

Abstract
Background:
The rate of entry of cocaine into the brain is a critical factor that influences neuronal plasticity and the development of cocaine addiction. Until now, passive diffusion has been considered the unique mechanism known by which cocaine crosses the blood-brain barrier.
Methods:
We reassessed mechanisms of transport of cocaine at the blood-brain barrier using a human cerebral capillary endothelial cell line (hCMEC/D3) and in situ mouse carotid perfusion.
Results:
Both in vivo and in vitro cocaine transport studies demonstrated the coexistence of a carrier-mediated process with passive diffusion. At pharmacological exposure level, passive diffusion of cocaine accounted for only 22.5% of the total cocaine influx in mice and 5.9% in hCMEC/D3 cells, whereas the carrier-mediated influx rate was 3.4 times greater than its passive diffusion rate in vivo. The functional identification of this carrier-mediated transport demonstrated the involvement of a proton antiporter that shared the properties of the previously characterized clonidine and nicotine transporter. The functionnal characterization suggests that the solute carrier (SLC) transporters Oct (Slc22a1-3), Mate (Slc47a1) and Octn (Slc22a4-5) are not involved in the cocaine transport in vivo and in vitro. Diphenhydramine, heroin, tramadol, cocaethylene, and norcocaine all strongly inhibited cocaine transport, unlike benzoylecgonine. Trans-stimulation studies indicated that diphenhydramine, nicotine, 3,4-methylenedioxyamphetamine (ecstasy) and the cathinone compound 3,4-methylenedioxypyrovalerone (MDPV) were also substrates of the cocaine transporter.
Conclusions:
Cocaine transport at the BBB involves a proton-antiporter flux that is quantitatively much more important than its passive diffusion. The molecular identification and characterization of this transporter will provide new tools to understand its role in addictive mechanisms.
Keywords: blood-brain barrier, biological transport, cocaine, drug of abuse, pharmacokinetics.
Introduction
Cocaine is a psychoactive drug that acts predominantly as an inhibitor of solute carrier family (SLC)6A3, also known as the dopamine transporter (DAT), and is involved in dopamine uptake at the synaptic cleft. This inhibition is considered to be the main contributor to the reinforcement and behavioral properties of cocaine (Volkow et al., 1997b). Cocaine has been shown to be more addictive when smoked (crack use) or intravenously injected compared with nasal inhalation (or snorting) (Gossop et al., 1992; Cone, 1995). Neurobehavioral and molecular studies have demonstrated the critical role of the rate of cocaine delivery to the brain in the neuronal plasticity and reward effects induced by cocaine (Samaha et al., 2004; Samaha and Robinson, 2005; Volkow et al., 2012; Minogianis et al., 2013). Increasing the rapidity of cocaine administration leads to an increase of its abuse liability (Volkow et al., 2000; Abreu et al., 2001; Samaha et al., 2004). Variability in the rate of brain cocaine uptake may thus have significant consequences on its acute and chronic effects in the central nervous system (CNS).
The rate at which drugs reach the brain parenchyma depends not only on their route of administration but also on their ability to cross the cerebral endothelium, also called the blood-brain barrier (BBB), which constitutes the main brain interface modulating exchanges of compounds between the brain and blood (Abbott et al., 2010). Passive diffusion is the primary route by which solutes cross membranes. According to the pH partition theory, nonionized drugs cross cell membranes, and neuropharmacodynamic events depend solely on their neutral unbound fraction in the plasma. The relative “sufficient” lipophilicity (estimated by log P) of the neutral form has been historically correlated with the rate of drug diffusion at the BBB. As 6% of cocaine is uncharged (clog P 3.1) at pH 7.4 (clog D7.4 1.5; pKa 8.6), it has been assumed that this 6% crosses the BBB by passive diffusion and fast enough to produce its CNS effects, whereas the 94% of positively charged cocaine cannot cross the BBB. Although the pH partition theory is physicochemically relevant, it leads to the misinterpretation of the CNS effects of many psychotropic drugs, such as ecstasy (3,4-methylenedioxyamphetamine [MDMA]; clog P 1.8; clog D7.4 −0.9; pKa 10.1), which are 99.8% cationic at neutral pH. An alternative hypothesis is that a carrier-mediated transport process, capable of carrying at least the cationic form of these psychostimulant substances across the BBB, may also be involved.
The question of how CNS drugs are transferred to the brain parenchyma has been poorly addressed, mainly because of these redundant mechanistic assumptions concerning passive diffusion. Although some pharmacokinetic studies suggest that passive diffusion is not the only mechanism by which cocaine crosses the BBB (Benuck et al., 1987; Gatley et al., 1990; Telang et al., 1999; Raje et al., 2003), this mechanism of transport has not yet been elucidated at the BBB. Although our knowledge of carrier-mediated systems is still incomplete, it is known that ABC transporters, such as P-glycoprotein (P-gp; ABCB1) or BCRP (ABCG2) could be involved in the unidirectional transport of their substrates by limiting their brain uptake and/or increasing their efflux from the brain into the blood. Some bidirectional SLC transporters are also known to be involved in the transport of compounds at the BBB (Abbott et al., 2010; Giacomini et al., 2010; Tournier et al., 2011). Functional studies have previously established that a drug/proton-coupled antiporter present at the mouse BBB speeds up the transport of nicotine or clonidine and that it can unlikely be saturated by their in vivo pharmacological plasma concentrations (Andre et al., 2009; Cisternino et al., 2013). Cocaine has also been shown previously to modulate the clonidine transporter at the mouse BBB (Andre et al., 2009).
Based on these preliminary results, we have characterized cocaine transport at the mouse luminal BBB by in situ brain perfusion (Dagenais et al., 2000) and in hCMEC/D3 cells used as an in vitro model of the human BBB (Dauchy et al., 2009; Weksler et al., 2013). Both models permit the qualitative and quantitative properties of drug transport to be determined (Takasato et al., 1984; Dagenais et al., 2000) and can be used to evaluate the contribution of carrier-mediated systems by assessing the concentration dependency of the transport through exposure of these models to cocaine concentrations high enough to surpass the concentration at the half-maximal carrier velocity (K m) of the transporter (Kell et al., 2013). Our results show that the transport of cocaine across the BBB mainly involves a carrier-mediated system functionally identified as a proton/cocaine-antiporter, in addition to a relative minor passive component in the pharmacological vascular/extracellular concentration range. The features of this molecularly unknown transporter highlight its importance in governing the delivery of cocaine to the mouse and possibly to the human brain.
Methods
Drugs and Chemicals
[3H]-(−)Cocaine (35.3 Ci/mmol) and [14C]-sucrose (435 mCi/mmol) were purchased from Perkin Elmer (Courtaboeuf, France). Opioids and levogyre cocaine-related compounds were obtained from Euromedex (Souffelweyersheim, France). (−)-Cocaine and morphine were obtained from Francopia (Paris, France). MDMA and 3,4-methylenedioxypyrovalerone (MDPV) (purity >99%) were synthetized by the chemistry departments of Paris Descartes University (Dr. H. Galons and Dr. H. Chen). PSC833 was donated by Novartis (Basel, Switzerland). All other chemicals were from Sigma (Saint Quentin-Fallavier, France).
Cell Culture
hCMEC/D3 cells were seeded at 25000 cells/cm2 on collagen-coated culture flask in EBM-2 medium (Lonza, Basel, Switzerland) supplemented as described in Dauchy et al. (2009). For experiments, cells seeded in multiwell dishes (25000 cells/cm2) were used 3 to 4 days after seeding and between passages 28 and 35.
In Vitro Transport Experiment
hCMEC/D3 cells prepared as described above were first preincubated 30 minutes with the Krebs N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES) (KH) buffer (in mM: 128, NaCl; 24, NaHCO3; 4.2, KCl; 2.4, NaH2PO4; 1.5, CaCl2; 0.9, MgSO4; 10, HEPES; and 9, d-glucose) adjusted to pH 7.40. In some experiments, iso-osmotic substitution of sodium in KH buffer was performed with N-methyl-d-glucamine chloride (NMDG). The experiment was initiated by adding KH buffer containing [3H]-cocaine (0.1 µCi/mL, ~3nM) in presence or absence of another drug. After 5 minutes of incubation at 37°C, cells were placed on ice, and ice-cold Dulbecco’s phosphate buffered saline was added to stop the transport. Cells were then rapidly washed twice with ice-cold Dulbecco’s phosphate buffered saline and lysed with sodium dodecyl sulfate 10% (30 minutes at 37°C under agitation). In trans-stimulation studies, cells were loaded 5 minutes with [3H]-cocaine (0.1 µCi/mL; KH buffer) before incubation with KH buffer with or without (control) an unlabeled compound (10 µM) before stopping the transport and washing the cells as describe. Cell lysates were placed in vials and mixed with Ultima gold (Perkin Elmer) before being counted in a Tri-Carb counter (Perkin Elmer) to measure disintegrations per minute (dpm). Cell lysate was kept to quantify proteins (micro BCA protein assay kit, Pierce, Sigma).
Manipulation of pH
To bring KH buffer to pH 5.40 or 6.40, hydrochloric acid was added. NH4Cl prepulse condition leads to cytosol acidification and was achieved first by preincubating cells with KH buffer containing NH4Cl (30mM) for 15 minutes, which was subsequently replaced by NH4Cl-free KH buffer for 5 minutes before initiating the transport of [3H]-cocaine as described above. Alkalinization of the cytosol was obtained by NH4Cl pulse condition, which consists of studying the transport of [3H]-cocaine in a KH buffer (pHe 7.40) also containing NH4Cl (30mM) for 5 minutes.
Animals
In vivo experiments were performed with Swiss mice (25–40g; Janvier, Genest, France) unless otherwise specified. In some experiments, DBA, C57bl/6, Fvb (Janvier), and Fvb double knockout organic cation transporter 1 and 2 [Oct1,2(−/−)] (The Netherlands Cancer Institute, Amsterdam) male mice were used. All mice (7–11 weeks old) were housed in a controlled environment (22±3°C; 55±10% relative humidity) and a 12-hour–dark/–light cycle with access to food and tap water ad libitum. All experiments complied with the ethical rules of the Agriculture French Ministry for experimentation with laboratory animals and were approved by the ethics review committee (Paris Descartes University no. 12–183).
In Situ Carotid Perfusion
Surgical procedure and perfusion
The transport of [3H]-cocaine at the luminal BBB was measured by in situ carotid perfusion allowing to totally substitute brain vascular composition with an artificial perfusion fluid (Dagenais et al., 2000). Briefly, mice were anesthetized with ketamine-xylazine (140-8mg/kg, intraperitoneally), and a catheter was inserted into the right carotid artery. Before perfusion, the thorax was opened and the heart was cut. Perfusion was started immediately at a constant flow rate of 2.5mL/min, reaching a carotid pressure of ~120 mmHg. Each mouse was perfused with [3H]-cocaine (0.3 µCi/mL, ~9nM) and [14C]-sucrose (0.1 µCi/mL) as a vascular marker. Perfusion was terminated after 60 seconds by decapitating the mouse. The right cerebral hemisphere and 2 aliquots of perfusion fluid were placed in tared vials, weighted, digested with Solvable (Perkin Elmer), and mixed with Ultima gold before dual-labeled dpm counting.
Perfusion fluid
The perfusion fluid was Krebs-carbonate similar to KH buffer without HEPES gassed with 95% O2/5% CO2 to bring pH to 7.40 unless otherwise specified. Hydrochloric acid was added in some experiments to bring the pH to 5.40 or 6.40. Krebs-carbonate perfusate was changed to remove Na+ and/or Cl− by iso-osmotic replacement. Sodium was replaced by lithium chloride, and sodium and chloride were replaced by mannitol. Mice were also perfused with carbonate-free HEPES-buffered saline (in mM: 152, NaCl; 4.2, KCl; 1.5, CaCl2; 0.9, MgSO4; 10, HEPES; and 9, d-glucose), gassed with O2, and brought to pH 7.40. The perfusion fluid was warmed to 37°C and checked with a digital pH meter (±0.05 pH units) before perfusion.
[3H]-Cocaine transport study
The brain accumulation of [3H]-cocaine was measured under trans-influx zero to determine the kinetic conditions required to measure transport solely across the membrane separating the sucrose (vascular) space from the non-sucrose (brain parenchyma) space. The perfusion time adopted (60 seconds) ensured that the tissue distribution of the drug was that of the initial linear part of the distribution kinetics. The brain apparent distribution did not reach (pseudo-) equilibrium during this initial linear phase (data not shown). The first membrane (luminal) delimiting the sucrose space is the only kinetic interface that affected the distribution of [3H]-cocaine. Cis-inhibition was performed by co-perfusion of [3H]-cocaine (60 seconds) with an unlabeled drug.
Apparent tissue distribution volume and initial transport parameters
Calculations were done as previously described (Dagenais et al., 2000). The brain “vascular” volume was estimated using the [14C]-sucrose distribution volume (Vv; µL/g):
| (Equation 1) |
where X * (dpm/g) is the amount of [14C]-sucrose in the right brain hemisphere, and C * perf (dpm/µL) is the [14C]-sucrose concentration in the perfusion fluid. The data for any mouse whose Vv was above the normal value (Cattelotte et al., 2008) was excluded from the study.
The apparent tissue distribution volume (V tissue, µL/g) was calculated as:
| (Equation 2) |
where X tissue (dpm/g) is the calculated amount of [3H]-cocaine from the right brain hemisphere and C perf (dpm/µL) is the [3H]-cocaine concentration in the perfusion fluid:
| (Equation 3) |
where X tot (dpm/g) is the total quantity of [3H]-cocaine measured in the sample tissue. The amount of [3H]-cocaine in the vascular “[14C]-sucrose” space ( ) was calculated and subtracted from the total (X tot) (Equation 3).
The initial transport rate, also called brain clearance, expressed as a K in (µL/sec/g), was calculated from:
| (Equation 4) |
where T is the perfusion time (seconds).
The permeability-surface area product (PS, µL/sec/g) was calculated as
| (Equation 5) |
where F (vascular flow rate; µL/sec/g) has been previously measured to be 42.3 µL/sec/g (Cattelotte et al., 2008).
The flux (J in; nmol/sec/g of brain) is given by:
| (Equation 6) |
The cocaine brain flux (J in) or cellular uptake velocity (nmol/min/mg) is described as a saturable (Michaelis-Menten term) and a passive unsaturable component:
| (Equation 7) |
where C tot (mM) is the total cocaine concentration in the perfusate or incubation buffer, V max (nmol/sec/g or nmol/min/mg) is the maximal velocity of transport, and K m (mM) represents the concentration at the half-maximal carrier velocity. K passive (µL/sec/g or µL/min/mg) is an unsaturable component that represents the rate of transport by passive diffusion. The data were fitted using nonlinear regression analysis.
Data Analysis
Data are means±standard deviation (SD). One way analysis of variance and post hoc test (Dunnett) were used to identify significant differences, unless specified otherwise, and statistical significance was set at P<0.05. The transport parameters (K m, V max, K passive) were estimated by plotting cocaine brain or cell flux data against total concentration using Equation 7 and nonlinear regression with WinNonlin software. The errors associated with these parameters are asymptotic standard errors returned by the nonlinear regression routine.
Results
Brain Transport of Cocaine Measured by in Situ Brain Perfusion in Selected Transport-Deficient or Wild-Type Mouse Strains
The transport rate of [3H]-cocaine was measured by in situ brain perfusion to check for differences in cocaine transport between mouse strains and to test the hypothesis of the involvement of specific transporters. Brain transport (K in; n=4–5 mice) measured in selected mouse strains perfused with [3H]-cocaine (9nM) for 60 seconds in Krebs-carbonate buffer (pH 7.40) was as follows: DBA (21±2 µL/sec/g), C57Bl/6 (19±2 µL/sec/g), Fvb (20±2 µL/sec/g), and Swiss (20±2 µL/sec/g). These rates were not significantly different. The transport of [3H]-cocaine in wild-type Fvb did not differ significantly from that in [Oct1,Oct2(−/−)] mice (19±2 µL/sec/g; n=4). P-gp was not involved in [3H]-cocaine transport, since its transport rate was not significantly modified by co-perfusion of the highly potent P-gp inhibitor PSC833 (10 µM) (23±3 µL/sec/g; n=4) in Swiss mice.
Passive and Carrier-Mediated Cocaine Transport Components
Concentration dependence of cocaine transport in vitro
The concentration dependence of cocaine uptake was assessed in hCMEC/D3 cells over a range of concentrations high enough to surpass the K m for xenobiotic transporters. Cells were incubated for 5 minutes in KH buffer (pH 7.40, 37°C) containing cocaine (0.05–10mM) and [3H]-cocaine (0.1 µCi/mL). The total cocaine uptake velocity (nmol/min/mg of protein) was plotted against the total cocaine concentration in the incubation buffer. This global cocaine uptake velocity (dashed line) resulted in both a saturable uptake component fitted by a Michaelis-Menten equation in addition to an unsaturable component (Equation 7) (Figure 1a-b). Kinetic analysis provided a K m of 0.123±0.023mM, a V max of 4.27±0.26 nmol/min/mg, and a K passive of 1.09±0.09 µL/min/mg for the transport of cocaine in hCMEC/D3 cells. At concentrations (<10 µM) far less than the K m, this passive flux represents 5.9% of the total flux of cocaine (Figure 1b).
Figure 1.
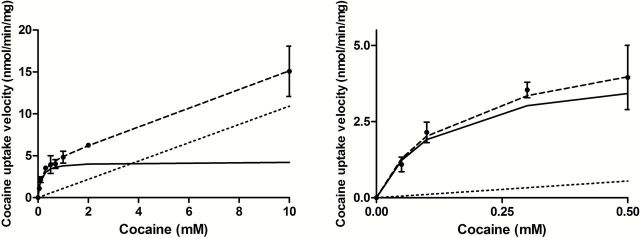
Passive and carrier-mediated flux of cocaine in hCMEC/D3 cells. A, Total uptake (nmol/min/mg; dashed line) was measured in hCMEC/D3 cells and plotted against total cocaine concentration in the KH incubation buffer at pHe 7.40. The straight dotted line represents the passive transport of cocaine (K passive of 1.09±0.09 µL/min/mg at pH 7.40). The solid line represents the curve obtained by subtracting the passive flux from the total flux and fitting to the carrier-mediated Michaelis-Menten term (see Equation 7) by nonlinear least-square regression. Estimated parameters for cocaine transport in hCMEC/D3 cells are: K m, 0.123±0.023mM; V max, 4.26±0.26 nmol/min/mg. Data represent means±SD of experiments performed in triplicate. B, Total (dashed line) and individual passive (dotted line) and carrier-mediated (solid line) cocaine transport into hCMEC/D3 cells fitted according to Equation 7 for concentrations <0.5mM. Data represent means±SD of experiments performed in triplicate.
Concentration dependence of cocaine transport in vivo
The experimentally determined net cocaine flux (J in) was plotted against its concentration (Figure 2a-b). J in was fitted to Equation 7 (dashed line), which yielded both a saturable (Michaelis-Menten) and an unsaturable component, as shown by the shape of the curve (Figure 2a). Regression analysis of the unsaturated component of cocaine flux into the brain yielded a K passive of 4.5±1.0 µL/sec/g (pHe 7.40). The unsaturated flux (dotted line) was subtracted from the total cocaine flux measured (J in) to obtain the carrier-mediated cocaine component (solid line), which was plotted against the total (unbound) cocaine concentration. Modeling this carrier-mediated brain cocaine flux yielded an apparent K m of 4.5±1.8mM and a V max of 127.2±34.5 nmol/sec/g of brain (solid line; Figure 2a). Using Equation 7, we found that the minimal experimental concentration at which passive diffusion and carrier-mediated flux are equal is around 24mM of cocaine. Above this theoretical vascular cocaine concentration, the passive diffusion flux will surpass the carrier-mediated one. The net brain cocaine transport rate (K in) under unsaturated conditions (which also reflect the pharmacological concentration range) is about 20 µL/sec/g. This represents a total brain extraction of about 47% as compared with the vascular flow marker diazepam (see Methods); passive diffusion (K passive 4.5±1.0 µL/sec/g) represents 22.5% of the total cocaine flux at the mouse BBB.
Figure 2.

Passive and carrier-mediated flux of cocaine at the mouse luminal BBB. A, Total flux (J in; nmol/sec/g; dashed line) measured in the right brain hemisphere of male Swiss mice and plotted against total cocaine concentration in the perfusion buffer (Krebs-carbonate) at pHe 7.40 (Equation 6). The straight dotted line represents the passive diffusion flux of cocaine (K passive of 4.5±1.0 µL/sec/g at pH 7.4). The solid line represents the curve obtained by subtracting the passive flux from the total flux and fitted to the carrier-mediated Michaelis-Menten term (see Equation 7) by nonlinear least-square regression. Estimated parameters for the brain transport of cocaine are: K m, 4.5±1.8mM; V max, 127.2±34.5 nmol/sec/g. Data represent means±SD of 4 to 5 mice. B, Total (dashed line), carrier-mediated (solid line), and passive (dotted line) cocaine fluxes at pharmacological concentrations at the mouse BBB, fitted according to Equation 7 for concentrations <0.1mM.
Effect of cis-Inhibition of Selected Compounds on [3H]-Cocaine Transport in vitro and in vivo
We performed drug-cocaine interaction studies on hCMEC/D3 cells and mice using cis-inhibition of [3H]-cocaine transport to discriminate between the effects of known transporters possibly involved in drug influx and to gain more insight into the molecular features of drugs that could interact with the cocaine transporter. Many compounds inhibit/modulate transporters, although they are not necessarily transported themselves, as is known for cocaine and SLC6A3. The concentrations used do not reflect or predict in vivo drug-drug interactions, as the unbound concentrations used can never be reached by their systemic administration in mice or humans.
We used broad-range inhibitors like tetraethylammonium (TEA), choline, and carnitine, which inhibit a wide range of known organic transporters, including Octs (Slc22a1-3), Mate (Slc47a1), and Octn (Slc22a4-5), both in vitro and in vivo. These compounds used at concentrations surpassing the cocaine transporter K m did not significantly modulate the transport of [3H]-cocaine in vitro or in vivo, suggesting that these SLC transporters are not involved in cocaine transport at the BBB (Tables 1A and 2). It has previously been demonstrated that Oct1-3, Pmat, and Mate1 are not expressed or functional at the mouse BBB (Agarwal et al., 2012; Andre et al., 2012). However, the hCMEC/D3 cell line has been shown to express some OCTs that could affect its functional properties (Weksler et al., 2013). In parallel experiments in the hCMEC/D3 cell line, we found that MPP+, a known substrate of the OCTs, was significantly inhibited by TEA (2mM), a substrate/inhibitor for OCTs (data not shown). However, the same TEA concentration did not affect [3H]-cocaine transport in these cells (Table 1A). Previous reports have also identified a range of substrates of a drug/proton-antiporter, such as clonidine, nicotine, diphenhydramine, oxycodone, and naloxone; these significantly affected [3H]-cocaine transport with varying potencies (Tables 1 and 2). Some cocaine metabolites were also tested: benzoylecgonine and ecgonine altered [3H]-cocaine transport in vivo and/or in vitro but with low potencies, whereas a few other cocaine-related compounds such as norcocaine, cocaethylene, and ecgonine methylester significantly decreased the BBB transport of [3H]-cocaine at lower concentrations (Tables 1B and 2).
Table 1.
Effects of Selected Organic Compounds, Psychoactive Drugs, and Their Metabolites (A) and Cocaine Metabolites (B) on [3H]-Cocaine Uptake in hCMEC/D3 Cellsa
| Compound | Concentration, mM | Relative uptake, % |
|---|---|---|
| A | ||
| Carnitine | 5 | 96.1±15.0 |
| Choline | 5 | 102.0±14.2 |
| TEA | 5 | 120.6±9.8 |
| Nicotine | 0.05 | 73.8±2.6* |
| MDMA | 0.05 | 29.7±0.7*** |
| MDPV | 0.05 | 18.1±0.4*** |
| Clonidine | 0.05 | 17.3±1.3*** |
| Diphenhydramine | 0.05 | 8.3±0.4*** |
| Verapamil | 0.05 | 3.7±0.2*** |
| Imipramine | 0.05 | 1.6±0.4*** |
| Oxycodone | 0.05 | 68.7±4.4*** |
| Codeine | 0.05 | 63.4±5.0** |
| Morphine | 0.05 | 62.9±14.7** |
| Hydrocodone | 0.05 | 49.2±5.8*** |
| Oxymorphone | 0.05 | 36.3±1.1*** |
| Norbuprenorphine | 0.05 | 31.7±6.3** |
| 6-Acetylcodeine | 0.05 | 31.3±1.7*** |
| Naloxone | 0.05 | 28.3±0.9*** |
| Heroin | 0.05 | 20.6±0.6*** |
| Tramadol | 0.05 | 19.3±2.0*** |
| Methadone | 0.05 | 6.1±0.3*** |
| B | ||
| Benzoylecgonine | 0.05 | 104.1±3.0 |
| 0.1 | 94.2±14.9 | |
| 1 | 72.0±11.4* | |
| Ecgonine | 0.05 | 103.9±8.7 |
| 0.1 | 93.2±2.5 | |
| 1 | 75.3±5.0* | |
| Ecgoninemethylester | 0.01 | 62.9±4.0*** |
| 0.05 | 27.7±2.2*** | |
| 0.1 | 18.3±3.4*** | |
| Cocaethylene | 0.01 | 35.4±4.4*** |
| 0.05 | 15.3±0.3*** | |
| 0.1 | 12.0±0.6*** | |
| Norcocaine | 0.01 | 29.5±1.1*** |
| 0.05 | 18.2±0.1*** | |
| 0.1 | 13.5±0.3*** | |
Abbreviations: MDMA, 3,4-methylenedioxyamphetamine; MDPV, 3,4-methylenedioxypyrovalerone; TEA, tetraethylammonium.
aThe inhibition of the transport of [3H]-cocaine (3nM) by the above compounds was evaluated in hCMEC/D3 cells with Krebs-HEPES buffer (pHe 7.40) and an incubation time of 5min. The uptake rate for controls set at 100±10% was 0.105±0.01 µL/min/µg. Data are expressed as means ± SD (performed in triplicate). * P<0.05, ** P<0.01, *** P<0.001 compared with the control group.
Table 2.
Effects of Selected Organic Compounds, Psychoactive Drugs, and Their Metabolites on [3H]-Cocaine Transport in the Mouse Braina
| Compound | Concentration, mM | Relative transport rate, % |
|---|---|---|
| Carnitine | 15 | 107.4±9.2 |
| Choline | 10 | 109.8±14.5 |
| TEA | 15 | 115.1±13.9 |
| Benzoylecgonine | 5 | 110.3±12.6 |
| Norcocaine | 5 | 59.7±3.7*** |
| Cocaethylene | 5 | 55.1±3.0*** |
| Morphine | 5 | 96.8±11.9 |
| MDMA | 5 | 82.1±7.1* |
| Nicotine | 10 | 76.7±6.9** |
| Methadone | 5 | 76.3±7.6** |
| MDPV | 5 | 59.8±7.7*** |
| Diphenhydramine | 5 | 46.6±2.4*** |
Abbreviations: MDMA, 3,4-methylenedioxyamphetamine; MDPV, 3,4-methylenedioxypyrovalerone; TEA, tetraethylammonium.
aThe effect of the above compounds on the luminal BBB transport of [3H]-cocaine (9nM) was measured by in situ brain co-perfusion (cis-inhibition) in Krebs-carbonate perfusion fluid (pHe 7.40) for 60sec in Swiss mice. Brain transport (K in) for controls, set at 100%, was 20.1±3.0 µL/sec/g. Data are expressed as means±SD (n=4–6 mice per condition). * P<0.05, ** P<0.01, *** P<0.001 compared with the control group.
Effect of trans-Stimulation by Organic Compounds on Intracellular [3H]-Cocaine Content in hCMEC/D3 Cells
The trans-stimulation technique provides a qualitative indication of whether a given compound is a substrate, but it does not provide kinetic information to quantify active vs passive transport rate. We assessed the ability of some compounds present in the extracellular compartment to further stimulate the exit of accumulated [3H]-cocaine from hCMEC/D3 cells that could occur with a bidirectional transporter. The trans-efflux zero or exit of [3H]-cocaine was strongly stimulated (P<0.001) by MDMA, MDPV, nicotine, and diphenhydramine (Figure 3), suggesting that they may also be substrates of the cocaine transporter. The trans-stimulation study also suggests that the transporter is bidirectional and could transport cocaine in or out of the cell according to its concentration gradient. Cells not exposed to the 5-minute washout with KH buffer had about 4.6-fold more intracellular [3H]-cocaine than control cells that were washed out (P<0.001, n=4).
Figure 3.

Trans-stimulation studies of cocaine transport in hCMEC/D3 cells. hCMEC/D3 cells were loaded with [3H]-cocaine for 5 minutes and then incubated with KH buffer alone (control) or with 10 µM of unlabeled compound (TEA, benzoylecgonine, cocaine, MDPV, MDMA, diphenhydramine, or nicotine) in KH buffer. Data represent means±SD performed in quadruplicate. *** P<0.001 compared with controls.
Effects of Inorganic Ions on [3H]-Cocaine Transport
To study the force driving the cocaine carrier and gain further biochemical information to better assess its functional identity, the intracellular pH and extracellular proton or sodium concentrations were manipulated in both in vitro and in vivo models.
Modulations of the extracellular/vascular pHe
The proportion of neutral and cationic species of cocaine depends on the pHe, which can be estimated using a basic pK a value of 8.6 for cocaine in water (Lu et al., 2007). Cocaine is 0.06% neutral at pHe 5.40, 0.6% at pHe 6.40, and 5.9% at pHe 7.40. The incubation of hCMEC/D3 cells with KH buffer set to yield a pHe of 5.40, 6.40, or 7.40 shows a 2.5- and 4.6-fold increase (P<0.001) in [3H]-cocaine uptake at pHe 6.40 and 7.40, respectively, as compared with pHe 5.40 (Figure 4a). Similarly, the brain transport of [3H]-cocaine measured for 60 seconds at pHe 6.40 and 7.40 in mice was significantly increased (P<0.001) by 4.7- and 16.9-fold, respectively, as compared with its transport measured at pHe 5.40 (Figure 5a). This increase could be related to the higher neutral cocaine fraction in the perfusate and/or to a change in the transport rate according to the proton extracellular concentration. Adding unlabeled cocaine (10mM) led to a significant 1.8-, 2.0-, and 1.9-fold decrease in [3H]-cocaine transport at the mouse BBB at pHe 5.40, 6.40, and 7.40, respectively, confirming that a carrier-mediated process is involved (Figure 5a). This inhibition effect by unlabeled cocaine shown at pHe 5.40 or 6.40 could suggest that the cationic form (>99.4%) of cocaine is a substrate of the transporter. However, with these functional approaches, we cannot reject that the neutral form of cocaine is not a substrate.
Figure 4.

Effect of the modulation of extracellular (pHe) and intracellular (pHi) pH on cocaine transport in hCMEC/D3 cells. a, Effects of changes in the incubation buffer pHe. [3H]-Cocaine uptake was measured for 5 minutes in KH buffer adjusted to pH 5.40, 6.40, or 7.40. *** P<0.001 compared with the pH 5.4 group, and ### P<0.001 for a comparison between pH 6.40 and 7.40 (n=3–4). b, Modulation of pHi with NH4Cl. In pulse condition (cellular alkalinization): after 30 minutes of the usual preincubation, a solution (pHe 7.40) with NH4Cl (30mM) and containing [3H]-cocaine was added for 5 minutes. The NH4Cl prepulse condition (cellular acidification) was obtained by preincubating cells with the incubation buffer (pHe 7.40) plus NH4Cl (30mM) for 15 minutes. Then, the incubation medium was replaced by the usual KH buffer without NH4Cl for 5 minutes. [3H]-Cocaine uptake was measured in the usual KH incubation buffer for 5 minutes (n=3–4).
Figure 5.

Effects of changes in the vascular perfusion medium pHe, alteration of the BBB pHi, and sodium dependency of [3H]-cocaine transport at the BBB in Swiss mice. a, Effects of the Krebs-carbonate perfusion buffer at a pHe of 5.40, 6.40, or 7.40 on [3H]-cocaine brain transport (K in; µL/sec/g), with (black column) or without (white column) co-perfusion with unlabeled cocaine (10mM), measured by in situ mouse brain perfusion for 60 seconds. Data represent means±SD (n=4 mice). * P<0.05, ** P<0.01, *** P<0.001 compared with the pH 5.4 group; + P<0.05, +++ P<0.001 for comparison (Student t test) between with and without unlabeled cocaine at the same pH; and ### P<0.001 for a comparison of pH 6.40 and 7.40 without unlabeled cocaine. b, Effects of altering pHi (white columns) and removing sodium (grey column). The vascular perfusion media were Krebs-carbonate buffer plus NH4Cl (30mM; “NH4Cl pulse”), carbonate-free HEPES-buffered solution, and “mannitol” (sodium-free and chloride-free) Krebs-carbonate buffer to alter pHi (white columns). The effect of Na+-free perfusion buffer (pHe 7.40) was studied by replacing sodium with lithium (Li+; grey column). Data represent means±SD of 4 to 7 animals. * P<0.05, ** P<0.01, *** P<0.001 compared with the control group.
Alteration of the intracellular pHi
To limit the confounding effect of the proportion of neutral cocaine in the extracellular/vascular compartment and thus to better illustrate the effect of proton on the transporter activity, [3H]-cocaine transport (pHe 7.40) was also evaluated with protocols that only increased or decreased the pHi (Taylor et al., 2006; Andre et al., 2009).
Alkalinization protocols
In hCMEC/D3 cells, a NH4Cl pulse (30mM) significantly decreased [3H]-cocaine uptake into the cells (Figure 4b). In vivo, mouse brains were perfused for 60 seconds with Krebs-carbonate buffer containing [3H]-cocaine (9nM) with or without NH4Cl (30mM), adjusted to pHe 7.40. This increase in pHi by NH4Cl led to a significant 1.8-fold reduction (P<0.001) in the brain transport of [3H]-cocaine (Figure 5b) in accordance with a proton-antiporter mechanism.
Physiologically, the blood acts as a carbonate buffer, and replacing it abruptly by a vascular solution without carbonate (eg, HEPES-medium) rapidly and transiently increases the pHi of endothelial cells. The brain transport of [3H]-cocaine was decreased 1.8-fold with HEPES-buffer compared with Krebs-carbonate buffer (Figure 5b).
Acidification protocols
NH4Cl prepulse treatment resulted in a significant 1.85-fold (P<0.001) increase in cocaine uptake in hCMEC/D3 cells (Figure 4b). This protocol could not be easily transposed to in situ brain perfusion. Instead, we used a pHi acidification protocol in which sodium was removed from the vascular solution that rapidly and transiently decreased the pHi. The [3H]-cocaine K in of mice perfused with the mannitol perfusion buffer was increased 1.35-fold (P<0.05) compared with control Krebs-carbonate–perfused mice (Figure 5b) in accordance with a proton-antiporter mechanism.
Effect of sodium on [3H]-cocaine transport
When hCMEC/D3 cells were incubated with NMDG-buffer (pHe 7.40), no significant change (97.0±9.7%, n=5) in [3H]-cocaine transport was observed after 5 minutes of incubation compared with control in KH buffer (100.0±9.0%). It can be difficult to study the influence of ions such as sodium on a putative H+-coupled transport system in situ, because acute exposure to an Na+-free extracellular medium can alter the Na+-dependent transporters involved in controlling the pHi and thus indirectly alter the activity of proton-dependent transporters. We then perfused the brains of mice with [3H]-cocaine in lithium buffer, which does not significantly affect the pHi at the BBB (Figure 5b) (Andre et al., 2009), and compared this with [3H]-cocaine in regular Krebs-carbonate buffer. There was no significant change in the brain transport of [3H]-cocaine, suggesting that sodium is not involved in [3H]-cocaine transport at the BBB (Figure 5b).
Discussion
Cocaine saturability studies of brain endothelial cells in vivo and in vitro demonstrate the coexistence of an additive carrier-mediated process along with the passive diffusion flux of cocaine at the BBB. The highest plasma cocaine concentrations reported in human or rodent studies are <10 µM (Benuck et al., 1987; Booze et al., 1997; Volkow et al., 2000). Therefore, according to the apparent K m, an unbound extracellular/vascular cocaine concentration <4.5mM in mice or 0.12mM in human cells would allow a cocaine flux across the BBB that is always proportional to its vascular/extracellular concentration. At the pharmacotoxicological range of cocaine concentrations (which are far less than the transporter K m), the passive diffusion flux of cocaine accounts for about 22.5% of the total cocaine influx in mice and 5.9% of the flux in hCMEC/D3 cells. A comparison of the in vivo passive (~4.5 µL/sec/g) and carrier-mediated (~15.5 µL/sec/g) transport rates suggests that the carrier-mediated influx of cocaine is 3.4 times greater than its passive diffusion when concentrations are less than the apparent carrier-mediated K m. This carrier-mediated component could be responsible for the fast rise in cocaine concentration in the brain parenchyma. Although both in vitro and in vivo experiments show that the proportion of passive cocaine flux is minor compared with its active flux, quantitative differences do exist between the 2 types of models. Some in vitro-in vivo studies have identified a few factors that could affect these proportions. The presence of an unstirred water layer in static in vitro models, but not in in situ perfusion model, could contribute to a boundary layer of water that decreases passive diffusion in vitro (Avdeef et al., 2004). The greatest biophysical difference between models comes from the lack of a pressure gradient in in vitro systems, which could allow passive diffusion to be described simply by Fick’s law. The existence of a hydrostatic pressure gradient in vivo/in situ permits a greater convective solvent effect that also affects and increases the convective/passive transport of the solute in accordance with the Navier-Stokes equations. This also contributes to a greater passive diffusion rate in vivo compared with in vitro experiments. Although speculative, differences between the K m and V max measured in vitro/human and in vivo/mouse models could possibly be linked to interspecies differences and expression levels, respectively. Better molecular tools are needed to understand the differences between models in this polyspecific, high-capacity, carrier-mediated system.
Our studies suggest that P-gp is not involved but reveal a carrier-mediated system that favors the influx of cocaine at the BBB, which could result from its ability to interact with an SLC transporter. The lack of in vitro or in vivo inhibition by known transporter inhibitors and the lack of differences in Oct1,2-knockout mice suggest that at least Octs, Mate, and Octn are not likely to be involved in the active handling of cocaine in these models. The diverse chemical or biochemical approaches used in in vitro and in vivo protocols altered the transport of cocaine at the BBB in a way that suggests the presence of a sodium-independent cocaine/proton-antiporter with features similar to those found for nicotine, diphenhydramine, tramadol, and clonidine transport at the mouse BBB (Andre et al., 2009; Cisternino et al., 2013). A transporter with similar biochemical features has also been found in some rodent BBB cell lines, where it transports oxycodone, diphenhydramine, and codeine (Okura et al., 2008; Fischer et al., 2010; Sadiq et al., 2011). The trans-stimulation of cocaine transport in hCMEC/D3 cells by nicotine, diphenhydramine, MDMA, or MDPV suggests that they share the same transporter. Interestingly, an analogous MDMA/proton-antiporter, which interacts with some other amphetamines, has also been functionally characterized in Caco-2 cells (Kuwayama et al., 2008). The lack of a trans-stimulation effect of benzoylecgonine could also indicate that it is not a substrate, in accordance with certain studies showing that benzoylecgonine has no significant brain uptake (Gatley et al., 1994; Chow et al., 2001). To obtain more information on the chemical structures able to interact with this transporter, we performed cis-inhibition experiments with substrate-related compounds that shared a similar DAT and with other pharmacological substances targeting the CNS, such as opioids, diphenhydramine, and nicotine. Compounds like heroin, diphenhydramine, or imipramine were shown in vitro to exhibit a high potency of inhibition. In the smaller set of compounds tested in vivo, diphenhydramine was shown to be the strongest. However, the concentration needed to significantly inhibit cocaine transport precluded any potential for drug-drug interactions in vivo. The inhibitory ability of several of these compounds suggests, however, that this transporter interacts with specific chemical features but with different potencies within the same chemical family, as illustrated by the slight inhibitory potency of benzoylecgonine in vitro and in vivo. The methoxy moiety missing only in benzoylecgonine and ecgonine compared with the other cocaine-related structures could represent a critical chemical moiety in this recognition and inhibition. The quite wide chemical diversity of interacting compounds suggests that computational chemical approaches with a wider range of structures could better assess the critical chemical moieties involved in such interactions.
Neurobiological studies have recently confirmed that the BBB transport rate of drugs of abuse like nicotine, morphine, or cocaine is a critical factor that affects the neurobehavioral plasticity involved in addiction (Samaha and Robinson, 2005; Minogianis et al., 2013; Seleman et al., 2014). A higher rate of cocaine brain delivery is associated with a greater risk of addiction in rodents (Minogianis et al., 2013). Positron emission tomography (PET) studies have also shown that the pharmacokinetic profiles of drugs of abuse like nicotine and cocaine, and in particular the speed/rate at which drugs enter the human brain, are crucial for their reward effects (Volkow et al., 2012). Indeed, these PET studies have shown that the rate of cocaine entry into the brain is the key factor associated with the subjective “high” effect rather than the overall presence or extent of drug distribution into the brain (Volkow et al., 1997a, 1997b, 2000). Interestingly, some differences in pharmacokinetics and cocaine conditioning have been shown depending on the strain of mice used and suggest that the DAB strain is more resistant to cocaine conditioning in the place preference paradigm (Niculescu et al., 2005; Eisener-Dorman et al., 2011). However, our experiments did not reveal a difference in cocaine transport at the BBB of DAB mice, suggesting that such cocaine conditioning differences are more likely linked to a neural mechanism (Eisener-Dorman et al., 2011). Remarkably, a clinical PET study performed in cocaine abusers abstinent for 3 months (detoxification unit) and who were defined by a history of cocaine use of >4g/wk of snorting and/or crack for at least 6 months has shown a decreased brain uptake of [11C]-cocaine without any change for DAT availability (Volkow et al., 1996). In light of our results, this decrease in BBB cocaine uptake in recent cocaine abusers could be related to decreased activity of this cocaine transporter, possibly representing an endothelial cell-related brain mechanism involved in tolerance.
In conclusion, cocaine transport at the mouse BBB involves a proton-antiporter flux that is quantitatively much more important than its passive diffusion. This carrier-mediated system could be responsible for the faster uptake of cocaine by the brain, which in turn may be involved in high risk of addiction. Although the CNS effects of cocaine have been shown to be dependent on its rate of access to its CNS biological targets, which depends on its route of administration, our study adds a new rate-regulatory component within the BBB. The cocaine/proton-antiporter could be a new pharmacological target whose inhibition could be used to limit drug effects and the risk of relapse. The knowledge of its molecular identity is thus critical to exploit this potential.
Statement of Interest
None.
Acknowledgments
We thank Dr. A. H. Schinkel for supplying the knockout mice, Dr. H. Galons and Dr. H. Chen for generously supplying MDMA and MDPV, and Novartis for generously supplying PSC833. We thank Dr. S. Rasika at Gap-Junction.com for editing the English text. We also thank the Paris Descartes University foundation and Servier Laboratories for the financial support granted to Hélène Chapy.
References
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. (2010). Structure and function of the blood-brain barrier. Neurobiol Dis 37:13–25. [DOI] [PubMed] [Google Scholar]
- Abreu ME, Bigelow GE, Fleisher L, Walsh SL. (2001). Effect of intravenous injection speed on responses to cocaine and hydromorphone in humans. Psychopharmacology (Berl) 154:76–84. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Uchida Y, Mittapalli RK, Sane R, Terasaki T, Elmquist WF. (2012). Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug Metab Dispos 40:1164–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre P, Debray M, Scherrmann JM, Cisternino S. (2009). Clonidine transport at the mouse blood-brain barrier by a new H+ antiporter that interacts with addictive drugs. J Cereb Blood Flow Metab 29:1293–1304. [DOI] [PubMed] [Google Scholar]
- Andre P, Saubamea B, Cochois-Guegan V, Marie-Claire C, Cattelotte J, Smirnova M, Schinkel AH, Scherrmann JM, Cisternino S. (2012). Transport of biogenic amine neurotransmitters at the mouse blood-retina and blood-brain barriers by uptake1 and uptake2. J Cereb Blood Flow Metab 32:1989–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdeef A, Nielsen PE, Tsinman O. (2004). PAMPA: a drug absorption in vitro model 11. Matching the in vivo unstirred water layer thickness by individual-well stirring in microtitre plates. Eur J Pharm Sci 22:365–374. [DOI] [PubMed] [Google Scholar]
- Benuck M, Lajtha A, Reith ME. (1987). Pharmacokinetics of systemically administered cocaine and locomotor stimulation in mice. J Pharmacol Exp Ther 243:144–149. [PubMed] [Google Scholar]
- Booze RM, Lehner AF, Wallace DR, Welch MA, Mactutus CF. (1997). Dose-response cocaine pharmacokinetics and metabolite profile following intravenous administration and arterial sampling in unanesthetized, freely moving male rats. Neurotoxicol Teratol 19:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattelotte J, Andre P, Ouellet M, Bourasset F, Scherrmann JM, Cisternino S. (2008). In situ mouse carotid perfusion model: glucose and cholesterol transport in the eye and brain. J Cereb Blood Flow Metab 28:1449–1459. [DOI] [PubMed] [Google Scholar]
- Chow HH, Anavy N, Villalobos A. (2001). Direct nose-brain transport of benzoylecgonine following intranasal administration in rats. J Pharm Sci 90:1729–1735. [DOI] [PubMed] [Google Scholar]
- Cisternino S, Chapy H, Andre P, Smirnova M, Debray M, Scherrmann JM. (2013). Coexistence of passive and proton antiporter-mediated processes in nicotine transport at the mouse blood-brain barrier. AAPS J 15:299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone EJ. (1995). Pharmacokinetics and pharmacodynamics of cocaine. J Anal Toxicol 19:459–478. [DOI] [PubMed] [Google Scholar]
- Dagenais C, Rousselle C, Pollack GM, Scherrmann JM. (2000). Development of an in situ mouse brain perfusion model and its application to mdr1a P-glycoprotein-deficient mice. J Cereb Blood Flow Metab 20:381–386. [DOI] [PubMed] [Google Scholar]
- Dauchy S, Miller F, Couraud PO, Weaver RJ, Weksler B, Romero IA, Scherrmann JM, De Waziers I, Decleves X. (2009). Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem Pharmacol 77:897–909. [DOI] [PubMed] [Google Scholar]
- Eisener-Dorman AF, Grabowski-Boase L, Tarantino LM. (2011). Cocaine locomotor activation, sensitization and place preference in six inbred strains of mice. Behav Brain Funct 7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Bernhagen J, Neubert RH, Brandsch M. (2010). Uptake of codeine into intestinal epithelial (Caco-2) and brain endothelial (RBE4) cells. Eur J Pharm Sci 41:31–42. [DOI] [PubMed] [Google Scholar]
- Gatley SJ, MacGregor RR, Fowler JS, Wolf AP, Dewey SL, Schlyer DJ. (1990). Rapid stereoselective hydrolysis of (+)-cocaine in baboon plasma prevents its uptake in the brain: implications for behavioral studies. J Neurochem 54:720–723. [DOI] [PubMed] [Google Scholar]
- Gatley SJ, Yu DW, Fowler JS, MacGregor RR, Schlyer DJ, Dewey SL, Wolf AP, Martin T, Shea CE, Volkow ND. (1994). Studies with differentially labeled [11C]cocaine, [11C]norcocaine, [11C]benzoylecgonine, and [11C]- and 4’-[18F]fluorococaine to probe the extent to which [11C]cocaine metabolites contribute to PET images of the baboon brain. J Neurochem 62:1154–1162. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, et al. (2010). Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossop M, Griffiths P, Powis B, Strang J. (1992). Severity of dependence and route of administration of heroin, cocaine and amphetamines. Br J Addict 87:1527–1536. [DOI] [PubMed] [Google Scholar]
- Kell DB, Dobson PD, Bilsland E, Oliver SG. (2013). The promiscuous binding of pharmaceutical drugs and their transporter-mediated uptake into cells: what we (need to) know and how we can do so. Drug Discov Today 18:218–239. [DOI] [PubMed] [Google Scholar]
- Kuwayama K, Inoue H, Kanamori T, Tsujikawa K, Miyaguchi H, Iwata Y, Miyauchi S, Kamo N, Kishi T. (2008). Uptake of 3,4-methylenedioxymethamphetamine and its related compounds by a proton-coupled transport system in Caco-2 cells. Biochim Biophys Acta 1778:42–50. [DOI] [PubMed] [Google Scholar]
- Lu H, Chen X, Zhan CG. (2007). First-principles calculation of pKa for cocaine, nicotine, neurotransmitters, and anilines in aqueous solution. J Phys Chem B 111:10599–10605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogianis EA, Levesque D, Samaha AN. (2013). The speed of cocaine delivery determines the subsequent motivation to self-administer the drug. Neuropsychopharmacology 38:2644–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niculescu M, Ehrlich ME, Unterwald EM. (2005). Age-specific behavioral responses to psychostimulants in mice. Pharmacol Biochem Behav 82:280–288. [DOI] [PubMed] [Google Scholar]
- Okura T, Hattori A, Takano Y, Sato T, Hammarlund-Udenaes M, Terasaki T, Deguchi Y. (2008). Involvement of the pyrilamine transporter, a putative organic cation transporter, in blood-brain barrier transport of oxycodone. Drug Metab Dispos 36:2005–2013. [DOI] [PubMed] [Google Scholar]
- Raje S, Cao J, Newman AH, Gao H, Eddington ND. (2003). Evaluation of the blood-brain barrier transport, population pharmacokinetics, and brain distribution of benztropine analogs and cocaine using in vitro and in vivo techniques. J Pharmacol Exp Ther 307:801–808. [DOI] [PubMed] [Google Scholar]
- Sadiq MW, Borgs A, Okura T, Shimomura K, Kato S, Deguchi Y, Jansson B, Bjorkman S, Terasaki T, Hammarlund-Udenaes M. (2011). Diphenhydramine active uptake at the blood-brain barrier and its interaction with oxycodone in vitro and in vivo. J Pharm Sci 100:3912–3923. [DOI] [PubMed] [Google Scholar]
- Samaha AN, Mallet N, Ferguson SM, Gonon F, Robinson TE. (2004). The rate of cocaine administration alters gene regulation and behavioral plasticity: implications for addiction. J Neurosci 24:6362–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaha AN, Robinson TE. (2005). Why does the rapid delivery of drugs to the brain promote addiction? Trends Pharmacol Sci 26:82–87. [DOI] [PubMed] [Google Scholar]
- Seleman M, Chapy H, Cisternino S, Courtin C, Smirnova M, Schlatter J, Chiadmi F, Scherrmann JM, Noble F, Marie-Claire C. (2014). Impact of P-glycoprotein at the blood-brain barrier on the uptake of heroin and its main metabolites: behavioral effects and consequences on the transcriptional responses and reinforcing properties. Psychopharmacology (Berl) 231:3139–3149. [DOI] [PubMed] [Google Scholar]
- Takasato Y, Rapoport SI, Smith QR. (1984). An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol 247:H484–493. [DOI] [PubMed] [Google Scholar]
- Taylor CJ, Nicola PA, Wang S, Barrand MA, Hladky SB. (2006). Transporters involved in regulation of intracellular pH in primary cultured rat brain endothelial cells. J Physiol 576:769–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telang FW, Volkow ND, Levy A, Logan J, Fowler JS, Felder C, Wong C, Wang GJ. (1999). Distribution of tracer levels of cocaine in the human brain as assessed with averaged [11C]cocaine images. Synapse 31:290–296. [DOI] [PubMed] [Google Scholar]
- Tournier N, Decleves X, Saubamea B, Scherrmann JM, Cisternino S. (2011). Opioid transport by ATP-binding cassette transporters at the blood-brain barrier: implications for neuropsychopharmacology. Curr Pharm Des 17:2829–2842. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemannn R, Gatley SJ, MacGregor RR, Wolf AP. (1996). Cocaine uptake is decreased in the brain of detoxified cocaine abusers. Neuropsychopharmacology 14:159–168. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS. (1997a) Imaging studies of cocaine in the human brain and studies of the cocaine addict. Ann N Y Acad Sci 820:41–54 [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley SJ, Pappas N, Hitzemann R, Shea CE. (1997b) Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature 386:827–830. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fischman MW, Foltin R, Fowler JS, Franceschi D, Franceschi M, Logan J, Gatley SJ, Wong C, Ding YS, Hitzemann R, Pappas N. (2000). Effects of route of administration on cocaine induced dopamine transporter blockade in the human brain. Life Sci 67:1507–1515. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Tomasi D. (2012). Addiction circuitry in the human brain. Annu Rev Pharmacol Toxicol 52:321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksler B, Romero IA, Couraud PO. (2013). The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids and barriers of the CNS 10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]