequency range of NPTX2−/−/NPTXR−/− mice compared to wild types (Fig. 6I). Indeed further analysis confirmed a significant reduction in peak gamma power of NPTX2−/−/NPTXR−/− mice that was accompanied by a modest decrease in peak gamma frequency (Fig. 6J–K). In contrast peak theta power was similar between wild type and NPTX2−/−/NPTXR−/− mice, though the mutants exhibited a small reduction in peak theta frequency (Fig. 6L–M).
In addition to theta and gamma oscillations we also observed sharp wave-associated ripple events (SWRs) during periods of quiet wakefulness in wild type and NPTX2−/−/NPTXR−/− mice (eg. Fig. 7A–B). These high frequency oscillations (125–250Hz) are thought to drive memory consolidation during periods of immobility, consummatory behaviours, and slow wave sleep by allowing rapid network replay of exploratory driven activity patterns over compressed time periods (Buzsaki, 1986; Foster and Wilson, 2006). Of particular relevance to the current study, PVFSIs are implicated in coordinating the ensembles of PCs that participate in SWRs (Csicsvari et al., 1999a, b; Ellender et al., 2010). Indeed among the hippocampal oscillatory rhythms examined in awake behaving mice, SWRs induce the greatest depth of PVFSI firing modulation (Varga et al., 2012; Varga et al., 2014). Overall, we found that the incidence of SWRs in NPTX2−/−/NPTXR−/− mice was reduced compared to wild types (Fig. 7C). Analysis of individual SWRs using Morlet wavelets (eg. Fig. 7D–E) revealed that SWRs in wild type and NPTX2−/−/NPTXR−/− mice had similar durations, peak amplitudes and peak power, but that SWRs in NPTX2−/−/NPTXR−/− mice were reduced in peak frequency (Fig 7F–I). Importantly, the laminar profiles of sharp waves and associated ripples were normal in NPTX2−/−/NPTXR−/− mice (eg Fig. 7A–B), excluding the possibility that the deficits observed related to changes in the depth profiles of the recordings.
Figure 7. Altered SWRs and behavioral deficits in NPTX2−/−/NPTXR−/− mice.

(A,B) Depth profiles (inter-electrode distance of 50μm) of representative recordings illustrating SWRs in (A) wild type and (B) NPTX2−/−/NPTXR−/− mice in vivo. (C) The incidence of SWRs was significantly reduced in NPTX2−/−/NPTXR−/− mice (WT (n=6) vs NPTX2−/−/NPTXR−/− (n=6): 0.26 ± 0.05 Hz vs 0.13 ± 0.02 Hz; p=0.0387; Student’s t test). (D,E) Examples of sharp wave ripples from (D) wild type and (E) NPTX2−/−/NPTXR−/− mice, recorded from stratum pyramidale, bandpass filtered (130 to 250Hz, top) with corresponding wavelet transforms of the raw traces (bottom). (F) NPTX2−/−/NPTXR−/− mice did not display a significant difference in the SWR duration (WT vs NPTX2−/−/NPTXR−/− : 59.7 ± 2.96 ms vs 58.8 ± 5.5 ms; p=0.8880; Student’s t test). (G) NPTX2−/−/NPTXR−/− mice did show a significant reduction in the peak frequency of SWRs (WT vs NPTX2−/−/NPTXR−/− : 157 ± 2.47 Hz vs 142 ± 3.05 Hz; p=0.0037; Student’s t test). NPTX2−/−/NPTXR−/− mice did not display a significant difference in either (H) SWR amplitude (WT vs NPTX2−/−/NPTXR−/− : 348 ± 48.2 μV vs 326 ± 36.6 μV; t=0.3622; p=0.7247; Student’s t test) or (I) SWR power (peak power, mV2/Hz, WT vs NPTX2−/−/NPTXR−/− : 0.31 ± 0.04 vs 0.28 ± 0.03; t=0.4777; p=0.6431; Student’s t test). (J) NPTX2−/−/NPTXR−/− showed early open field locomotor hyperactivity and late hypoactivity (n=15 WT and 13 NPTX2−/−/NPTXR−/− mice; genotype x time interaction: F1,26=7.8620, p=0.0094; 2-way ANOVA). (K-L) NPTX2−/−/NPTXR−/− displayed increased anxiety-like behavior in (K) the novel open field (t=2.3810, p=0.0248; Student’s t test). (L) Anxiety-like behavior was confirmed in the elevated O-maze (n=15 mice per genotype; t=2.1624, p=0.0393; Student’s t test). (M) NPTX2−/−/NPTXR−/− mice made fewer correct choices in a reward alternation task (n=7 WTs and 11 mutants; main effect of genotype; F1,16=7.7876, p=0.0131; 2-way ANOVA) *, p <0.05 **, p < 0.01, ***, p < 0.001 vs wild type. In vivo recordings were performed in P32-34 mice and behavior was assessed in P70-91 mice.
In general our in vitro and in vivo network findings are consistent with previous reports that mouse mutants with reduced glutamatergic drive onto PVFSIs display altered hippocampal oscillations (Fuchs et al., 2007; Racz et al., 2009; Belforte et al., 2010; Korotkova et al., 2010; Caputi et al., 2012; Carlen et al., 2012). In particular, mice with targeted deletion of GluA4 exhibit compromised kainate induced gamma oscillations in vitro similar to our observations for CCh-induced gamma oscillations in NPTX2−/−/NPTXR−/− mice (Fuchs et al., 2007). Moreover, ablation of GluA4 selectively in hippocampal PVFSIs reduces SWR occurrence and decreases peak frequency similar to our current findings in NPTX2−/−/NPTXR−/− mice (Caputi et al., 2012). Thus, NPTX2−/−/NPTXR−/− mice phenocopy several network features of GluA4−/− knockouts. However, in contrast to our findings in NPTX2−/−/NPTXR−/− mice, PVFSI GluA4−/− mice did not exhibit in vivo gamma oscillation deficits and displayed decreases in SWR peak amplitude and power (Caputi et al., 2012).
To investigate potential behavioral consequences of the circuit defects in NPTX2−/−/NPTXR−/− mice, we examined their performance in a series of tasks designed to probe general exploration, anxiety, and spatial working memory. In the novel open field test, NPTX2−/−/NPTXR−/− mice exhibited increased exploratory activity relative to wild types during the first 5 min of the test, but reduced exploration towards the end of a 60 min session, indicating more rapid habituation (Fig. 7J). This habituation phenotype of NPTX2−/−/NPTXR−/− mice yielded an overall decrease in exploration compared to wild types throughout the entire session consistent with findings in GluA4−/− mice (Fuchs et al., 2007). Despite their increased initial locomotion NPTX2−/−/NPTXR−/− mice spent less time than controls exploring the center area of the open field during the first 10 min, consistent with an anxiety-like phenotype (Fig. 7K). Indeed subsequent evaluation in the elevated O-maze revealed that mutant mice spent less time than controls in the open arms confirming anxiety-like behavior (Fig. 7L). Finally, we found that NPTX2−/−/NPTXR−/− made significantly fewer correct choices across multiple trials in the rewarded T-maze alternation task, suggesting impaired hippocampus-dependent working memory (Fig. 7M). This deficit in spatial working memory of NPTX2−/−/NPTXR−/− mice likely relates directly to the loss of GluA4 within PVFSIs as several prior studies have reported deficits in working memory following genetic manipulations that compromise PVFSI function (Fuchs et al., 2007; Belforte et al., 2010; Korotkova et al., 2010; Murray et al., 2011; Caputi et al., 2012; Carlen et al., 2012).
Previously we found that NPTX2−/− mice display increased sensitivity to kindling-induced epilepsy (Chang et al., 2010). As NPTX2−/− mice cannot homeostatically increase PVFSI GluA4 we postulated that this phenotype reflects failure of NPTX2−/− circuits to rebalance network I/E dynamics and suppress epileptogenesis early in the kindling protocol. Importantly, NPTX2−/− mice showed identical initial responses to kindling but diverged from wild types as the kindling process evolved (Chang et al., 2010), consistent with normal baseline GluA4 content and circuit integration of PVFSIs in NPTX2−/− mice. In contrast, the severe basal deficit of PVFSI synaptic recruitment in NPTX2−/−/NPTXR−/− mice predicts a basal epilepsy phenotype. However, NPTX2−/−/NPTXR−/− mice did not exhibit overt seizures, nor did we detect any electrophysiological features of epilepsy during hippocampal in vivo recordings. To further assess acute epileptiform sensitivity of wild type, NPTX2−/−, NPTXR−/−, and NPTX2−/−/NPTXR−/− mice we monitored hippocampal network activity while challenging slices with a modest increase in extracellular potassium (to 8.5 mM; Traynelis and Dingledine, 1988). In adult rat slices this challenge typically promotes spontaneous recurring interictal bursting, with a minority of slices (~20%) progressing to electrographic seizure-like activity comprised of ictal events reminiscent of discharges recorded in vivo during tonic-clonic seizures (Traynelis and Dingledine, 1988). Similarly, in our recordings wild type, NPTX2−/−, and NPTXR−/− slices all consistently exhibited spontaneous interictal bursting within 7–10 minutes of increased [K+]o with only a minority of slices yielding ictal events (Fig. 8A,C,D). In contrast, slices from NPTX2−/−/NPTXR−/− consistently displayed full-blown epileptiform activity producing spontaneous ictal activity with characteristic tonic and clonic bursts at regular intervals (Figure 8B–D). Pooling of data across all recordings revealed that ictal frequency is significantly enhanced in NPTX2−/−/NPTXR−/− compared to wild type mice while interictal frequency is significantly lower in NPTX2−/−/NPTXR−/− due to the excess time spent in ictal discharges (Fig 8C–D). While data from NPTX2−/− and NPTXR−/− single knockouts trended towards intermediate phenotypes slices from these lines were not significantly different from wild types.
Figure 8. Increased susceptibility to epileptiform activity in NPTX2−/−/NPTXR−/− mice.

(A) Representative recordings of epileptiform activity induced following bath application of 8.5 mM [K+]o in slices from wild type mice: (i) representative trace from WT mouse showing rhythmic clusters of interictal events; (ii) two clusters of interictal events from boxed region of i on an expanded time scale; (iii) expanded time scale trace from boxed area in ii, showing rhythmicity of interictal events; (iv) single interictal event from boxed area of iii on expanded time scale. (B) Similar to A but for a representative NPTX2−/−/NPTXR−/− recording: (i) representative trace from NPTX2−/−/NPTXR−/− mouse showing rhythmic clusters of ictal events; (ii) single ictal event from boxed region in i on expanded time scale; (iii) boxed event from ii on expanded time scale with interictal events preceding the onset of an ictal event; (iv) beginning of ictal event on an expanded time scale from boxed region in iii. (C) NPTX2−/−/NPTXR−/− mice displayed significantly more ictal events during high K+ epileptiform activity (ictal events per hour, WT (n=9) vs NPTX2−/− (n=7) vs NPTXR−/− (n=7) vs NPTX2−/−/NPTXR−/− (n=11): 0.78 ± 0.39 vs 8.77 ± 6.05 vs 4.60 ± 2.48 vs 19.12 ± 4.64; H=13.85, p=0.0031, Kruskal-Wallis test with post hoc Dunn’s multiple comparisons test). (D) Interictal events in NPTX2−/−/NPTXR−/− mice were significantly less frequent (interictal event frequency, WT vs NPTX2−/− vs NPTXR−/− vs NPTX2−/−/NPTXR−/−: 0.70 ± 0.07 Hz vs 0.73 ± 0.15 Hz vs 0.35 ± 0.10 Hz vs 0.41 ± 0.04 Hz; p=0.0033, Kruskal-Wallis test with post hoc Dunn’s multiple comparisons test). (E) Representative recording from an NPTX2−/−/NPTXR−/− slice showing the effect of indiplon on high K+ induced ictal activity. Shown is a full time course of indiplon-mediated attenuation of ictal events (i) with a single ictal event prior to indiplon application (ii) and remaining interictal events (iii) after indiplon from the boxed regions in i shown on expanded time scales. (F) Bath application of indiplon significantly reduced the frequency of ictal events in NPTX2−/−/NPTXR−/− mice (ictal events per hour, baseline vs indiplon (n=6): 30.0 ± 4.3 vs 10.3 ± 5.9; p = 0.0141; Paired t test. **, p < 0.01 vs WT). Recordings throughout were performed in P15-30 mice.
Considering our findings of GluA4 loss and deficient synaptic drive in NPTX2−/−/NPTXR−/− PVFSIs, the increased epileptiform sensitivity of slices from these mice likely stems from deficient recruitment of perisomatic inhibition leading to network hyperexcitability. This interpretation is consistent with evidence that GluA4 mutant mice serve as an animal model of absence epilepsy (Paz et al., 2011). However, our synaptic findings indicate that despite the reduced circuit integration of PVFSIs in NPTX2−/−/NPTXR−/− mice these cells still exhibit some capacity for afferent recruitment and once engaged provide efficient GABAergic input to PCs (cf. Figs. 4–5). Thus, we reasoned that the epilepsy-like phenotype of NPTX2−/−/NPTXR−/− mice might be sensitive to an increase in the efficacy of GABAergic tone from the PVFSIs engaged in a blunted fashion during the epileptiform activity. To test this we utilized the GABAA receptor positive allosteric modulator indiplon that is selective for alpha1-containing receptors which are preferentially, though not solely, localized postsynaptic to PVFSI inputs (Klausberger et al., 2002; Petroski et al., 2006). Following initiation of epileptiform activity in NPTX2−/−/NPTXR−/− slices, indiplon eliminated ictal activity in 50% of recordings tested and reduced ictal frequency in the remaining slices (Fig 8E–F). Following indiplon interictal events remained as the dominant activity similar to wild type, NPTX2−/−, and NPTXR−/− mice indicating that the underlying pacemaker synaptic drive engaged by high [K+]o was not disrupted (eg. Fig. 8E). Rather indiplon reversal of epileptiform activity likely proceeds by enhancing circuit inhibition, including, but not limited to, PVFSI mediated perisomatic inhibition to improve I/E balance. This rescue of the NPTX2−/−/NPTXR−/− epileptiform phenotype is consistent with the ability of optogenetic mediated recruitment of PVFSIs to arrest spontaneous seizures in vivo (Krook-Magnuson et al., 2013) and further validates PVFSI-mediated inhibition as an effective therapeutic target for disorders precipitated by circuit I/E imbalance.
In summary we have uncovered an essential role for NPTX2 and NPTXR in orchestrating the basic developmental program for synaptic integration and network function of hippocampal PVFSIs. The coincidental temporal relation of NPTX2, NPTXR, and GluA4 expression, combined with loss of GluA4 in NPTX2−/−/NPTXR−/− mice suggests these NPTXs regulate PVFSI circuit integration by controlling expression and synaptic localization of GluA4-containing AMPARs. Although GluA4 levels are upregulated in concert with a number of other proteins that are also critical for promoting speed and precision in PVFSI function such as PV, Kv3 channels, Nav1.1 sodium channels, and 2-pore domain potassium leak channels (TWIK1 and TASK1/3), our findings suggest normal developmental progression of these other proteins in NPTX2−/−/NPTXR−/− mice. Thus, within PVFSIs pentraxins serve to selectively drive maturation of AMPAR function by facilitating the production of GluA4. Given the reduction in PVFSI sEPSC frequency it is also possible that NPTXs serve a synaptogenic function in PVFSIs and that NPTX2−/−/NPTXR−/− mice have reduced numbers of excitatory synaptic contacts onto them. However, overall dendritic architectures of PVFSIs were similar in wild type and NPTX2−/−/NPTXR−/− mice and the mutants exhibited only modest reductions in synaptic density (supplemental Fig. 2C–J). Thus, the deficits in NPTX2−/−/NPTXR−/− PVFSI recruitment relate primarily to reduced AMPAR content at synapses rather than dramatic changes in the numbers of afferents impinging upon PVFSIs.
Prior studies have focused on the role of neuronal pentraxins as AMPAR clustering proteins without evidence of a larger role in coordinating both overall protein expression and synaptic localization (O’Brien et al., 1999; Mi et al., 2002; O’Brien et al., 2002; Xu et al., 2003; Sia et al., 2007; Chang et al., 2010). Interestingly, GluA4 transcription proceeds normally in the absence of NPTX2 and NPTXR. Given the direct association of neuronal pentraxins with the N-terminal domain of GluA4 it is possible that this interaction stabilizes GluA4 to prevent rapid degradation. Alternatively, pentraxins may regulate GluA4 mRNA translation. Future studies into the cellular mechanisms of NPTX mediated GluA4 regulation will be necessary to resolve these possibilities. Concerning the sources of NPTX2 and NPTXR for regulating PVFSI GluA4, evidence to date implicates presynaptically derived NPTXs in driving synaptic AMPAR accumulation (Chang et al., 2010; Sia et al., 2007). While NPTX2 is secreted, the transmembrane domain of NPTXR may serve to anchor NPTX complexes at presynaptic terminals innervating PVFIs. Such heterocomplexes may then be released to interact with postsynaptic PVFSIs following activity dependent cleavage of NPTXR by extracellular matrix metalloproteases (Cho et al., 2008). An alternate possibility would place NPTXR in the postsynaptic membrane of PVFIs to bind presynaptically derived NPTX2. However, while activity dependent NPTX2 expression has recently been demonstrated in somatostatin interneurons (Spiegel et al., 2014), NPTX expression has not yet been localized within PVFIs.
Previously, NPTX2 alone was implicated in homeostatic adaptations in GluA4 levels and AMPAR function (Chang et al., 2010). However, NPTX2−/− PVFSIs exhibit normal basal excitatory drive. Thus, NPTX2−/−/NPTXR−/− mice provided a unique opportunity to assess the impact of NPTXs on cellular and circuit function, without a requirement for perturbed activity. Indeed we found that NPTX2−/−/NPTXR−/− mice exhibited basal deficits in PVFSI AMPAR function yielding a circuit with reduced feedforward inhibition, disrupted network oscillations, and prone to epileptiform activity. In association with these circuit changes NPTX2−/−/NPTXR−/− mice exhibited increased anxiety-like behavior as well as deficits in exploration and spatial working memory. These cellular, network, and behavioral deficiencies are similar to findings in GluA4 mutants (Fuchs et al., 2007; Beyer et al., 2008; Paz et al., 2011) indicating that the reduction of GluA4 in NPTX2−/−/NPTXR−/− mice is sufficient in magnitude to phenocopy mice with targeted deletion of GluA4.. Our current findings extend the role of NPTXs to coordinating the basic synaptic developmental program of PVFSIs, and reveal the essential role of NPTXs in controlling hippocampal network properties. The restricted expression of GluA4 to PVFSIs, combined with the selective synaptic regulation by NPTXs point to the importance of these targets for understanding and treating disorders associated with I/E imbalance due to PVFSI dysfunction.
Experimental Procedures
Detailed experimental procedures are provided in Supplemental Materials.
IHC, cell culture, and Post-embedding EM
Fixed brains from mice P2-P60 were removed, cryoprotected, and sectioned then stained for the indicated proteins with visualization using either fluorescently labeled secondary Abs or through immunoperoxidase labeling. Images of the stained sections were obtained and labeled cells were manually counted with the results converted to cell density measurements. Hippocampal cell cultures were prepared from P0-P5 mice and fixation and staining was performed after 15–30 days in vitro. Post-embedding immunogold EM was performed on tissue from 2 month old mice with excitatory synapse density and synaptic GluA4 content evaluated in PV immunopositve processes.
Biochemistry, qPCR, and ISH
Western blot analyses were performed on hippocmapal synaptosomes and membrane fractions or cerebellar lysates obtained from P22-P30 mice as indicated. For quantification densitometry of protein bands of interest were normalized to those of beta-actin. Immunoprecipitations were performed from SPM fractions run through GluA4 conjugated beads. qPCR was carried out on P20 hippocampi using a StepOne instrument from Applied Biosystems and target gene levels were normalized to the endogenous control gene gadph. ISH was carried out on fresh frozen sections from P30 mice using the RNAscope Fluorescent Miltiplex kit (Advanced Cell Diagnostics Inc.).
Electrophysiology and Behavior
Whole-cell patch recordings from PVFSIs and CA1/CA3 pyramids were performed in submerged hippocampal slices from P5-30 mice to evaluate PVFSI synaptic/intrinsic/spiking properties, feedforward inhibition, and GDPs as indicated. For in vitro gamma oscillations and epileptiform activity, CA3 local field potentials (LFPs) were recorded in slices maintained in an interface chamber. In vivo recordings were performed in awake head fixed mice (P32-34) using a 32 channel electrode inserted into the hippocampus through a craniotomy to monitor CA1 LFPs. For behavior, adult (10–13 weeks old) mice were assessed for novelty-induced locomotor activity in a novel square arena, anxiety-like behavior in an elevated O-maze, and spatial working memory using rewarded alternation in a T-maze.
Supplementary Material
Highlights.
GluA4 is undetectable in neonatal PVFSIs then increases and plateaus by P15.
NPTX2−/−/NPTXR−/− mice have profound loss of GluA4.
PVFSI AMPAR function and recruitment are compromised in NPTX2−/−/NPTXR−/− mice.
I/E imbalances in NPTX2−/−/NPTXR−/− mice impair rhythmogenesis and working memory.
Acknowledgments
We thank Dr. Ya-Xian Wang (NIH/NIDCD IR) for expert technical assistance with immunogold EM experiments. Work supported by a PRAT fellowship to M.S.W., an NICHD intramural award to C.J.M., NIDCD intramural research program funding to R.S.P., an NIMH intramural award to H.A.C., NIH grants (PAR-02-059, NS 039156) to P.F.W., and an NIH grant (EY022730) to M.T.C.4
Footnotes
Author Contributions:
K.A.P., P.F.W. and C.J.M. conceived of and designed the study. E.B. performed IHC and ISH. R.C. examined PVFSI dendritic architecture. X.Y. carried out western blots, cell culture, IHC, and qPCR. R.W.M. performed biochemical fractionation and IP experiments. M.S.W. and R.S.P. performed electron microscopy. K.A.P., G.A.V. and M.S. performed synaptic physiology experiments. M.T.C.1 performed gamma oscillation and epileptiform activity experiments. M.T.C.1, M.T.C.4, and Y.M. carried out in vivo physiology experiments. R.-M.K. and H.A.C. performed behavioral experiments. Each author contributed to manuscript writing/editing.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barad Z, Shevtsova O, Arbuthnott GW, Leitch B. Selective loss of AMPA receptors at corticothalamic synapses in the epileptic stargazer mouse. Neuroscience. 2012;217:19–31. doi: 10.1016/j.neuroscience.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Bernard C, Milh M, Morozov YM, Ben-Ari Y, Freund TF, Gozlan H. Altering cannabinoid signaling during development disrupts neuronal activity. Proc Natl Acad Sci U S A. 2005;102:9388–9393. doi: 10.1073/pnas.0409641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjartmar L, Huberman AD, Ullian EM, Renteria RC, Liu X, Xu W, Prezioso J, Susman MW, Stellwagen D, Stokes CC, Cho R, Worley P, Malenka RC, Ball S, Peachey NS, Copenhagen D, Chapman B, Nakamoto M, Barres BA, Perin MS. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci. 2006;26:6269–6281. doi: 10.1523/JNEUROSCI.4212-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G. Hippocampal sharp waves: their origin and significance. Brain Res. 1986;398:242–252. doi: 10.1016/0006-8993(86)91483-6. [DOI] [PubMed] [Google Scholar]
- Caputi A, Fuchs EC, Allen K, Le Magueresse C, Monyer H. Selective reduction of AMPA currents onto hippocampal interneurons impairs network oscillatory activity. PLoS One. 2012;7:e37318. doi: 10.1371/journal.pone.0037318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Ruhlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17:537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MC, Park JM, Pelkey KA, Grabenstatter HL, Xu DS, Linden DJ, Sutula TP, McBain CJ, Worley PF. Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nat Neurosci. 2010;13:1090–U1083. doi: 10.1038/nn.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RW, Park JM, Wolff SB, Xu D, Hopf C, Kim JA, Reddy RC, Petralia RS, Perin MS, Linden DJ, Worley PF. mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron. 2008;57:858–871. doi: 10.1016/j.neuron.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurko A, Mamiya A, Buzsaki G. Fast network oscillations in the hippocampal CA1 region of the behaving rat. J Neurosci. 1999a;19:RC20. doi: 10.1523/JNEUROSCI.19-16-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurko A, Mamiya A, Buzsaki G. Oscillatory coupling of hippocampal pyramidal cells and interneurons in the behaving Rat. J Neurosci. 1999b;19:274–287. doi: 10.1523/JNEUROSCI.19-01-00274.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doischer D, Hosp JA, Yanagawa Y, Obata K, Jonas P, Vida I, Bartos M. Postnatal differentiation of basket cells from slow to fast signaling devices. J Neurosci. 2008;28:12956–12968. doi: 10.1523/JNEUROSCI.2890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhang L, Weiser M, Rudy B, McBain CJ. Developmental expression and functional characterization of the potassium-channel subunit Kv3.1b in parvalbumin-containing interneurons of the rat hippocampus. J Neurosci. 1996;16:506–518. doi: 10.1523/JNEUROSCI.16-02-00506.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Nissen W, Colgin LL, Mann EO, Paulsen O. Priming of hippocampal population bursts by individual perisomatic-targeting interneurons. J Neurosci. 2010;30:5979–5991. doi: 10.1523/JNEUROSCI.3962-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K, Lerma J, Marin O, Rico B. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature. 2010;464:1376–1380. doi: 10.1038/nature08928. [DOI] [PubMed] [Google Scholar]
- Foster DJ, Wilson MA. Reverse replay of behavioural sequences in hippocampal place cells during the awake state. Nature. 2006;440:680–683. doi: 10.1038/nature04587. [DOI] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JN, Monyer H. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53:591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, Scanziani M. Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron. 2005;48:315–327. doi: 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Lubke J, Roth A, Frotscher M, Jonas P. Submillisecond AMPA receptor-mediated signaling at a principal neuron-interneuron synapse. Neuron. 1997;18:1009–1023. doi: 10.1016/s0896-6273(00)80339-6. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Caroni P, Luthi A, Herry C. Perineuronal nets protect fear memories from erasure. Science. 2009;325:1258–1261. doi: 10.1126/science.1174146. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Jeong HY, Kruglikov I, Tremblay R, Lazarenko RM, Rudy B. Rapid developmental maturation of neocortical FS cell intrinsic excitability. Cereb Cortex. 2011;21:666–682. doi: 10.1093/cercor/bhq138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Huang S, Chang MC, Worley P, Kirkwood A, Quinlan EM. Obligatory role for the immediate early gene NARP in critical period plasticity. Neuron. 2013;79:335–346. doi: 10.1016/j.neuron.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Roberts JD, Somogyi P. Cell type- and input-specific differences in the number and subtypes of synaptic GABA(A) receptors in the hippocampus. J Neurosci. 2002;22:2513–2521. doi: 10.1523/JNEUROSCI.22-07-02513.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch SM, Ullian EM. Neuronal pentraxins mediate silent synapse conversion in the developing visual system. J Neurosci. 2010;30:5404–5414. doi: 10.1523/JNEUROSCI.4893-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Oijala M, Soltesz I. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun. 2013;4:1376. doi: 10.1038/ncomms2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Foldy C, Soltesz I. Distinct endocannabinoid control of GABA release at perisomatic and dendritic synapses in the hippocampus. J Neurosci. 2010;30:7993–8000. doi: 10.1523/JNEUROSCI.6238-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari A, Nahm WK, Noebels JL. Paradoxical proepileptic response to NMDA receptor blockade linked to cortical interneuron defect in stargazer mice. Front Cell Neurosci. 2013;7:156. doi: 10.3389/fncel.2013.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann EO, Suckling JM, Hajos N, Greenfield SA, Paulsen O. Perisomatic feedback inhibition underlies cholinergically induced fast network oscillations in the rat hippocampus in vitro. Neuron. 2005;45:105–117. doi: 10.1016/j.neuron.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Mi R, Tang X, Sutter R, Xu D, Worley P, O’Brien RJ. Differing mechanisms for glutamate receptor aggregation on dendritic spines and shafts in cultured hippocampal neurons. J Neurosci. 2002;22:7606–7616. doi: 10.1523/JNEUROSCI.22-17-07606.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov YM, Freund TF. Postnatal development and migration of cholecystokinin-immunoreactive interneurons in rat hippocampus. Neuroscience. 2003;120:923–939. doi: 10.1016/s0306-4522(03)00409-3. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, Bartos M, Wisden W, Wulff P. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci. 2011;14:297–299. doi: 10.1038/nn.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien R, Xu D, Mi R, Tang X, Hopf C, Worley P. Synaptically targeted narp plays an essential role in the aggregation of AMPA receptors at excitatory synapses in cultured spinal neurons. J Neurosci. 2002;22:4487–4498. doi: 10.1523/JNEUROSCI.22-11-04487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P. Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron. 1999;23:309–323. doi: 10.1016/s0896-6273(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Okaty BW, Miller MN, Sugino K, Hempel CM, Nelson SB. Transcriptional and electrophysiological maturation of neocortical fast-spiking GABAergic interneurons. J Neurosci. 2009;29:7040–7052. doi: 10.1523/JNEUROSCI.0105-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen SF, Tuncdemir SN, Bader PL, Tirko NN, Fishell G, Tsien RW. Oxytocin enhances hippocampal spike transmission by modulating fast-spiking interneurons. Nature. 2013;500:458–462. doi: 10.1038/nature12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, Huguenard JR. A new mode of corticothalamic transmission revealed in the Gria4(−/−) model of absence epilepsy. Nat Neurosci. 2011;14:1167–1173. doi: 10.1038/nn.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski RE, Pomeroy JE, Das R, Bowman H, Yang W, Chen AP, Foster AC. Indiplon is a high-affinity positive allosteric modulator with selectivity for alpha1 subunit-containing GABAA receptors. J Pharmacol Exp Ther. 2006;317:369–377. doi: 10.1124/jpet.105.096701. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 2002;298:1248–1251. doi: 10.1126/science.1072699. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feedforward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Racz A, Ponomarenko AA, Fuchs EC, Monyer H. Augmented hippocampal ripple oscillations in mice with reduced fast excitation onto parvalbumin-positive cells. J Neurosci. 2009;29:2563–2568. doi: 10.1523/JNEUROSCI.5036-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto-Ohshima A, Aoki E, Semba R, Emson PC, Heizmann CW. Appearance of parvalbumin-specific immunoreactivity in the cerebral cortex and hippocampus of the developing rat and gerbil brain. Histochemistry. 1990;94:579–589. doi: 10.1007/BF00271984. [DOI] [PubMed] [Google Scholar]
- Shevtsova O, Leitch B. Selective loss of AMPA receptor subunits at inhibitory neuron synapses in the cerebellum of the ataxic stargazer mouse. Brain Res. 2012;1427:54–64. doi: 10.1016/j.brainres.2011.10.022. [DOI] [PubMed] [Google Scholar]
- Sia GM, Beique JC, Rumbaugh G, Cho R, Worley PF, Huganir RL. Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron. 2007;55:87–102. doi: 10.1016/j.neuron.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, Greenberg ME. Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell. 2014;157:1216–1229. doi: 10.1016/j.cell.2014.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Chen YJ, Shen C, Luo Z, Bates CR, Lee D, Marchetto S, Gao TM, Borg JP, Xiong WC, Mei L. Erbin interacts with TARP gamma-2 for surface expression of AMPA receptors in cortical interneurons. Nat Neurosci. 2013;16:290–299. doi: 10.1038/nn.3320. [DOI] [PubMed] [Google Scholar]
- Ting AK, Chen YJ, Wen L, Yin DM, Shen CY, Tao YM, Liu XH, Xiong WC, Mei L. Neuregulin 1 Promotes Excitatory Synapse Development and Function in GABAergic Interneurons. J Neurosci. 2011;31:15–25. doi: 10.1523/JNEUROSCI.2538-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Tricoire L, Pelkey KA, Daw MI, Sousa VH, Miyoshi G, Jeffries B, Cauli B, Fishell G, McBain CJ. Common origins of hippocampal Ivy and nitric oxide synthase expressing neurogliaform cells. J Neurosci. 2010;30:2165–2176. doi: 10.1523/JNEUROSCI.5123-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui CC, Copeland NG, Gilbert DJ, Jenkins NA, Barnes C, Worley PF. Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. J Neurosci. 1996;16:2463–2478. doi: 10.1523/JNEUROSCI.16-08-02463.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga C, Golshani P, Soltesz I. Frequency-invariant temporal ordering of interneuronal discharges during hippocampal oscillations in awake mice. Proc Natl Acad Sci U S A. 2012;109:E2726–2734. doi: 10.1073/pnas.1210929109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga C, Oijala M, Lish J, Szabo GG, Bezaire M, Marchionni I, Golshani P, Soltesz I. Functional fission of parvalbumin interneuron classes during fast network events. Elife. 2014:3. doi: 10.7554/eLife.04006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O’Brien RJ, Worley P. Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron. 2003;39:513–528. doi: 10.1016/s0896-6273(03)00463-x. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.