

Abstract
Background:
Recent studies demonstrate that the rapid antidepressant ketamine increases spine number and function in the medial prefrontal cortex (mPFC), and that these effects are dependent on activation of glutamate α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors and brain-derived neurotrophic factor (BDNF). In vitro studies also show that activation of AMPA receptors stimulates BNDF release via activation of L-type voltage-dependent calcium channels (VDCC).
Methods:
Based on this evidence, we examined the role of BDNF release and the impact of L-type VDCCs on the behavioral actions of ketamine.
Results:
The results demonstrate that infusion of a neutralizing BDNF antibody into the mPFC blocks the behavioral effects of ketamine in the forced swim test (FST). In addition, we show that pretreatment with nifedipine or verapamil, two structurally-different L-type calcium channel antagonists, blocks the behavioral effects of ketamine in the FST. Finally, we show that ketamine treatment stimulates BDNF release in primary cortical neurons and that this effect is blocked by inhibition of AMPA receptors or L-type VDCCs.
Conclusions:
Taken together, these results indicate that the antidepressant effects of ketamine are mediated by activation of L-type VDCCs and the release of BDNF. They further elucidate the cellular mechanisms underlying this novel rapid-acting antidepressant.
Keywords: BDNF, depression, glutamate, ketamine, L-type VDCC
Introduction
Depression is a debilitating and costly illness that affects approximately 17% of the population at some point in life (Kessler et al., 2003). Although there are several different classes of antidepressants available, these medications can take several weeks to months to produce a therapeutic response, and approximately one-third of depressed patients are nonresponsive and considered treatment resistant (Trivedi et al., 2006). Recent clinical studies demonstrate that a single, low dose of ketamine, an N-methyl-D-asparate (NMDA) receptor antagonist, produces a rapid and long-lasting antidepressant response in treatment-resistant patients (Berman et al., 2000; Zarate et al., 2006; Price et al., 2009). Preclinical studies in rodent models have also shown that a subanesthetic dose of ketamine produces antidepressant behavioral effects, which are dependent on activation of the mammalian target of rapamycin complex 1 (mTORC1) pathway (Li et al., 2010). Furthermore, activation of mTORC1 signaling and the subsequent antidepressant behavioral responses require activation of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors (Maeng et al., 2008; Li et al., 2010).
Recent studies also demonstrate a role for brain-derived neurotrophic factor (BDNF) in the actions of ketamine. BDNF has been shown to play an important role in neuropsychiatric disorders, including the pathophysiology and treatment response to typical antidepressants (Chen et al., 2001; Duman and Monteggia, 2006; Martinowich et al., 2007). The rapid and long-lasting behavioral effects of ketamine are blocked in conditional BDNF-deletion mutant mice (Autry et al., 2011) and in BDNF Val66Met knock-in mice (Liu et al., 2012). BDNF Val66Met is a human polymorphism present in approximately 25% of the population. The Met allele impairs activity-dependent BDNF release (Egan et al., 2003; Chen et al., 2004), and is associated with increased susceptibility to depression (Sen et al., 2003; Gatt et al., 2009). The finding that BDNF Val66Met knock-in mice do not respond to ketamine suggests that BDNF release is required for the actions of ketamine (Liu et al., 2012). This contrasts with the actions of typical antidepressants, which require chronic administration and only regulate expression, not release, of BDNF (Duman and Aghajanian, 2012).
In the current study we directly test the role of BDNF release by infusion of a BDNF neutralizing antibody in the medial prefrontal cortex (mPFC). A previous study indicated a key role for mPFC, demonstrating that blockade of mTORC1 in this region blocks the behavioral actions of systemic ketamine (Li et al., 2010). We examine the interaction between glutamate-AMPA depolarization and BDNF release. This is based, in part, on a recent study in cultured cells demonstrating that activation of AMPA receptors increases BDNF release and subsequently stimulates mTORC1 signaling, and that these effects require activation of L-type voltage-dependent calcium channels (VDCC; Jourdi et al., 2009). Based on these findings, we also test the role of L-type VDCCs using pharmacological inhibitors. Finally, we directly measure the ability of ketamine to stimulate the release of BDNF in primary neuronal cultures.
Materials and methods
Animals and Drug Administration
Adult male Sprague-Dawley rats (Charles River Laboratories; 275–300g) were pair-housed and maintained on a 12h light/dark cycle with food and water available ad libitum. Pregnant female rats were used as a source of embryonic tissue for primary neuronal cultures. All procedures were done in accordance with NIH guidelines and the Yale University Institutional Animal Care and Use Committee. Rats received a single i.p. injection of either nifedipine (Sigma-Aldrich; 10mg/kg) dissolved in 100% polyethylene glycol, verapamil (Sigma-Aldrich; 10mg/kg) dissolved in 0.9% saline, or 0.9% saline 30min prior to a single injection of either ketamine (Hospira; 10mg/kg i.p.) or 0.9% saline. Animals were tested in the forced swim test (FST) 24 hours following the second injection.
mPFC Cannulation and Infusion Procedure
Animals were anaesthetized with 50mg/kg pentobarbital i.p., and bilateral guide cannulae (22 G) were implanted at the following coordinates from bregma: +3.5 anterior/posterior; +/- 1 medial/lateral; and -4 dorsal/ventral. Following one week of recovery, rats were bilaterally infused with a function-blocking anti-BDNF antibody (Chemicon; 0.5 µg/side) at a rate of 0.25 µl/min. The function-blocking anti-BDNF antibody was diluted in 0.9% saline at a working concentration of 1µg/ul; control rats were infused with 0.9% saline. Rats were injected with either ketamine (10mg/kg i.p.) or 0.9% saline 30min following the antibody infusion. Animals were tested in the FST 24 hours following the injection.
Forced Swim Test
Behavioral testing in the FST was conducted as previously described (Li et al., 2010). Rats were exposed to a 15 minute pre-swim in 25°C water in a clear Plexiglas cylinder (65cm height, 30cm diameter). Twenty-four hours following the pre-swim, rats were infused with the function-blocking anti-BDNF antibody into mPFC or injected with either nifedipine or verapamil 30 minutes prior to a ketamine injection (i.p.) Twenty-four hours after the ketamine injection, rats were re-exposed to the swim tanks for a 10 minute period, which was video-recorded for analysis. Data were analyzed by scoring the total time immobile during the entire 10 minute swim by a blinded experimenter. Significance was determined at p < 0.05, and the data was plotted by total seconds immobile.
Locomotor Activity
Locomotor activity was measured using automated activity meters (Omnitech Electronics), which consisted of two parallel rows of photosensors, with 16 sensors per row. Twenty-four hours after drug administration, rats were placed in clear plastic boxes that were fitted to the activity meters and locomotion was recorded for a total of 30 minutes.
Primary Cortical Culture
Pregnant females were euthanized and cortices from E18 embryos were dissected. After incubation in trypsin-EDTA (0.25%; Gibco) for 10min, cortices were dissociated and neurons were plated at 0.6 million cells per well in 6-well polylysine-coated plates in DMEM (Gibco) containing 10% fetal bovine serum and 1% penicillin-streptomycin. The following day, the medium was changed to a serum-free medium containing neurobasal and B27 (Gibco), which was changed every 5 days. Cells were maintained at 37 °C, 5% CO2, and 95% humidity.
Drug Treatment and BDNF ELISA
After 10 days in vitro the medium was changed to a neurobasal medium containing an anti-BDNF antibody (2 μg/mL; Santa Cruz Biotechnology Inc.). Four hours following the medium change, cultured neurons were incubated with 0.5 µM ketamine for 15min, 60min, or 6hr. For blockade of ketamine studies, neurons were incubated with 50µM NBQX or 10µM verapamil 20min prior to ketamine treatment (15 minutes). Following the incubation with ketamine, the medium was carefully collected and the secreted BDNF captured by the antibody was immunoprecipitated. Immunoprecipitation was carried out using protein G-sepharose beads (GE Healthcare). Briefly, culture media was incubated in the protein G-sephorose beads and then the beads were washed and boiled at 100°C for 5 minutes in an elution buffer. BDNF in the immunoprecipitates was detected by an immuno assay (BDNF-ELISA E-max; Promega) according to the manufacturer’s instructions. Briefly, 96-well plates (Corning) were coated with monoclonal antibody and incubated at 4°C for 18 hours. The plates were incubated in a block and sample buffer at room temperature for 1 hour, followed by an incubation of the immobilized monoclonal antibody to BDNF with standards. Samples were maintained at room temperature for 2 hours. Then the plates were incubated with polyclonal antibody for 2 hours at room temperature, washed, and incubated at room temperature with a secondary anti-IgY antibody conjugated to horseradish peroxidase for 1 hour. Next, the plates were incubated in peroxidase substrate and tetramethylbenzidine solution to produce the color reaction. The reaction was stopped with 1M hydrochloric acid and the absorbance at 450nm was measured with an automated microplate reader. Standard curves were plotted for each plate. Protein concentrations in each immunoprecipitate were measured using a BCA kit (Thermo Scientific) and values of BDNF were corrected for the total amount of protein in the sample.
Results
Antidepressant Actions of Ketamine in the FST
Recent evidence suggests that the behavioral effects of ketamine require the release of BDNF (Liu et al., 2012). To further test the importance of BDNF release in the mPFC, rats were infused with a function-blocking anti-BDNF antibody 30min prior to ketamine administration, and tested in the FST 24hr after ketamine. The antibody and infusion conditions were based on a previous study examining the role of BDNF in learning and memory (Slipczuk et al., 2009); they show that anti-BDNF antibody infusion completely blocked learning-induced mTORC1 signaling, demonstrating the function-blocking efficacy of this antibody-infusion paradigm. Ketamine administration significantly decreased the immobility time in the FST compared to the vehicle-treated rats, as previously described (Li et al., 2010). In addition, infusion of the function-blocking anti-BDNF antibody into mPFC completely blocked the behavioral actions of ketamine in the FST (F1,37 = 4.158, p < 0.05; Figure 1A–C). These effects were observed over the total 10min of the FST, as well as in the first (0–5min) and second (5–10min) time blocks. Infusions of the antibody alone, in the absence of ketamine, had no effect on immobility when compared to vehicle-treated controls. Furthermore, control studies demonstrate that a heat inactivated IgG antibody had no effects on the behavioral actions of ketamine in the FST, indicating that the blockade of ketamine was due to neutralization of BDNF (not shown). There was no effect on locomotor activity across all groups, indicating that the effects seen in the FST were not due to altered ambulatory activity (Figure 1D).
Figure 1.
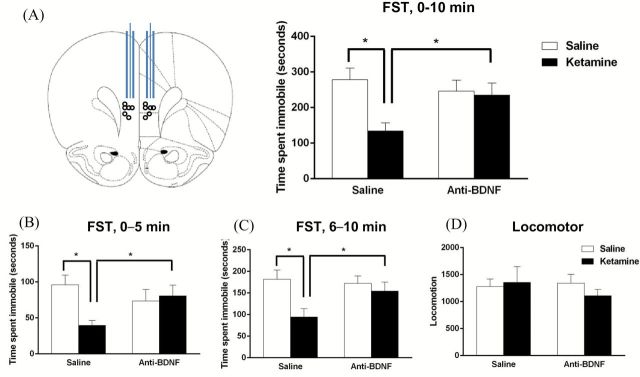
Infusion of anti-BDNF antibody into the mPFC blocks the behavioral effects of ketamine in the FST. Rats received a bilateral infusion into the mPFC of a function-blocking anti-BDNF antibody (1µg/µl) 30min prior to a ketamine injection (10mg/kg i.p.), and immobility in the FST was determined 24 hours following the ketamine injection. (A) Ketamine significantly reduced the immobility time compared to vehicle-treated rats, and pretreatment with the function-blocking anti-BDNF antibody completely blocked this effect. Values are the mean ± SEM (n = 7–11; antibody x drug interaction, F1,37 = 4.158, *p < 0.05). (B and C) Total swim time was divided into two epochs: the first 5 minutes and the second 5 minutes. Ketamine significantly reduced immobility time, which was blocked by the neutralizing antibody in both the first 5 minutes (ANOVA, F3,37 = 3.357, *p < 0.05) and the second 5 minutes (ANOVA, F3,37 = 3.630, *p < 0.05). (D) There was no effect on locomotor activity.
L-Type VDCCs are Required for the Behavioral Effects of Ketamine in the FST
The antidepressant actions of ketamine are blocked by pretreatment with a glutamate-AMPA receptor antagonist (Maeng et al., 2008; Li et al., 2010). Furthermore, an in vitro study demonstrated that activation of the mTORC1 pathway by stimulation of AMPA receptors is dependent on activation of L-type VDCC (Jourdi et al., 2009). We hypothesized that this cellular mechanism would also be required for the antidepressant effects of ketamine. To test this hypothesis, rats were pretreated with nifedipine (10mg/kg) or verapamil (10mg/kg), two structurally different VDCC blockers, 30min prior to a ketamine injection; the following day (24hr after ketamine), rats were examined in the FST. Doses for L-type VDCC blockers were chosen based on previous studies of these antagonists in learning and memory (Woodside et al., 2004; Seoane et al., 2009). Rats injected with ketamine had reduced mobility in the FST compared to vehicle-treated rats, and pretreatment with nifedipine completely blocked the antidepressant effect of ketamine, but had no effect alone (two-way ANOVA, F1,20 = 7.023, p < 0.05; Figure 2A–C). Pretreatment with verapamil also completely blocked the effects of ketamine in the FST and had no effect alone (ANOVA, F3,28 = 3.936, p < 0.05; Figure 2D–F,). These effects were observed over the total 10min of the FST, as well as in the first (0–5min) and second (5–10min) time blocks for both nifedipine and verapamil. Previous studies have shown that both nifedipine (24 hour following injection) and verapamil (30 and 60min following injection) do not have an effect on locomotor activity, indicating the effects seen in the FST were not due to a generalized decrease in ambulation for the groups receiving a channel blocker (Borroni et al., 2000; Cain et al., 2002).
Figure 2.

L-type channel antagonists block the antidepressant behavioral effects of ketamine in the FST. Rats were injected i.p. with either nifedipine (10mg/kg) or verapamil (10mg/kg) 30min prior to a systemic ketamine injection (10mg/kg). 24 hours after the ketamine injection, immobility was measured in the FST. (A) Ketamine produced a significant decrease in immobility time over the entire 10min test that was completely blocked by pretreatment with nifedipine (n = 6; drug x drug interaction, F1,20 = 7.023, *p < 0.05). (D) Verapamil pretreatment also completely blocked the effects of ketamine over the entire 10min test (n = 8; ANOVA, F3,28 = 3.936, *p < 0.05). Fisher’s post-hoc least significant difference tests revealed a significant difference between vehicle-treated and ketamine-treated rats, pretreatment with verapamil and ketamine (p < 0.05), and verapamil and ketamine alone (p < 0.01). (B, C, E, and F) Immobility was also examined during the first (0–5min) and second (6–10min) time blocks of the 10min test. Ketamine significantly decreased immobility time compared to controls in the first and second epochs, and these effects was blocked by nifedipine (B and C) or verapamil (E and F) in the second epoch: (B) ANOVA, F3,20 = 5.581, *p < 0.05; (C) ANOVA, F3,20 = 10.067, *p < 0.05; (E) ANOVA, F3,28 = 4.857, *p < 0.05; and (F) ANOVA, F3,28 = 3.130, *p < 0.05. All values are the means ± SEM.
Ketamine Treatment Increases BDNF Release in Primary Neuronal Cultures: Requirement for AMPA and L-Type VDCC
Blockade by function-blocking BDNF antibody and in Val66Met mice (Liu et al., 2012) suggests that the actions of ketamine require the release of BDNF. In vivo analysis of extracellular BDNF is technically difficult because of the size and limited diffusion of BDNF. Therefore, to directly test this hypothesis, we examined the influence of ketamine incubation on the release of BDNF in primary neuronal cultures. Cortical neurons were stimulated with a low dose of ketamine (0.5 µM) for different periods of time (15min, 60min, or 6hr). Media was collected at the indicated time points and BDNF was enriched by immunoprecipitation. ELISA analysis revealed a significant increase (~50%) in BDNF following a 15min (t[22] = 3.10, p < 0.01), 60min (t[10] = 3.33, p < 0.01), or 6hr incubation with ketamine (t[4] = 3.14, p < 0.05; Figure 3).
Figure 3.
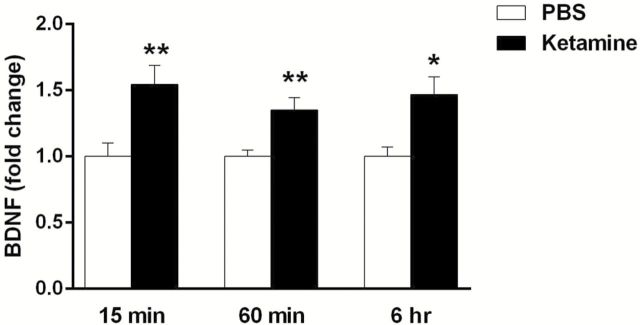
Incubation of primary neuronal cultures with ketamine rapidly increases BDNF release. Primary cortical neuronal cultures were stimulated with 0.5 µM ketamine for 15 and 60min and for 6hr, and culture medium was collected for BDNF analysis. Ketamine significantly increased BDNF release into the culture media following 15min (n = 12; t[22] = 3.10,**p < 0.01), 60min (n = 6; t[10] = 3.33, **p < 0.01), and 6 hour (n = 3; t[4] = 3.14, *p < 0.05) incubations. All values are expressed as fold change compared to the control and are shown as means ± SEM.
A previous study in cultured neurons reported that AMPA-stimulation of BDNF release is blocked by L-type VDCC antagonists (Jourdi et al., 2009). Additionally, the antidepressant effects of ketamine are dependent on stimulation of AMPA receptors (Maeng et al., 2008; Li et al., 2010), and here we show that the behavioral actions of ketamine require activation of L-type VDCCs (Figure 2). These results suggest that both AMPA and VDCCs are required for ketamine-stimulated release of BDNF. To test this hypothesis, cortical neurons were pretreated with either NBQX (50 µM; an AMPA receptor antagonist) or verapamil (10 µM) 20min prior to ketamine stimulation. These doses were chosen based on previous studies examining the role of AMPA receptor activation on protein synthesis in cultured cells (Jourdi et al., 2009). ELISA analysis revealed that pretreatment with either NBQX (F1,20 = 13.209, p < 0.01) or verapamil (F1,44 = 14.809, p < 0.001) completely blocked ketamine-induced BDNF release (Figure 4A and B).
Figure 4.
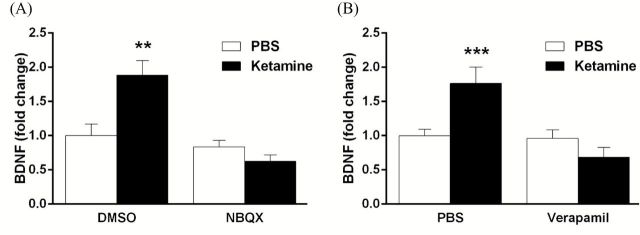
Ketamine-induced BDNF release is dependent on activation of glutamate-AMPA receptors and L-type VDCCs. (A) Cortical neurons were incubated with NBQX (50 µM) 20min prior to ketamine, and medium was collected 15min later (after ketamine). Incubation with the AMPA receptor antagonist completely blocked ketamine-induced BDNF release (n = 6; drug x drug interaction, F1,20 = 13.209, *p < 0.01). (B) Pretreatment with verapamil (10 µM) also blocked ketamine-induced BDNF release (n = 9–10; drug x drug interaction, F1,44 = 14.809, **p < 0.001). All values are expressed as fold change compared to the control and are shown as means ± SEM.
Discussion
The results demonstrate that the antidepressant actions of ketamine are dependent on BDNF release and activation of L-type VDCCs. While previous studies have found that the behavioral actions of BDNF are blocked in BDNF-conditional deletion mutants (Autry et al., 2011) and in BDNF Val66Met knock-in mice (Liu et al., 2012), we wanted to further examine the role of BDNF release in the antidepressant effects of ketamine. Our results show that neutralizing BDNF within the extracellular space of the mPFC was sufficient to block the behavioral actions of ketamine. This indicates that the release of BDNF is necessary for the antidepressant actions of ketamine in the FST. BDNF in the extracellular space would then bind to membrane tropomyosin related kinase B (TrkB) receptors and stimulate intracellular pathways that lead to activation of mTORC1 signaling.
Previous studies have also demonstrated that the rapid and long-lasting effects of ketamine require activation of glutamate-AMPA receptors and mTORC1 (Maeng et al., 2008; Li et al., 2010). In addition, activation of AMPA receptors stimulates mTORC1 signaling, and this effect requires stimulation of L-type VDCCs (Jourdi et al., 2009). Here, we find that pretreatment with nifedipine, a dyhydropyridine calcium channel antagonist, or verapamil, a phenylakylamine calcium channel antagonist, blocks the behavioral actions of ketamine in the FST. These findings demonstrate a requirement for L-type VDCC activation using two structurally different calcium channel antagonists.
Our in vivo studies indicate that the behavioral actions of ketamine are dependent on the release of BDNF, which was then directly examined using primary neuronal cultures. We found that incubation with a low dose of ketamine stimulates the release of BDNF into the culture medium at 15min, 60min, and 6hr. The increase of BDNF release is most likely due to an activity-dependent release caused by ketamine stimulation. This is supported by studies demonstrating that the antidepressant behavioral actions of ketamine require activation of AMPA receptors (Maeng et al., 2008; Li et al., 2010). A requirement for AMPA receptors was confirmed by studies demonstrating that incubation with the antagonist NBQX completely blocks ketamine-stimulated release of BDNF. In addition, pretreatment with the VDCC blocker verapamil completely blocked the release of BDNF caused by ketamine. Together, these studies demonstrate that activation of both AMPA and VDCCs are necessary for ketamine-induced BDNF release.
The cellular localization of NMDA receptors that mediate the actions of ketamine is still under investigation. One theory holds that ketamine causes activation of AMPA receptors via disinhibition of gamma-aminobutyric acid (GABA) neurons and increased glutamatergic transmission. The primary neuronal cultures contain a mixed population of cells, including both glutamatergic and GABAergic neurons. Blockade of ketamine-stimulated BDNF release by AMPA and VDCC blockers is consistent with the possibility that ketamine causes a glutamate burst via disinhibition of GABA neurons in culture. However, it is also possible that ketamine acts on NMDA receptors located on glutamatergic neurons, although the cellular mechanisms are less clear.
Taken together, these results demonstrate that BDNF release and activation of L-type VDCCs are necessary for the antidepressant effects of ketamine. To understand if these mechanisms are unique for the rapid and long-lasting effects of ketamine, it will be important to test if typical antidepressants act through these mechanisms. In addition, other rapid-acting antidepressants must be examined. This includes antagonists of the mGlu2/3 receptor (e.g., LY 341495) and muscarinic receptor (e.g., scopolamine) that produce behavioral effects similar to ketamine and that are also dependent on mTORC1 activation (Dwyer et al., 2012; Voleti et al., 2013). Studies are underway to test whether the antidepressant effects of these drugs are also mediated by BDNF release and activation of L-type VDCCs.
Statement of Interest
There are no competing financial interests in relation to the work described.
Acknowledgments
Supported by National Institutes of Health Grant MH093897 (R.S.D.) and the State of Connecticut.
References
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. (2000). Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–354. [DOI] [PubMed] [Google Scholar]
- Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ. (2000). Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J Neurosci 20:9272–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CK, Blouin AM, Barad M. (2002). L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditional fear in mice. J Neurosci 22:9113–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. (2001). Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 50:260–265. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, Lee FS. (2004). Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci 24:4401–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Aghajanian GK. (2012). Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. (2006). A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59:1116–1127. [DOI] [PubMed] [Google Scholar]
- Dwyer JM, Lepack AE, Duman RS. (2012). mTOR activation is required for the antidepressant effects of mGluR(2)/(3) blockade. Int J Neuropsychop 15:429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112:257–269. [DOI] [PubMed] [Google Scholar]
- Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, Gordon E, Kemp AH, Williams LM. (2009). Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry 14:681–695. [DOI] [PubMed] [Google Scholar]
- Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, Baudry M. (2009). Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci 29:8688–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, Rush AJ, Walters EE, Wang PS. (2003). The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 289:3095–3105. [DOI] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. (2012). Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry 71:996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. (2008). Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 63:349–352. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Manji H, Lu B. (2007). New insights into BDNF function in depression and anxiety. Nat Neurosci 10:1089–1093. [DOI] [PubMed] [Google Scholar]
- Price RB, Nock MK, Charney DS, Mathew SJ. (2009). Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry 66:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Nesse RM, Stoltenberg SF, Li S, Gleiberman L, Chakravarti A, Weder AB, Burmeister M. (2003). A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology 28:397–401. [DOI] [PubMed] [Google Scholar]
- Seoane A, Massey PV, Keen H, Bashir ZI, Brown MW. (2009). L-type voltage-dependent calcium channel antagonists impair perirhinal long-term recognition memory and plasticity processes. J Neurosci 29:9534–9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. (2009). BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLOS ONE 4:e6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M. (2006). Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psych 163:28–40. [DOI] [PubMed] [Google Scholar]
- Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, Sanacora G, Eid T, Aghajanian G, Duman RS. (2013). Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry 74:742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodside BL, Borroni AM, Hammonds MD, Teyler TJ. (2004). NMDA receptors and voltage-dependent calcium channels mediate different aspects of acquisition and retention of a spatial memory task. Neurobiol Learn Mem 81:105–114. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856–864. [DOI] [PubMed] [Google Scholar]