

Abstract
For accurate and reliable gene expression analysis using quantitative real-time reverse transcription PCR (qPCR), the selection of appropriate reference genes as an internal control for normalization is crucial. We hypothesized that non-coding, small nucleolar RNAs (snoRNAs) would be stably expressed in different barley varieties and under different experimental treatments, in different tissues and at different developmental stages of plant growth and therefore might prove to be suitable reference genes for expression analysis of both microRNAs (miRNAs) and mRNAs. In this study, we examined the expression stability of ten candidate reference genes in six barley genotypes under five experimental stresses, drought, fungal infection, boron toxicity, nutrient deficiency and salinity. We compared four commonly used housekeeping genes; Actin (ACT), alpha-Tubulin (α-TUB), Glycolytic glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ADP-ribosylation factor 1-like protein (ADP), four snoRNAs; (U18, U61, snoR14 and snoR23) and two microRNAs (miR168, miR159) as candidate reference genes. We found that ADP, snoR14 and snoR23 were ranked as the best of these candidates across diverse samples. For accurate and reliable gene expression analysis using quantitative real-time reverse transcription PCR (qPCR), the selection of appropriate reference genes as an internal control for normalization is crucial. We hypothesized that non-coding, small nucleolar RNAs (snoRNAs) would be stably expressed in different barley varieties and under different experimental treatments, in different tissues and at different developmental stages of plant growth and therefore might prove to be suitable reference genes for expression analysis of both microRNAs (miRNAs) and mRNAs. In this study, we examined the expression stability of ten candidate reference genes in six barley genotypes under five experimental stresses, drought, fungal infection, boron toxicity, nutrient deficiency and salinity. We compared four commonly used housekeeping genes; Actin (ACT), alpha-Tubulin (α-TUB), Glycolytic glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ADP-ribosylation factor 1-like protein (ADP), four snoRNAs; (U18, U61, snoR14 and snoR23) and two microRNAs (miR168, miR159) as candidate reference genes. We found that ADP, snoR14 and snoR23 were ranked as the best of these candidates across diverse samples. Additionally, we found that miR168 was a suitable reference gene for expression analysis in barley. Finally, we validated the performance of our stable and unstable candidate reference genes for both mRNA and miRNA qPCR data normalization under different stress conditions and demonstrated the superiority of the stable candidates. Our data demonstrate the suitability of barley snoRNAs and miRNAs as potential reference genes for miRNA and mRNA qPCR data normalization under different stress treatments.
Introduction
The study of gene expression has become increasingly widespread in numerous organisms. Real time quantitative (reverse transcription) polymerase chain reaction (qPCR) is a commonly used technique due to its high sensitivity, accuracy and reproducibility. Among several quantification strategies, a commonly used method is relative quantification where data are normalized to an internal control gene [1]. The internal control gene is called a reference or house-keeping gene (HKG) and, while subjected to the same experimental factors during sampling and cDNA preparation as the gene(s) of interest, is not differentially expressed, providing a constant for relative quantification of differences in expression of the gene(s) of interest. Hence, the selection of appropriate reference genes is important for obtaining valid results and proper interpretation from the analysis [2]. The most frequently used HKGs in plant qPCR analysis are protein-coding genes, such as Actin (ACT), alpha-Tubulin (α-TUB), Cyclophilin, Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ADP-ribosylation factor 1-like protein (ADP) and ribosomal RNAs [3–9]. However, several studies have shown that the expression of these genes varies considerably in different cells and tissues under different experimental conditions and, in these comparisons, they become unsuitable for qPCR data normalization [9–16].
Until two decades ago it was believed that the DNA between protein-coding genes was nothing more than junk DNA [17]. This idea was challenged by the discovery of small regulatory RNAs in eukaryotes. MicroRNAs (miRNAs) are a class of small RNAs, approximately 18–24 nucleotides (nt) long, non-coding and single-stranded molecules. miRNAs play pivotal roles in cellular homeostasis by the alteration of gene expression under stress conditions [18, 19]. Recent advances in high-throughput sequencing and bioinformatics analyses have enhanced the discovery of miRNAs in different plant species [20–23]. However, the regulatory function of miRNAs is just beginning to be understood [24–26]. miRNAs negatively regulate their target messenger RNAs (mRNAs) and the regulation is subject to various levels of control [18]. To determine the function of miRNAs, the expression levels of miRNAs and their target mRNA in vivo must be precisely compared. To investigate the differential expression of miRNAs, sequencing and computational analyses require the experimental validation of expression profiles. However, miRNA experimental validation is difficult because of their short length (∼ 18–24 nt) and lack of common sequence features (e.g. polyA) [27].
Barley (Hordeum vulgare) is one of the most important cereal crops cultivated in the world and, with a large amount of genetic and genomic data available, including its full genome sequence [28], is a model for cereal genomics studies. Although several reports have suggested suitable reference genes for RNA quantification in cereals, all show some variation in expression amongst plant tissues, species and experimental conditions so that there is no known reference genes suitable for all experiments [29–32]. Additionally, inadequate information is available on suitable reference genes for miRNA-qPCR. Studies of miRNA expression using qPCR often provide limited information on the stability of reference genes, but, where investigated, lower expression stability of some commonly used HKGs has been found in miRNA-qPCR experiments [2, 33, 34]. The use of miRNAs [1, 27, 35–37] and small nucleolar RNAs (snoRNAs) [38, 39] as reference genes has been proposed for better normalization in miRNA-qPCR expression analysis in both plants and animals. However, barley miRNAs and snoRNAs have never been assessed for their expression stability as reference genes under biotic and abiotic stresses.
The present work was designed to identify and evaluate suitable reference genes in barley under drought, salinity, boron toxicity, low nutrient stress and fungal infection. The selected internal controls were used for miRNA and mRNA expression profiling in barley to validate the performance of stable reference genes under drought stress treatment. Candidate reference genes were examined and compared with commonly used HKGs and ranked with three statistical algorithms. We found that the expression of four commonly used HKGs varied with experimental conditions. By contrast, snoRNAs and miRNAs were suitable internal controls for miRNA and mRNA-qPCR expression analysis in barley.
Materials and Methods
Plants and Growth Environments
Drought treatment
H. vulgare cvs. ‘Fleet’ and ‘Commander’ were grown in a glasshouse at 22–23°C day (d)/16°C night temperatures, with a day length of 12 hours (h). Plants were grown in pots with coco-peat soil. 30 individual plants of each genotype were grown. Drought treatment was applied as follows: plants were initially well-watered, then water withheld until visible wilting. Samples were collected after 21 d, after flag leaf emergence and 10 d after anthesis. The sampled materials corresponded to three developmental stages: tillering, booting and grain filling. Consistently well-watered plants were sampled as control plants. Leaf and root materials were sampled at tillering and booting stages and grains were harvested at the grain filling stage, 18 d post-anthesis.
Boron treatment
H. vulgare cv. ‘Clipper’ plants were grown hydroponically for 21 d in sufficient and excessive concentrations of boron with temperatures and day length as above. Seedlings were grown in a nutrient solution containing 5 mM NH4NO3, 5 mM KNO3, 2 mM Ca(NO3)2, 2 mM MgSO4, 100 μM KH2PO4, 50 μM NaFe(III) EDTA, 50 μM B(OH)3, 5 μM MnCl2, 10 μM ZnSO4, 0.5 μM CuSO4 and 0.1 μM Na2MoO3. After 7 d, half of the seedlings were transferred to a nutrient solution with an additional 3 mM H3BO3. Leaves were harvested after 14 d.
Fungal infection
H. vulgare cv. ‘Sloop’ seeds were sown in pots with coco-peat soil in a growth chamber at 18°C with a 14/10 h day/night light period and 60±10% relative humidity (RH). After three weeks, three individual plants were infected with Rhynchosporium commune. After inoculation the seedlings were maintained in darkness for 24 h at 100% RH, then returned to the 14/10 h day/night light period with 80±10% RH until the leaf samples were harvested, 14 d after inoculation. Same age uninfected plant leaves from three individual plants were harvested as the control group.
Salt treatment
H. vulgare cv. ‘WI4330’ seedlings were grown hydroponically as described for the boron treatment. At the appearance of the third leaf (approx. 10 d), half of the seedlings were subjected to the treatment by transferring them to a nutrient solution with an additional 50 mM NaCl. NaCl was added to the nutrient solution twice daily in increments of 50 mM, to a final concentration of 250 mM. Roots were harvested for RNA extraction when the third leaf was fully expanded approximately after a further 12 d.
Nitrate treatment
H. vulgare cv. ‘Golden Promise’ seedlings were grown in a fully-supported hydroponics set-up [40] in a glasshouse with a temperatures ranging between 19–23°C. Sufficient and low nitrate treatments were achieved by supplying 5 mM and 0.5 mM nitrate to the nutrient solution, respectively. The nutrient solution contained (in mM): 2.0 MgSO4.7H2O, 0.1 KH2PO4, 0.5 Na2Si307, 0.05 NaFe(III)EDTA, 0.05 H3BO3, 0.005 MnCl2.4H2O, 0.01 ZnSO4.7H2O, 0.0005 CuSO4.7H2O and 0.0001 Na2MoO4.2H2O with 2 KNO3 and 1.5 Ca(NO3)2.4H2O in the sufficient NO3 − treatment (5 mM NO3 −) and 0.25 KNO3 and 0.125 Ca(NO3)2.4H2O in the low NO3 - treatment (0.5 mM NO3 −). In order to maintain similar K+ and Ca2+ levels to the sufficient NO3 − treatment, the low NO3 − treatment also comprised (in mM): 0.875 K2SO4 and 1.375 CaCl2.2H2O. The treatments were established at the start of the experiment and the nutrient solution was replaced every 10 d to ensure that nutrients were not depleted. Following 21 d of sufficient and low nitrate treatment, the 2nd leaf blade was collected.
All harvested materials were immediately frozen in liquid N2 and stored at −80°C.
Candidate Reference Gene Selection and Primer Design
Four common HKGs (ACT, α-TUB, GAPDH and ADP), four snoRNAs (U18, U61, snoR14 and snoR23) and two miRNAs (miR168 and miR159) were selected. The sequences of Hvu-Actin (AY145451.1: http://www.ncbi.nlm.nih.gov/nuccore/AY145451), Hvu-Tubulin (U40042.1: http://www.ncbi.nlm.nih.gov/nuccore/U40042.1), Hvu-GAPDH (X60343.1: http://www.ncbi.nlm.nih.gov/nuccore/X60343.1) and Hvu-ADP (AJ508228.2: http://www.ncbi.nlm.nih.gov/nuccore/AJ508228.2) were downloaded from the NCBI Genbank while the sequences of all barley snoRNAs were downloaded from the plant snoRNA database (http://bioinf.scri.sari.ac.uk/cgi-bin/plant_snorna/home). The sequences of miR168 (accession number MIMAT0018216) and miR159 (accession number MIMAT0018210) were obtained from miRBase (http://www.mirbase.org/index.shtml). The selection of these two miRNAs was based on our previous miRNA expression studies in drought-treated H. vulgare cv. ‘Golden Promise’, where these two miRNAs were relatively invariant across drought treated and well-watered samples (data not shown).
Primers for ACT, α-TUB, GAPDH, ADP and snoRNAs were designed using AlleleID software (Premier Biosoft International, Palo Alto, CA, USA) considering amplicon sizes in the range of 60–80 bases. To eliminate the possibility of an inhibiting effect of the RNA secondary structure during cDNA synthesis, the primers for snoRNAs were designed in their loop regions [41]. miRNA specific stem-loop RT primers and appropriate forward and reverse primers for individual miRNA were designed following the previously described method [42, 43]. A minimum of three primer pairs were designed and tested for each gene. Primer pairs for 10 genes were selected on the basis of their amplification efficiency and specificity and are listed in Table 1. All primers used in this study were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA).
Table 1. Ten candidate reference genes and primer sequences used for their qPCR.
| Candidate reference genes | Accession number | Annotation | Primer (5′–3′) | Amplicon (bp) | PCR efficiency | Regression coefficient (R2) |
|---|---|---|---|---|---|---|
| Hvu-U61 | - | Small nucleolar RNA | Fw GAGGAAACGAAACCTGTGC | 65 | 93.94 | 0.999 |
| Rev ACTTCTTAGAGGGTTGTGTTAC | ||||||
| Hvu-U18 | - | Small nucleolar RNA | Fw GTGATGAAGAAAAGTTGGTC | 67 | 89.25 | 0.999 |
| Rev AGAAGTTTATTAAGGATGGTTATC | ||||||
| Hvu-snoR14 | - | Small nucleolar RNA | Fw GATGTTTATGTATGATAGTCTGTC | 67 | 95.55 | 0.999 |
| Rev GTCGGGATGTATGCGTGTC | ||||||
| Hvu-snoR23 | - | Small nucleolar RNA | Fw TCGGCAGTGGTGTCATC | 64 | 98.31 | 0.999 |
| Rev CTCAGTGGAAAGAGAAGTCG | ||||||
| Hvu-ADP | AJ508228.2 | ADP-ribosylation factor 1-like protein | Fw GCTCTCCAACAACATTGCCAAC | 77 | 100.79 | 0.999 |
| Rev GAGACATCCAGCATCATTCATTCC | ||||||
| Hvu-α-TUB | U40042.1 | Tubulin alpha-2 chain | Fw GTCCACCCACTCCCTCCTTG | 78 | 106.49 | 0.999 |
| Rev CGGCGGCAGATGTCATAGATG | ||||||
| Hvu-ACT | AY145451.1 | Actin | Fw CCACGAGACGACCTACAAC | 80 | 102.36 | 0.999 |
| Rev CACTGAGCACGATGTTTCC | ||||||
| Hvu-GAPDH | X60343.1 | Glycolytic glyceraldehyde-3-phosphate dehydrogenase | Fw GCCAAGACCCAGTAGAGC | 78 | 92.12 | 0.999 |
| Rev CACATTTATTCCCATAGACAAAGG | ||||||
| Hvu-miR168 | MIMAT0018216 | microRNA | Fw CTCACGTCGCTTGGTGCAGAT | 60 | 107.31 | 0.997 |
| Rev GAGCTGGGTCCGAGGT | ||||||
| Stem-loop RT primer GTCGTATCCAGAGCTGGGTCCGAGGTATTCGCTCTGGATACGACGTCCCG | ||||||
| Hvu-miR159 | MIMAT0018210 | microRNA | Fw CGTGGGTTTGGATTGAAGGGA | 61 | 107.21 | 0.999 |
| Rev GTGCAGGGTCCGAGGT | ||||||
| Stem-loop RT primer GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCAGAGC |
Specific qPCR amplification for all candidate internal controls was confirmed by a single, distinct melt peak in melt curve analysis (S1 Fig.) and a single band of desired size in 3% agarose gel electrophoresis (S2 Fig.). Further confirmation of these qPCR amplified products was done by sequencing using the respective reverse primers each containing M13 reverse primer sequence and a spacer (S1 Table), and all of these qPCR products showed correct sequences.
qPCR Analysis
Total RNA was extracted from each of the samples using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, except for grain RNA extraction, which included additional 2% (w/v) polyvinylpyrrolidone (PVP-40) (Sigma). From each of the five experiments RNA was isolated from a minimum of three biological replicates from each of the treated and control conditions.
The isolated RNA samples were treated with DNA-free reagents (Ambion, Life Technologies, Grand Island, NY, USA) twice according to the manufacturer’s instruction in order to completely remove genomic DNA (gDNA). A polymerase chain reaction (PCR) was carried out to ensure no gDNA contamination in the DNAse-treated RNA samples (S3 Fig.). The concentration and integrity of the DNA-free RNA was determined by Agilent-2100 Bioanalyzer using RNA 6000 NanoChips (Agilent Technologies, Santa Clara, CA, USA). RNA samples with RNA integrity numbers (RIN) ≥ 5 were used for cDNA synthesis. cDNAs were synthesized in a final volume of 40 μl. In each 40 μl reaction, the reaction mixture contained 2 μg RNA, 2 μl (10 mM) dNTP mix and a cocktail of 2 μl (1 μM) appropriate stem loop primer, 2 μl (10 mM) appropriate snoRNA specific reverse primers and 2 μl 50 μM Oligo (dT)20 (Life Technologies, Carlsbad, CA, USA) (Table 1). cDNA synthesis was carried out using SuperScript III RT (Life Technologies, Carlsbad, CA, USA). All investigated samples were transcribed by the pulsed RT method recommended for stem-loop primers [43]. In brief, the samples were incubated at 16°C for 30 minutes (min) followed by pulsed RT of 60 cycles at 30°C for 30 seconds (s), 42°C for 30 s and 50°C for 1 s. Finally, the reaction was terminated by incubating the samples at 85°C for 5 min.
To provide a template for the standard curve, four (three technical replicates and one no template control) 20-μL PCR reaction mixtures were combined and purified using illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare). The purified product was then quantified using the QUBIT fluorometer and the Qubit dsDNA HS assay kit (Life Technologies) according to the manufacturer’s instructions. An aliquot of this solution was diluted to produce a stock solution containing109 copies of the PCR product per μL. A dilution series covering six orders of magnitude was prepared from the 109 stock solution to produce solutions covering 107 to 101 copies per μL to derive a standard curve (S4 Fig.) for each assay. Reactions were prepared in triplicates and a no template control.
qPCR assays were assembled using the fluid-handling robotic platform CAS-1200 (QIAgility, Qiagen, Valencia, CA, USA). The cDNA samples were diluted 20 times in sterile milli Q water and the diluted cDNA solution was used in the qPCR reaction. Each assay contained: 2 μl of cDNA solution (or water as a no template control), 5 μl of KAPA Sybr Fast qPCR Universal Readymix (Geneworks, Adelaide, Australia), 1.2 μl of each of the forward and reverse primers at 4 μM and 0.6 μl water to make up the total reaction volume of 10 μl. Amplifications were performed in an RG 6000 Rotor-Gene Real-Time Thermal Cycler (Qiagen, Valencia, CA, USA) with 2 min at 95°C followed by 50 cycles of 1 s at 95°C, 1 s at 60°C, 25 s at 72°C, and fluorescent acquisition at 72°C, followed by melt curve analysis of temperature increasing from 60°C to 95°C with fluorescence readings acquired at 0.5°C increments (S1 Fig.).
Gene Expression Stability Ranking of Candidate Reference Genes
The stability of candidate reference genes’ expression was analysed using three software packages, geNorm (version 3.5) ([3] http://www.biogazelle.com/genormplus/website), Normfinder (version 0.953) ([44] http://www.mdl.dk/publicationsnormfinder.htm webcite) and BestKeeper (version 1) ([4] http://genequantification.com/bestkeeper.html). Raw expression values of candidate reference genes obtained from the qPCR under five experimental treatments were analysed by geNorm and NormFinder according to the instruction manual. geNorm was used to make pairwise comparisons and exclude values stepwise until the most stable pair of genes remained [3]. NormFinder was used to examine the expression stability of each candidate independently [44]. Both software packages were used to calculate the average expression stability (M) of candidate reference genes, with the highest M value indicating the least stable candidate reference genes and the lowest M value the most stable [3] and to rank their performance accordingly. BestKeeper analysis was used to calculate the geometric mean of the Cycle threshold (Ct) values of the candidate reference genes [4] and estimate gene expression stability based on the standard deviation of the Ct value, coefficient of correlation (r) and percentage covariance, to rank the candidates from the most to least stably expressed.
Determination of the Optimal Number of Reference Genes
To determine the optimal number of reference genes, the pairwise variation, V, for all datasets was estimated using geNorm (as before). V was calculated between two sequential normalization factors NFn and NFn+1 to determine the optimal number of reference genes [3]. If V was below 0.15, the addition of a third gene did not result in a noticeable improvement for the normalization. In particular, if Vn/n+1 was less than 0.15, using n+1 reference genes did not significantly improve the normalization [3].
Validation of Candidate Reference Genes’ Utility
To validate the reliability of putatively more stable candidate reference genes, the relative expression levels of an mRNA (Superoxide dismutase) and an miRNA (miR5048) (S2 Table) were also quantified for all the samples described above by qPCR (as before). Melt curves and standard curves in qPCR for Superoxide dismutase (AK363344.1) and miR5048 (MIMAT0020544) are given in S5 Fig. and S6 Fig., respectively. Expression of miR5048 was compared with previously described deep sequencing data [45].
Results
Gene Expression Stability and Ranking of Candidate Reference Genes by the geNorm analysis
Average expression stabilities (M) for candidate reference genes using pairwise comparison are shown in Fig. 1. snoR14 and ADP had the lowest M value (0.399), that is the most stable expression amongst all 10 candidate reference genes across all tissue samples. In contrast, miR159 had the highest M value (2.056) that is the least stable expression across all samples (Fig. 1A). Under drought stress, the most stable reference genes were ADP and snoR14, and the least stable was again miR159 (Fig. 1B). Under boron treatment the most stable reference genes were snoR23 and ADP, and the least stable reference gene was GAPDH (Fig. 1C). Under both fungal infection (Fig. 1D) and nitrate treatment (Fig. 1E) snoR14 and snoR23 were the most stable candidates. α-TUB was the least stable gene under both fungal infection (Fig. 1D) and nitrate treatment (Fig. 1E). Under salt treatment, U61 and snoR14 were the most stable candidates while ACT was the least stable candidate (Fig. 1F).
Fig 1. geNorm analysis of average expression stability values and ranking of ten candidate reference genes based on pairwise comparison.

Genes on the x-axis in order of increasing stability (y-axis M value) for (A) all samples, (B) drought-treated samples, (C) boron-treated samples, (D) fungal-infected samples, (E) salt-treated samples and (F) nitrate-treated samples. Under individual treatment condition, reference genes’ expression stability was obtained comparing to the untreated (control) condition.
Gene Expression Stability and Ranking of Candidate Reference Genes by the Norm Finder analysis
The most stable reference gene identified by NormFinder across all samples was snoR14. ADP and snoR23 were ranked second and third, respectively. snoR14, ADP and snoR23 also performed well under boron, fungal and nitrate stresses (Table 2). miR159 was the least stable (M >1) when used as a universal reference gene across all experimental samples. Though the M value was < 1, α-TUB was the most unstable candidate in fungal and nitrate treatment conditions (Table 2). ACT and GAPDH were the least stable candidates in salinity and boron treatments respectively. These results were broadly consistent with the pairwise analysis using the geNorm algorithm (Fig. 1).
Table 2. Expression stability ranking of candidate reference genes as calculated by NormFinder and BestKeeper.
| Experimental conditions | |||||||
|---|---|---|---|---|---|---|---|
| Rank | All samples | Drought | Boron | Fungus | Salt | Nitrate | |
| NormFinder (Stability value, M) | 1 | snoR14 (0.380) | U61 (0.154) | snoR23 (0.044) | ADP (0.041) | snoR14 (0.119) | snoR23 (0.04) |
| 2 | ADP (0.554) | snoR14(0.184) | snoR14 (0.061) | snoR14 (0.078) | U61 (0.141) | snoR14 (0.045) | |
| 3 | snoR23 (0.641) | miR168 (0.223) | ADP (0.063) | snoR23 (0.081) | snoR23 (0.151) | ADP (0.066) | |
| 4 | miR168 (0.708) | ADP (0.309) | U61 (0.102) | miR168 (0.104) | miR168 (0.200) | miR168 (0.238) | |
| 5 | U61 (0.765) | snoR23 (0.481) | miR168 (0.185) | U61 (0.106) | ADP (0.233) | U61 (0.305) | |
| 6 | U18 (0.801) | ACT (0.567) | α-TUB (0.242) | miR159 (0.112) | miR159 (0.259) | GAPDH (0.323) | |
| 7 | ACT (0.883) | α-TUB (0.587) | U18 (0.254) | ACT (0.129) | U18 (0.269) | ACT (0.325) | |
| 8 | α-TUB (0.976) | GAPDH (0.899) | ACT (0.291) | GAPDH (0.245) | GAPDH (0.294) | miR159 (0.353) | |
| 9 | GAPDH (1.88) | U18 (1.244) | miR159 (0.346) | U18 (0.260) | α-TUB (0.311) | U18 (0.455) | |
| 10 | miR159 (2.205) | miR159 (2.165) | GAPDH (0.435) | α-TUB (0.326) | ACT (0.380) | α-TUB (0.657) | |
| BestKeeper (SD) | 1 | ADP (0.573) | snoR23 (0.39) | ADP (0.27) | snoR14 (0.075) | miR168 (0.155) | miR168 (0.518) |
| 2 | snoR14 (0.772) | ADP (0.45) | snoR14 (0.28) | snoR23 (0.111) | U61 (0.2) | snoR14 (0.813) | |
| 3 | snoR23 (0.816) | snoR14 (0.61) | snoR23 (0.313) | ADP (0.177) | snoR23 (0.222) | snoR23 (0.871) | |
| 4 | miR168 (0.972) | miR168 (0.7) | U61 (0.328) | U61 (0.184) | snoR14 (0.23) | ADP (0.935) | |
| 5 | GAPDH (1.074) | U61 (0.82) | miR168 (0.333) | miR168 (0.185) | ADP (0.259) | GAPDH (1.35) | |
| 6 | ACT (1.094) | GAPDH (0.89) | GAPDH (0.4) | GAPDH (0.193) | U18 (0.273) | U61 (1.48) | |
| 7 | U61 (1.377) | ACT (0.93) | U18 (0.46) | miR159 (0.222) | GAPDH (0.298) | α-TUB (1.64) | |
| 8 | α-TUB (1.557) | α-TUB (1) | α-TUB (0.51) | ACT (0.307) | α-TUB (0.31) | ACT (1.745) | |
| 9 | U18 (1.913) | U18 (1.41) | ACT (0.56) | U18 (0.439) | miR159 (0.36) | U18 (1.75) | |
| 10 | miR159 (3.046) | miR159 (2.73) | miR159 (0.752) | α-TUB (0.563) | ACT (0.449) | miR159 (1.8) | |
Gene Expression Stability and Ranking of Candidate Reference Genes by Bestkeeper analysis
Expression of the 10 candidate reference genes was measured and Ct values are shown in Fig. 2. Average Ct values for the 10 candidates among all experimental samples varied between 14.98 and 24.35 (Fig. 2); in which higher and lower abundance of mRNA is represented by lower and higher Ct values, respectively. It should be noted that the Ct values obtained for the most stable genes (∼ 20) make them suitable for the normalization of a wide range of experimental targets, but they would be less suitable for certain targets with very high or low expression.
Fig 2. Gene expression of ten candidate internal controls in all samples.
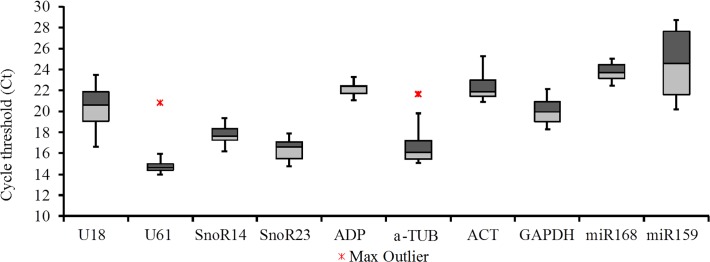
Data points are cycle threshold (Ct) values for each candidate reference gene in all samples. Boxes indicate the 25th (light shading) and 75th (dark shading) percentiles, the line indicates the median and whiskers depict the maximum and minimum values.
The Bestkeeper analysis suggested that ADP (0.573) was the most stable reference gene followed by snoR14 (0.772) and snoR23 (0.816), while miR159 (3.046) was the least stable across all samples (Table 2). SnoR14, snoR23 and ADP also performed well in drought, boron and fungal treatment experiments (Table 2). Under salt and nitrate treatments miR168 was the most stable reference gene. miR159 was the least stable candidate under drought, boron and nitrate treatments, while α-TUB and ACT were the least stable reference genes under fungal and salt treatments, respectively.
Consensus ranking performance of each candidate reference gene in all samples (n = 44) was evaluated by the three algorithms (Fig. 3). For each candidate, the consensus ranking was calculated as geometric mean of three rankings suggested by three algorithms; geNorm, Norm Finder and Bestkeeper.
Fig 3. Consensus ranking of ten reference genes.

Each candidate gene was assessed in the composite of all samples (n = 44). For each candidate, the consensus ranking was calculated as geometric mean of all rankings suggested by three algorithms; geNorm, Norm Finder and Bestkeeper.
The Optimal Number of Reference Genes
The optimal number of reference genes under individual treatment was determined by geNorm analysis (Fig. 4). Under boron, fungal, salt and nitrate treatments, the V2/3 values were less than 0.15 (Fig. 4). This analysis suggested that, apart from the two most stable reference genes found in the respective treatment conditions, the addition of a third, reference genes did not improve the normalization so that the use of the two most stable reference genes would be sufficient for normalization. Under drought treatment, V2/3-V7/8 was ≤ 0.15, while V8/9 and V9/10 was > 0.15 (Fig. 4). In such a situation, the use of between two and seven reference genes resulted in reliable normalization. It is worth noting that the V value is only a guideline for the determination of an optimal number of reference genes and thus should not be considered as an exact cut-off point.
Fig 4. Determination of the optimal number of reference genes for normalization of qPCR data in each experimental condition.

If the V (variation) value is below 0.15, the addition of a further reference gene does not result in any improvement of normalization.
Validation of Stable Reference Genes for mRNA and miRNA Expression Under Various Stress Conditions
To validate the stability of reference genes, we used two groups for normalization. Group 1 denotes the three putatively stable reference genes, ADP, snoR14 and snoR23, suggested by the consensus ranking, and group 2 denotes the three commonly used HKGs, ACT, α-TUB and GAPDH. For each experimental condition, the expression of SUPEROXIDE DISMUTASE was normalized to group 1 and group 2 in the treated and control samples (Fig. 5). The formation of reactive oxygen species (ROS) in plants is triggered by various environmental stresses, such as drought, nutrient deficiency, nutrient toxicity, salinity and pathogen attack [46, 47]. SUPEROXIDE DISMUTASE is one of the first line of defence antioxidant enzymes. Though its expression is not broadly reported for each particular stress condition, it is well-known to be up-regulated under drought stress in plant species including barley [48–55]. As expected, normalized expression of SUPEROXIDE DISMUTASE to group 1 and group 2 resulted in lower expression under well-watered (control) than under drought-treated samples (Fig. 5A). However, the expression difference between the treated and control samples was greater when normalized with group 1 candidates (Fig. 5A). In the boron treatment experiment, both normalizer groups showed similar results where SUPEROXIDE DISMUTASE was significantly up-regulated in the treated samples (Fig. 5B). In the fungal infected samples the expression level of SUPEROXIDE DISMUTASE was reduced compared to the control samples when normalized to group 1. However, when normalized to group 2 the expression level of SUPEROXIDE DISMUTASE was not significantly different (Fig. 5C). In the nitrate and salt treatment samples, group 1 normalization resulted in significantly higher SUPEROXIDE DISMUTASE expression than in the control samples (Fig. 5D & E). However, in both of these experimental conditions, group 2 normalization did not show any significant difference of SUPEROXIDE DISMUTASE expression between the treated and control samples (Fig. 5D & E).
Fig 5. Validation of putatively stable reference genes.

Comparison of relative expression of (A, B, C, D, E) SUPEROXIDE DISMUTASE (AK363344.1) and (F, G, H, I, J) miR5048 (MIMAT0020544) in drought-treated, boron treated, fungal infected, nitrate treated and salt treated samples and their respective controls by qPCR when normalized to group1 (a combined group of three stable reference genes; snoR14, ADP, snoR23) and group 2 (a combined group commonly used housekeeping genes; ACT, α-TUB, GAPDH). The error bars indicate the standard deviation of the mean. Statistical analysis by t-test. (Inset in 5F) The relative expression of miR5048 from deep sequencing of drought-stressed and well-watered barley leaves. Reads per million (RPM) of 1 was the highest number of counts.
We also quantified the relative expression of a barley miRNA, miR5048, normalizing with the same groups of candidates. The normalization against group 1 reference genes resulted in down-regulation of miR5048 under drought treatment compared with the control conditions (Fig. 5F) which was consistent with our pre-existing deep sequencing data (see inset in Fig. 5F) [45]. In contrast, normalizing the expression level of miR5048 to group 2 candidates resulted in comparatively higher expression of miR5048 with no significant difference between the treated and control samples. This result was inconsistent with the normalized expression using group 1 candidate reference genes as well as inconsistent with our pre-existing deep sequencing result (Fig. 5F and inset). These data indicated an increased sensitivity of detection of differential expression using the putatively more stable group 1 candidates.
We also took advantage of the available boron treated, fungal infected, nitrate treated and salt treated samples to compared miR5048 expression in these conditions normalizing the expression level to the two normalizer groups to check for the groups’ comparative sensitivity.
Under the boron treatment, miR5048 expression normalized to group 1 showed that miR5048 was down-regulated in the treated samples compared to the control samples. However, normalization to group 2 did not show any difference between the treated and control samples (Fig. 5G). Under the fungal infection, miR5048 expression was not abundant and there was no significant difference in expression with treatment detected using either normalizing group of reference genes (Fig. 5H). Under the nitrate treatment, upon normalization to both normalizer groups, miR5048 was significantly up-regulated compared to the control samples (Fig. 5I). The salt treatment did not result in a significant up-regulation of miR5048 expression with either normalizing group (Fig. 5J).
Discussion
Normalization is an important requirement for the study of gene expression by qPCR. Random selection of reference genes, which may be influenced by experimental treatments, could cause the misinterpretation of results [1, 11]. An appropriate reference gene should have an invariant level of expression regardless of experimental conditions; however, such genes may be hard to find as plant gene expression is affected by environmental conditions [56–59]. It is clear from recent studies that internal reference genes for qPCR assays should be specifically selected for the experimental treatment of interest in both plants and animals [32, 33, 60, 61].
In our study, we evaluated the expression stabilities of ten candidate reference genes, ACT, α-TUB, GAPDH, ADP, snoR14, snoR23, U61, U18, miR159 and miR168, in samples from barley plants grown in five stressed conditions. There is no universally accepted method for reference gene selection and stability analysis. We hypothesized that small, non-coding RNAs could be more suitable reference genes for qPCR normalization of miRNAs when compared with commonly used protein-coding reference genes using three popular algorithms in different computer programs such as geNorm, NormFinder and BestKeeper. Using these different analytical algorithms the selected snoRNAs (snoR14 and snoR23) consistently ranked amongst the best and most suitable reference genes, whether individually or in combination, with only ADP amongst the more commonly used protein-coding reference genes ranked more highly than other snoRNAs in a composite of all samples by two of the three algorithms.
It is necessary to validate reference genes under each set of experimental conditions [62]. geNorm, NormFinder and BestKeeper analyses were performed separately for each experimental treatment and indicated that two of the candidate snoRNA reference genes, snoR14 and snoR23, were steadily stable in an experiment specific manner. snoR14, in particular and in combination with at least one other reference gene, had potential as a universal reference gene in all the studied sample conditions. Amongst protein-coding genes, ADP was indicated as a good potential internal control under drought treatment. This result is consistent with a previous study in barley [9]. Under salt stress, the snoRNA U61 showed stable expression and consistently ranked in the top two potential reference genes by three algorithms. U61 also ranked as the fourth most stable reference gene under boron treatment, miRNAs were also proposed as reference genes for qPCR data normalization of both miRNA and protein-coding genes in an experiment in soybean [1]. The miRNAs, miR168 and miR159, behaved very differently from each other as reference genes in the experiments reported here, underlining the importance of empirical selection and knowledge of the individual gene’s differential expression with treatment. miR168 was selected as a candidate because it was reportedly not significantly affected by drought in Arabidopsis [63] and rice [64]. Genome-wide deep sequencing of peach also indicated that there was no change in the expression of miR168 in drought-stressed leaf and root tissues [65]. Our results confirmed the comparatively consistent stability of miR168 as a reference gene under each of the experimental conditions, where it ranked in the top five using the three algorithms.
geNorm and BestKeeper specify a gene expression stability threshold value above which a candidate reference gene should be considered as an unreliable internal control. The threshold value is M = 1.5 in geNorm, a value exceeded by miR159 and GAPDH in a composite of all samples. The reliability threshold of SD = 1 in BestKeeper was also a value exceeded by miR159 and GAPDH, as well as U61, U18, and α-TUB in a composite of all samples.
Though the selection of miR159 was based on our previous miRNA expression studies in drought-treated barley ‘Golden Promise’ (data not shown), miR159 was ranked as the least stable candidate in drought-treated samples by three algorithms. An explanation of this inconsistency under drought treatment may reside in the use of different varieties, developmental stages and the method of quantification which may lead to changes in miRNA expression as well as impact on gene regulation. Additionally, miR159 was ranked as the least stable candidate under boron and nitrate treatments by BestKeeper. miR159 expression also varied in roots under drought stress in peach [65] and showed variable patterns in rice between young and old leaves [66]. Hence we do not consider miR159 a suitable reference gene.
Conventionally used HKGs, those involved in basic cellular mechanisms, have been used extensively for quantification of transcript expression. However, it has also been reported that their expression levels are not completely independent of the exogenous conditions [9, 13–16]. In our study, we found that some of the commonly used housekeeping genes such as ACT, α-TUB and GAPDH were not the most suitable HKGs under specific experimental conditions as in most cases they were ranked lower than the other candidates (Table 2). Additionally, in BestKeeper analysis, the reliability threshold SD = 1 was exceeded by ACT, GAPDH and α-TUB when used as HKGs for nitrate-treated samples demonstrating the importance of selecting reference genes specific to the experimental treatments. We found that the non-coding RNAs generally outperformed ACT, GAPDH and α-TUB. These results suggest that the common HKGs for qPCR data normalization should be used carefully, demanding a thorough evaluation for every experimental set of samples before use. Additionally, comparison of different algorithms for reference genes selection may facilitate a reliable evaluation of stably expressed genes as well as precise data normalization.
In our analysis of the stability of candidate reference genes, we validated their performance by normalizing the relative expression of an mRNA and an miRNA in barley under five experimental conditions. For evaluating mRNA expression, we selected SUPEROXIDE DISMUTASE whose enzymatic product rapidly scavenges reactive oxygen species in plants under various environmental stresses [46, 47, 67–70]. Increased expression of SUPEROXIDE DISMUTASE has been identified under drought stress in many plant species including cotton [48], pea [49], Coffea [50], common bean [51], rice [52], Populus [53] and wheat [54, 55]. To validate the performance of stable candidate internal controls for expression data normalization, we used the combination of ADP, snoR14 and snoR23, the top three stable reference genes suggested by the consensus ranking. In comparison to this normalization to these putatively stable candidates, we also considered the combination of relatively unstable candidates ACT, α-TUB, GAPDH suggested by the consensus ranking. With both sets of normalizers, SUPEROXIDE DISMUTASE expression increased in the drought treatment but the difference of expression between drought and well-watered samples was greater when the stable group of normalizers was used. This increased sensitivity of detection of differential expression was also evident in the fungal infection, low and high nitrate and salt stress experimental samples using the putatively stable group of reference genes. This demonstrated that the choice of appropriate and stable reference genes does increase the sensitivity of experimental assays in qPCR and validated our comparative ranking of the candidates.
To evaluate the putatively stable candidates for normalizing miRNA expression, we selected miR5048 for comparison with our pre-existing deep sequencing data for expression of this miRNA in the barley variety ‘Golden Promise’ under drought and well-watered conditions. Deep sequencing is well-recognised to reflect RNA presence and quantity from a genome at a given stage [71] and should correlate with the expression level identified through qPCR [72]. However, it should be noted that different methods of library construction including choice of adapters and even barcodes could generate significant bias during RNA sequencing profiling of miRNAs [73, 74]. Using the same set of putatively more stable reference genes for miR5048 expression data normalization, miR5048 was down-regulated under drought treatment consistent with the deep sequencing result. However, normalization with the putatively unstable, commonly used ACT, α-TUB and GAPDH failed to detect statistically significant down-regulation of miR5048 under drought stress, conflicting with our deep sequencing result. Thus, the validation of candidate reference genes for normalizing miRNA relative expression profiles increased our confidence in the analysis of stability of ADP, snoR14 and snoR23 in drought-stressed barley and highlighted how the use of inappropriate reference genes might lead to erroneous results.
Apart from the drought treatment, as there is lack of reference for miR5048 expression, we could not compare our validation results under individual experimental conditions. Nonetheless, for miR5048 as well as for SUPEROXIDE DISMUTASE, there was an increase in the sensitivity of detection of differential expression for boron, fungal and salt treatments when our higher-ranked stable candidates were used, once again validating their superiority.
Conclusion
We evaluated ten candidate reference genes in different tissues, genotypes and experimental treatments for their ability to normalize miRNA qPCR data. The expression stability of these candidate genes was evaluated across a set of 44 samples using the computer programs geNorm, NormFinder and BestKeeper. We found that two putative snoRNAs, snoR14 and snoR23, outperformed the generally used HKGs in barley. To our knowledge, this work is the first stability evaluation of a set of commonly used and novel putative reference genes for qPCR in barley under different experimental conditions. This study suggests our set of evaluated candidates ADP, snoR14 and snoR23 as potential reference genes in barley. Additionally, this work proposes a list of potential candidate reference genes under five, different experimental conditions. We recommend to use a combination of ADP, snoR14 and snoR23 as potential reference genes for miRNA and mRNA qPCR data normalization under the experimental conditions mentioned in this study. As the arbitrary selection of internal controls, or the use pre-identified reference genes, may yield inaccurate results, reference gene selection needs to be optimized for individual assays counting a number of candidate reference genes for evaluation. We also recommend the use of multiple reference genes for more reliable and valid normalization of gene expression in barley.
Supporting Information
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(DOCX)
(DOCX)
Acknowledgments
The authors thank and acknowledge Julie Hayes, Jessica Hilary Bovill, Rhiannon Schilling and Jamil Chowdhury for providing boron, salt, nitrate and fungal stress experiment RNA samples, respectively. The authors would specially like to thank Lorenzo Giusti for early discussion and his helpful advice on qPCR experiments.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Funded by the Australian Centre for Plant Functional Genomics.
References
- 1. Kulcheski FR, Marcelino-Guimaraes FC, Nepomuceno AL, Abdelnoor RV, Margis R (2010) The use of microRNAs as reference genes for quantitative polymerase chain reaction in soybean. Analytical Biochemistry 406: 185–192. 10.1016/j.ab.2010.07.020 [DOI] [PubMed] [Google Scholar]
- 2. Bustin S (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. Journal of Molecular Endocrinology 29: 23–39. [DOI] [PubMed] [Google Scholar]
- 3. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology 3: research0034.0031—research0034.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnology Letters 26: 509–515. [DOI] [PubMed] [Google Scholar]
- 5. Radonić A, Thulke S, Mackay IM, Landt O, Siegert W, Nitsche A (2004) Guideline to reference gene selection for quantitative real-time PCR. Biochemical and Biophysical Research Communications 313: 856–862. [DOI] [PubMed] [Google Scholar]
- 6. Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible W-R (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiology 139: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tong Z, Gao Z, Wang F, Zhou J, Zhang Z (2009) Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Molecular Biology 10: 71 10.1186/1471-2199-10-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meng Y, Moscou MJ, Wise RP (2009) Blufensin1 negatively impacts basal defense in response to barley powdery mildew. Plant Physiology 149: 271–285. 10.1104/pp.108.129031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rapacz M, Stepien A, Skorupa K (2012) Internal standards for quantitative RT-PCR studies of gene expression under drought treatment in barley (Hordeum vulgare L.): the effects of developmental stage and leaf age. Acta Physiologiae Plantarum 34:1723–1733. [Google Scholar]
- 10. Zhong H, Simons JW (1999) Direct comparison of GAPDH, beta-actin, cyclophilin, and 28S rRNA as internal standards for quantifying RNA levels under hypoxia. Biochemical and Biophysical Research Communications 259: 523–526. [DOI] [PubMed] [Google Scholar]
- 11. Selvey S, Thompson E, Matthaei K, Lea RA, Irving MG, Griffiths LR (2001) β-Actin—an unsuitable internal control for RT-PCR. Molecular and Cellular Probes 15: 307–311. [DOI] [PubMed] [Google Scholar]
- 12. Glare EM, Divjak M, Bailey MJ, Walters EH (2002) beta-actin and GAPDH housekeeping gene expression in asthmatic airways is variable and not suitable for normalising mRNA levels. Thorax 57: 765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A (2004) Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 37: 112–119. [DOI] [PubMed] [Google Scholar]
- 14. Ohl F, Jung M, Xu C, Stephan C, Rabien A, Burkhardt M, et al. (2005) Gene expression studies in prostate cancer tissue: which reference gene should be selected for normalization? Journal of Molecular Medicine 83: 1014–1024. [DOI] [PubMed] [Google Scholar]
- 15. Jain M, Nijhawan A, Tyagi AK, Khurana JP (2006) Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochemical and biophysical research communications 345: 646–651. [DOI] [PubMed] [Google Scholar]
- 16. Li QQ, Skinner J, Bennett JE (2012) Evaluation of reference genes for real-time quantitative PCR studies in Candida glabrata following azole treatment. BMC Molecular Biology 13: 22 10.1186/1471-2199-13-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brosius J (2005) Waste not, want not–transcript excess in multicellular eukaryotes. Trends in Genetics 21: 287–288. [DOI] [PubMed] [Google Scholar]
- 18. Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP (2002) MicroRNAs in plants. Genes & Development 16: 1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annual Review of Plant Biology. pp. 19–53. [DOI] [PubMed]
- 20. Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu J-K (2008) Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biology 8: 25 10.1186/1471-2229-8-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barrera-Figueroa BE, Gao L, Diop NN, Wu Z, Ehlers JD, Roberts P A, et al. (2011) Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC Plant Biology 11: 127 10.1186/1471-2229-11-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carnavale Bottino M, Rosario S, Grativol C, Thiebaut F, Rojas CA, Farrineli L, et al. (2013) High-throughput sequencing of small RNA transcriptome reveals salt stress regulated microRNAs in sugarcane. PLoS ONE 8:e59423 10.1371/journal.pone.0059423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang T, Chen L, Zhao M, Tian Q, Zhang W-H (2011) Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genomics 12: 367 10.1186/1471-2164-12-367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kantar M, Unver T, Budak H (2010) Regulation of barley miRNAs upon dehydration stress correlated with target gene expression. Functional & Integrative Genomics 10: 493–507. [DOI] [PubMed] [Google Scholar]
- 25. Kantar M, Lucas SJ, Budak H (2011) miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 233: 471–484. 10.1007/s00425-010-1309-4 [DOI] [PubMed] [Google Scholar]
- 26. Hackenberg M, Shi B-J, Gustafson P, Langridge P (2012) A Transgenic Transcription Factor (TaDREB3) in Barley Affects the Expression of MicroRNAs and Other Small Non-Coding RNAs. PloS One 7(8): e42030 10.1371/journal.pone.0042030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benes V, Castoldi M (2010) Expression profiling of microRNA using real-time quantitative PCR, how to use it and what is available. Methods 50: 244–249. 10.1016/j.ymeth.2010.01.026 [DOI] [PubMed] [Google Scholar]
- 28. Mayer KF, Waugh R, Brown JW, Schulman A, Langridge P, Platzer M, et al. (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491: 711–716. 10.1038/nature11543 [DOI] [PubMed] [Google Scholar]
- 29. Huang L, Yan H, Jiang X, Yin G, Zhang X, Qi X, et al. (2014) Identification of Candidate Reference Genes in Perennial Ryegrass for Quantitative RT-PCR under Various Abiotic Stress Conditions. PloS One 9: e93724 10.1371/journal.pone.0093724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paolacci AR, Tanzarella OA, Porceddu E, Ciaffi M (2009) Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC molecular biology 10: 11 10.1186/1471-2199-10-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Giménez MJ, Pistón F, Atienza SG (2011) Identification of suitable reference genes for normalization of qPCR data in comparative transcriptomics analyses in the Triticeae. Planta 233. [DOI] [PubMed] [Google Scholar]
- 32. Lin Y, Zhang C, Lan H, Gao S, Liu H, Liu J, et al. (2014) Validation of Potential Reference Genes for qPCR in Maize across Abiotic Stresses, Hormone Treatments, and Tissue Types. PloS One 9: e95445 10.1371/journal.pone.0095445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmittgen TD, Zakrajsek BA (2000) Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. Journal of biochemical and biophysical methods 46: 69–81. [DOI] [PubMed] [Google Scholar]
- 34. Huggett J, Dheda K, Bustin S, Zumla A (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes and Immunity 6: 279–284. [DOI] [PubMed] [Google Scholar]
- 35. Davoren PA, McNeill RE, Lowery AJ, Kerin MJ, Miller N (2008) Identification of suitable endogenous control genes for microRNA gene expression analysis in human breast cancer. BMC Molecular Biology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peltier HJ, Latham GJ (2008) Normalization of microRNA expression levels in quantitative RT-PCR assays: Identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 14: 844–852. 10.1261/rna.939908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lardizábal MN, Nocito AL, Daniele SM, Ornella LA, Palatnik JF, Veggi LM (2012) Reference genes for real-time PCR quantification of microRNAs and messenger RNAs in rat models of hepatotoxicity. PloS One 7: e36323 10.1371/journal.pone.0036323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matoušková P, Bártíková H, Boušová I, Hanušová V, Szotáková B, Skálová L (2014) Reference Genes for Real-Time PCR Quantification of Messenger RNAs and MicroRNAs in Mouse Model of Obesity. PloS One 9: e86033 10.1371/journal.pone.0086033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brattelid T, Aarnes E-K, Helgeland E, Guvaåg S, Eichele H, Jonassen AK (2011) Normalization strategy is critical for the outcome of miRNA expression analyses in the rat heart. Physiological Genomics 43: 604–610. 10.1152/physiolgenomics.00131.2010 [DOI] [PubMed] [Google Scholar]
- 40. Genc Y, McDonald GK, Tester M (2007) Reassessment of tissue Na+ concentration as a criterion for salinity tolerance in bread wheat. Plant Cell and Environment 30: 1486–1498. [DOI] [PubMed] [Google Scholar]
- 41. Shen J, Xie K, Xiong L (2010) Global expression profiling of rice microRNAs by one-tube stem-loop reverse transcription quantitative PCR revealed important roles of microRNAs in abiotic stress responses. Molecular Genetics and Genomics 284: 477–488. 10.1007/s00438-010-0581-0 [DOI] [PubMed] [Google Scholar]
- 42. Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, et al. (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33: e179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Research 64:5245–5250. [DOI] [PubMed] [Google Scholar]
- 45. Schreiber AW, Shi BJ, Huang CY, Langridge P, Baumann U (2011) Discovery of barley miRNAs through deep sequencing of short reads. BMC Genomics 12 (1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cervilla LM, Blasco B, Rios JJ, Romero L, Ruiz JM (2007) Oxidative stress and antioxidants in tomato (Solanum lycopersicum) plants subjected to boron toxicity. Annals of Botany 100 (4): 747–756 10.1093/aob/mcm156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tripathy BC, Oelmuller R (2012) Reactive oxygen species generation and signalling in plants. Plant Signaling & Behavior 7: 1621–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burke JJ, Gamble PE, Hatfield JL, Quisenberry JE (1985) Plant morphological and biochemical responses to field water deficits I. Responses of glutathione reductase activity and paraquat sensitivity. Plant Physiology 79: 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mittler R, Zilinskas BA (1994) Regulation of pea cytosolic ascorbate peroxidase and other antioxidant enzymes during the progression of drought stress and following recovery from drought. The Plant Journal 5: 397–405. [DOI] [PubMed] [Google Scholar]
- 50. Pinheiro HA, DaMatta FM, Chaves ARM, Fontes EPB, Loureiro ME (2004) Drought tolerance in relation to protection against oxidative stress in clones of Coffea canephora subjected to long-term drought. Plant Science 167: 1307–1314. [Google Scholar]
- 51. Turkan I, Bor M, Ozdemir F, Koca H (2005) Differential responses of lipid peroxidation and antioxidants in the leaves of drought-tolerant P-acutifolius Gray and drought-sensitive P-vulgaris L. subjected to polyethylene glycol mediated water stress. Plant Science 168: 223–231. [Google Scholar]
- 52. Sharma P, Dubey RS (2005) Drought induces oxidative stress and enhances the activities of antioxidant enzymes in growing rice seedlings. Plant Growth Regulation 46: 209–221. [Google Scholar]
- 53. Lei YB, Yin CY, Li CY (2006) Differences in some morphological, physiological, and biochemical responses to drought stress in two contrasting populations of Populus przewalskii. Physiologia Plantarum 127: 182–191. [Google Scholar]
- 54. Lascano HR, Antonicelli GE, Luna CM, Melchiorre MN, Gómez LD, Racca RW, et al. (2001) Antioxidant system response of different wheat cultivars under drought: field and in vitro studies. Functional Plant Biology 28: 1095–1102. [Google Scholar]
- 55. Tian X, Lei YB (2007) Physiological responses of wheat seedlings to drought and UV-B radiation. Effect of exogenous sodium nitroprusside application. Russian Journal of Plant Physiology 54: 676–682. [Google Scholar]
- 56. Gutierrez L, Mauriat M, Guenin S, Pelloux J, Lefebvre JF, Louvet R, et al. (2008) The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnology Journal 6: 609–618. 10.1111/j.1467-7652.2008.00346.x [DOI] [PubMed] [Google Scholar]
- 57. Nicot N, Hausman JF, Hoffmann L, Evers D (2005) Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. Journal of Experimental Botany 56:2907–2914. [DOI] [PubMed] [Google Scholar]
- 58. Olbrich M, Gerstner E, Welzl G, Fleischmann F, Osswald W, Bahnweg G, et al. (2008) Quantification of mRNAs and housekeeping gene selection for quantitative real-time RT-PCR normalization in European beech (Fagus sylvatica L) during abiotic and biotic stress. Z Naturforsch 63c:574–582. [DOI] [PubMed] [Google Scholar]
- 59. Schmidt GW, Delaney SK (2010) Stable internal reference genes for normalization of real-time RT-PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Molecular Genetics and Genomics 283:233–241. 10.1007/s00438-010-0511-1 [DOI] [PubMed] [Google Scholar]
- 60. Ma S, Niu H, Liu C, Zhang J, Hou C, Wang D (2013) Expression stabilities of candidate reference genes for RT-qPCR under different stress conditions in soybean. PloS One 8: e75271 10.1371/journal.pone.0075271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rueda-Martínez C, Lamas O, Mataró MJ, Robledo-Carmona J, Sánchez-Espín G, Jiménez-Navarro M, et al. (2014) Selection of Reference Genes for Quantitative Real Time PCR (qPCR) Assays in Tissue from Human Ascending Aorta. PloS One 9: e97449 10.1371/journal.pone.0097449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guénin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Gutierrez L (2009) Normalization of qPCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. Journal of Experimental Botany 60: 487–493. 10.1093/jxb/ern305 [DOI] [PubMed] [Google Scholar]
- 63. Liu H-H, Tian X, Li Y-J, Wu C-A, Zheng C-C (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana . RNA 14: 836–843. 10.1261/rna.895308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou L, Liu Y, Liu Z, Kong D, Duan M, Luo L (2010) Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa . Journal of Experimental Botany 61: 4157–4168. 10.1093/jxb/erq237 [DOI] [PubMed] [Google Scholar]
- 65. Eldem V, Akçay UÇ, Ozhuner E, Bakır Y, Uranbey S, Unver T (2012) Genome-wide identification of miRNAs responsive to drought in peach (Prunus persica) by high-throughput deep sequencing. PloS One 7: e50298 10.1371/journal.pone.0050298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xie K, Shen J, Hou X, Yao J, Li X, Xiao J, et al. (2012) Gradual increase of miR156 regulates temporal expression changes of numerous genes during leaf development in rice. Plant Physiology 158: 1382–1394. 10.1104/pp.111.190488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McKersie BD, Chen Y, de Beus M, Bowley SR, Bowler C, Inzé D, et al. (1993) Superoxide dismutase enhances tolerance of freezing stress in transgenic alfalfa (Medicago sativa L.). Plant Physiology 103: 1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mckersie BD, Bowley SR, Harjanto E, Leprince O (1996) Water-deficit tolerance and field performance of transgenic alfalfa overexpressing superoxide dismutase. Plant Physiology 111: 1177–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hernández JA, Ferrer MA, Jiménez A, Barceló AR, Sevilla F (2001) Antioxidant Systems and O2.−/H2O2 Production in the Apoplast of Pea Leaves. Its Relation with Salt-Induced Necrotic Lesions in Minor Veins. Plant Physiology 127: 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang F-Z, Wang Q-B, Kwon S-Y, Kwak S-S, Su W-A (2005) Enhanced drought tolerance of transgenic rice plants expressing a pea manganese superoxide dismutase. Journal of Plant Physiology 162: 465–472. [DOI] [PubMed] [Google Scholar]
- 71. Chu Y, Corey DR (2012) RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic Acid Therapeutics 22: 271–274. 10.1089/nat.2012.0367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li H, Lovci MT, Kwon Y-S, Rosenfeld MG, Fu X-D, Yeo GW (2008) Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model. Proceedings of the National Academy of Sciences 105: 20179–20184. 10.1073/pnas.0807121105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Linsen SE, de Wit E, Janssens G, Heater S, Chapman L, Parkin RK (2009) Limitations and possibilities of small RNA digital gene expression profiling. Nature Methods 6: 474–476. 10.1038/nmeth0709-474 [DOI] [PubMed] [Google Scholar]
- 74. Raabe CA, Tang T-H, Brosius J, Rozhdestvensky TS (2013) Biases in small RNA deep sequencing data. Nucleic Acids Research. 10.1093/nar/gkt1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(TIF)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.