

SUMMARY
Stroke remains a significant problem despite decades of work on neuroprotective strategies. NMDA receptor (NMDAR) antagonists are neuroprotective in preclinical models, but have been clinically unsuccessful, in part due to side effects. Here we describe a prototypical GluN2B-selective antagonist with an IC50 value that is 10-fold more potent at acidic pH 6.9 associated with ischemic tissue compared to pH 7.6, a value close to the pH in healthy brain tissue. This should maximize neuroprotection in ischemic tissue while minimizing on-target side-effects associated with NMDAR blockade in non-injured brain regions. We have determined the mechanism underlying pH-dependent inhibition and demonstrate the utility of this approach in vivo. We also identify dicarboxylate dimers as a novel proton sensor in proteins. These results provide insight into the molecular basis of pH-dependent neuroprotective NMDAR block, which could be beneficial in a wide range of neurological insults associated with tissue acidification.
Keywords: NMDA receptors, GluN2B selective antagonists, NR2B, neuroprotection, pH, proton, ischemia, PCP, MCAo
INTRODUCTION
The high mortality and morbidity of patients suffering from neurological damage as a result of cerebral ischemia, stroke, subarachnoid hemorrhage, or traumatic brain injury (TBI) constitute an enormous burden on society. Despite decades of research and multiple clinical strategies, there are few therapeutic alternatives for stroke apart from dissolution of the vessel clot in a subset of patients. A number of studies have shown that glutamate rises in injured tissue in animal models and human patients (Table S1). Virtually all neurons in the CNS express NMDA receptors (NMDARs), and without exception, overactivation of NMDARs is neurotoxic (Olney, 1969; Choi et al., 1988; Ellren and Lehmann 1989; Mody and MacDonald 1995; Wroge et al., 2012). Although numerous studies have confirmed the neuroprotective efficacy of NMDAR antagonists in animal models of injury (Table S2), multiple clinical trials with NMDAR antagonists failed to improve patient outcome in stroke or TBI (Table S3). Thus, a dichotomy exists between the lack of clinical efficacy and unequivocal preclinical data showing that NMDAR blockade within hours of ischemia reduces neuronal death in multiple species (Table S2). Post-hoc analyses of clinical trials involving NMDAR antagonists have identified several factors that may account for their failures (Morris et al., 1999, Albers et al., 2001, Sacco et al., 2001; Gladstone et al., 2002; Farin and Marshall 2004). The most commonly discussed shortcomings include (1) the presence of dose-limiting side effects that prompted reduced therapeutic exposures, (2) the administration of NMDAR antagonists after an effective temporal window (several hours post-injury) recognized in animal studies, (3) the heterogeneity of ischemic injuries, and (4) the lack of quantifiable clinical endpoints. The latter could be addressed with careful management of the clinical trial, inclusion criteria and post-injury analysis (Kidwell et al., 2001; Narayan et al., 2002). Advances in clinical trial design have dramatically shortened the time to administer neuroprotectants to patients (Saver, 2013), bringing delivery in line with therapeutic windows for efficacy demonstrated in preclinical studies. If clinical treatments can be delivered within two hours by first responders, NMDAR antagonists with expanded tolerability margins could become useful therapeutic agents. To improve the tolerability profile of NMDAR antagonists, we sought to limit GluN2B receptor antagonism to the affected regions and times at which brain injury occurs, thereby reducing overall side-effects.
It has long been known that ischemia is associated with tissue acidification as a result of altered energy production and failure to clear tissue CO2 (Kaplan et al., 1987; Matsumoto et al., 1990; Katsura et al., 1992). Tissue acidification can reach pH 6.9 in penumbral regions that are protected by NMDAR antagonists in animal models of ischemia, and pH 6.5 in the ischemic core (Mutch and Hansen 1984; Smith et al., 1986; Nedergaard et al., 1991; Katsura and Seisjo 1998). We studied pH-dependence of GluN2B-selective NMDAR antagonists (Pahk and Williams 1997; Mott et al., 1998, Wang et al., 2014) using medicinal chemistry to synthesize compounds that were maximally potent and effective in ischemic tissue, yet minimally potent in healthy brain. Our goal was to identify a novel class of clinically-relevant NMDAR antagonists with expanded tolerability/safety margins compared to previous generations of NMDAR antagonists. Here we describe the mechanistic and structural basis for pH-sensitive GluN2B-selective antagonists and demonstrate their utility as neuroprotectants in vivo.
RESULTS
pH-sensitive GluN2B-selective antagonists
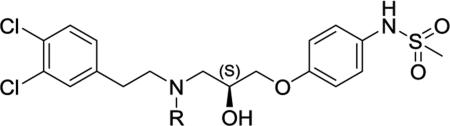
We evaluated the pH sensitivity of multiple GluN2B-selective non-competitive antagonists identified by bioinformatic criteria as di-aryl compounds with a nitrogen-containing linker and >10Å separation between terminal aromatic rings (Chenard and Menniti 1999). Among these, the S-enantiomeric analogues of the propanolamine AM-92016 (93-2, Table 1) emerged as a potent, synthetically-tractable class of molecules that showed systematic variations in pH sensitivity. We subsequently evaluated the pH-dependence of over 200 analogues on rat GluN1-1a/GluN2B receptors (hereafter GluN1/GluN2B) expressed in Xenopus laevis oocytes (Tahirovic et al., 2008; Mosley et al., 2009), including the series of N-alkyl substituted analogues summarized in Table 1. Within this subseries, an optimal length of the N-alkyl chain, compound 93-31 (4 carbons) maximized the pH sensitivity and showed an order of magnitude shift in potency per half-log unit change in pH in Xenopus oocytes (Table 1). Figure 1A,B shows similar effects of pH on concentration-effect data for 93-31 inhibition of GluN1/GluN2B receptors expressed in HEK293 cells recorded under voltage clamp. Although inhibition observed in mammalian cells is more potent (pH 6.9 IC50=0.040 μM) compared to Xenopus oocytes, the potency is enhanced 9.0-fold at acidic pH. We subsequently tested the effects of 93-31 on triheteromeric NMDARs that contain one copy each of GluN2A and GluN2B (Hansen et al., 2014). Although triheteromeric receptors show reduced sensitivity to GluN2B-selective inhibitors (Hatton and Paoletti 2005, Hansen et al., 2014), compound 93-31 still showed 4.4-fold enhanced potency at acidic pH at GluN1/GluN2A/GluN2B receptors (Figure S1), suggesting it will retain pH sensitivity at triheteromeric receptors expressed in adult cortex. We selected compound 93-31 as a prototype to evaluate the mechanism of pH-sensitive NMDAR inhibition.
Table 1.
Optimization of pH dependence of GluN1/GluN2B antagonists as a function of N-alkyl chain length and steric bulk.
| |||||||
|---|---|---|---|---|---|---|---|
| Ra | # | Chain amine pKab | Fold increase in ionized speciesc | N-substituent Volume Angstroms3 | IC50 pH 6.9 H.M (N) d | IC50 pH 7.6 HM (N) d | IC50(7.6) /IC50(6.9) Ratio e |
| -H | (S) 93-4 | 8.6 | 1.1 | -- | 0.036 (10) | 0.045 (11) | 1.3 |
| -CH3 | (S) 93-2 | 8.0 | 1.3 | 19.8 | 0.050 (29) | 0.13 (22) | 2.6 |
| -CH2CH3 | (S) 93-5 | 8.1 | 1.2 | 33.4 | 0.021 (22) | 0.067 (14) | 3.2 |
| -CH2CH2CH3 | (S) 93-6 | 8.1 | 1.2 | 47.9 | 0.094 (17) | 0.48 (16) | 5.1 |
| -CH2CH2CH2CH3 | (S) 93-31 | 8.1 | 1.2 | 62.3 | 0.19 (32) | 1.8 (23) | 9.4 |
| -CH2CH2CH2CH2CH3 | (S) 93-87 | 8.1 | 1.2 | 77.5 | 0.16 (9) | 0.60 (6) | 3.8 |
| -CH(CH3)2 | (S) 93-115 | 8.2 | 1.2 | 47.9 | 0.19 (11) | 0.62 (12) | 3.3 |
| -CH2CH(CH3)2 | (S) 93-97 | 8.0 | 1.3 | 63.6 | 1.1 (21) | 3.2 (31) | 2.9 |
Compound synthesis described in Tahirovic et al., (2008); all compounds were the S enantiomer.
The pKa of the chain nitrogen was calculated using ACD/pKa DB 12.00, www.acdlabs.com.
Fold increase ionized species when reducing pH from 7.6 (pH1) to 6.9 (pH2) was calculated using equation (3): the Henderson-Hasselbach equation as (1 + 10(pH2 - pKa)) / (1 + 10(pH1 - pKa))
IC50 values for inhibition of GluN1/GluN2B expressed in Xenopus oocytes were determined as described in the Methods from composite inhibition curves. N is the number of oocytes recorded; measurements made for oocytes at both pH values in the same experiment. The slope varied between −0.70 to −1.09; maximum inhibition 79-100%.
The pH-dependent potency ratio for inhibition of GluN1/GluN2B receptors.
See also Figure S1 and Table S6.
Figure 1. Proton sensitive inhibition of GluN1/GluN2B NMDARs.

A. Representative whole-cell current recordings from HEK cells transiently expressing rat GluN1/GluN2B receptors. Current responses were elicited by 100 μM glutamate (open bar) in the absence of (black) or presence of 0.3 μM 93-31 (gray) at pH 7.6 (left panel) and pH 6.9 (right panel) (VHOLD=−60 mV). Glycine (30 μM) was present in all the recording solutions. B. Composite concentration-response curves at pH 7.6 and pH 6.9 for compound 93-31 determined by whole-cell currents recorded from HEK cells transiently expressing GluN1/GluN2B receptors. C. Representative whole-cell currents recorded from HEK cells transiently expressing rat GluN1/GluN2B receptors were elicited by 100 μM glutamate and 30 μM glycine (open bar), followed by co-application of glutamate and glycine plus increasing concentrations of 93-31 (filled bar) at pH 6.9 (VHOLD=−60 mV). D. Plot of the reciprocal of the fitted time constant describing the onset of inhibition as a function of 93-31 concentration at pH 6.9 (filled circles) and pH 7.6 (open squares) is shown with fitted linear regression superimposed. Insets display the calculated kon, koff, and KD values at each pH. All data presented as mean±SEM. See also Figure S1 and Table S4.
Mechanism underlying pH sensitivity of GluN2B antagonists
One potential explanation for the pH sensitivity of 93-31 could be a systematic change in the pKa of the tertiary amine in the diaryl linker that could alter the fractions of ionized and unionized ligand, and thus alter potency. This occurs with phosphono-containing competitive antagonists, leading to lower concentrations of the more active species, which reduces potencies for these competitive antagonists under acidic conditions (Benveniste and Mayer 1992). Table 1 summarizes the predicted free solution pKa values of the tertiary amine in this series, and shows that the relative abundance of ionized or unionized species does not account for pH-dependent potency. In addition, we also examined the branched chain analogues iso-propyl (93-115) and iso-butyl (93-97), which show still less pH sensitivity (Table 1). For example, compound 93-97, the iso-butyl isomer of 93-31, showed a similar pKa but only a 2.9-fold increase in potency at lower pH values, compared to a 9.4-fold increase for the straight chain isomer 93-31. These data exclude the possibility that pH-induced changes in ligand ionization account for the proton sensitivity for inhibition of GluN1/GluN2B receptors.
To investigate the mechanism underlying proton sensitivity, we evaluated whether extracellular pH alters the rates at which 93-31 binds to and dissociates from the receptor. We expressed GluN1/GluN2B receptors in HEK cells and performed rapid agonist/modulator concentration jumps while recording the whole-cell current. The onset of inhibition following application of compound 93-31 (Figure 1C) could be well described by a single exponential function, allowing us to determine the association and dissociation rate constants for 93-31 at two different pH values by analyzing the concentration-dependence for onset of inhibition (Figure 1D).
Analysis of these data suggests that increasing the proton concentration slowed the dissociation rate kOFF and increased the association rate kON (Figure- 1D, Table S4). This decreased the KD by 10.3-fold at pH 6.9 compared to pH 7.6. We interpret these data to suggest that the pH sensitivity of the association and dissociation rates (and thus binding) for compound 93-31 accounts for the proton sensitivity of the IC50 (Table S4). Given the minimal effect of pH on ligand ionization (Table 1), agonist EC50 (Traynelis et al., 1995), and response time course (Banke et al., 2005), we hypothesized that the proton-sensitive inhibition reflects altered protein conformation, or an altered set of side-chain rotamers and/or ionization states at acidic pH that renders compound 93-31 a more potent inhibitor.
Binding of pH-sensitive antagonists
To explore the structural basis for the pH sensitivity of the 93-series compounds, we docked the compounds in Table 1 into a model derived from crystallographic data for a structure of Xenopus GluN1-ATD dimerized with rat GluN2B-ATD (3QEL, Karakas et al., 2011). Figure 2 shows a high scoring pose of 93-31 docked into the ifenprodil binding site (Figure 2A-D). From these docking studies it is clear that 93-31 can adopt a pose similar to that determined for ifenprodil (Figure 2B-E) with molecular contacts analogous to those described for other propanolamines (Burger et al., 2012). For example, the chlorophenyl ring of 93-31 overlays the phenyl ring of ifenprodil in a hydrophobic pocket defined by GluN1-Tyr109 and GluN2B-Phe114/Ile82 (Figure 2C,D), while the phenylether moiety of 93-31 is superimposed on the phenol ring of ifenprodil and the sulfonamide matches the phenol hydroxyl group (Figure 2E). The tertiary amine of 93-31 occupies the same region as its ifenprodil piperidine counterpart (Figure 2B,E). When docking with the central tertiary nitrogen protonated (S-enantiomer), the ammonium group is predicted to make favorable contact with GluN2B-Gln110 (Figure 2C), which is also simultaneously able to interact with the ether moiety of 93-31. The hydroxyl group is able to form a hydrogen bond with the side-chain carboxyl of GluN2B-Glu106. All ligands shown in Table 1 docked in a similar fashion.
Figure 2. Binding of 93-31 to the GluN1/GluN2B ATD heterodimers.

A. A model of the GluN1/GluN2B NMDAR built from crystallographic data for the NMDAR (Karakas and Furukawa, 2014). The red ellipse indicates the ifenprodil binding site within the amino terminal domain at the interface of GluN1 and GluN2B. B. Superimposed conformations of 93-31 (cyan) and ifenprodil (magenta) docked into the ATD dimer (Karakas et al., 2011; see Methods). C. The bi-chlorophenyl ring sits in a hydrophobic pocket defined by GluN1-Tyr109 and GluN2B-Ile82/Phe114. The amino group is predicted to interact with GluN2B-Gln110, and Glu106 hydrogen bonds with the hydroxyl moiety. D. Reversed view of 93-31 showing interactions with GluN1 Tyr109, and GluN2B-Asp113, which is predicted to have an elevated pKa within the protein compared to free solution. E. Juxtaposed functional groups of ifenprodil (3QEL) and 93-31 (from left to right: chlorophenyl/phenyl, tertiary amine and sulfonamide/phenol); R is n-butyl.
Examination of the binding pockets represented in structures for ifenprodil and Ro25-6981-bound ATD heterodimer (3QEL and 3QEM) reveals a small 8Å tunnel leading away from the binding site interface towards the GluN1 subunit that is lined primarily by hydrophobic side chains (Figure 3A). This passageway is close to the chain nitrogen of the N-substituted propanolamines, raising the possibility that it might be involved in the differential properties observed for the different sized alkyl substituents on this nitrogen. Extensive induced-fit docking suggested that the N-alkyl derivatives can occupy and exploit the space created by this tunnel (Figure 3A). We therefore tested whether inhibition of receptors by 93-series compounds is altered by mutations at residues forming this tunnel, choosing side chains for substitutions that occlude the tunnel but do not otherwise perturb the overall protein structure (confirmed by molecular dynamics simulations, Figure S2), such as GluN1(G112A). Modeling places the alanine side chain methyl within the tunnel, effectively occluding it. If the existence of this tunnel provides additional space for N-alkyl derivatives, then occlusion should alter IC50 for n-butyl-substituted 93-31 but not that of the unsubstituted analog 93-4 (Figure 3B,C). Concentration-effect curves for these two inhibitors against wild-type GluN1/GluN2B and GluN1(G112A)/GluN2B mutant receptors in oocytes showed that the IC50 for 93-31 was increased 6-fold at pH 6.9 from 0.19 to1.2 μM (n=23-29 oocytes) without a change in slope (0.8-0.9) or fitted minimum (17-20%), consistent with the idea that this tunnel altered the binding of this ligand. Further, the pH sensitivity of 93-31 was reduced to 3.8-fold by the GluN1(G112A) mutation (Figure 3D, Table 2). By contrast, 93-4 which lacks an N-linked substitution, showed only a 2-fold increase in the IC50 from 0.036 μM at wild type GluN1/GluN2B (n=10) compared to 0.070 μM at GluN1(G112A)/GluN2B (n=8) mutant receptors with no change of pH sensitivity (1.4-fold) for inhibition (Figure 3E). These results were not a reflection of a change in the receptor's inherent pH sensitivity, as the ratio of maximum receptor current at pH 6.9 to 7.6 for GluN1(G112A)/GluN2B (0.21±0.01 , n=8) was identical to that for wild type GluN1/GluN2B (0.21±0.01, n=8, p=0.92). These data are consistent with the idea that ligands with N-alkyl substitutions can take advantage of space afforded by this tunnel.
Figure 3. Exploitation of a GluN1 tunnel at the ATD heterodimer interface by 93-31.

A. Spacefill illustration of the tunnel entering into GluN1 (green arrows) with an induced fit docked pose of 93-31 showing that the n-butyl substituent on the chain nitrogen protrudes into and fills this space. B. Occlusion of the GluN1 tunnel by the mutation GluN1(G112A), shown as a bronze spacefill, clashes with the n-butyl group of 93-31 in this docked pose. C. The GluN1(G112A) mutation, shown as a bronze spacefill, does not interact with docked pose of 93-4, which lacks any substituent on the chain nitrogen. The bi-chlorophenyl ring is at bottom. D,E. Concentration effect curves for 93-31 (D) or 93-4 (E) inhibition of GluN1/GluN2B (left panel) and GluN1(G112A)/GluN2B (right panel) responses in oocytes. See text for fitted IC50 values. See also Figure S2.
Table 2.
pH-dependence for 93-31 IC50 for inhibition of selected binding site mutants of GluN1/GluN2B and estimates of the corresponding solution vs. intraprotein pKa values.
| Mutation | Solution pKa a | Intraprotein pKa* a | IC50 μM, Maximum % inhibition at pH 7.6 (N) b | IC50 μM, Maximum % inhibition at pH 6.9 (N) b | IC50(7.6) /IC50(6.9) |
|---|---|---|---|---|---|
| GluN2B c | -- | -- | 1.8 μM, 76% (32) | 0.19 μM, 83% (23) | 9.5 |
| GluN2B(D77A) | 3.9 | 4.1 | 68 μM, 100%d (6) | 5.9 μM, 93% (10) | 12 |
| GluN2B(K79A) | 10.5 | 11.1 | 2.7 μM, 70% (8) | 0.36 μM, 68% (10) | 7.5 |
| GluN2B(D104A) | 3.9 | 4.7 | 110 μM, 100% (6) | 19 μM, 100% (8) | 5.8 |
| GluN2B(E106A) | 4.3 | 7.0 | 11 μM, 96% (7) | 4.3 μM, 80% (9) | 2.6 |
| GluN2B(E106Q) | 4.3 | 7.0 | 8.7 μM, 70% (8) | 3.2 μM, 91 % (8) | 2.7 |
| GluN2B(E106D) | 4.3 | 7.0 | 4.4 μM, 91 % (7) | 1.1 μM, 85% (7) | 4.0 |
| GluN2B(D113A) | 3.9 | 5.8 | 6.2 μM, 55% (9) | 1.3 μM, 55% (7) | 4.8 |
| GluN2B(D136A) | 3.9 | 4.8 | 5.4 μM, 85% (10) | 0.55 μM, 74% (9) | 9.8 |
| GluN2B(K137A) | 10.5 | 7.8 | 28 μM, 95% (7) | 1.8 μM, 84% (10) | 16 |
| GluN2B(D138A) | 3.6 | 3.3 | 2.6 μM, 77% (10) | 0.18 μM, 86% (8) | 14 |
| GluN2B(D206A) | 3.6 | 3.1 | 82 μM, 100% d (9) | 83 μM, 100% d (7) | 1.0 |
| GluN2B(D210A) | 3.6 | 3.8 | 13 μM, 52% (7) | 3.9 μM, 69% (11) | 3.3 |
| GluN2B(D211A) | 3.6 | 2.9 | 7.1 μM, 75%, (9) | 2.5 μM, 89% (24) | 2.8 |
| GluN2B(K215A) | 10.5 | 9.7 | 3.3 μM, 63% (13) | 0.29 μM, 71% (8) | 11 |
| GluN2B(K234A) | 10.5 | 9.8 | 21 μM, 80% (6) | 3.9 μM, 89% (12) | 5.4 |
| GluN2B(E235A) | 4.3 | 5.1 | 120 μM, 100%d (13) | 80 μM, 100%d (7) | 1.5 |
| GluN2B(E235Q) | 4.3 | 5.1 | 1.2 μM, 67% (12) | 0.49 μM, 73% (8) | 2.4 |
| GluN2B(E235D) | 4.3 | 5.1 | 0.93 μM, 80% (7) | 0.22 μM, 76% (8) | 4.2 |
| GluN2B(E106Q,E235Q) | -- | -- | 9.2 μM, 71 % (7) | 4.7 μM, 98% (4) | 2.0 |
| GluN2B(E106D,E235D) | -- | -- | 2.2 μM, 79% (8) | 1.1 μM, 74% (10) | 2.0 |
| GluN2B(E236A) | 4.3 | 5.7 | 6.9 μM, 90% (10) | 0.85 μM, 93% (18) | 14 |
| GluN1(R115A) | 12.5 | 11.2 | 4.3 μM, 68% (15) | 2.8 μM, 83% (12) | 1.5 |
| GluN1(D130A) | 3.9 | 4.7 | 3.7 μM, 76% (8) | 2.4 μM, 96% (10) | 1.5 |
| GluN1(K131A) | 10.5 | 10.5 | 2.3 μM, 86% (7) | 0.51 μM, 89% (7) | 4.6 |
| GluN1(H134A) e | 6.0 | 3.2 | 0.7 μM, 157%e (17) | 0.8 μM, 147% e (14) | 0.9 |
| GluN1(G112A) | -- | -- | 4.6 μM, 73% (16) | 1.2 μM, 80% (29) | 3.8 |
| GluN1(G112V) | -- | -- | 18.7 μM, 85% f (7) | 18.5 μM, 85% f (5) | 1.0 |
pKa values in free solution were compared to those derived from the GluN1/GluN2B ATD heterodimer solved at pH 7.5 with coordinates from pdb code 3QEL.
Intraprotein pKa predicted with PropKA 3.1(Olsson 2011).
IC50 values to 2 significant figures for inhibition of GluN1/GluN2B expressed in Xenopus oocytes were determined as described in the Methods from composite 93-31 inhibition curves. The fitted maximal inhibition is given as percent, and the number of oocytes given in parentheses.
Data for compound 93-31 are reproduced from Table-1 and included here to facilitate comparison.
Maximum inhibition was fixed to be 100%.
This mutation converted 93-31 into a potentiator, with EC50 and maximal potentiation given.
Maximum inhibition was fixed to be 85%.
Identification of pH-sensitive binding-site residues
We next explored whether ionizable residues near the binding site might be involved in the pH-dependent change in association rate for 93-31. The pKa values for 20 ionizable residues in the GluN1/GluN2B dimer (3QEL) within 12Å of the ifenprodil binding pocket were calculated (Figure 4A, Table 2). Predicted pKa values (pKa*) for ionizable side chains caused by the surrounding microenvironment suggest that some residues may experience a locally-induced pKa shift, rendering side chain ionization(s) sensitive to a decrease in pH to 6.9, as occurs in the ischemic penumbra (Table 2). Among the 20 residues, several pKa* values were predicted to be substantially elevated relative to their free solution values, including a pair of closely spaced acidic residues (Figure 4A,B), GluN2B-Glu106 (N-terminus of helix-3, pKa* 7.0) and GluN2B-Glu235 (C-terminus of helix-8, pKa* 5.1). The side-chain rotamer of Glu235 in a crystal structure solved at pH 7.5 (3QEL) is oriented away from Glu106 (Figure 4B), and thus the latter only contributes a small increase to the predicted pKa of Glu235 (ΔpKa* 0.8). GluN2B-Glu106 is less solvent exposed than Glu235, and thus more likely to have an elevated pKa*. At pH 6.9, about half of Glu106 residues (pKa* 7.0) at any moment will be protonated. Protonation of either of these residues is predicted to allow a favorable interaction, enhanced by the formation of a carboxylic acid dimer, if both are protonated. This hypothesis is partially confirmed by a second crystal structure of GluN2B at pH 5.5, in which presumably protonated Glu235 interacts directly with Glu106 as a dimer (3JPW; Figure 4B). The local conformational reorganization is in striking contrast to structural data obtained at pH 7.5, which shows the side chain directed toward bulk solvent (3QEL). This difference in Glu235 orientation between the two structures was not previously noted (Karakas et al., 2009, 2011). We suggest that at lower pH, protonation of Glu106 and/or Glu235 is favored allowing for these residues to interact noncovalently. Glu235 and Glu106 reside at the N-termini of two helices, and ifenprodil (and presumably 93-31) inhibits GluN1/GluN2B by restricting domain movement. Thus, if Glu106 and Glu235 interact more strongly at lower pH, this could add a further restriction to intra-domain movement in addition to ligand binding. Rigid rotation of Glu235 within this structure (3QEL) so that it forms a bidentate interaction with Glu106 shifts the predicted pKa* values still further away from the free solution values to 6.8 (Glu235) and 7.9 (Glu106). To explore the effects of the dicarboxylate on the ligand, charge-neutral subsets of all nearby residues were used to generate electrostatic potential surfaces (see Methods). While there was no noticeable change on the predicted partial charges of 93-31, the electrostatic surface potential charge centered on the ammonium group of 93-31 was more positive in the presence of the Glu106:Glu235 carboxylic acid dimer (Figure 4C). Based on the structural differences in Glu106 and Glu235 for crystals solved at different pH values (3JPW vs 3QEL), and the predicted pKa* values, the amino acid pair Glu106:Glu235 is a strong candidate for constituting a pH sensor that influences 93-31 binding and inhibition.
Figure 4. Mutation of candidate residues for controlling the pH sensitivity of 93-31 binding.

A. Acidic (red) and basic residues (blue) are shown within 12Å of the ifenprodil (depicted as cyan sticks) binding site. We estimated the intraprotein pKa values using PropKa 3.1 (See Methods/Table 2), and subsequently mutated these residues to Ala to determine their effect on the pH sensitivity of the IC50 for 93-31. GluN1 is shown as grey, and GluN2B is orange. B. Glu106 and Glu235 are close enough to interact at the lower pH of 5.5 (light blue, 3JPW), but not at pH 7.5 (green, 3QEL) in GluN1/GluN2B crystal structures. C. (Top panel) Distances in Å are shown separating Glu235 and Glu106 in the crystal structure for GluN2B solved at pH 5.5 (PDB code 3JPW), implying O-protonation and O-H hydrogen bonds of ~1.6 Å and 1.9 Å, respectively. (Bottom panel) Electrostatic potential surfaces on a truncated 93-31 binding site (in red circle) were predicted with Spartan using the PM6 basis set. 93-31 is more positive (blue) in the presence of the carboxylic acid dimer between E106 and E235 (left) compared to the non-interacting charged state (right). Both PM6 models are charge neutral, each containing an equal number of cationic and anionic side chains surrounding the binding site. D. Occupation of the tunnel by n-butyl of 93-31 potentially orients the hydroxyl for interaction with Glu106. E. A similarly scoring docking pose of the R-enantiomer of 93-31 (93-88) orientates the hydroxyl away from Glu106 and towards the tunnel. See also Figure S3.
Carboxylic acid dimers: an unrecognized interaction within proteins
The interaction between Glu106 and Glu235 at pH 5.5 in 3JPW is somewhat unusual and suggestive of a hydrogen bonded carboxylic acid dimer (Figure 4 B,C). This bidendate dimeric form is well-represented in small molecule crystallography, with the Cambridge crystallographic database (Allen 2002) reporting ~2000 cases when queried with Mercury/Motif (Leiserowitz et al., 2002; Macrae et al., 2006). In addition, in the appropriate solvent and gas phase, carboxylic dimers are readily formed (Tzeli et al., 2011; Shipman et al., 2007). However, scanning the PDB for residues with the dimer geometry revealed 41 structures exhibiting a similar interaction between carboxylate side-chains, none of which were discussed with the exception of a carboxylic acid dimer between retinoic acid and Glu121 in a quintuple mutant of the cellular retinoic acid binding protein (3CWK, Vasileiou et al., 2009). The present results suggest that carboxylic acid dimers may represent a hitherto unrecognized functional interaction in proteins.
Potential role of Glu106:Glu235 in pH sensitive 93-31inhibition
Inspection of 93-31 docking poses in the GluN1/GluN2B ATD heterodimer at pH 7.5 suggests several possible roles for GluN2B-Glu106 and -Glu235 in terms of conferring pH sensitivity. The top scoring docking results suggest that Glu106 can hydrogen-bond with 93-31, while Glu235 is close enough to interact electrostatically with the ligand (Figure 4B,C). Occupation of the tunnel by n-butyl orients the hydroxyl for a favorable interaction with Glu106, the orientation of which we propose is pH sensitive (Figure 4D). Interestingly, docking of the R-isomer of 93-31(93-88, Figure 4E) places the n-butyl chain outside of the tunnel and orients the hydroxyl group away from Glu106 in similarly high scoring poses. This docking result is consistent with the significantly reduced pH sensitivity of 93-88 inhibition (3.8±0.7 fold shift in IC50 at pH 6.9 compared to the IC50 at pH 7.6) versus 93-31 (10±2.7 fold shift, n=9-10, p<0.05, unpaired t-test) when IC50 values were assessed for both compounds in the same experiment. The exact nature of the hydrogen-bonding environment is difficult to predict using docking programs, due to the absence of solvent and the multiplicity of protonation states. However, these results suggest that a favorable network of interactions may exist under acidic conditions between 93-31, Glu106 and Glu235. Consistent with this idea, NP10075, a pH-sensitive GluN2B-selective antagonist recently reported by Wang et al. (2014) docked in poses similar to those of 93-31 (Figure S3). Whereas NP10075 lacks the side-chain of the 93-series that exploits the GluN1 tunnel, the bulky piperazine moiety nevertheless appears to orient the hydroxyl to interact with Glu106 in similar fashion to 93-31. This raises the idea that the pH sensitivity of NP10075 also reflects an interaction with the pH-sensitive dicarboxylate Glu106:Glu235 dimer.
Glu235 also lies in close proximity to Glu106 in the ligand-bound structure such that it could be responsible for raising the pKa of Glu106, leading to protonation at pH 6.9. To directly assess this prediction, we mutated all residues within 12Å of the ifenprodil binding pocket to alanine, and measured the pH sensitivity of 93-31 potency. A large number of these mutations modestly perturbed the pH sensitivity of 93-31 inhibition (Table 2). However, among these, three GluN2B mutations (E106A, E206A, E235A) eliminated the pH-dependence of inhibition by 93-31, two of which show pH-sensitive conformations. The third residue (Glu206) is close enough to interact with Glu235, and replacement with a neutral residue may render the local microenvironment favorable to maintenance of a negative charge on Glu106 and Glu235 at both pH values, preventing carboxylic acid dimer formation. Furthermore, both E206A and E106A substantially increased IC50 values at both pH values, perhaps reflective of a larger structural rearrangement that reduces 93-31 binding, thereby obscuring the pH effect.
Mutations at Glu106 and Glu235 produce modest effects on inhibition by 93-31 at pH 7.6, but stronger effects at pH 6.9. For example, GluN2B(E106A) increased the IC50 by 6-fold at pH 7.6 (11 μM) and by 23-fold at pH 6.9 (IC50 4.3 μM). This is consistent with the idea that the potency of 93-31 could be dependent on the ionization state of Glu106, which given the shift of its pKa, can be influenced by modest reduction in pH to 6.9 in the ischemic penumbra. We mutated this glutamate to glutamine to remove the ionizable carboxylate, and found that GluN2B(E106Q) shifted IC50 values more at pH 6.9 (17-fold) than at pH 7.6 (5-fold). Isosteric replacement of ionizable Glu with the protonated Gln presumably allows for a weak interaction between these residues at both pH values, partially eliminating the pH sensitivity. The double mutant GluN2B(E106Q,E235Q) showed the same effect as GluN2B(E106Q) alone (Table 2), suggesting that the main effect is mediated by Glu106. We predicted that shortening the side chain by converting glutamate to aspartate should also diminish the pH sensitivity if there are tight geometrical constraints on interactions between the two closely spaced glutamates. Consistent with this prediction, introduction of an aspartate in GluN2B(E106D) diminished the pH sensitivity of 93-31 inhibition to 2.4- and 5.8-fold for pH 7.6 and 6.9, respectively. Again, introduction of the double mutation GluN2B(E106D,E235D) resulted in an effect similar to GluN2B(E106D), suggesting Glu106 was the primary determinant. In summary, the differential position of Glu235 in the crystal structures solved at two pH values, the predicted shift in the Glu106:Glu235 dimer pKa*'s to values that change ionization as pH drops to 6.9, and the mutagenesis-induced change in pH sensitivity of 93-31 all suggest that Glu106 and Glu235 are candidates for controlling the pH sensitivity of 93-31 binding.
Potential role of other pH-sensitive binding-site residues
Several other acidic residues revealed substantially altered pKa values (Asp113, Lys137, His134) or pH sensitivity (Asp206, Asp 210, Asp211). GluN2B-Asp113 (pKa 5.8, Figure 2D) is close enough to form hydrogen bonds with the backbone amide hydrogen of GluN2B-Asp138 and the side chain of GluN2B-Ser141, and form a water-bridged interaction with GluN1-Tyr109. The predicted increase in pKa* for Asp113 could lead to fractional protonation under acidic conditions, which should alter the hydrogen bonding networks associated with GluN2B-Asp138 and GluN1-Tyr109. If the interaction is reciprocal and favors protonation of a fraction of Asp113 side chains, binding of 93-31 may be facilitated and stabilize the protonated state (i.e. raising the pKa). In this context, it may be noted that a string of Glu and Asp side chains form a negatively charged layer above the ifenprodil binding site (Figure 4A, GluN2B residues 236, 235, 104, 106, 136, 113). Each of these residues experiences an increase in predicted pKa* relative to the free solution. The proposed Glu106:Glu235 pH sensor resides at one end of the layer. Any change in the protonation state of the dicarboxylate sensor could significantly influence this cluster of residues, allowing them to serve as a trigger for perturbation of local conformation and ionization state as part of the binding event. The close clustering of acidic residues has been implicated in the pH-sensing mechanism of the acid sensing ion channel-1 protein (Jasti et al., 2007). Similarly, there is a cluster of cationic GluN1 residues (343, 115, 134, Figure 3A), the intraprotein pKa*'s of which are predicted to decrease relative to free solution. Thus, the ligand resides between electrostatic layers of opposite charge, is a situation that may render ligand binding sensitive to changes in both ligand configuration and pH. The complexity of this region is highlighted by the mutation of GluN1-His134 to Ala, which converts 93-31 into a pH-insensitive positive allosteric modulator (Table 2).
Three mutations at residues near the GluN1/GluN2B interface (Asp206, Asp210, Asp211) substantially altered the pH sensitivity of 93-31 inhibition (Table 2). GluN2B-Asp210 is close enough to interact with GluN1-Arg323. Similarly, GluN2B-Asp206 can interact with GluN1-Lys322, although the large decrease in potency at both pH 6.9 and 7.5 for GluN2B(D206A) suggest a larger disturbance of 93-31 binding. Lastly, Asp211 interacts with the positive N-terminus of an alpha-helix. Whereas the pKa predictions suggest pH-induced effects are unlikely at pH 6.9, protonation of any of these residues could weaken or abolish their respective interactions.
Neuroprotection by pH-dependent GluN2B-selective antagonists
In order to test the hypothesis that a pH drop associated with ischemic tissue engages the pH-sensitive enhanced potency of GluN2B antagonists in vivo for neuroprotection, we evaluated the efficacy of the pH-sensitive inhibitors on infarct volume following transient occlusion of the middle cerebral artery (MCAo) in mice. We selected two structural isomers that differed only in the N-alkyl chain, compound 93-31 which contains an n-butyl and is pH sensitive, and compound 93-97 with less pH sensitivity and an iso-butyl substituent (Table 1). Each compound was administered by intracerebroventricular (ICV) injection directly into the lateral ventricle 20 minutes prior to initiation of transient focal ischemia by MCAo. Figure 5A,B shows both the method of determining the infarct volume and that ICV administration of 93-31 produced dose-dependent and robust 59% reduction in infarct volume compared to vehicle. However, compound 93-97 was ineffective as a neuroprotectant with no reduction in infarct volume, consistent with our in vitro studies showing that the potency of 93-97 is relatively insensitive to pH. That is, 93-97 potency is similar to that of 93-31 at physiological pH 7.35 (93-31 IC50=2.2 μM, 93-97 IC50=4.8 μM, n=5-6), but not enhanced sufficiently at pH 6.9 (Table 1) to render it efficacious in penumbral regions (Figure 5A,B).
Figure 5. 93-31 is neuroprotective against transient ischemia without side effects.

A. (Left panel) A single mouse brain section (2 mm) obtained 24 hours post-ischemia and stained with TTC as described in the Methods. (Right panel) The same mouse section is shown with areas for which the TTC intensity that is 20% below threshold selected from contralateral cortex (blue circle) is shown as red. B. Summary of MCAo infarct volume results from treatment groups injected with (in pmols) 93-31 (50, 500), or 93-97 (500) compound by ICV, or an equivalent PBS volume (vehicle) 20 mins before MCAo. *p<0.005 (ANOVA, Tukey post hoc test), number of mice/group indicated. C. Dose-response curve for 93-31 administered IP 20 min preceding MCAo in mice (solid symbols) or 30 minutes post MCAo reperfusion (open symbol). Infarct volume was determined by TTC staining as described in A and Methods. # mice/group indicated; all measurements were significantly different (p<0.05) from vehicle (ANOVA and Dunnett's test). D. Horizontal locomotor activity (cumulative 2 hr period) following IP administration of the indicated drug and dose in rats is shown. n=6-8. Higher doses of Ro 25-6981 and ketamine were not tested. E. 93-31 failed to produce PCP-associated lever responding in rats trained to discriminate PCP from saline. n=6 for all except 4 mg/kg PCP (n=3) and 30 mg/kg 93-31 (n=5). F. The highest dose tested in the PCP discrimination study, 56 mg/kg, significantly suppressed response rates in the same subjects suggesting behaviorally active doses were tested (see Methods). n=6 for all. All data presented as mean±SEM. See also Figure S4 and Table S5.
Prior generations of competitive and noncompetitive NMDAR antagonists cause a variety of off-target and on-target adverse effects that prevented clinical development, including motor dysfunction, cognitive impairment, and psychotomimetic effects including hallucinations, paranoia, disorganized thought, and blunted affect (Lees et al., 2000; Sacco et al., 2001; Diener et al., 2002; Rowland 2005; Wood 2005; Muir 2006; Blagrove et al., 2009). Although the GluN2B-selective class of antagonists are better tolerated than competitive antagonists or channel blockers, they are not necessarily free from on-target side effects (Chaperon et al., 2003; DeVry and Jentzsch 2003; Yurkewicz et al., 2005; Nicholson et al., 2007; Preskorn et al., 2008; Nutt et al., 2008). Our working hypothesis is that enhanced pH sensitivity should increase the therapeutic ratio to enable efficacious dosing for neuroprotection in ischemic penumbral regions surrounding the infarct (pH 6.9), with reduced inhibition of GluN2B receptors in normal brain (pH 7.4). That is, the pH sensitivity of the NMDAR antagonist should lower the potential for on-target side effects. To determine a therapeutic ratio (TR; the ratio of ED50 for efficacy vs side-effect studies) for 93-31 as a neuroprotectant, we completed additional MCAo studies in which 93-31 showed potent neuroprotection during ischemia, reducing infarct volumes up to 50% with an apparent ED50 value of <1 mg/kg (Figure 5C). For these studies, 93-31 was dosed intraperitoneally (IP) 20 minutes prior to transient ischemia by MCAo. Further, administration of 93-31 (10 mg/kg IP) 30 min post MCAo also reduced infarct volume to a similar extent (open symbol, Figure 5C). PK studies in surrogate mice support drug exposures in brain consistent with potency at low pH (Table S5, Supplemental Experimental Procedures).
We subsequently made three measurements of potential on-target dose-limiting side effects. First, we evaluated effects of 93-31 on locomotor activity and found no significant effects on horizontal activity at doses up to 300 mg/kg compared to vehicle (4415±479, n=24) over two hours following 1 hour habituation, in contrast to the channel blockers aptiganel and ketamine. The GluN2B-selective antagonists ifenprodil and Ro 25-6981 reduced and increased locomotor activity, respectively, at high doses (Figure 5D). Second, we evaluated the effects of 93-31 in a rotorod test. Both the non-competitive NMDAR antagonist (+)MK-801 and ifenprodil impaired performance, exhibiting strong reductions in latency to fall (Vehicle 140±13 sec, n=12; 0.6 mg/kg (+)MK-801 3±2 sec, n=5; 30 mg/kg ifenprodil 35±6 sec, n=5). By contrast, there was no significant effect of 93-31 (108±14 sec, n=6) nor 93-97 (117±19 sec, n=6) on rotorod performance high doses of 30 mg/kg (Figure S4). Third, we tested the ability of GluN2B-selective NMDAR blockers to be recognized by rodents trained to discriminate the non-competitive channel blocker PCP from saline. Trained animals were administered 93-31, PCP, or saline on test days. Interestingly, 93-31 failed to substitute for PCP in this paradigm (Figure 5E), suggesting this compound is devoid of psychotomimetic effects at the doses tested. At the highest dose (56 mg/kg), there was a significant depression in lever-pressing behavior, supporting that behaviorally active doses were tested and therefore the lack of PCP-like effects was not attributable to an inadequate dose range (Figure 5F). Of these four behaviors (locomotor activity, rotorod, PCP discrimination, lever pressing), we identified only one in which 93-31 caused significant effects at doses of 30 mg/kg or higher. From the ED50 for neuroprotection (<1 mg/kg) we calculate a TR for neuroprotection in rodents following IP administration of >30 for the pH-sensitive 93-31 GluN2B inhibitor, a value considerably higher than that estimated for neuroprotection in the literature for ifenprodil and dizolcilpine (TR≤1, Hatfield et al., 1992; Bertorelli et al., 1998; Dawson et al., 2001; Xiao et al., 2004) and CP-101,606 (TR ~4, Yang et al., 2003, Nicholson et al., 2007).
DISCUSSION
Two important findings emerge from this study. First, these data provide molecular insight into the nature of inhibition produced by therapeutically-relevant GluN2B-selective modulators that bind to the GluN1/GluN2B heterodimer interface of the amino terminal domain (Karakas et al., 2011; Burger et al., 2012; Karakas and Furukawa 2014; Lee et al., 2014). Our data suggest that changes in the 93-31 association/dissociation rates at acidic pH reflect an altered configuration around the binding pockets when ionizable residue(s) in helix-3 and helix-8 are protonated. The juxtaposition of ionizable side chains by tertiary protein structure has been suggested to shift intra-protein pKa values (Harris and Turner 2002; Jasti et al., 2007; Ritschel et al., 2009; Bombarda and Ullmann 2010; Sussman et al., 2013), creating a carboxylate-based pH sensor for ligand binding within the amino terminal domain that docking studies suggest directly interacts with 93-31. Interestingly, an interacting carboxylate pair has recently been suggested to form the pH sensor of a Ca2+ leak channel (Chang et al., 2014). While the Ca2+ leak channel dicarboxylate pair does not form the classic reciprocal hydrogen bonding of a carboxylic acid dimer, it nonetheless alternates between multiple states with varying pH, including protonation of both side-chains. In addition to ionization of the GluN2B glutamate residues impacting 93-31 binding rates, it is also possible that occupancy of the binding site by different propanolamines (Table 1) can differentially impact position and pKa of these residues. Whereas data suggest that most of the pH sensitivity is derived from changes in binding rates of 93-31, it is possible that some pH sensitivity reflects the ability of 93-31 binding to bring about down-stream protein conformation changes that govern efficacy (Low et al., 2003, Gielen et al., 2008).
Second, the development of GluN2B inhibitors with pH-dependent potency is important considering problems with past clinical programs centered on the neuroprotection by NMDAR antagonists. Data presented here show that NMDAR antagonists can be designed that provide enhanced receptor blockade during a decrease in extracellular pH, resulting in neuroprotection in acidic ischemic tissue without untoward side effects. This is because the increased potency in ischemic tissue reduces the concentration needed for neuroprotection below that which will cause effects at normal pH in healthy brain. Decreased extracellular pH universally accompanies ischemic injury and TBI (Mutch and Hansen 1984; Smith et al., 1986; Nedergaard et al., 1991; Katsura et al., 1992; Katsura and Siesjo 1998) suggesting this strategy may be applicable to acute neurodegeneration under a wide range of insults. Our application of this concept to the well-tolerated class of GluN2B-selective NMDAR antagonists further increases the likelihood that antagonists with these properties will be clinically viable for indications with synaptic or interstitial acidic pH changes. This context-dependence enhances the TR for such pH-sensitive compounds. We calculate from the effects on infarct volume following ischemia and from the dose-effect relationship in healthy animals using PCP discrimination studies that 93-31 has a TR greater than 30, which is higher than that can be calculated from the literature for GluN2B-NMDAR antagonists acting as neuroprotectants (see above).
In light of these data, the results of clinical studies with CP-101,606, which nearly reached significance for a TBI trial despite a lengthy time window for drug administration (8 hours) post-injury, are encouraging. CP-101,606 (Mott et al., 1998) and other GluN2B antagonists lack substantial pH sensitivity (Table S6) and thus it is intriguing to speculate that a compound with similar actions as CP-101,606 but with the additional enhanced pH sensitivity might be clinically successful. If the concept of context-dependent inhibition is coupled with modern clinical trial designs that allow rapid, blinded trials by first responders within hours of ischemic onset or traumatic insult (Saver 2013), there is excellent likelihood that the well-known neuroprotective potential of NMDAR antagonists can impact clinical outcomes in ischemia and TBI. This proof-of-concept molecule demonstrates the utility of this strategy, and creates a strong precedent for exploiting the pH-dependence of the GluN2B class of NMDAR antagonists for brain injury involving overactivation of NMDARs and extracellular acidification.
EXPERIMENTAL PROCEDURES
Voltage clamp recordings from transfected HEK cells
HEK 293 cell preparation and transfection were conducted (see Supplemental Experimental Procedures) , as described previously (Yuan et al., 2009). Following transfection of GluN1-1a (hereafter GluN1, Genbank U11418) and GluN2B (Genbank U11419), whole cell voltage-clamp recordings were conducted on transiently transfected HEK cells using an Axopatch 200B amplifier (Molecular Devices, Union City, CA, USA). Current responses were digitized at 40 kHz by pClamp10 software (Molecular Devices). Recordings were filtered at 8 kHz using an eight-pole Bessel filter (−3 dB; Frequency Devices, Haverhill, MD, USA). Thin-walled borosilicate glass capillary tubes (World Precision Instruments, Cat. No. TW-150F-4, Hamden, CT, USA) were used to form recording micropipettes for whole cell currents. All recording micropipettes were filled with an internal solution containing (in mM) 110 D-gluconic acid, 110 CsOH, 30 CsCl, 5 HEPES, 5 BAPTA, 4 NaCl, 2 MgCl2, 2 NaATP, 0.5 CaCl2, and 0.3 NaGTP (pH 7.35). Cells were bathed at 23°C in external solution that contained (in mM) 150 NaCl, 10 HEPES, 3 KCl, 0.5 CaCl2, 0.01 EDTA, and 30 D-mannitol at pH 7.6 or pH 6.9. All recording solutions were made from external solution and recordings were performed at holding potentials of −60 mV. Rapid solution exchange for macroscopic recordings was accomplished with a two-barrel theta glass pipette controlled by a piezoelectric translator (Burleigh Instruments, Fishers, NY); 10 to 90% open tip solution exchange times were under 1 ms, and solution exchange around the whole cell was under 5 ms (Vance et al., 2011). Assuming 93-31 follows the law of mass action, the exponential time course for the onset of inhibition should have a tau described by equation (1)
| (1) |
where kON and kOFF are the microscopic association and dissociation rate constants. From this relationship we can determine the association rate (kON) from the slope of linear plot of 1/TauONSET and concentration (Figure 1D) and the dissociation rate (kOFF) from the intercept (KD= kOFF/kON).
Two electrode voltage clamp recordings from Xenopus oocytes
All protocols involving animals were approved by the Emory University IACUC. Stage V-VI Xenopus laevis unfertilized oocytes purchased from Ecocyte (Austin, TX), and were injected with 3-5 ng of GluN1 and 7-10 ng of GluN2B cRNAs, and incubated in Barth's solution at 18°C. Two electrode voltage-clamp recordings were made 2-7 days post injection at a holding potential of –40 mV using two Warner OC725B two-electrode voltage clamp amplifiers (23°C). Oocytes were perfused with a solution of (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.01 EDTA, and 0.5 BaCl2; pH was adjusted to 7.6 by addition of NaOH, and subsequently lower pH values by addition of HCl to ensure constant concentration of Na+ in all solutions. Concentration-response inhibition curves were obtained by bath application of increasing concentrations of antagonist in the presence of 50-100 μM L-glutamate and 30 μM glycine until steady state conditions were reached (1-6 min typically, 15 min in some cases). Recordings were made from 3-10 oocytes per experiment from 2 to 6 experiments. The percent responses for the composite data were fit by the equation (2),
| (2) |
where minimum is the residual percent response in saturating concentration (constrained to ≥0) of the experimental compounds, IC50 is the concentration of antagonist that causes half maximal inhibition, and nH is a slope factor of the inhibition curve.
Triheteromeric GluN1/2A/2B receptors were expressed in Xenopus oocytes using the constructs and method described in Hansen et al. 2014 (see Supplemental Experimental Procedures).
Docking of 93-series compounds
Compounds 93-31, 93-97, and NP10075 were built in Maestro 9.2 (Schrodinger suite) and docked using the OPLS2005 force field and Glide Induced Fit docking protocol (Sherman et al., 2006). The atomic coordinates for GluN1/GluN2B amino terminal domain co-crystallized with ifenprodil was used as the input structure (3QEL, Karakas et al., 2011). The binding site was defined by residues within 5 Å of ifenprodil, which were treated as flexible. A maximum of 100 poses per ligand was generated. Both N-protonated stereoisomers and the unprotonated form were docked. The unprotonated forms were docked with N-inversion enabled. All poses were also subsequently scored with MM-GBSA binding energy calculations (Prime ver 3.1). VMD 1.9 (Humphrey et al., 1996) was used for visualization, while prediction of altered pKa values was carried out with the VMD PropKa plugin (Olsson 2011). The semi-empirical PM6 electrostatic potential evaluation of the GluN1/GluN2B ligand binding site and the associated protein side chains was performed with Spartan 10, version 1.1.0. from Wavefunction Inc. (Young 2001, http://www.wavefun.com/products/products.html; accessed 10/9/2013).
In vivo models of transient focal ischemia
Transient focal cerebral ischemia (30 min) was induced in mice by intraluminal middle cerebral artery occlusion (MCAO; Junge et al., 2003). Male C57BL/6 mice (3–5 months old, Jackson Labs) were anesthetized with 2% isoflurane in 98% O2. We avoided the use of ketamine given its confounding neuroprotective channel-blocking actions on NMDARs (Hamill et al., 2009). The rectal temperature was monitored and mouse temperature maintained at 37°C (range 36.5-37.5) with a homeothermic blanket. Blood pressure was not monitored or controlled. Relative changes in regional cerebral blood flow were monitored with a laser Doppler flowmeter (Perimed); the Doppler probe was glued to the skull 2 mm posterior and 4-6 mm lateral of the bregma. Only animals with reduction in blood flow to 20% and clear recovery of blood flow were included in the study. An 11-mm 5-0 Dermalon or Look (SP185) black nylon non-absorbable suture with the tip flame-rounded was introduced for 30 min into the left internal carotid artery through the external carotid artery stump until monitored cerebral blood flow was reduced <20% (at 10.5 – 11 mm of suture insertion). Only animals with sustained reduction in blood flow to <20% for 30 min and clear recovery of blood flow to >90% following removal of the intraluminal suture were included. After 24 hour, the brain was removed and cut into 2 mm sections. The lesion was identified with 2% 2,3,5-triphenyltetrazolium chloride (TTC) in PBS at 37°C for 20 min and the infarct area measured using NIH IMAGE (Scion Corporation, Beta 4.0.2 release). The area of the lesion, as identified by digital threshold reductions in TTC staining 20% lower than intensity in contralateral cortex, was manually outlined and the cubic volume of the infarct size determined. A ratio of the contralateral to ipsilateral hemisphere section volume was multiplied by the corresponding infarct section volume to correct for edema. Drug was administered by intraperitoneal (IP, 3 mL/kg) or by intracerebroventricular (ICV, 1 ul) injection 10 min before initiation of surgery (i.e. 20 min before vessel occlusion) or 30 min after removal of the intraluminal suture. ICV injections were made into the right ventricle (2 mm post., 1 mm lat. of the bregma, needle inserted 3 mm). Compound was formulated in 1:1 DMSO:phosphate buffered saline (pH 7.4) for IP injection and 1:10 DMSO:phosphate buffered saline (pH 7.4) for ICV. Those performing the surgical procedure and analysis were blinded to the identity of the treatment.
Measurement of locomotor activity, rotorod activity, and PCP discrimination
Male Sprague-Dawley rats (100-150 g) were placed in activity monitoring boxes for a one hour habituation period prior to drug testing. At one hour, monitoring was interrupted to inject animals IP with various doses of drug in 50% v/v DMSO:saline. Animals were returned to the cage and total locomotor activity was monitored for the next 2 hours. The total number of light beam breaks in the cage (horizontal and vertical) was automatically determined by a computer and results averaged for each drug. An ANOVA followed by Dunnett's test used to compare total horizontal activity of drug doses to vehicle controls.
For rotorod, male C57BL/6 mice (>90 days old) were tested using a four-chamber Rotamax 4/8 rotorod (Columbus Instruments, Columbus, OH). The test was initiated by placing mice on a rotating rod (5 rpm) that was 3.8 cm diameter by 8 cm wide and suspended 30 cm from the floor of a chamber. After 10 sec the rotation was accelerated from 5 to 35 rpm over a 5 min period. The latency to fall (sec) was recorded automatically with a light-activated sensor in the bottom of the chamber. Animals were trained four times with an inter-trial interval of 25 min, each day for two days. On day three mice were randomly assigned to groups and administered compound or vehicle (IP; 47.5% DMSO: 47.5% saline:5% DMA). Twenty minutes post drug administration the mice were tested as before with an inter-trial interval of 25 min and latency to fall recorded. Those performing the study were blinded to the identity of the treatment group.
To evaluate the psychotomimetic effects of the test compounds, six adult male Sprague Dawley rats (Charles River, Wilmington, DE) were trained to discriminate administration of 2.0 mg/kg PCP from saline injections (Balster and Willetts 1988). They were singly housed with free access to water under a 12-h light/dark cycle. Subjects were maintained at 85 to 90% free feeding weight with food access (Harlan Teklad Rodent Diet, Williamston, IL) limited to post-session periods to motivate lever-pressing for food. The subjects were trained daily (Monday - Friday) in 30 min sessions in standard two-lever operant conditioning chambers (MedAssociates, St. Alban, VT). Completion of 32 consecutive responses (fixed ratio 32; FR 32) on the appropriate lever resulted in reinforcement with delivery of a 45-mg food pellet (P.J. Noyes Company, Inc., Lancaster, NH). Responding on the incorrect lever reset the FR count towards pellet delivery. PCP and saline were administered IP under a double alternation schedule (eg. drug, drug, saline, saline) 15 min prior to the beginning of the session. During sessions, a white stimulus light located centrally above each lever was illuminated. Test sessions were conducted up to twice weekly when the subjects met the following criteria on the four preceding training sessions (two PCP and two saline): (1) first FR completed on the correct lever, and (2) greater than 85% correct-lever responding over the entire session. During test sessions, completion of 32 consecutive responses on either lever resulted in the delivery of food.
Rats were tested at 93-31 doses of 1, 3, 10, 30, and 56 mg/kg administered IP, 30 min pre-session; various doses of PCP were tested in the same animals for comparison. Data were evaluated by calculating the mean percentage of responses on the PCP-associated lever and the overall mean response rate. For sessions in which a subject's responding was less than 0.05 resp/sec, the percent PCP-lever responding was excluded from determination of the group mean. Full substitution for PCP was defined as greater than 80% PCP-lever responding while partial substitution was defined as producing between 20% and 80% PCP lever responding.
Chemistry
The synthetic reaction schemes for all 93-series compounds can be found in Tahirovic et al. (2008).
Statistics
Values presented are mean±SEM, and are compared using Student pair or unpaired t-test, and ANOVA where appropriate. The level of significance was chosen as 0.05.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NINDS (NS036654 ST), by ATDC (ST), EmTech Bio (RD). We thank Hiro Furukawa for critical comments on the manuscript, and Phuong Le, Jing Zhang, Anel Tankovic for excellent technical assistance. We thank Ray Dingledine (NS36604) for early contributions and discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
HY designed and performed electrophysiology experiments, analyzed data and wrote the paper. GW, JPS performed molecular dynamic simulations, ligand docking, homology modeling, and wrote the paper. SJM designed and performed pharmacology experiments in Xenopus oocytes and in vivo experiments, analyzed the data, and wrote the paper. KLN designed and performed behavioral experiments, analyzed the data, and wrote the paper. SAS designed and performed pharmacology experiments in Xenopus oocytes, analyzed the data, and wrote the paper. PL performed pharmacology experiments in Xenopus oocytes and in vivo experiments, and analyzed the data. YAT, TG, DM synthesized compounds and wrote the paper. DCL supervised the compounds synthesis and wrote the paper. SFT designed experiments, performed in vivo experiments, analyzed data, wrote the paper and supervised the project. All authors discussed the results and implications and commented on the manuscript.
SUPPLEMENTAL INFORMATION
Supplemental information includes four figures, six tables, and methods, and can be found with this article online at http://www.cell.com/neuron/home.
REFERENCES
- Albers GW, Goldstein LB, Hall D, Lesko LM. Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA. 2001;286:2673–2682. doi: 10.1001/jama.286.21.2673. [DOI] [PubMed] [Google Scholar]
- Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. B. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- Balster RL, Willetts J. Receptor mediation of the discriminative stimulus properties of phencyclidine and sigma-opioid agonists. Psychopharmacol. Ser. 1988;4:122–135. doi: 10.1007/978-3-642-73223-2_10. [DOI] [PubMed] [Google Scholar]
- Banke TG, Dravid SM, Traynelis SF. Protons trap NR1/NR2B NMDA receptors in a nonconducting state. J. Neurosci. 2005;25:42–51. doi: 10.1523/JNEUROSCI.3154-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste M, Mayer ML. Effect of extracellular pH on the potency of N-methyl-D-aspartic acid receptor competitive antagonists. Mol. Pharmacol. 1992;42:679–686. [PubMed] [Google Scholar]
- Bertorelli R, Adami M, Di Santo E, Ghezzi P. MK 801 and dexamethasone reduce both tumor necrosis factor levels and infarct volume after focal cerebral ischemia in the rat brain. Neurosci. Lett. 1998;246:41–44. doi: 10.1016/s0304-3940(98)00221-3. [DOI] [PubMed] [Google Scholar]
- Blagrove M, Morgan CJA, Curran HV, Bromley L, Brandner B. The incidence of unpleasant dreams after sub-anaesthetic ketamine. Psychopharmacol. 2009;203:109–120. doi: 10.1007/s00213-008-1377-3. [DOI] [PubMed] [Google Scholar]
- Bombarda E, Ullmann GM. pH-dependent pKa values in proteins--a theoretical analysis of protonation energies with practical consequences for enzymatic reactions. J. Phys. Chem. B. 2010;114:1994–2003. doi: 10.1021/jp908926w. [DOI] [PubMed] [Google Scholar]
- Bordi F, Pietra C, Ziviani L, Reggiani A. The glycine antagonist GV150526 protects somatosensory evoked potentials and reduces the infarct area in the MCAo model of focal ischemia in the rat. Exp. Neurol. 1997;145:425–433. doi: 10.1006/exnr.1997.6442. [DOI] [PubMed] [Google Scholar]
- Burger PB, Yuan H, Karakas E, Geballe M, Furukawa H, Liotta DC, Snyder JP, Traynelis SF. Mapping the binding of GluN2B-selective N-methyl-D-aspartate receptor negative allosteric modulators. Mol. Pharmacol. 2012;82:344–359. doi: 10.1124/mol.112.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Bruni R, Kloss B, Assur Z, Kloppmann E, Rost B, Hendrickson WA, Liu Q. Structural basis for a pH-sensitive calcium leak across membranes. Science. 2014;344:1131–1135. doi: 10.1126/science.1252043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaperon F, Müller W, Auberson YP, Tricklebank MD, Neijt HC. Substitution for PCP, disruption of prepulse inhibition and hyperactivity induced by N-methyl-D-aspartate receptor antagonists: preferential involvement of the NR2B rather than NR2A subunit. Behav. Pharmacol. 2003;14:477–487. doi: 10.1097/01.fbp.0000091471.79060.ed. [DOI] [PubMed] [Google Scholar]
- Chenard BL, Menniti FS. Antagonists selective for NMDA receptors containing the NR2B subunit. Curr. Pharm. Des. 1999;5:381–404. [PubMed] [Google Scholar]
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J. Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Wadsworth G, Palmer AM. A comparative assessment of the efficacy and side-effect liability of neuroprotective compounds in experimental stroke. Brain Res. 2001;892:344–350. doi: 10.1016/s0006-8993(00)03269-8. [DOI] [PubMed] [Google Scholar]
- De Vry J, Jentzsch KR. Role of the NMDA receptor NR2B subunit in the discriminative stimulus effects of ketamine. Behav. Pharmacol. 2003;14:229–235. doi: 10.1097/00008877-200305000-00007. [DOI] [PubMed] [Google Scholar]
- Diener HC, AlKhedr A, Busse O, Hacke W, Zingmark PH, Jonsson N, Basun H. Treatment of acute ischaemic stroke with the low-affinity, use-dependent NMDA antagonist AR-R15896AR. A safety and tolerability study. J. Neurol. 2002;249:561–568. doi: 10.1007/s004150200065. [DOI] [PubMed] [Google Scholar]
- Ellren K, Lehmann A. Calcium dependency of N-methyl-D-aspartate toxicity in slices from the immature rat hippocampus. Neurosci. 1989;32:371–379. doi: 10.1016/0306-4522(89)90085-7. [DOI] [PubMed] [Google Scholar]
- Farin A, Marshall LF. Lessons from epidemiologic studies in clinical trials of traumatic brain injury. Acta. Neurochir. Suppl. 2004;89:101–107. doi: 10.1007/978-3-7091-0603-7_14. [DOI] [PubMed] [Google Scholar]
- Gielen M, Le Goff A, Stroebel D, Johnson JW, Neyton J, Paoletti P. Structural rearrangements of NR1/NR2A NMDA receptors during allosteric inhibition. Neuron. 2008;57:80–93. doi: 10.1016/j.neuron.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladstone DJ, Black SE, Hakim AM, Heart, and Ontario Centre of Excellence in Stroke Recovery Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–2136. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- Hamill CE, Mannaioni G, Lyuboslavsky P, Sastre AA, Traynelis SF. Protease-activated receptor 1-dependent neuronal damage involves NMDA receptor function. Exp. Neurol. 2009;217:136–146. doi: 10.1016/j.expneurol.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TK, Turner GJ. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- Hatfield RH, Gill R, Brazell C. The dose-response relationship and therapeutic window for dizocilpine (MK-801) in a rat focal ischaemia model. Eur. J. Pharmacol. 1992;216:1–7. doi: 10.1016/0014-2999(92)90201-e. [DOI] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron. 2005;46:261–274. doi: 10.1016/j.neuron.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449:316–323. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- Junge CE, Sugawara T, Mannaioni G, Alagarsamy S, Conn PJ, Brat DJ, Chan PH, Traynelis SF. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13019–13024. doi: 10.1073/pnas.2235594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J, Dimlich RV, Biros MH, Hedges J. Mechanisms of ischemic cerebral injury. Resuscitation. 1987;15:149–169. doi: 10.1016/0300-9572(87)90012-8. [DOI] [PubMed] [Google Scholar]
- Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009;28:3910–3920. doi: 10.1038/emboj.2009.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukawa H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 2011;475:249–253. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura K, Asplund B, Ekholm A, Siesjö BK. Extra-and Intracellular pH in the Brain During Ischaemia, Related to Tissue Lactate Content in Normo- and Hypercapnic rats. Eur. J. Neurosci. 1992;4:166–176. doi: 10.1111/j.1460-9568.1992.tb00863.x. [DOI] [PubMed] [Google Scholar]
- Katsura K, Siesjo B. Acid-base Metab. In: Kalia K, Ransom BR, editors. ischemia. Wiley-Liss; 1998. [Google Scholar]
- Kidwell CS, Liebeskind DS, Starkman S, Saver JL. Trends in acute ischemic stroke trials through the 20th century. Stroke. 2001;32:1349–1359. doi: 10.1161/01.str.32.6.1349. [DOI] [PubMed] [Google Scholar]
- Lee CH, Lü W, Michel JC, Goehring A, Du J, Song X, Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees KR, Asplund K, Carolei A, Davis SM, Diener HC, Kaste M, Orgogozo JM, Whitehead J. Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: a randomised controlled trial. GAIN International Investigators. Lancet. 2000;355:1949–1954. doi: 10.1016/s0140-6736(00)02326-6. [DOI] [PubMed] [Google Scholar]
- Leiserowitz L, Harris TK, Turner GJ. Molecular packing modes. Carboxylic acids. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 2002;53:85–98. [Google Scholar]
- Low CM, Lyuboslavsky P, French A, Le P, Wyatte K, Thiel WH, Marchan EM, Igarashi K, Kashiwagi K, Gernert K, et al. Molecular determinants of proton-sensitive N-methyl-D-aspartate receptor gating. Mol. Pharmacol. 2003;63:1212–1222. doi: 10.1124/mol.63.6.1212. [DOI] [PubMed] [Google Scholar]
- Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J. Mercury: visualization and analysis of crystal structures. J. Appl. Cryst. 2006;39:453–457. [Google Scholar]
- Matsumoto T, Obrenovitch TP, Parkinson NA, Symon L. Cortical activity, ionic homeostasis, and acidosis during rat brain repetitive ischemia. Stroke. 1990;21:1192–1198. doi: 10.1161/01.str.21.8.1192. [DOI] [PubMed] [Google Scholar]
- Mody I, MacDonald JF. NMDA receptor-dependent excitotoxicity: the role of intracellular Ca2+ release. Trends Pharmacol. Sci. 1995;16:356–359. doi: 10.1016/s0165-6147(00)89070-7. [DOI] [PubMed] [Google Scholar]
- Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF. Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J. Neurosurg. 1999;91:737–743. doi: 10.3171/jns.1999.91.5.0737. [DOI] [PubMed] [Google Scholar]
- Mosley CA, Myers SJ, Murray EE, Santangelo R, Tahirovic YA, Kurtkaya N, Mullasseril P, Yuan H, Lyuboslavsky P, Le P, et al. Synthesis, structural activity-relationships, and biological evaluation of novel amide-based allosteric binding site antagonists in NR1A/NR2B N-methyl-D-aspartate receptors. Bioorg. Med. Chem. 2009;17:6463–6480. doi: 10.1016/j.bmc.2009.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, Traynelis SF, Dingledine R. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat. Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr. Opin. Pharmacol. 2006;6:53–60. doi: 10.1016/j.coph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Mutch WA, Hansen AJ. Extracellular pH changes during spreading depression and cerebral ischemia: mechanisms of brain pH regulation. J. Cereb. Blood Flow Metab. 1984;4:17–27. doi: 10.1038/jcbfm.1984.3. [DOI] [PubMed] [Google Scholar]
- Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, Bullock MR, Choi SC, Clifton GL, Contant CF, et al. Clinical trials in head injury. J. Neurotrauma. 2002;19:503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Kraig RP, Tanabe J, Pulsinelli WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 1991;260:R581–588. doi: 10.1152/ajpregu.1991.260.3.R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson KL, Mansbach RS, Menniti FS, Balster RL. The phencyclidine-like discriminative stimulus effects and reinforcing properties of the NR2B-selective N-methyl-D-aspartate antagonist CP-101 606 in rats and rhesus monkeys. Behav. Pharmacol. 2007;18:731–743. doi: 10.1097/FBP.0b013e3282f14ed6. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Gunzler SA, Kirchhoff T, Hogarth P, Weaver JL, Krams M, Jamerson B, Menniti FS, Landen JW. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and Parkinsonism. Mov. Disord. 2008;23:1860–1866. doi: 10.1002/mds.22169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–721. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- Olsson M. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Compu. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Pahk AJ, Williams K. Influence of extracellular pH on inhibition by ifenprodil at N-methyl-D-aspartate receptors in Xenopus oocytes. Neurosci. Lett. 1997;225:29–32. doi: 10.1016/s0304-3940(97)00176-6. [DOI] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J. Clin. Psychopharmacol. 2008;28:631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Prime, version 3.1. Schrödinger, LLC; New York, N.Y.: 2012. [Google Scholar]
- Reggiani A, Pietra C, Arban R, Marzola P, Guerrini U, Ziviani L, Boicelli A, Sbarbati A, Osculati F. The neuroprotective activity of the glycine receptor antagonist GV150526: an in vivo study by magnetic resonance imaging. Eur. J. Pharmacol. 2001;419:147–153. doi: 10.1016/s0014-2999(01)00948-7. [DOI] [PubMed] [Google Scholar]
- Ritschel T, Hoertner S, Heine A, Diederich F, Klebe G. Crystal structure analysis and in silico pKa calculations suggest strong pKa shifts of ligands as driving force for high-affinity binding to TGT. Chembiochem. 2009;10:716–27.. doi: 10.1002/cbic.200800782. [DOI] [PubMed] [Google Scholar]
- Rowland LM. Subanesthetic ketamine: how it alters physiology and behavior in humans. Aviat. Space Environ. Med. 2005;76:C52–C58. [PubMed] [Google Scholar]
- Sacco RL, DeRosa JT, Haley EC, Jr, Levin B, Ordronneau P, Phillips SJ, Rundek T, Snipes RG, Thompson JL, Glycine Antagonist in Neuroprotection Americas Investigators Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: a randomized controlled trial. JAMA. 2001;285:1719–1728. doi: 10.1001/jama.285.13.1719. [DOI] [PubMed] [Google Scholar]
- Saver JL. The 2012 Feinberg Lecture: treatment swift and treatment sure. Stroke. 2013;44:270–277. doi: 10.1161/STROKEAHA.112.671354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006;49:534–53. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Shipman ST, Douglass PC, Yoo HS, Hinkle CE, Mierzejewski EL, Pate BH. Vibrational dynamics of carboxylic acid dimers in gas and dilute solution. Phys. Chem. Chem. Phys. 2007;9:4572–4586. doi: 10.1039/b704900e. [DOI] [PubMed] [Google Scholar]
- Smith ML, von Hanwehr R, Siesjö BK. Changes in extra-and intracellular pH in the brain during and following ischemia in hyperglycemic and in moderately hypoglycemic rats. J. Cereb. Blood. Flow Metab. 1986;6:574–583. doi: 10.1038/jcbfm.1986.104. [DOI] [PubMed] [Google Scholar]
- Sussman F, Villaverde MC, Domínguez JL, Danielson UH. On the Active Site Protonation State in Aspartic Proteases: Implications for Drug Design. Curr. Pharm. Des. 2013;19:4257–4275. doi: 10.2174/1381612811319230009. [DOI] [PubMed] [Google Scholar]
- Tahirovic YA, Geballe M, Gruszecka-Kowalik E, Myers SJ, Lyuboslavsky P, Le P, French A, Irier H, Choi WB, Easterling K. Enantiomeric propanolamines as selective N-methyl-D-aspartate 2B receptor antagonists. J. Med. Chem. 2008;51:5506–5521. doi: 10.1021/jm8002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Hartley M, Heinemann SF. Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science. 1995;268:873–876. doi: 10.1126/science.7754371. [DOI] [PubMed] [Google Scholar]
- Tzeli D, Theodorakopoulos G, Petsalakis ID, Ajami D, Rebek J. Theoretical study of hydrogen bonding in homodimers and heterodimers of amide, boronic acid, and carboxylic acid, free and in encapsulation complexes. J. Am. Chem. Soc. 2011;133:16977–16985. doi: 10.1021/ja206555d. [DOI] [PubMed] [Google Scholar]
- Vance KM, Simorowski N, Traynelis SF, Furukawa H. Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat. Commun. 2011;2:294. doi: 10.1038/ncomms1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileiou C, Lee KS, Crist RM, Vaezeslami S, Geiger JH, Borhan B. Dissection of the critical binding determinants of cellular retinoic acid binding protein II by mutagenesis and fluorescence binding assay. Proteins. 2009;76:281–290. doi: 10.1002/prot.22334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, James ML, Venkatraman TN, Wilson LJ, Lyuboslavsky P, Myers SJ, Lascola CD, Laskowitz DT. pH-sensitive NMDA inhibitors improve outcome in a murine model of SAH. Neurocrit Care. 2014;20:119–131. doi: 10.1007/s12028-013-9944-9. [DOI] [PubMed] [Google Scholar]
- Wood PL. The NMDA receptor complex: a long and winding road to therapeutics. IDrugs. 2005;8:229–235. [PubMed] [Google Scholar]
- Wroge CM, Hogins J, Eisenman L, Mennerick S. Synaptic NMDA receptors mediate hypoxic excitotoxic death. J. Neurosci. 2012;32:6732–6742. doi: 10.1523/JNEUROSCI.6371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F, Pardue S, Arnold T, Carden D, Alexander JS, Monroe J, Sharp CD, Turnage R, Conrad S. Effect of ifenprodil, a polyamine site NMDA receptor antagonist, on brain edema formation following asphyxial cardiac arrest in rats. Resuscitation. 2004;61:209–219. doi: 10.1016/j.resuscitation.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Yang Y, Li Q, Yang T, Hussain M, Shuaib A. Reduced brain infarct volume and improved neurological outcome by inhibition of the NR2B subunit of NMDA receptors by using CP101,606-27 alone and in combination with rt-PA in a thromboembolic stroke model in rats. J. Neurosurg. 2003;98:397–403. doi: 10.3171/jns.2003.98.2.0397. [DOI] [PubMed] [Google Scholar]
- Young D. Comput. Chem. A Pract. Guid. Appl. Tech. to Real World Probl. Vol. 330. Wiley-Interscience; 2001. [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci. 2009;29:12045–12058. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurkewicz L, Weaver J, Bullock MR, Marshall LF. The effect of the selective NMDA receptor antagonist traxoprodil in the treatment of traumatic brain injury. J. Neurotrauma. 2005;22:1428–1443. doi: 10.1089/neu.2005.22.1428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.