

Abstract
Background:
Impaired stress resilience and a dysfunctional hypothalamic-pituitary-adrenal (HPA) axis are suggested to play key roles in the pathophysiology of illness progression in bipolar disorder (BD), but the mechanisms leading to this dysfunction have never been elucidated. This study aimed to examine HPA axis activity and underlying molecular mechanisms in patients with BD and unaffected siblings of BD patients.
Methods:
Twenty-four euthymic patients with BD, 18 siblings of BD patients, and 26 healthy controls were recruited for this study. All subjects underwent a dexamethasone suppression test followed by analyses associated with the HPA axis and the glucocorticoid receptor (GR).
Results:
Patients with BD, particularly those at a late stage of illness, presented increased salivary post-dexamethasone cortisol levels when compared to controls (p = 0.015). Accordingly, these patients presented reduced ex vivo GR responsiveness (p = 0.008) and increased basal protein levels of FK506-binding protein 51 (FKBP51, p = 0.012), a co-chaperone known to desensitize GR, in peripheral blood mononuclear cells. Moreover, BD patients presented increased methylation at the FK506-binding protein 5 (FKBP5) gene. BD siblings presented significantly lower FKBP51 protein levels than BD patients, even though no differences were found in FKBP5 basal mRNA levels.
Conclusions:
Our data suggest that the epigenetic modulation of the FKBP5 gene, along with increased FKBP51 levels, is associated with the GR hyporesponsiveness seen in BD patients. Our findings are consistent with the notion that unaffected first-degree relatives of BD patients share biological factors that influence the disorder, and that such changes are more pronounced in the late stages of the illness.
Keywords: bipolar disorder, cortisol, FKBP51, glucocorticoid receptor, HPA axis.
Introduction
Bipolar disorder (BD) affects around 0.6% of the population and is associated with clinical comorbidities and increased standardized mortality ratios (Osby et al., 2001; Merikangas et al., 2011). Particularly, progression of BD has been associated with several unfavorable clinical outcomes, including reduced interepisode intervals, functional impairments and inferior responsiveness to treatment in late-stage BD patients when compared with early-stage ones (Kessing, 2005; Berk et al., 2011; Rosa et al., 2012). In addition, evidence suggests that patients with BD present reduced resilience to stress and an increased vulnerability to new mood episodes as the illness progresses (Fries et al., 2012). In fact, stress resilience and coping mechanisms are primarily mediated by the HPA axis, which appears to be dysfunctional in patients with BD (Daban et al., 2005). BD patients have been shown to have a hyperactive HPA axis, in some cases with high levels of cortisol (Cervantes et al., 2001) and non-suppression of its levels in the dexamethasone suppression test or the dexamethasone/cortisol releasing hormone test (Watson et al., 2004). In addition, high-risk subjects, such as first-degree relatives, have also been shown to present elevated baseline cortisol levels (Ellenbogen et al., 2006; Mannie et al., 2007) and abnormal responses to the dexamethasone/cortisol releasing hormone test (Holsboer et al., 1995; Modell et al., 1998). In this sense, HPA axis abnormalities seem to be a trait conferring vulnerability to mood disorders (Deshauer et al., 1999).
One of the main players in HPA axis modulation is the glucocorticoid receptor (GR), which translocates from the cytosol to the nucleus upon hormone binding and acts as a transcription factor. The function of GR depends on a large molecular complex entailing several chaperones and co-chaperones, including FK506-binding protein 51 (FKBP51; Binder, 2009). In vitro overexpression of human FKBP51 reduces hormone binding affinity and nuclear translocation of GR (Wochnik et al., 2005), and high levels of FKBP51 have been shown to lead to GR insensitivity, accompanied by increased blood cortisol levels in New World monkeys (Scammell et al., 2001). Interestingly, glucocorticoids can induce the expression of FKBP51 as part of an intracellular ultra-short negative feedback loop for GR activity (Vermeer et al., 2003).
Given the reported familial and genetic component of the pathophysiology of BD (Willour et al., 2009), it is likely that most of these stress-related features reflect a particular genetic background. In this vein, several studies have suggested that the genetic contribution to BD operates mostly through gene-environment interactions (Petronis, 2003; McGowan and Kato, 2008). Mechanistically, environmental impact reprograms gene activity by changing epigenetic modifications, thus increasing the risk for the disease in susceptible subjects and interfering with the course of illness. Among such epigenetic modifications, alterations in DNA methylation have been consistently reported in patients with BD (Connor and Akbarian, 2008; Huzayyin et al., 2013). Of note, chronic exposure to glucocorticoids has been shown to induce alterations in DNA methylation at the murine FKBP5 gene and at the human Fkbp5 gene in patients with post-traumatic stress disorder (Yang et al., 2012; Klengel et al., 2013). Therefore, FKBP5 methylation may be one of the mechanisms by which stress plays its role in BD pathophysiology.
Stress resilience and coping mechanisms are believed to be key elements in the development and progressive course of BD. However, the mechanisms behind the associated HPA axis dysfunction are still poorly understood, as is the role they play in determining the risk for the disease in susceptible subjects. Therefore, this study aimed to examine HPA axis activity and underlying molecular mechanisms in patients with BD, first-degree relatives, and healthy controls, with a focus on identifying clinical and epigenetic mechanisms associated with the development and progression of BD.
Methods
Subjects
The present study was approved by the Research Ethics Committee of Hospital de Clínicas de Porto Alegre (HCPA), Brazil, under protocol no. 12–0102. Subjects received a detailed description of the study, and gave written informed consent. All participants were at least 18 years old.
Twenty-four euthymic patients diagnosed with BD type I according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) Axis I criteria were recruited at an outpatient program of HCPA. Euthymia was confirmed using the Hamilton Depression Rating Scale (HDRS) and the Young Mania Rating Scale (YMRS; scores < 7 for each scale). In order to evaluate whether the mechanisms under investigation were also involved in BD progression, we divided the BD patients into early (stages I and II) and late (stages III and IV) stages of the illness, according to a previously published staging model of BD (Kapczinski et al., 2009). For this purpose, a series of clinical parameters were collected using a semi-structured interview, including data on course of illness, functioning, and comorbidities, as previously described (Pfaffenseller et al., 2014; Rosa et al., 2014).
Eighteen siblings of patients with BD were also included in the study: each had at least one sibling with the diagnosis of BD type I. Siblings recruited for this study were not necessarily related to the 24 BD patients enrolled; therefore, both groups were considered independent in the analyses. Moreover, 26 nonrelated healthy controls without any history of psychiatric illnesses or neurological disorders were also recruited. Subjects were matched by frequency, so that age and sex would not differ between groups by the end of recruitment. Control subjects were selected at the Blood Bank at HCPA after screening for exclusion criteria. All of the participants were volunteers, with no specific compensation provided.
The exclusion criteria for BD patients, siblings, and controls were history of autoimmune diseases, chronic infection/inflammatory disorders, or any severe systemic disease, and use of immunosuppressive therapy. All subjects were clinically interviewed by a trained psychiatrist and evaluated using the Functioning Assessment Short Test (FAST; Rosa et al., 2007; Cacilhas et al., 2009) and the Childhood Trauma Questionnaire (CTQ; Bernstein et al., 2003; Grassi-Oliveira et al., 2006) for the assessment of functioning and early childhood adverse events, respectively. Twenty milliliters of peripheral venous blood were collected from each subject for further analysis. All blood collections were performed at the same period of the day (late afternoon).
Dexamethasone Suppression Test
All subjects underwent a low-dose dexamethasone suppression test with 1.0mg dexamethasone for the assessment of HPA axis activity. All subjects were informed that no foods or drinks should be taken for at least 30min prior to saliva collection. The first saliva sample was collected at 08:00 hours on the first day (T1). A second saliva sample was collected on the same day, at 23:00 hours (T2), followed by the oral administration of dexamethasone. On the next day, a third saliva sample was collected, around 08:00 hours, immediately after waking up (T3), and the last sample was collected at 16:00 hours (T4). All samples were kept at 4°C until arrival in the laboratory, where they were centrifuged, aliquoted, and stored at -20°C until further analysis.
Cortisol and Adrenocorticotropic Hormone
Salivary cortisol levels were measured using the Cortisol Enzyme Immunoassay kit (Arbor Assay), according to the manufacturer’s instructions. A standard curve ranging from 3200 to 0 pg/ml was used to calculate cortisol levels for each sample. Plasma samples were used for the measurement of adrenocorticotripic hormone (ACTH) levels using an ELISA kit (Calbiotech), according to the manufacturer’s instructions. A standard curve ranging from 0 to 517 pg/ml was used to determine ACTH levels in the samples.
Ex Vivo GR Responsiveness
Measurement of dexamethasone-induced FKBP5 mRNA expression in peripheral blood mononuclear cells (PBMCs) was used to estimate GR responsiveness, as previously described (Vermeer et al., 2003, 2004; Chun et al., 2011). PBMCs were isolated from total heparinized blood by Ficoll-Hypaque density-gradient centrifugation (GE Healthcare). Following counting, cells were resuspended to a concentration of 0.5 x 106 cells/ml in RPMI 1640 medium supplemented with 10% charcoal-treated fetal bovine serum, 10 µg/ml gentamycin, and 0.25 µg/ml amphotericin B. PBMCs were then seeded onto a 24-well plate in 0.5ml aliquots (0.25 x 106 cells/well). Following overnight stabilization in culture, cells were treated for 24 hours with 10–9, 10–8, or 10–7 M dexamethasone or vehicle. Total RNA was isolated using the Illustra RNAspin Mini RNA Isolation kit (GE Healthcare) and reverse transcribed using the Omniscript Reverse Transcriptase kit (Qiagen), according to the manufacturers’ instructions. The newly synthesized complementary DNA (cDNA) was then amplified by real-time polymerase chain reaction (PCR) performed on the LightCycler system (Roche Applied Science), using the QuantiFast SYBR Green PCR kit (Qiagen). The following primers were used: 5’-ccattgctttattggcctct-3’ (forward) and 5’-ggatatacgccaacatgttcaa-3’ (reverse) for FKBP5 (Menke et al., 2013); and 5’-agatgagtatgcctgccgtg-3’ (forward) and 5’-tgcggcatcttcaaacctcc-3’ (reverse) for beta-2 microglobulin.
Western Blot Analysis
For protein analysis, isolated PBMCs were lysed in 50 µL of lysis buffer (60mM Tris-HCl [pH 6.8], 2% SDS, and 10% saccharose), supplemented with 1:100 protease (Sigma) and phosphatase (Roche Applied Science) inhibitors, followed by sonication and determination of protein concentration using a BCA kit (Thermo Fisher Scientific). For immunoblot detection, 15 µg of cell lysate total protein were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis under denaturing conditions, followed by transfer onto nitrocellulose membranes. After blocking, membranes were incubated overnight with a monoclonal antibody against FKBP51 (1:1000, Bethyl), followed by a horseradish peroxidase-conjugated antibody against rabbit IgG (Cell Signaling). Signals were visualized using ECL detection reagent (Millipore) and monitored using the ChemiDoc MP Imaging System (BioRad). Band intensities were normalized by the intensity of a reference sample (pooled PBMCs) loaded onto each gel to ensure accurate comparison of samples loaded on different gels.
FKBP5 DNA Methylation
FKBP5 methylation was analyzed by bisulfite pyrosequencing, as previously described (Blair et al., 2013; Klengel et al., 2013). Genomic DNA was isolated from total blood using the Illustra blood genomicPrep Mini Spin kit (GE Healthcare), followed by spectrophotometric quantification (NanoDrop, Thermo Fisher). Approximately 300ng of DNA were converted with bisulfite using the EZ DNA Methylation kit (Zymo Research). Bisulfite-converted DNA samples were used as templates in PCR reactions for the amplification of intron 2, intron 7, and the putative promoter region of the FKBP5 gene (Klengel et al., 2013). All sites analyzed were located close to glucocorticoid response elements (GREs).
Statistical Analysis
Statistical analyses were performed using PASW Statistics version 18.0. Descriptive statistics were used to report demographic and clinical characteristics of the sample. Normality of data distribution was assessed with Shapiro-Wilk’s test and histogram visualization. Categorical variables were compared using chi-square or Fisher’s exact tests. One-way analysis of variance (ANOVA), followed by Tukey’s post hoc test, were performed to compare parametric variables between BD patients, siblings, and controls, and independent t-tests were performed to analyze differences in clinical and demographic data between early- and late-stage BD patients. Analyses of covariance were conducted, adjusting for the potential confounding variables of age, sex, body mass index (BMI), and years of education. The Kruskal-Wallis test was followed by the Mann-Whitney test; they were used to compare non-parametric variables between independent samples. Bonferroni correction was applied to control for multiple comparisons. Correlations between continuous variables were assessed using Pearson’s or Spearman’s correlation tests. Significance was set at p < 0.05, except when otherwise specified.
Results
Sample
Characteristics of BD patients, siblings, and healthy controls are shown in Table 1. Groups did not differ with regard to age, gender, BMI, smoking, years of education, or CTQ scores. Even though all participants were euthymic at enrollment, BD patients and siblings scored higher in mania and depression scales when compared to controls (YMRS: F(2.65) = 4.9, p = 0.01; HDRS: F(2.65) = 17.9, p < 0.001). Moreover, BD patients presented higher FAST scores when compared with siblings and controls [F(2) = 22.24, p < 0.001], whereas no significant differences in FAST scores were found between siblings and controls (p = 0.138). BD patients also presented a higher prevalence of hypothyroidism (χ2 = 15.6, p < 0.001). Patterns of medication use in the BD patients recruited for this study were very similar to those previously described for international and Brazilian standards, as assessed by a multi-national, multi-center, observational cohort study (Vieta et al., 2013).
Table 1.
Characteristics of controls, siblings, and patients with bipolar disorder
| Characteristic | Controls (n = 26) | Siblings (n = 18) | Patients (n = 24) | p |
|---|---|---|---|---|
| Age (years)a,d | 46.9 (7.08) | 51.1 (13.2) | 46.9 (7.08) | 0.453 |
| Gender (male/female)c | 8/18 | 6/12 | 7/17 | 0.959 |
| HDRSa,d | 0.65 (1.3) | 3.56 (3.11) | 4.17 (2.14) | <0.001 |
| YMRSa,d | 0.08 (0.27) | 1.06 (1.3) | 0.96 (1.6) | 0.01 |
| Body mass indexa,d | 28.1 (4.7) | 27.3 (5.8) | 30.3 (6.12) | 0.183 |
| Smokingc | 33.3% | 7.7% | 25% | 0.094 |
| Years of educationa,d | 11.54 (3.95) | 10.33 (4.34) | 11.13 (3.6) | 0.611 |
| FAST scorea,d | 5.92 (6.37) | 11.93 (12.3) | 23.7 (9.92) | <0.001 |
| CTQ scorea,d | 33.04 (9.7) | 35.12 (7.9) | 39.04 (11.0) | 0.131 |
| Age at illness onseta | 25.67 (10.7) | |||
| Length of euthymia (months)b | 11 (35.3) | |||
| Length of illness (years)a | 21.04 (10.8) | |||
| Number of manic/mixed episodesa | 5.3 (5.8) | |||
| Number of depressive episodesa | 8.5 (8.4) | |||
| Total number of episodesa | 13.8 (11.5) | |||
| Number of hospitalizationsa | 5.38 (8.7) | |||
| Comorbidities | ||||
| Hypothyroidismc | 0% | 11.1% | 41.6% | < 0.001 |
| Hypertensionc | 0% | 16.6% | 25% | 0.03 |
| Diabetes mellitusc | 0% | 0% | 16.6% | 0.02 |
| Dyslipidemiac | 3.8% | 33.3% | 20.8% | 0.036 |
| Obesityc | 0% | 0% | 4.17% | 0.394 |
| Otherc | 7.7% | 27.7% | 33.3% | 0.073 |
| Medications | ||||
| Mood stabilizers | 66.6% | |||
| Lithium | 37.5% | |||
| Valproate | 25% | |||
| Carbamazepine | 8.3% | |||
| Lamotrigine | 4.2% | |||
| Antidepressants | 4.16% | |||
| Atypical antipsychotics | 33.3% | |||
| Typical antipsychotics | 12.5% | |||
| Benzodiazepines | 8.33% | |||
aMean (standard deviation); bMedian (interquartile range); cChi-square test; dOne-way ANOVA.
Cortisol and HPA Axis Activity
Results of the dexamethasone suppression test are shown in Table 2. All groups presented significantly lower post-dexamethasone salivary cortisol levels compared to basal levels (T1 vs. T3; p < 0.05 for all groups). Moreover, the cortisol suppression ratio did not differ between the groups. Basal salivary cortisol levels (T1) were similar in all groups, as were bedtime cortisol levels (T2). However, patients with BD presented higher post-dexamethasone cortisol levels (T3) when compared with controls [F(2) = 4.21, p = 0.015; Figure 1A]. The findings remained similar when age, sex, BMI, and education were included as covariates. A positive correlation was also found between post-dexamethasone cortisol levels and total number of episodes in BD patients (r = -0.329, p = 0.017), and no significant correlation between FAST scores and post-dexamethasone cortisol levels was found (r = 0.156, p = 0.284), neither when considering the entire sample, nor when analyzing each group individually. No differences were found for the last time point (T4). The mean decline of cortisol levels throughout the day (diurnal slope) did not differ between the groups, nor did plasma ACTH levels.
Table 2.
Cortisol and HPA axis parameters
| Characteristic | Controls (n = 26) | Siblings (n = 18) | Patients (n = 24) | p |
|---|---|---|---|---|
| Cortisol (pg/µl) | ||||
| T1, basal levelsa | 2.84 (2.16) | 3.13 (2.24) | 3.1 (1.62) | 0.881 |
| T2, bedtime levelsa | 1.16 (0.66) | 0.98 (0.73) | 1.89 (1.94) | 0.102 |
| T3, post-dexamethasone levelsa | 0.23 (0.11) | 0.32 (0.22) | 0.41 (0.21) | 0.02 |
| T4a | 0.26 (0.08) | 0.38 (0.08) | 0.37 (0.2) | 0.087 |
| Cortisol suppression ratioa,b | 13.47 (11.13) | 11.55 (8.86) | 9.64 (6.96) | 0.462 |
| Diurnal slope, mean declinea,c | 0.15 (0.13) | 0.22 (0.13) | 0.13 (0.08) | 0.163 |
| Plasma ACTH levels (pg/ml)a | 18.99 (11.57) | 15.8 (7.25) | 20.32 (12.2) | 0.469 |
aMean (standard deviation); bSuppression ratio = cortisol T1 / cortisol T3; cDiurnal slope = (cortisol T1 – cortisol T2) / (time T2 – awakening time). T1 = 08:00 hours on day 1; T2 = 23:00 hours on day 2 (pre-dexamethasone); T3 = 08:00 hours on day 2; T4 = 16:00 hours on day 2.
Figure 1.

HPA axis activity and FKBP51 alterations in bipolar disorder. (A) Post-dexamethasone salivary cortisol levels; one-way ANOVA followed by Tukey’s post hoc test. (B) Ex vivo GR responsiveness assay. Isolated peripheral blood mononuclear cells were incubated with increasing concentrations of dexamethasone, with assessment of FKBP5 mRNA levels after 24 hours; Kruskall-Wallis followed by Mann-Whitney test and Bonferroni correction. (C) Basal FKBP5 mRNA levels; one-way ANOVA followed by Tukey’s post hoc test. (D) FKBP51 protein levels; Kruskall-Wallis followed by Mann-Whitney test and Bonferroni correction. (E–F) Methylation status of the FKBP5 gene at two specific CpG dinucleotides; one-way ANOVA followed by Tukey’s post hoc test. *p = 0.008 (BD patients vs. controls). RQ: relative quantification.
GR Responsiveness
BD patients showed significantly lower FKBP5 mRNA levels in response to the lower concentration of dexamethasone when compared to the control group (U = 22, Z = -2.66, p = 0.008; Figure 1B). No differences were found for the two other dexamethasone concentrations. Siblings were not statistically different from controls or BD patients. No significant correlations were found between GR responsiveness and salivary cortisol levels at any of the assessed time points, nor in relation to number of episodes, length of illness, or mood symptoms.
Baseline FKBP5 mRNA Levels
Vehicle-treated cells from the previous experiment were also analyzed for baseline FKBP5 mRNA levels. After log-transformation of data, no differences were found in any of the comparisons (p > 0.05; Figure 1C). No significant correlations were found between baseline FKBP5 mRNA levels and clinical parameters.
FKBP51 Protein Levels
Considering that FKBP51 is a negative regulator of GR, we assessed basal FKBP51 protein levels in the PBMCs isolated from all subjects. Patients with BD presented increased FKBP51 protein levels when compared to siblings (U = 62, Z = -2.505, p = 0.012); a trend was also observed in the comparison with controls (U = 113, Z = -1.959, p = 0.05, Figure 1D). A significant correlation was found between FKBP51 and post-dexamethasone cortisol levels (Spearman’s ρ = 0.313, p = 0.049). No correlations were found between FKBP51 protein levels and basal FKBP5 mRNA levels, GR responsiveness, or clinical parameters.
FKBP5 Methylation
Based on previous evidence of epigenetic mechanisms modulating the FKBP51 ultra-short feedback loop (Klengel et al., 2013), we assessed DNA methylation of the FKBP5 gene at three different loci (intron 7, intron 2, and promoter) in all subjects. Differences were found at two CpG sites (dinucleotides composed of a cytosine next to a guanine in the same strand of the DNA) in intron 7 and one CpG in intron 2 of the FKBP5 gene, but only the differences at CpG 6 of intron 7 and CpG 2 of intron 2 remained significant after post hoc tests (Table 3). BD patients showed an increased percentage of methylation at intron 7 when compared with siblings [CpG 6, F(2,57) = 5.58; p = 0.007; Figure 1E], and a trend was also observed in the comparison with controls (p = 0.054). Patients with BD also showed increased methylation at CpG 2 of intron 2 when compared with controls [F(2,53) = 3.7, p = 0.04; Figure 1F]. No other differences regarding individual CpG sites were found in our sample. Significant negative correlations were found between the number of previous manic episodes and methylation status at two CpG sites of intron 7 (CpG 1: r = -0.444, p = 0.034; CpG5: r = -0.426, p = 0.043).
Table 3.
FKBP5 methylation in patients, siblings, and controls (%)
| Controls (n = 26) | Siblings (n = 18) | Patients (n = 24) | p | |
|---|---|---|---|---|
| Intron 7 | ||||
| P1_CpG1a,c | 69.6 (3.6) | 70.7 (5.7) | 68.3 (3.9) | 0.239 |
| P1_CpG2a,c | 90.6 (3.4) | 93.2 (3.8) | 91.3 (4.1) | 0.12 |
| P1_CpG3b,d | 100 (3.4) | 99.9 (2.7) | 97.6 (4.6) | 0.041 |
| P1_CpG4b,d | 79.6 (6.2) | 78.2 (3.7) | 79.9 (4.6) | 0.249 |
| P1_CpG5a,c | 47.8 (4.2) | 48.5 (2.3) | 49.0 (2.9) | 0.482 |
| P1_CpG6a,c | 56.9 (4.3) | 55.5 (3.7) | 60.1 (5.1) | 0.006 |
| Intron 2 | ||||
| P4_CpG1a,c | 66.6 (2.4) | 66.7 (3.05) | 66.9 (1.9) | 0.893 |
| P4_CpG2a,c | 59.8 (2.1) | 59.9 (2.05) | 61.6 (2.6) | 0.031 |
| P4_CpG3a,c | 48.6 (1.9) | 50.4 (2.8) | 50.4 (3.0) | 0.058 |
| P4_CpG4a,c | 61.2 (1.9) | 61.4 (1.8) | 60.6 (1.3) | 0.322 |
| Promoter | ||||
| GRE3_CpG1b,d | 94.8 (6.2) | 94.7 (4.4) | 94.4 (5.5) | 0.235 |
| GRE3_CpG2a,c | 47.1 (1.5) | 46.9 (1.6) | 46.8 (1.9) | 0.801 |
| GRE3_CpG3a,c | 100 | 100 | 100 | 1.000 |
| GRE3_CpG4a,c | 92.5 (3.5) | 90.9 (2.8) | 92.3 (3.5) | 0.315 |
| GRE3_CpG5a,c | 84.2 (1.6) | 84.3 (2.2) | 85.0 (2.1) | 0.427 |
| GRE3_CpG6a,c | 62.2 (1.9) | 62.4 (1.7) | 63.5 (1.7) | 0.059 |
aMean (standard deviation); bMedian (interquartile range); cOne-way ANOVA test; dKruskal-Wallis test.
As previously reported (Klengel et al., 2013), further analyses were performed after summarizing the percentage of the 16 CpG sites into seven bins, according to their spatial proximity to the consensus GRE sites. No differences were found between the groups for percentage of methylation in the seven bins. Results remained unaltered after controlling for potential confounders. No correlations were found between FKBP5 methylation and CTQ or FAST scores.
Analyses According to Staging
In order to assess whether the mechanisms above are involved in BD progression, we divided BD patients into early and late stages of illness, and further analyzed our data comparing the two staging groups. Late-stage patients presented higher results than early-stage ones for total number of episodes [t(14.7) = -4.27, p = 0.001], number of manic/mixed episodes [t(13.6) = -3.57, p = 0.003], number of depressive episodes [t(15.8) = -2.91, p = 0.01], number of hospitalizations [t(13.3) = -2.73, p = 0.017], and FAST scores [t(21) = -3.32, p = 0.003]; conversely, the two groups did not differ with regard to length of illness, age at illness onset, or any other sociodemographic or clinical variable (Table 4).
Table 4.
Characteristics of patients with early- vs. late-stage bipolar disorder
| Characteristic | Early-stage bipolar disorder (n = 10) | Late-stage bipolar disorder (n = 14) | p |
|---|---|---|---|
| Age (years)a,c | 44.4 (7.38) | 48.79 (6.51) | 0.138 |
| Gender (male/female)d | 3 / 7 | 4 / 10 | 0.993 |
| HDRSa,c | 3.4 (2.22) | 4.71 (1.97) | 0.141 |
| YMRSa,c | 1.2 (1.93) | 0.79 (1.13) | 0.544 |
| Body mass indexa,c | 27.8 (4.95) | 32.2 (6.3) | 0.081 |
| Smokingd | 10% | 35.7% | 0.151 |
| Years of educationa,c | 12.4 (3.83) | 10.21 (3.37) | 0.154 |
| Age at illness onseta,c | 23.22 (10.87) | 26.07 (10.27) | 0.532 |
| Length of euthymia (months)b,f | 10 (62.5) | 12 (33.3) | 0.815 |
| Length of illness (years)a,c | 19.7 (12.59) | 22 (9.78) | 0.619 |
| Number of manic/mixed episodesa,c | 1.7 (0.82) | 7.93 (6.45) | 0.003 |
| Number of depressive episodesa,c | 3.9 (2.72) | 11.79 (9.59) | 0.01 |
| Total number of episodesa,c | 5.6 (2.63) | 19.71 (11.95) | 0.001 |
| Number of hospitalizationsa,c | 0.9 (0.99) | 8.57 (10.4) | 0.017 |
| FAST scorea,c | 17.2 (8.16) | 28.69 (8.27) | 0.003 |
| CTQ scorea,c | 40.8 (11.9) | 37.7 (10.6) | 0.516 |
| Comorbidities | |||
| Hypothyroidisme | 30% | 50% | 0.421 |
| Hypertensione | 10% | 35.7% | 0.341 |
| Diabetes mellituse | 20% | 14.3% | 1.000 |
| Dyslipidemiae | 30% | 21.4% | 0.615 |
| Obesitye | 0% | 7.14% | 1.000 |
| Othere | 30% | 28.6% | 0.673 |
| Medications | |||
| Mood stabilizerse | 90% | 50% | 0.079 |
| Lithiume | 40% | 35.7% | 1.000 |
| Valproatee | 30% | 21.4% | 0.665 |
| Carbamazepinee | 20% | 0% | 0.163 |
| Lamotriginee | 10% | 0% | 0.417 |
| Antidepressantse | 10% | 0% | 0.417 |
| Atypical antipsychoticse | 30% | 35.7% | 1.000 |
| Typical antipsychoticse | 0% | 21.4% | 0.239 |
| Benzodiazepinese | 10% | 7.14% | 1.000 |
aMean (standard deviation); bMedian (interquartile range); cStudent t-test; dChi-square test; eFisher’s exact test; fMann-Whitney test.
Late-stage BD patients showed increased post-dexamethasone cortisol levels when compared with controls [F(3) = 2.96, p = 0.033; Figure 2A], differently from early-stage BD patients. Accordingly, late-stage patients presented reduced GR responsiveness to 10-9M dexamethasone when compared with controls (U = 6, Z = -2.603, p = 0.009; Figure 2B); no difference was found between early-stage BD patients and controls (U = 16, Z = -1.854, p = 0.064), nor between early- and late-stage BD patients (U = 16, Z = -0.714, p = 0.475). The groups did not differ in terms of basal FKBP5 mRNA or FKBP51 protein levels (Figure 2C and D). However, early-stage patients showed increased methylation at intron 7 when compared with BD siblings [CpG 6, F(3.56) = 4.33; p = 0.008; Figure 2E] and controls (p = 0.046).
Figure 2.
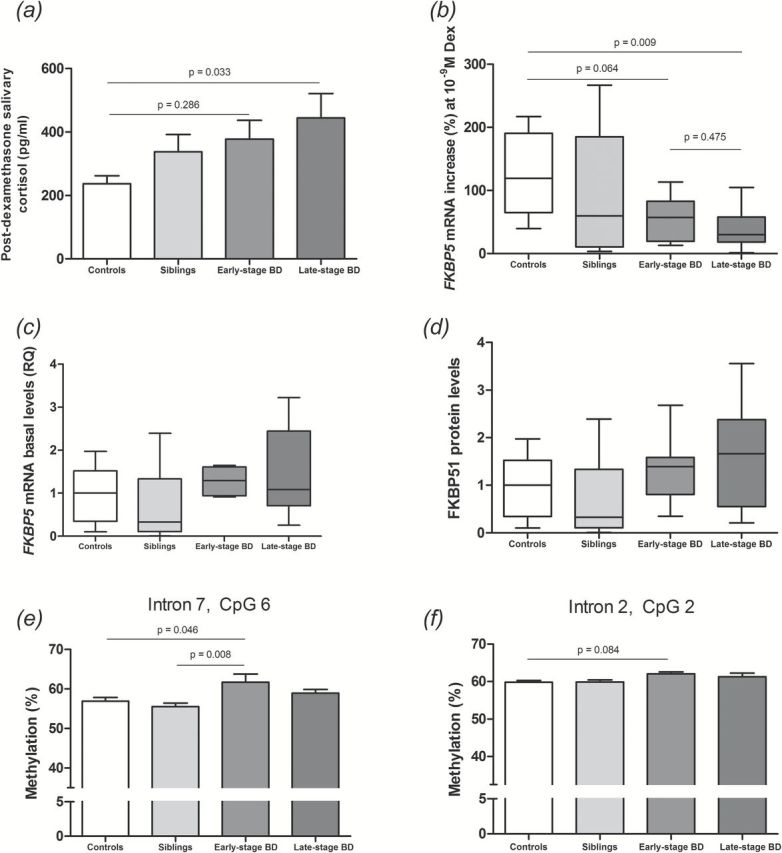
HPA axis activity and FKBP51 alterations in patients with early- vs. late-stage bipolar disorder. (A) Post-dexamethasone salivary cortisol levels; one-way ANOVA followed by Tukey’s post hoc test. (B) Ex vivo GR responsiveness assay; Kruskall-Wallis followed by Mann-Whitney test and Bonferroni correction. (C) Basal FKBP5 mRNA levels; Kruskall-Wallis followed by Mann-Whitney test and Bonferroni correction. (D) FKBP51 protein levels; Kruskall-Wallis followed by Mann-Whitney test and Bonferroni correction. (E–F) Methylation status of the FKBP5 gene at two specific CpG dinucleotides; one-way ANOVA followed by Tukey’s post hoc test. BD: bipolar disorder.
Discussion
The present study showed that patients with BD, particularly at a late stage of illness, presented increased salivary post-dexamethasone cortisol levels when compared to controls. Consistent with these findings, BD patients presented reduced ex vivo GR responsiveness and increased basal protein levels of FKBP51. Moreover, patients presented increased intronic methylation at the FKBP5 gene, which may be responsible for the reduced induction of FKBP5 mRNA expression by GR. In addition, siblings of patients with BD presented significantly lower FKBP51 protein levels than BD patients, even though no differences were found in FKBP5 basal mRNA levels. As far as we are aware, this is the first study to assess alterations in FKBP51 levels and epigenetic mechanisms in patients with BD and first-degree relatives. Taken together, our results provide evidence for a FKBP51-mediated and epigenetically-induced modulation of the stress axis in BD.
Increased post-dexamethasone cortisol levels reflect a diminished ability of dexamethasone to reduce cortisol levels overnight, possibly accounting for a dysfunctional HPA axis in patients with BD. This seems to be the case especially in late-stage BD patients, and the positive correlation found between post-dexamethasone cortisol levels and total number of episodes suggests that the dysfunction is related to the progression of BD. High post-dexamethasone cortisol levels had been previously associated with persistent deficits in executive performance in patients with mood disorders (Watson et al., 2006), as well as with depression severity (Osuch et al., 2001). As also reported for patients with major depressive disorder (MDD) (Binder et al., 2004), post-dexamethasone cortisol levels could be one of the mediators of increased recurrence of episodes in patients with late-stage BD: the number of previous mood episodes has already been shown to predict risk of recurrence in patients with BD (Kessing and Andersen, 2005). In this scenario, our findings point to a prognostic value of dexamethasone suppression test results and HPA axis activity in BD.
BD patients, siblings, and controls did not show significant differences in ACTH levels. Therefore, we hypothesized that the HPA axis dysfunction seen in BD patients could be mostly due to the role of GR in the cortisol-induced negative feedback. Taking the ex vivo induction of FKBP5 mRNA by dexamethasone as a measure of GR responsiveness, our results show that PBMCs from patients with BD—especially late-stage ones—induce significantly less FKBP5 mRNA expression after dexamethasone stimulation when compared with controls. Among the mechanisms possibly implicated in this GR resistance, we found that patients with BD showed increased basal FKBP51 protein levels. In fact, we found a positive correlation between basal FKBP51 protein expression and post-dexamethasone cortisol levels. Increased FKBP51 protein levels have also been associated with increased recurrence of depressive episodes in patients with MDD (Binder et al., 2004), which corroborates the current evidence of their role in increased recurrence of mood episodes and BD progression. Interestingly, these differences were not found in mRNA levels, suggesting that the increased protein levels found in BD patients were independent of mRNA expression levels. A discrepancy between mRNA and protein levels had been previously reported for a sample of patients with MDD (Binder et al., 2004), possibly explained by an enhanced protein translation or stability in patients.
Taking into account the methylation status of FKBP5, patients with BD presented increased methylation levels when compared to controls and siblings, especially in distal intronic GREs (introns 2 and 7). Distal intronic elements are known to significantly contribute to the transcriptional regulation of FKBP5 by glucocorticoids (Hubler and Scammell, 2004); therefore, increased methylation in these regions may reduce the inducibility of FKBP5 expression after dexamethasone stimulation. Of note, no correlation was found between DNA methylation and basal FKBP5 mRNA and protein levels, suggesting that methylation at those regions may be important for GR-mediated FKBP5 expression, but not for basal expression (for which transcription factors other than GR may be relevant). Interestingly, illness progression seems to correlate with reduced intronic methylation, based on negative correlations found between number of episodes and FKBP5 methylation status, and the fact that methylation was increased only in early-stage BD patients. This result may be secondary to chronic stress and hypercortisolemia, based on the fact that GR activation can induce alterations in Fkbp5 methylation, and chronic exposure to dexamethasone can lead to DNA demethylation in intronic CpGs in the FKBP5 gene (Klengel et al., 2013).
Our results also suggest that some of the parameters analyzed regarding HPA axis activity and its modulation may be altered in siblings of patients with BD. Our post-dexamethasone cortisol results indicate that the HPA axis activity of BD siblings lies somewhere between that of controls and patients with BD, as also observed for GR responsiveness and FAST scores. Moreover, BD siblings presented significantly lower FKBP51 protein levels than BD patients, even though no differences were found in FKBP5 basal mRNA levels. These results are consistent with the notion that non-affected siblings share biological underpinnings of BD, which could place them in a group of latent-stage BD (Berk et al., 2007; Kapczinski et al., 2009). However, our sample consisted purely of adult BD siblings with an average age of 51.1±13.2, whereas the average age of illness onset for BD is estimated to be much lower, around 18 years old (Price and Marzani-Nissen, 2012). In fact, we believe siblings of BD patients may be considered a resilient group of individuals, because they have remained nonsymptomatic even though they may present specific BD endophenotypes. In this sense, only a longitudinal follow-up of siblings will adequately assess if they are more likely to develop BD due to the subtle alterations in functioning, HPA axis regulation, and FKBP51-related mechanisms.
Some limitations of our study need to be mentioned. Our relatively small sample size reduced the statistical power of secondary analyses; it would be interesting to see the effects of a larger sample on our results, especially in comparisons that came close to significance. In this sense, the small sample size precluded meaningful results in matched-pair analyses between related BD patient-siblings pairs. Moreover, even though the groups did not differ with regard to the use of medications, it is possible that different medications may have had an impact on the parameters analyzed (type I error). Conversely, the strengths of this study are the assessment of multiple cortisol parameters, which enabled us to discuss HPA axis activity from different points of view. Moreover, the inclusion of both BD patients and siblings in the analysis allowed us to discuss the role of environmental effects on the disorder. Improvements to the present study include a larger sample, the recruitment of acutely ill patients, assessment of neuropsychological data, analysis of FKBP5 polymorphisms, and a longitudinal approach of the current analyses.
In summary, our findings draw attention to the role of HPA axis activity in the pathophysiology of BD from a novel perspective: namely, that of dysfunctional negative feedback of the HPA axis and impaired GR responsiveness due to increased FKBP51 levels and increased FKBP5 intronic methylation. These results add substantial support to previous evidence of altered cortisol levels in mood disorders and may be crucial to improving our understanding of BD pathophysiology, development, and progression. A clearer understanding of the environmental and physiological stimuli leading to these alterations is essential to the development of effective strategies aiming at preventing the development of BD in high-risk subjects. Our data is also consistent with the notion that a set of biological markers have more pronounced changes in late-stage BD (Grande et al., 2014; Pfaffenseller et al., 2014) and may help to explain the changes in resilience to stress as well as physical comorbidities found among these patients.
Statement of Interest
Dr Kapczinski has received grant/research support from AstraZeneca, Eli Lilly, Janssen-Cilag, Servier, CNPq, CAPES, NARSAD, and the Stanley Medical Research Institute; has been a member of the speaker’s boards of AstraZeneca, Eli Lilly, Janssen, and Servier; and has served as a consultant for Servier. Dr Kauer-Sant’Anna has received research grants from CNPq-INCT-TM, CNPq Universal, CAPES, SMRI, NARSAD, AstraZeneca, and Eli Lilly. All other authors declare no conflicts of interest.
Acknowledgments
This work was supported by Fundo de Incentivo à Pesquisa–Hospital de Clínicas de Porto Alegre (FIPE-HCPA) and the National Science and Technology Institute for Translational Medicine–Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq (573671/2008-7). The funding agencies did not have any role in study design, data collection and analysis, the decision to publish, or manuscript preparation.
Dr Fries was supported by a doctoral scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and a sandwich doctoral scholarship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the German Academic Exchange Service (DAAD).
References
- Berk M, Hallam KT, McGorry PD. (2007). The potential utility of a staging model as a course specifier: a bipolar disorder perspective. J Affect Disord 100:279–281. [DOI] [PubMed] [Google Scholar]
- Berk M, Brnabic A, Dodd S, Kelin K, Tohen M, Malhi GS, Berk L, Conus P, McGorry PD (2011) . Does stage of illness impact treatment response in bipolar disorder? Empirical treatment data and their implication for the staging model and early intervention. Bipolar Disord 13:87–98. [DOI] [PubMed] [Google Scholar]
- Bernstein DP, Stein JA, Newcomb MD, Walker E, Pogge D, Ahluvalia T, Stokes J, Handelsman L, Medrano M, Desmond D, Zule W. (2003). Development and validation of a brief screening version of the Childhood Trauma Questionnaire. Child Abuse Negl 27:169–190. [DOI] [PubMed] [Google Scholar]
- Binder EB. (2009). The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34(Suppl 1):S186–195. [DOI] [PubMed] [Google Scholar]
- Binder EB, et al. (2004). Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet 36:1319–1325. [DOI] [PubMed] [Google Scholar]
- Blair LJ, Nordhues BA, Hill SE, Scaglione KM, O’Leary JC, Fontaine SN, Breydo L, Zhang B, Li P, Wang L, Cotman C, Paulson HL, Muschol M, Uversky VN, Klengel T, Binder EB, Kayed R, Golde TE, Berchtold N, Dickey CA. (2013). Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J Clin Invest 123:4158–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacilhas AA, Magalhães PV, Ceresér KM, Walz JC, Weyne F, Rosa AR, Vieta E, Kapczinski F. (2009). Validity of a short functioning test (FAST) in Brazilian outpatients with bipolar disorder. Value Health 12:624–627. [DOI] [PubMed] [Google Scholar]
- Cervantes P, Gelber S, Kin FN, Nair VN, Schwartz G. (2001). Circadian secretion of cortisol in bipolar disorder. J Psychiatry Neurosci 26:411–416. [PMC free article] [PubMed] [Google Scholar]
- Chun E, Lee HS, Bang BR, Kim TW, Lee SH, Kim JH, Cho SH, Min KU, Kim YY, Park HW. (2011). Dexamethasone-induced FKBP51 expression in peripheral blood mononuclear cells could play a role in predicting the response of asthmatics to treatment with corticosteroids. J Clin Immunol 31:122–127. [DOI] [PubMed] [Google Scholar]
- Connor CM, Akbarian S. (2008). DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics 3:55–58. [DOI] [PubMed] [Google Scholar]
- Daban C, Vieta E, Mackin P, Young AH. (2005). Hypothalamic-pituitary-adrenal axis and bipolar disorder. Psychiatr Clin North Am 28:469–480. [DOI] [PubMed] [Google Scholar]
- Deshauer D, Grof E, Alda M, Grof P. (1999). Patterns of DST positivity in remitted affective disorders. Biol Psychiatry 45:1023–1029. [DOI] [PubMed] [Google Scholar]
- Ellenbogen MA, Hodgins S, Walker CD, Couture S, Adam S. (2006). Daytime cortisol and stress reactivity in the offspring of parents with bipolar disorder. Psychoneuroendocrinology 31:1164–1180. [DOI] [PubMed] [Google Scholar]
- Fries GR, Pfaffenseller B, Stertz L, Paz AV, Dargél AA, Kunz M, Kapczinski F. (2012). Staging and neuroprogression in bipolar disorder. Curr Psychiatry Rep 14:667–675. [DOI] [PubMed] [Google Scholar]
- Grande I, Magalhães PV, Chendo I, Stertz L, Panizutti B, Colpo GD, Rosa AR, Gama CS, Kapczinski F, Vieta E. (2014). Staging bipolar disorder: clinical, biochemical, and functional correlates. Acta Psychiatr Scand 129:437–444. [DOI] [PubMed] [Google Scholar]
- Grassi-Oliveira R, Stein LM, Pezzi JC. (2006). Translation and content validation of the Childhood Trauma Questionnaire into Portuguese language. Rev Saude Publica 40:249–255. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Lauer CJ, Schreiber W, Krieg JC. (1995). Altered hypothalamic-pituitary-adrenocortical regulation in healthy subjects at high familial risk for affective disorders. Neuroendocrinology 62:340–347. [DOI] [PubMed] [Google Scholar]
- Hubler TR, Scammell JG. (2004). Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones 9:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huzayyin AA, Andreazza AC, Turecki G, Cruceanu C, Rouleau GA, Alda M, Young LT. (2013). Decreased global methylation in patients with bipolar disorder who respond to lithium. Int J Neuropsychop 17:561–569. [DOI] [PubMed] [Google Scholar]
- Kapczinski F, Dias VV, Kauer-Sant’Anna M, Frey BN, Grassi-Oliveira R, Colom F, Berk M. (2009). Clinical implications of a staging model for bipolar disorders. Expert Rev Neurother 9:957–966. [DOI] [PubMed] [Google Scholar]
- Kessing LV, Andersen PK. (2005). Predictive effects of previous episodes on the risk of recurrence in depressive and bipolar disorders. Curr Psychiatry Rep 7:413–420. [DOI] [PubMed] [Google Scholar]
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TW, Mercer KB, Mayberg HS, Bradley B, Nemeroff CB, Holsboer F, Heim CM, Ressler KJ, Rein T, Binder EB. (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 16:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannie ZN, Harmer CJ, Cowen PJ. (2007). Increased waking salivary cortisol levels in young people at familial risk of depression. Am J Psych 164:617–621. [DOI] [PubMed] [Google Scholar]
- McGowan PO, Kato T. (2008). Epigenetics in mood disorders. Environ Health Prev Med 13:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A, Klengel T, Rubel J, Brückl T, Pfister H, Lucae S, Uhr M, Holsboer F, Binder EB. (2013). Genetic variation in FKBP5 associated with the extent of stress hormone dysregulation in major depression. Genes Brain Behav 12:289–296. [DOI] [PubMed] [Google Scholar]
- Merikangas KR, Jin R, He JP, Kessler RC, Lee S, Sampson NA, Viana MC, Andrade LH, Hu C, Karam EG, Ladea M, Medina-Mora ME, Ono Y, Posada-Villa J, Sagar R, Wells JE, Zarkov Z. (2011). Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch Gen Psychiatry 68:241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell S, Lauer CJ, Schreiber W, Huber J, Krieg JC, Holsboer F. (1998). Hormonal response pattern in the combined DEX-CRH test is stable over time in subjects at high familial risk for affective disorders. Neuropsychopharmacology 18:253–262. [DOI] [PubMed] [Google Scholar]
- Osby U, Brandt L, Correia N, Ekbom A, Sparén P. (2001). Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry 58:844–850. [DOI] [PubMed] [Google Scholar]
- Osuch EA, Cora-Locatelli G, Frye MA, Huggins T, Kimbrell TA, Ketter TA, Callahan AM, Post RM. (2001). Post-dexamethasone cortisol correlates with severity of depression before and during carbamazepine treatment in women but not men. Acta Psychiatr Scand 104:397–401. [DOI] [PubMed] [Google Scholar]
- Petronis A. (2003). Epigenetics and bipolar disorder: new opportunities and challenges. Am J Med Genet C Semin Med Genet 123C:65–75. [DOI] [PubMed] [Google Scholar]
- Pfaffenseller B, Wollenhaupt-Aguiar B, Fries GR, Colpo GD, Burque RK, Bristot G, Ferrari P, Ceresér KMM, Rosa AR, Klamt F, Kapczinski F.(2014) Impaired endoplasmic reticulum stress response in bipolar disorder: cellular evidence of illness progression. Int J Neuropsychop 17:1453–1463. [DOI] [PubMed] [Google Scholar]
- Price AL, Marzani-Nissen GR. (2012). Bipolar disorders: a review. Am Fam Phys 85:483–493. [PubMed] [Google Scholar]
- Rosa AR, Sánchez-Moreno J, Martínez-Aran A, Salamero M, Torrent C, Reinares M, Comes M, Colom F, Van Riel W, Ayuso-Mateos JL, Kapczinski F, Vieta E. (2007). Validity and reliability of the Functioning Assessment Short Test (FAST) in bipolar disorder. Clin Pract Epidemiol Ment Health 3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa AR, González-Ortega I, González-Pinto A, Echeburúa E, Comes M, Martínez-Àran A, Ugarte A, Fernández M, Vieta E (2012) One-year psychosocial functioning in patients in the early vs. late stage of bipolar disorder. Acta Psychiatr Scand 125:335–341. [DOI] [PubMed] [Google Scholar]
- Rosa AR, Magalhães PVS, Czepielewski L, Sulzbach MV, Goi PD, Vieta E, Gama CC, Kapczinski F.(2014) Clinical staging in bipolar disorder: focus on cognition and functioning. J Clin Psychiatry 75:e450–e456. [DOI] [PubMed] [Google Scholar]
- Scammell JG, Denny WB, Valentine DL, Smith DF. (2001). Overexpression of the FK506-binding immunophilin FKBP51 is the common cause of glucocorticoid resistance in three New World primates. Gen Comp Endocrinol 124:152–165. [DOI] [PubMed] [Google Scholar]
- Vermeer H, Hendriks-Stegeman BI, van der Burg B, van Buul-Offers SC, Jansen M. (2003). Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: a potential marker for glucocorticoid sensitivity, potency, and bioavailability. J Clin Endocrinol Metab 88:277–284. [DOI] [PubMed] [Google Scholar]
- Vermeer H, Hendriks-Stegeman BI, van Suylekom D, Rijkers GT, van Buul-Offers SC, Jansen M. (2004). An in vitro bioassay to determine individual sensitivity to glucocorticoids: induction of FKBP51 mRNA in peripheral blood mononuclear cells. Mol Cell Endocrinol 218:49–55. [DOI] [PubMed] [Google Scholar]
- Vieta E, Langosch JM, Figueira ML, Souery D, Blasco-Colmenares E, Medina E, Moreno-Manzanaro M, Gonzalez MA, Bellivier F. (2013). Clinical management and burden of bipolar disorder: results from a multinational longitudinal study (WAVE-bd). Int J Neuropsychop 16:1719–1732. [DOI] [PubMed] [Google Scholar]
- Watson S, Gallagher P, Ritchie JC, Ferrier IN, Young AH. (2004). Hypothalamic-pituitary-adrenal axis function in patients with bipolar disorder. Br J Psychiatry 184:496–502. [DOI] [PubMed] [Google Scholar]
- Watson S, Thompson JM, Ritchie JC, Nicol Ferrier I, Young AH. (2006). Neuropsychological impairment in bipolar disorder: the relationship with glucocorticoid receptor function. Bipolar Disord 8:85–90. [DOI] [PubMed] [Google Scholar]
- Willour VL, Chen H, Toolan J, Belmonte P, Cutler DJ, Goes FS, Zandi PP, Lee RS, MacKinnon DF, Mondimore FM, Schweizer B, DePaulo JR, Gershon ES, McMahon FJ, Potash JB, Group BDP, Consortium NGIBD. (2009). Family-based association of FKBP5 in bipolar disorder. Mol Psychiatry 14:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wochnik GM, Rüegg J, Abel GA, Schmidt U, Holsboer F, Rein T. (2005). FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem 280:4609–4616. [DOI] [PubMed] [Google Scholar]
- Yang X, Ewald ER, Huo Y, Tamashiro KL, Salvatori R, Sawa A, Wand GS, Lee RS. (2012). Glucocorticoid-induced loss of DNA methylation in non-neuronal cells and potential involvement of DNMT1 in epigenetic regulation of Fkbp5. Biochem Biophys Res Commun 420:570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]