

Abstract
Δ-distance maps can detect local remodeling that is difficult to accurately determine using superimpositions. TMSs 11 in both LacY and XylE of the Major Facilitator Superfamily (MFS) uniquely contribute the greatest amount of mobile surface area in the outward occluded state, and undergo analogous movements. The intracellular part of TMS11 moves away from the C-terminal domain and into the substrate cavity during the conformational change from the outward occluded to the inward occluded state. Upon releasing the substrate to the inside and assuming the inward open state, a difference was noted between LacY and XylE where TMS11 of LacY moved further into the substrate release space whereas in XylE, TMS11 slightly retracted into the C-terminal domain. Independent movement of the N-terminal half of TMS11 suggests that it is flexible in the middle. Repeat-swapped homology modeling was used to discover that a loop connecting TMSs 10 and 11 in LacY probably moves during the transition between the yet to be solved outward open state and the outward occluded state. TMSs 11 and the other elements displaying a notable domain-independent movement colocalize with the interdomain linker, suggesting that these elements could drive the alternating access movement between the domain halves. Preliminary evidence indicates that analogous movements occur in other members of the MFS.
Keywords: TMS11, LacY, XylE, MFS, contact maps, occluded conformational states
Introduction
The Major Facilitator Superfamily (MFS) is the largest family of secondary carriers, containing 83 families within TC# 2.A.1 of the Transporter Classification Database, TCDB (www.tcdb.org;1), and an additional 7 families within the extended superfamily. Most MFS proteins have a 6+6 α-helical transmembrane segment (TMS) topology (but some have 6+2+6) where the two halves are connected by an interdomain linker that allows movement of the halves relative to each other during the alternating access transport cycle. The term “rocker switch” is specifically used to define a kinetic behavior of secondary carriers where transport rate is temperature dependent, but the affinity is temperature independent2, and the term can also be used to describe the alternating access mechanism3.
The interdomain linker of LacY contains three cytoplasmic helical segments that colocalize in space when the outward open conformation is stabilized. The cycle involves at least four major states: outward open, outward and inward occluded, and inward open. Only the outward open states of LacY and XylE have not been determined by x-ray crystallography. However, this state has been determined for the MFS homologue, FucP4. We attempt here to examine a model relying on the available x-ray data, a repeat-swapped homology model5.
Many MFS proteins use a proton gradient, typically created by primary active transporters, to allow intracellular accumulation of solutes such as lactose and xylose. The E. coli lactose transporter, LacY, is one of the most intensely studied secondary carriers, having had the effects of point mutations on transport and counterflow investigated for each of its residues. The xylose transporter, XylE, provides a model system of an MFS belonging to the sugar porter family, which includes the human glucose transporters, the GLUTs; although some GLUTs have been structurally determined6. XylE was the first MFS transporter to have crystal structures determined for both inward and outward-facing occluded states7,8.
Δ-distance or difference distance mapping offers advantages over superimposition because it allows detection of local differences that superimposition of parts of homologous structures cannot reveal. We have previously published a Δ-distance map comparing the occluded states of XylE, showing that the majority of the inter-domain distance remodeling is localized to the C-terminal bundle9. Publication of the crystal structure of a sugar-bound state of LacY confirmed these observations10. Note, however, that this finding does not contradict unfolding experiments11, because they measure other physical properties, not distance remodeling.
In this article, we use an established technique, Δ-distance mapping, to compare structures in different conformational states. The concept of calculating Δ-distance maps is not new12–14, but the recent availability of multiple conformational states for MFS homologues, LacY and XylE, has created a new application area. In fact, the most recent version of Chimera, release 1.1014, contains a new function called RR distance maps. While this function can be used to compare two contact maps, it does not visualize whether distances increase or decrease, and it only shows distance remodeling as a color scale of standard deviations. Δ-distance mapping provides higher precision in localizing conformational changes compared to superimposition-based methods. This approach reveals the main differences between LacY and XylE as they transition between their four main conformational states: outward open, outward occluded, inward occluded, and inward open.
Since crystal structures of LacY and XylE are not yet available in the outward open state, we used a repeat-swapped homology model to estimate the missing state. Common patterns and differences in internal distance remodeling patterns exhibited in these two transporters were identified. To complement the results from the Δ-distance mapping, we provide a detailed description of the different conformational states and the changes occurring between them. In particular, the movements of full-length and segmental TMSs displaced by the alternating access movement between domain halves were examined, and they were correlated with the placement of these elements in relation to the interdomain linker.
While this study is primarily limited to making comparisons between LacY and XylE, the only two proteins to have availability of three determined conformational states, we attempt to generalize to other MFS proteins by creating homology models. This approach has identified conserved movements, also involving TMS11 for the fucose porter, FucP4, and a member of the peptide transporting POT/PTR family, NRT115. This study is the first of its kind, because it uses three recent developments: the repeat-swapped model of the outward open state of LacY5, the recently published crystal structure of the sugar-bound state of LacY10, and the use of Δ-distance mapping to compare conformational states9
Methods
Δ-distance maps
We downloaded the LacY PDB files for the inward open conformation, with an affinity inactivator (2Y5Y) and in the sugar bound, outward occluded state (4OAA). The range of residues was restricted to the greatest common range, from 4 (LEU)-403 (PRO). We selected only ’CA’ (backbone α-carbon) atoms from the ‘A’ chains. There was a 2 line discrepancy in line count between the files, indicating missing atoms in 4OAA. Residues 191–206 were missing in 4OAA, and residues 189–202 were missing in 2Y5Y. We had to delete segments so that corresponding segments encompassing residues 189–206 were missing in both files. As a result, both files had 382 lines, running from residue 4 to residue 403 but missing 18 residues (189–206).
The CA atoms of XylE chain A of 4JA3 (8–465) and of 4GBY (5–479, no atoms missing between 8–465) were selected, yielding 426 and 475 lines, respectively. 4JA3 was missing 265–275, 304–309, 398–406 and 434–439 (inclusive). In total, 32 residues were missing in 4JA3 (8–465). To compensate for the missing atoms in 4JA3, we removed all atoms from 4GBY that were not present in 4JA3.
The data were analyzed using a Perl script that calculated all pairwise Euclidian distances in Angstroms, rounding the numbers to two decimal places. The resulting matrices were loaded as a tab-delimited spreadsheet in Microsoft Excel. The distance map of LacY (2Y5Y) was subtracted from 4OAA, generating a Δ-distance map for the comparison. Conditional formatting was applied, using the Blue – White – Red color scale. The quadrants of the plot representing the alternating access movements between the N- and C-terminal halves were not considered when applying conditional formatting, making the detection of small remodeling of helix distances within each half possible.
Repeat-swapped homology model of the outward open states
A theoretical model of the outward open conformation of LacY has previously been created using the repeat-swapping methodology5. The model of the C-terminal half of the sequence was based on the N-terminal half of the inward open structure and vice versa. In Modeller 9.7, LacY in the inward facing state was used as the template. A sequence alignment from AlignMe16 was used to guide the homology modeling. The model was energy minimized using Charmm 3.4. The repeat-swapped model of the outward open state of LacY is available in the Protein DataBank (PDB): PM0076824 (repeat-swapped model). Modeller was used to emulate the above experimental procedure and build a theoretical model of the outward open state of XylE that was limited to the C-terminal 6 TMS bundle.
Use of RR distance maps to generalize to other members of the MFS
We used AlignMe PST17 to align TMSs 1–6 and TMSs 7–12 of FucP. The resulting alignment was used to guide Modeller to create a homology model of TMSs 7–12 (in an inward open-like state) using TMSs 1–6 in the outward open state (3O7P) as the template. Thus, TMSs 7–12 in the outward open state could be superimposed on TMSs 7–12 in the inward open-like state (the model). RR distance maps were used to detect differences between the two structures and identified a cross-shaped region located near TMSs 11, which coincides with elevated standard deviation measurements.
With the outward occluded state of XylE (4GC0) as the template, a homology model of NRT1, also in the outward occluded state, was compared with the inward open state of NRT1 of the POT/PTR family (4CL5; TCID 2.A.17.3.1) using the standard deviation measurement function of RR distance maps in Chimera 1.10. A region corresponding to a loop connecting TMSs 11 and 12, was found that displayed the greatest distance remodeling in the C-terminal domain, approximately 10–15 standard deviations of change.
Schematic illustration of the substrate translocation pathway
We used the model clipping function in Chimera to create images representing cross-sections of the protein at different membrane-parallel planes. We used the command “mclip all offset 1” to move the clipping plane by one Angstrom. The cross-sections were exported to the Inkscape vector graphics drawing program where a vector path was placed around the edge of the channel in each cross-section. The images of the cross-sections were organized in a regular way such that the paths were also spaced regularly. The vector graphics file was parsed using a Perl script that converted the paths to coordinates viewable in molecular graphics programs. This PDB-like output could subsequently be opened in Chimera. By providing a reduced representation of the surface area, we were better able to view the shape of the channel.
Error-scaling of difference distance matrices
Difference distance matrices, which contain the absolute difference between Cartesian coordinate vectors of atoms, were first introduced in 197418, and can be used to reconstruct the geometry of a molecule except its handedness. To be able to objectively compare protein structures, we have employed the normalization proposed in Thomas R. Schneider’s error-scaled difference distance matrices19. According to Schneider, a very high noise can originate from coordinate errors, suggesting that difference distance matrices need to be normalized to their error levels. A heuristic approach to find the standard uncertainties has been developed in cases where the inversion of the full least-squares matrix is not available. These formulaes can be used to take the B values (temperature factors) in PDB files and put them on a common scale so that their influence can be factored into difference distance matrices. The following parameters can be obtained from PDB files: 1) the number of fully occupied atomic sites, 2) the number of reflections, 3) the completeness, 4) the resolution and 5) the Rfree. The positional error is . For 2Y5Y, Ni = 382, nobs = 35055, C = 97.7 (“completeness for range, %”), Rfree = 0.30 and dmin = 3.38. . For 4OAA, Ni = 382, nobs = 20954, C = 95.3 (“completeness for range, %”), Rfree = 0.29 and dmin = 3.50. . For 4JA3, Ni = 426, nobs = 17321, C = 82.3 (“completeness for range, %”), Rfree = 0.31 and dmin = 3.80. . For 4GBY, Ni = 426, nobs = 19517, C = 98.4 (“completeness for range, %”), Rfree = 0.27 and dmin = 2.81. . The estimated positional error is . For 2Y5Y, Bavg = 157.24. For 4OAA, Bavg = 157.54. For 4JA3, Bavg = 224.95. For 4GBY, Bavg = 75.72. Note that for XylE, 4GBY has particularly good accuracy, whereas 4JA3 has bad accuracy (panels c and d in Figure 1). The estimated standard deviation of an element of the difference distance matrix, , can be estimated by calculating: . The elements of the error-scaled matrix can be obtained by calculating: . Note that four separate B values go into the normalization of each cell. While we noted a marked global difference in the average B values, especially between inward occluded XylE (4JA3) and outward occluded XylE (4GBY), it does not affect the TMS11 region. Plotting the raw B-values for the four files also revealed nothing that would suggest that the B-values would be comparatively high in the TMS11 region.
Results
Δ-disance mapping
We calculated Δ-distance maps comparing the distance remodeling between outward and inward occluded states in the N- and C-terminal domains of LacY and XylE (Figure 1, Tables 1 and 2). This outward to inward occluded state transition, as discussed below, represents the largest conformational change that occurs between any of the four known states of these transporters. We also included Δ-distance maps comparing inward occluded and inward open states of LacY and XylE (Supplementary file 1) and analogous comparisons using theoretical models of outward open states (see “Repeat-swapped homology modeling” section). These are treated separately for two reasons: (1) they represent more limited conformational changes and (2) they represent usage of theoretical data and are more speculative. Together, these maps cover all four known conformational states that these MFS transporters can assume.
Figure 1. Δ-distance maps of the occluded states of LacY and XylE.
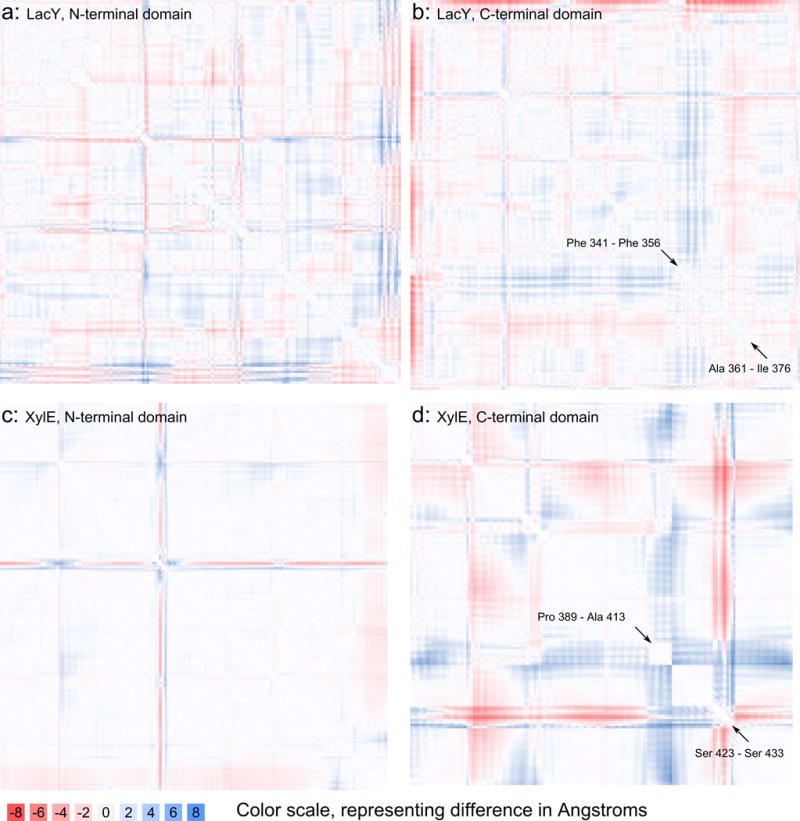
a and b: Δ-distance map (LacY), calculated by subtracting the distance map of 4OAA (outward occluded state) from that of 2Y5Y (inward affinity inactivator-occluded state); 2Y5Y-4OAA (“inward-outward” convention). Blue (positive difference) indicates that a distance between two backbone α-carbons (CA) increased as the protein transitioned to the inward occluded state, while red indicates a negative difference, meaning that the distance decreased. The first panel (a) shows quadrant 2 of the plot of the full structure and compares distances in the N-terminal domain of LacY, and the second panel (b) shows quadrant 4, comparing distances within LacY’s C-terminal domain.
c and d: Δ-distance map of 4JA3-4GBY (XylE in the inward occluded and outward occluded states, respectively), comparing distances in the N-terminal (c) and C-terminal (d) domains. TMS10B and TMS11 is located between the arrows with associated residue ranges presented in panels (b) and (d).
Table 1. Features of 2Y5Y and 4OAA (inward affinity inactivator-bound and outward occluded states of LacY, respectively).
The shift in the numbering between the location in the diagram (Figure 1) and the actual residue number in the protein is due to missing residues between the structures.
| Name | Residues in sequence | Location in structure |
|---|---|---|
| N-terminal cross 1 | Leu 70-Leu 72 | Border between TMSs 2–3 |
| N-terminal cross 2 | Tyr 113-Gly 115 | Mid-TMS4 |
| Edge of N-terminal quadrant | Leu 178-Lys 188 | C-terminal half of TMS6 |
| Edge of C-terminal quadrant | Ala 207-Ser 209 | Middle of interdomain linker; end of small helical segment protruding from TMS7* |
| Big blue cross (C-terminal quadrant) | Phe 341-Phe 356 | N-terminal half of TMS11 |
| Big red cross (C-terminal quadrant) | Ala 361-Ile 376 | C-terminal half of TMS11 |
) There are two additional helical units which are close together when the inward face of the transporter is closed.
Table 2.
Features of 4JA3 and 4GBY (inward and outward occluded states of XylE, respectively).
| Name | Residues in sequence | Location in structure |
|---|---|---|
| N-terminal cross | Ile 112-Thr 117 | Long loop between TMSs 3–4* |
| Edge of N-terminal quadrant | Thr 247-Gly 263 | Corresponds to third interdomain helical segment |
|
Big blue cross (C-terminal quadrant) |
Pro 389-Ala 413 | C-terminal 1/3 of TMS10 (“TMS10C”), N-terminal 1/3 of TMS11 (“TMS11N”)**,*** |
|
Big red cross (C-terminal quadrant) |
Ser 423-Ser 433 | C-terminal half of TMS11 |
) This extended loop is not found in LacY.
) The TMS10B region contains the E(X6)R motif located at 397–404, conserved across GLUTs 1–43. Arg 160-Glu 397 and Glu 153-Arg 404 form interdomain salt bridges that are broken in the inward open state. A third bridge, formed between Glu 397 and Arg 341 is missing in the inward occluded structure but present in the outward occluded and inward open states. Trp 392 forms part of the cytoplasmic lid of the substrate cavity in the outward occluded state3.
) Incorrectly identified as being restricted to TMS10B in Quistgaard et al. The differences identified by superimposition are the regions that could be identified from averaged superimposition solutions, and can differ from the more accurate localization of movement observed by using Δ-distance maps.
To clarify, the measurements taken, and the differences calculated between corresponding measurements, were not transformed before the values were pasted into an Excel spreadsheet, but normalization was carried out separately using ESCET 0.7. The color scale was normalized based on the current value distribution. We have indicated this fact by including a scale bar in Figure 1, translating values to Angstroms.
Repeat-swapped homology model of the outward open states
When we compared the theoretical models of the outward open states of both LacY and XylE with their X-ray crystallography-determined outward occluded counterparts, we noted that the differences in RMSD values (5.8 and 6.4 Angstroms for LacY and XylE, respectively) were larger than in the comparison between the inward occluded and the inward open state of XylE (1.8 Angstroms). This suggests that the biological differences that exist between the modeled outward open state and the outward occluded state are partly masked by differences that the methodology introduces.
Using Δ-distance maps we compared the outward occluded state of LacY with the repeat-swapped model of the outward open state5. A cross-shaped pattern in the fourth quadrant of the plot represents internal movement in the C-terminal 6 TMS bundle. The position of this cross corresponds to a short loop connecting TMSs 10 and 11 (Figure S1). The red color of the cross indicates that this part of the structure moves closer to the rest of the C-terminal domain when shifting from the outward open to the outward occluded state. We performed an analogous contact map comparison for the C-terminal domain of XylE but were unable to determine corresponding characteristic patterns.
Pairwise comparison of states: “4OAA” (outward occluded LacY)
Six TMSs contribute to the internal surface of the upper layers of the substrate translocation pathway: TMSs 1 & 7, TMSs 2 & 8, and TMSs 11 & 5. TMSs 1 & 7 make up the sides of the elongated cavity, whereas TMSs 8 & 5 and TMSs 2 & 11 connect the sides.
At the altitude of ~15 Angstroms from the base of the membrane (it is generally accepted that the fully dehydrated part of the bilayer is 15–25 Angstroms thick20, although the full thickness of the membrane is 70–80 Angstroms, including the head groups), the TMSs 2 & 8 channel area shrinks, while the area from TMSs 4 & 10 emerges to contribute substantially. TMSs 5 & 11 continue to comprise significant surface area. Although TMSs 1 & 7 also contribute channel surface area, they will cease to contribute 10–20 Angstroms further down.
The floor of the cavity is mainly composed of TMSs 10 and 11, but also of TMSs 4 and 5, which are slightly further up (Figure 2).
Figure 2. Topographical display of the substrate translocation pathways.
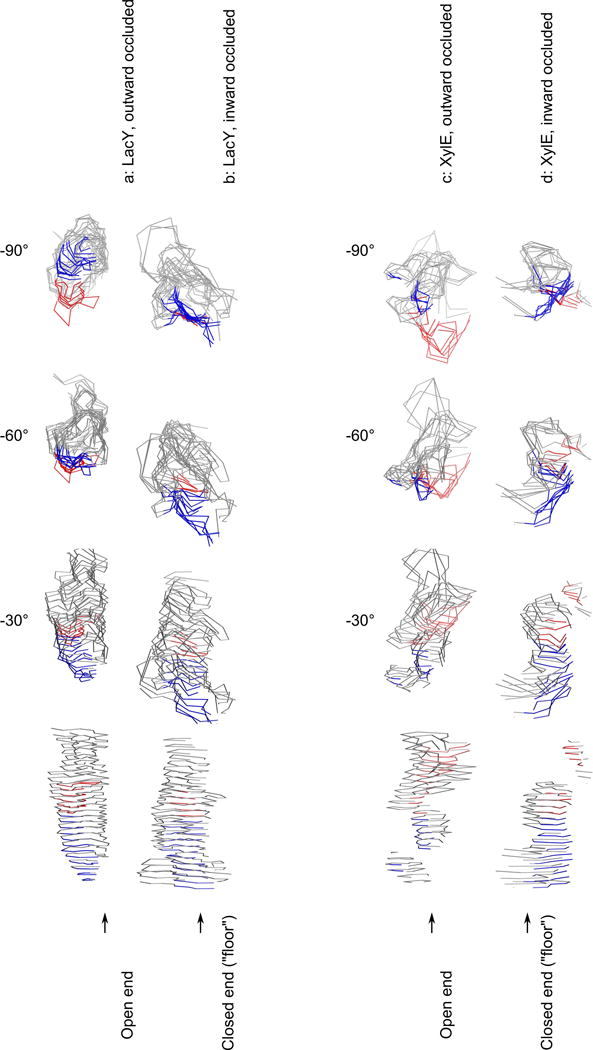
The pathway (in the central display, at 0° rotation), contained within the grey, blue and red outlines, is oriented with the extracellular side at the top of the diagram and the cytoplasmic side at the bottom. It is also shown at −90°, −60°,−30° rotations. (a) outward occluded LacY (4OAA); (b) inward affinity inactivator-occluded LacY (2Y5Y); (c) outward occluded XylE (4GBY); (d) inward occluded XylE (4JA3). The red and blue traces indicates where TMS11 contributes surface area, where red signifies that the distance to the C-terminal domain has decreased and blue that it has increased (when transitioning from the outward occluded to the inward occluded state; same meaning of color scheme as in Figure 1). TMS11 contributes the greatest amount of mobile surface area, and displays the greatest individual movement in relation to its domain half in both proteins. Each figure is made from 25 Angstrom steps, starting from the bottom of each channel. The molecular graphics file used to create this display is available on request.
“2Y5Y” (inward, affinity inactivator occluded LacY)
Six TMSs contribute to the internal surface of the lower cytoplasmic open end of the substrate translocation pathway: TMSs 10 & 4, TMSs 8 & 2, and TMSs 5 & 11. TMSs 10 & 4 make up the sides of the elongated cavity, whereas TMSs 8 & 5 and TMSs 11 & 2 connect the sides.
Towards the inner opening of the substrate translocation pathway, the surfaces of TMSs 8 and 2 are on opposite sides of the channel, as are those of TMSs 10 and 4, and TMSs 11 and 5. TMSs on the outskirts of the structure, such as TMSs 1, 6 and 7, do not seem to contribute much surface area to the channel. Towards the middle of the membrane, TMSs 1 and 7 steadily become more apparent and begin to converge with TMSs 2, 4, 8 and 10 as the opening gradually becomes more visible.
The TMS7 surface first emerges between TMSs 10 and 11, and the TMS1 surface emerges between TMSs 4 and 5, although TMSs 4, 5 and 11 are not found deep in the cavity. TMSs 1 and 7 contribute the largest surface area to the floor of the cavity, while TMSs 2 & 7 form narrow stripes on one side, and TMSs 8 & 10 does the same on the other side.
Comparison of the LacY outward occluded (4OAA) and putative inward occluded (2Y5Y) states: the tilting movement of TMS11
TMSs 1 and 7, the floor of the 2Y5Y transport pathway, form the sides of the open end of 4OAA. Conversely, the floor of 4OAA, mainly TMSs 10 & 11, but also TMSs 4 & 5, are the major contributors to the occluded “channel” surface at the open end of 2Y5Y.
Δ-distance maps have determined movement of TMS11 when switching from the outward occluded to the inward occluded state, in which the N-terminal half of the TMS moves away from the other parts of the C-terminal domain, and the C-terminal half of TMS11 approaches the rest of the C-terminal domain.
Differences between outward occluded LacY (4OAA) and outward occluded XylE (4GBY)
Close to the extracellular side, both structures appear similar; the same six TMSs contribute surface area lining of the “channel”: TMS2, TMS1, TMS5, TMS8, TMS7 and TMS11, in clockwise order. However, LacY has a better-defined cavity opening, whereas the cavity opening in XylE is a shallow valley.
Progressing further down the XylE channel, it turns towards the TMS2/TMS4 side and narrows until its surface area is composed almost exclusively of TMSs 2 and 4. At an intermediate point before the digression, TMSs 5 and 8 cease to contribute to the channel that leads towards the floor of the main cavity. In addition, TMSs 1 and 7 start to form a divider between two subsections of the channel: the shallower TMS5/TMS8 pair and the deeper TMS2/TMS4 pair subsections.
The channel space of XylE takes on a more complicated shape, potentially containing a “bubble” at the bottom that appears closed off from both ends, but the molecular dynamics of the structure likely permit passage of water. The TMSs contributing surface area to the bottom of the “bubble” are equivalent to the floor TMSs of LacY with the sole exception of TMS4.
Differences between inward inactivator bound (2Y5Y) LacY and inward occluded (4JA3) XylE
At the closed extracellular side, LacY and XylE both contain surface area from the same TMSs: TMSs 1, 7, 8 and 10. However, in XylE, there is surface area derived from TMSs 5 and 11 as well.
When observing the XylE channel opening from the cytoplasmic side, it is important to note that two entrances, divided by a thin column where TMSs 10 and 11 connect, merge into one slightly wider channel around 6 Angstroms down. Here, only 4 TMSs contribute surface area: TMSs 1, 4, 10 and 11. While both entrances are open to the cytoplasmic side of the molecule, only one is accessible from the hydrophobic wall of the membrane. 2–3 Angstroms further down, the trough that opens to the side closes, and the channel forms a narrower bottleneck made of TMSs 1, 5, 10 and 11. It widens into a chamber approximately in the middle of the molecule. The floor at this level is mainly composed of TMS7, and the walls derive from TMSs 1, 5, 7, 8, 10 and 11.
Discussion
TMS11 undergoes an analogous movement in LacY and XylE (Figure 1, Tables 1–2), in which the N-terminal halves of the TMSs become more distant from the rest of the structure in the inward state while the C-terminal halves of the TMSs are closer to the C-terminal 6 TMS bundle in the same state. It similarly appears that two other distantly related MFS permeases, FucP and NRT1, exhibit similar TMS11-associated movements.
We examined the Δ-distance maps representing the subsequent transitions between the inward occluded and the inward open states of both LacY and XylE, and discovered an important difference. We had previously reported9 that in XylE, the N-terminal portion of TMS11 “reverses” by moving back, closer towards the C-terminal 6 TMS bundle (Figure S3). However, we found that in LacY (comparing the affinity activator bound state with the inward open state), the N-terminal half of TMS11 in fact “strengthens” the pattern observed when going from the outward occluded to the inward occluded state. Thus, it moves even further away from the C-terminal 6 TMS bundle (Figure S2). This subtle but notable difference in movement does not mean that the N-terminal half of TMS11 in LacY moves inwards to close the cytoplasmic opening. The N-terminal half of TMS11 of XylE only bends slightly towards the rest of the C-terminal bundle, thus qualitatively constituting a reversal of the movement when going between the two occluded states without affecting the width of the substrate pathway. Though this difference may be substrate specificity-related, it nevertheless shows that the middle of TMS11 must be flexible to physically squeeze the substrate translocation pathway.
A conceptual difference introduced by Solcan et al. (2012)21 is that while there could be a “hinge” that can bend in the middle of TMSs 10 and 11, this movement could also be “teeter-like” containing a pivot, and these concepts are not mutually exclusive.
While Δ-distance maps are accurate at detecting which part of a sequence moves, detecting the difference between a mere “pivot point” and an actual “hinge”, where bending can occur, is difficult because it necessitates accurate detection of how the angle has changed. In an earlier publication, it had been suggested that the entire TMS 10–11 hairpin opened the gate to the inside in the PepT transporter of the POT family22. Solcan et al. (2012) reported that some bending/pivoting occured in the middle of TMSs 10 and 11 at Gly407 and Trp427, respectively. Solcan et al. tentatively extrapolated to other members of the MFS (GlpT, FucP, EmrD and LacY), showing that they also contain a bending point in TMS10 localized to a conserved glycyl residue. These authors discuss the movement of the extracellular end of TMS11 (which moves away from its domain half in Figure 1), and the authors report how it is packed closer to both TMS2 and TMS7. Solcan et al. discuss the transition from the occluded form to the inward open form without making a clear distinction as to how the transition between the two occluded forms differ from the transition between the inward occluded and inward open forms. Quistgaard et al. (2013)8,23 previously discussed the movement of TMS10B, and while we had initially thought that this was a mislocalization of the TMS11 movement, based on an incorrect superimposition, reexamination of the residue ranges highlighted in Figure 1 showed that there is evidence of TMS10B movement in XylE. This movement includes Pro389-Ser433 but is not observed in LacY where the movement involves Phe341-Ile376. This could indicate that the conserved glycyl residue identified by Solcan et al. may not be an actual bending point.
Careful examination of the structures also showed that while XylE has a discontinuity in TMS10, LacY has this type of discontinuity in TMS11. According to our view that the TMS11-associated movement is caused by movement of the interdomain linker, the movement of TMS11B might consequently be seen as more “indirect”. The conserved tryptophanyl residue in TMS11 has been shown to be substitutable by phenylalanine without loss of function. The question is whether or not TMS11 really bends in the middle or is just a pivot point. Tryptophanyl residues are not as flexible as glycyl residues, and we have demonstrated the coupled movement of the extracellular end of TMS11, suggesting that it might be more similar to a pivot than a hinge. We had previously suggested that there is a subtle difference between LacY and XylE in how they behave in the transition from the inward occluded state to the inward open conformation (see above). In Fig. S2 and S3, we can see evidence of bending, inasmuch as we detect independent movement of the intracellular halves of TMS11 (TMS11A) without coupled movement of TMS11B.
We would like to comment on the use of the structure 2Y5Y as a representation of the “inward occluded” conformation of LacY. This structure represents LacY bound to a substrate-like affinity inactivator, not a substrate, in the inward conformation, and this conformation is indeed similar to the inward open conformation of LacY (2V8N), displaying a global RMSD of 1.6 Angstrom between the two structures. In contrast, the inward occluded and inward open states of XylE display a global RMSD of 1.9. Strictly speaking, we think that 2Y5Y is an approximation of the inward occluded conformation, but cannot be considered the actual inward occluded state because of the presence of the inactivator. The subtle difference in biological movement between LacY or XylE as it goes from the inward occluded (or inward affinity inactivator-bound) state to the inward open state is probably not attributable to 2Y5Y not representing a genuine “inward occluded state”. Thus, 2Y5Y may not be identical to the substrate bound form, as noted above, but it may be closer to that form than it is to the inward open state.
It is a common misconception of Δ-distance maps that the values found are related to values obtained by superimposition. A superimposition of two structures will generally produce a globally averaged best solution, but the values in Δ-distance maps show individual distances independent of the rest of the structure. For example, if we superimpose the outward occluded (4GBY) and inward occluded (4JA3) conformations of XylE, we obtain a distance measurement of 3.26 Angstroms between the Cα atoms of Ala409 in the two conformations. However, the corresponding position in Fig. 1D is white, indicating a distance difference of zero (0). While this may seem contradictory, it is not, because all Δ-distance maps contain a diagonal of self-comparison distances. While the values in a Δ-distance map represent movement in relation to all other Cα atoms, the movement of a certain atom in relation to itself is always (by definition) going to be zero. The distance of 3.26 Angstroms found, using a superimposition based approach, does not represent and cannot be interpreted as a movement because it only represents a displacement that has resulted from the iterative process of finding the best global superimposition average. This is precisely why the use of Δ-distance maps should be strongly encouraged over superimposition-based methodologies. It also provides a likely explanation for why Quistgaard et al. only identified movement associated with TMS10B and not TMS118,23.
We have relied on theoretical models of the outward open states of LacY and XylE, created using repeat-swapped homology modeling. Only subtle differences exist between the outward open and outward occluded states because the major conformation changes occur between the two occluded states. However, we did note that a loop connecting TMSs 10 and 11 in LacY (but not in XylE) displayed a marked movement between the outward open and outward occluded states.
These findings illustrate the value of Δ-distance maps to facilitate interpretation of multi-state transport structures. This becomes particularly evident when noting that the movement of TMS10B, reported earlier7,8, in fact also includes the N-terminal third of TMS11.
In the outward occluded state of LacY, TMS11 lines the entire cavity, and while it is in principle equivalent to TMS5 in the first half of the protein, TMS11 uniquely contributes to both the side of the outward opening and the floor of the cavity, sealing the cavity from the cytoplasm. In the inward occluded state, however, TMS11 only contributes to the surface at the cytoplasmic opening of the cavity and does not line the side or floor of the cavity (Figure 2). The N-terminal part of TMS11 forms the floor in the outward occluded state, but an edge of the cytoplasmic open side in the inward occluded state. The C-terminal part of TMS11 forms a side of the extracellular opening in the outward occluded state (thus being tilted), but an outer scaffolding to the closed end in the inward occluded state. TMSs 2 and 7 do not contribute to the floor in the outward occluded state, but they may play a role in facilitating the tilting mechanism of TMS11, whose N-terminal part is located between these two TMSs, under the interdomain linker.
The tilting movement of TMS11 is related to the location of TMS11 under helices belonging to the interdomain linker. The displacement of TMS11 could either be an effect or a cause of the alternating access movement. TMS11 contains a charge pair whose polarity can be reversed without loss of activity and which is important for membrane insertion of LacY24. We have also noted that other portions of the structures displaying individual movement in relation to their domain halves, including the linker between TMSs 2 & 3 and the mid-TMS4, were in close contact with the interdomain linker, providing further evidence for this pattern (see Figure 3).
Figure 3. Overview of colocalization of the interdomain linker with structural elements.
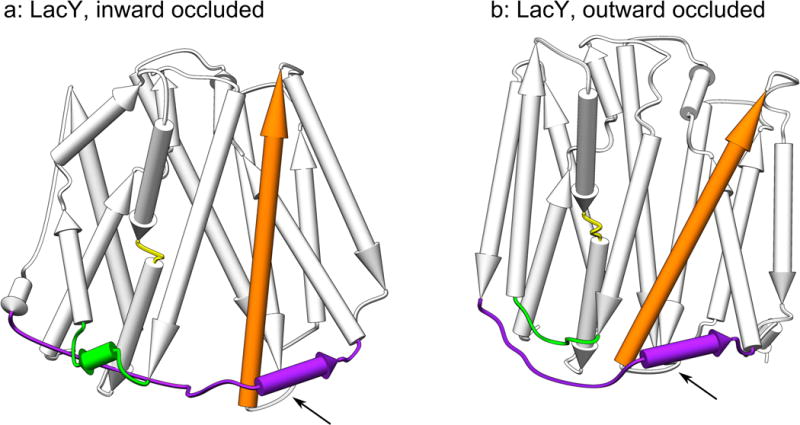
Panel (a) shows the inward occluded state, and (b) shows the outward occluded state of LacY. The interdomain linker, the loop between TMSs 2 & 3, the mid-TMS4 linker and TMS11 are highlighted in purple, green, yellow and orange, respectively. All of these elements generate strong signals on the Δ-distance maps (see Figure 1, Tables 1 and 2) and are colocalized with the interdomain linker. In general, the same pattern holds true for XylE, except that there is a long loop between TMSs 3 & 4 that is located on the extracellular side of the protein which displays independent movement (see Table 2).
To generalize the conclusion that a significant TMS11-associated movement is a common feature of MFS superfamily members beyond LacY and XylE, we have illustrated using RR distance maps (Figure S4) that when the fucose porter, FucP, switches from the outward to the inward state, a significant TMS11-associated distance remodeling occurs similar to that noted for LacY and XylE.
A similar effect was observed within a member of the POT/PTR peptide transporter family, where a comparison between the inward open and outward occluded states of NRT1 revealed a significantly elevated standard deviation measurement for a loop located between TMSs 11 and 12. These findings, revealed by examination of recently published MFS structures, show that TMS11-associated movement is not restricted to XylE and LacY. Since these additional findings are based on a homology modeling approach, any further generalizations would be unreliable. However, these initial homology modeling results give us increased confidence that the model presented for either LacY or XylE may be extended to the MFS superfamily in general.
There are four principal reasons to believe that the identified movement of TMS11 is a genuine biological effect and not an artefact (which could be the case when comparing X-ray structures of relatively low resolution): 1) the observed effect is on the order of magnitude of 4–6 Angstroms, greater than the approximate resolution limit used of 3 Angstroms, 2) the pattern of TMS11-associated movement is recurrent between multiple comparisons, 3) the similar movement (although incorrectly characterized as a movement limited to TMS10B) has been documented before for XylE8,23, FucP21 and POT, and 4) these observations are resilient to error-scaling19 (Figure S5). The PDB files for LacY and XylE used to draw the principal conclusions of this study were: 4GBY (2.81), 2Y5Y (3.38), 4OAA (3.50), 2V8N (3.60), 4JA3 (3.80), and 4JA4 (4.20), in order of decreasing accuracy, measured in Angstrom resolution, ranging from 2.81 to 4.20 (see Table 3). However, to make fully validated comparisons, the percentile ranks of Rfree, clashscore, Ramachandran outliers, sidechain outliers, and RSRZ outliers have to be considered. We thus consider that the differences detected in the Δ-distance maps represent true statistical differences, and do not result from inaccuracies in the modelling procedure. Our conclusions are adequately supported by the data, because, with the exception of the repeat-swapped model, the distances we detect are greater than the low resolution noise, and the same patterns reemerge between multiple comparisons.
Table 3.
Summary data of six principal structural models of conformational states of LacY and XylE used in this study.
| PDB ID | Protein | Conformation | Resolution* |
|---|---|---|---|
| 4OAA | LacY | Outward occluded | 3.50 |
| 2Y5Y | LacY | Inward, affinity inactivator-occluded | 3.38 |
| 2V8N | LacY | Inward open | 3.60 |
| 4GBY | XylE | Outward occluded | 2.81 |
| 4JA3 | XylE | Inward occluded | 3.80 |
| 4JA4 | XylE | Inward open | 4.20 |
) The magnitude of the TMS11 movement differs in panels b and d of Figure 1, being on the order of 4 Å for LacY, and 6 to 8 Å for XylE. The resolution limits are better on average for LacY, never reaching the 4 Å threshold, whereas for XylE in the inward open state, the resolution is only 4.2 Å.
Conclusions
Using superimposition-based approaches, the movement of TMS11 in XylE had not been characterized correctly7,8, because movement of the N-terminal half of this TMS appeared to be restricted to the C-terminal part of TMS10 (TMS10B), and a similar error in localization was noted for the N-terminal part of TMS11 in LacY. In this article, we have shown that more accurate positioning can be achieved using Δ-distance mapping. This established method12–14 could have wide applicability as more conformational state data become available. The majority of the TMS segments, including TMS11, that display intradomain movement, independent of the two domains’ alternating access movement, were found in close proximity to the interdomain linker helices. This commonality is interpreted as either a consequence of either the alternating access movement or a driving force causing it.
TMSs 11 in both LacY and XylE undergo a shared and crucial movement during the shift between outward and inward occluded states in which the N-terminal (intracellular) halves of TMSs 11 move away from the C-terminal (extracellular) 6 TMS bundles, while the C-terminal halves of TMSs 11 approach these bundles. TMSs 11 in both porters form the floor of the channel in the outward occluded state and contribute more mobile internal surface area than any of the other TMSs, indicating that TMSs 11 play a unique role in opening the substrate translocation pathway to the cytoplasmic side.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants GM077402 and GM094610 (to M.H.S.).
Footnotes
Contributions
Conceived and designed the experiments: AV, MHS. Performed the experiments: AV, AD, JL. Analyzed the data: AV, AD, JL. Contributed reagents/materials/analysis tools: AV, MHS. Contributed to the writing of the manuscript: AV, JL and MHS.
Competing financial interests
The author(s) declare no competing financial interests.
Supplementary materials
‘MFS_movement Supplementary file 1.pdf’: Figures S1–4, showing the comparison between the outward occluded state and repeat-swapped model of the outward open state of LacY (S1), the transitions between the inward occluded and inward open states (S2–3), RR distance maps to generalize the finding of TMS11-associated movement to other members of the MFS superfamily (S4A and S4B), as well as normalization data (S5).
References
- 1.Saier MH, Jr, Reddy VS, Tamang DG, Vastermark A. The transporter classification database. Nucleic Acids Res. 2014;42(Database issue):D251–258. doi: 10.1093/nar/gkt1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Law CJ, Yang Q, Soudant C, Maloney PC, Wang DN. Kinetic evidence is consistent with the rocker-switch mechanism of membrane transport by GlpT. Biochemistry. 2007;46(43):12190–12197. doi: 10.1021/bi701383g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forrest LR, Kramer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta. 2011;1807(2):167–188. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Dang S, Sun L, Huang Y, Lu F, Liu Y, Gong H, Wang J, Yan N. Structure of a fucose transporter in an outward-open conformation. Nature. 2010;467(7316):734–738. doi: 10.1038/nature09406. [DOI] [PubMed] [Google Scholar]
- 5.Radestock S, Forrest LR. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J Mol Biol. 2011;407(5):698–715. doi: 10.1016/j.jmb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510(7503):121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 7.Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature. 2012;490(7420):361–366. doi: 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- 8.Quistgaard EM, Low C, Moberg P, Tresaugues L, Nordlund P. Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat Struct Mol Biol. 2013;20(6):766–768. doi: 10.1038/nsmb.2569. [DOI] [PubMed] [Google Scholar]
- 9.Vastermark A, Lunt B, Saier M. Major Facilitator Superfamily Porters, LacY, FucP and XylE of Escherichia coli Appear to Have Evolved Positionally Dissimilar Catalytic Residues without Rearrangement of 3-TMS Repeat Units. J Mol Microbiol Biotechnol. 2014;24(2):82–90. doi: 10.1159/000358429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar H, Kasho V, Smirnova I, Finer-Moore JS, Kaback HR, Stroud RM. Structure of sugar-bound LacY. Proc Natl Acad Sci U S A. 2014;111(5):1784–1788. doi: 10.1073/pnas.1324141111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serdiuk T, Madej MG, Sugihara J, Kawamura S, Mari SA, Kaback HR, Muller DJ. Substrate-induced changes in the structural properties of LacY. Proc Natl Acad Sci U S A. 2014;111(16):E1571–1580. doi: 10.1073/pnas.1404446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider M, Brock O. Combining physicochemical and evolutionary information for protein contact prediction. PLoS One. 2014;9(10):e108438. doi: 10.1371/journal.pone.0108438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H, Tian R, Ni Z, Zhang Z, Guo Q, Saier MH., Jr Conformational Transition Pathway in the Inhibitor Binding Process of Human Monoacylglycerol Lipase. Protein J. 2014 doi: 10.1007/s10930-014-9572-z. [DOI] [PubMed] [Google Scholar]
- 14.Huang CC, Meng EC, Morris JH, Pettersen EF, Ferrin TE. Enhancing UCSF Chimera through web services. Nucleic Acids Res. 2014;42(Web Server issue):W478–484. doi: 10.1093/nar/gku377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker JL, Newstead S. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature. 2014;507(7490):68–72. doi: 10.1038/nature13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamm M, Staritzbichler R, Khafizov K, Forrest LR. AlignMe-a membrane protein sequence alignment web server. Nucleic Acids Res. 2014;42(Web Server issue):W246–251. doi: 10.1093/nar/gku291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stamm M, Staritzbichler R, Khafizov K, Forrest LR. AlignMe–a membrane protein sequence alignment web server. Nucleic Acids Res. 2014;42(Web Server issue):W246–251. doi: 10.1093/nar/gku291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishikawa K, Ooi T. Comparison of homologous tertiary structures of proteins. J Theor Biol. 1974;43(2):351–374. doi: 10.1016/s0022-5193(74)80066-4. [DOI] [PubMed] [Google Scholar]
- 19.Schneider TR. Domain identification by iterative analysis of error-scaled difference distance matrices. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2269–2275. doi: 10.1107/S0907444904023492. [DOI] [PubMed] [Google Scholar]
- 20.Gorter E, Grendel F. On Bimolecular Layers of Lipoids on the Chromocytes of the Blood. J Exp Med. 1925;41(4):439–443. doi: 10.1084/jem.41.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solcan N, Kwok J, Fowler PW, Cameron AD, Drew D, Iwata S, Newstead S. Alternating access mechanism in the POT family of oligopeptide transporters. Embo J. 2012;31(16):3411–3421. doi: 10.1038/emboj.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newstead S, Drew D, Cameron AD, Postis VL, Xia X, Fowler PW, Ingram JC, Carpenter EP, Sansom MS, McPherson MJ, Baldwin SA, Iwata S. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. Embo J. 2011;30(2):417–426. doi: 10.1038/emboj.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quistgaard EM, Low C, Moberg P, Nordlund P. Metal-mediated crystallization of the xylose transporter XylE from Escherichia coli in three different crystal forms. J Struct Biol. 2013;184(2):375–378. doi: 10.1016/j.jsb.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Dunten RL, Sahin-Toth M, Kaback HR. Role of the charge pair aspartic acid-237-lysine-358 in the lactose permease of Escherichia coli. Biochemistry. 1993;32(12):3139–3145. doi: 10.1021/bi00063a028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.