

Abstract
Background:
Dysregulation in the prefrontal cortex-nucleus accumbens pathway has been implicated in cocaine addiction. We have previously demonstrated that one intra-dorsomedial prefrontal cortex brain-derived neurotrophic factor (BDNF) infusion immediately following the last cocaine self-administration session caused a long-lasting inhibition of cocaine-seeking and normalized the cocaine-induced disturbance of glutamate transmission in the nucleus accumbens after extinction and a cocaine prime. However, the molecular mechanism mediating the brain-derived neurotrophic factor effect on cocaine-induced alterations in extracellular glutamate levels is unknown.
Methods:
In the present study, we determined the effects of brain-derived neurotrophic factor on cocaine-induced changes in the phosphorylation of synapsin (p-synapsin), a family of presynaptic proteins that mediate synaptic vesicle mobilization, in the nucleus accumbens during early withdrawal.
Results:
Two hours after cocaine self-administration, p-synapsin Ser9 and p-synapsin Ser62/67, but not p-synapsin Ser603, were increased in the nucleus accumbens. At 22 hours, only p-synapsin Ser9 was still elevated. Elevations at both time points were attenuated by an intra-dorsomedial prefrontal cortex brain-derived neurotrophic factor infusion immediately after the end of cocaine self-administration. Brain-derived neurotrophic factor also reduced cocaine self-administration withdrawal-induced phosphorylation of the protein phosphatase 2A C-subunit, suggesting that brain-derived neurotrophic factor disinhibits protein phosphatase 2A C-subunit, consistent with p-synapsin Ser9 dephosphorylation. Further, co-immunoprecipitation demonstrated that protein phosphatase 2A C-subunit and synapsin are associated in a protein-protein complex that was reduced after 2 hours of withdrawal from cocaine self-administration and reversed by brain-derived neurotrophic factor.
Conclusions:
Taken together, these findings demonstrate that brain-derived neurotrophic factor normalizes the cocaine self-administration–induced elevation of p-synapsin in nucleus accumbens that may underlie a disturbance in the probability of neurotransmitter release or represent a compensatory neuroadaptation in response to the hypofunction within the prefrontal cortex-nucleus accumbens pathway during cocaine withdrawal.
Keywords: cocaine self-administration, immunoblotting, nucleus accumbens, phosphatase, synapsin
Introduction
Susceptibility to drug relapse and other addictive behaviors is thought to depend on long-term neuroadaptations in the meso-cortico–basal ganglia circuitry (Koob et al., 1997; Wang and McGinty, 1997; White and Kalivas, 2001). Within this network, attention has focused on the prefrontal cortex (PFC) projection to the nucleus accumbens (NAc), because disruption of glutamate neurotransmission in this pathway by repeated cocaine exposure triggers relapse to drug-seeking (McFarland et al., 2003). Brain-derived neurotrophic factor (BDNF) is expressed in PFC-NAc neurons wherein it regulates presynaptic glutamate release. We have reported that a single BDNF infusion into dorsomedial (dm) PFC immediately following the final session of cocaine self-administration (SA) restores normal extracellular levels of glutamate in the NAc after cocaine SA and suppresses persistent cocaine-seeking (Berglind et al., 2007, 2009). In searching for potential mechanisms that underlie the ability of BDNF in the PFC to normalize glutamate transmission in the NAc, our attention became focused on synapsins for the following reasons. First, synapsins are members of a family of BDNF-regulated presynaptic proteins that control neurotransmitter exocytosis by reversibly tethering synaptic vesicles to each other and to the actin cytoskeleton in a phosphorylation-dependent manner (Benfenati et al., 1989: Jovanovic et al., 1996). Second, after stimulation, the facilitatory effect of BDNF on glutamate release is attenuated in mice lacking synapsins I and II (Jovanovic et al., 2000). Third, BDNF increases the number of docked synaptic vesicles at the active zone of excitatory synapses, facilitating release probability (Tyler and Pozzo-Miller, 2001).
Synapsin I(a,b), II(a,b), and III regulate trafficking of synaptic vesicles between the reserve pool and the active pool in terminals that release different transmitters (Gitler et al., 2004a). Synapsin I has been associated with regulating synaptic vesicles trafficking in GABAergic neurons (Baldelli et al., 2007), whereas synapsin IIa has been associated with synaptic vesicles trafficking in glutamatergic terminals (Gitler et al., 2004b, 2008). Synapsin III has been associated with synaptic vesicles trafficking in dopamine terminals in the striatum (Kile et al., 2010). Further, in mice lacking all 3 synapsins, the ability to mobilize the reserve pool of synaptic vesicles and release dopamine from striatal terminals was diminished following a cocaine challenge (Venton et al., 2006). In synapsin I-II mutant mice, reductions in vGlut1, vGlut2, and glutamate reuptake were observed (Bogen et al., 2006, 2009), and in vGlut knockout mice, there was a decrease of synapsin I expression and fewer synaptic vesicles in several brain regions (Fremeau et al., 2004). Mice with decreased synapsin I and II expression are prone to epileptiform seizures due to depletion of the reserve synaptic vesicle pool and enhanced docking in the readily releasable pool of excitatory neurons (Rosahl et al., 1995). For these reasons and because of similar molecular weights of synapsins Ia, Ib, and IIa that make it difficult to discriminate them consistently by immunoblotting, we focused attention only on cocaine-induced changes in phosphorylation of synapsin I and IIa, collectively termed here p-synapsin.
Unphosphorylated synapsins bind to and tether small synaptic vesicles to the actin cytoskeleton in the preterminal reserve pool, whereas phosphorylation of synapsins at specific sites causes release of synaptic vesicles, enabling them to traffic from the reserve pool to the readily releasable pool, increasing the probability of action potential-dependent neurotransmitter release. For example, high-frequency stimulation that evokes calcium-dependent glutamate release is associated with protein kinase A/calcium/calmodulin-dependent kinase (CAMK-mediated) phosphorylation of Ser9 and CaMKII-mediated phosphorylation of Ser603 (Chi et al., 2001; Hilfiker et al., 1999, 2005). At the termination of exocytotic release, protein phosphatase 2A (PP2A)-mediated dephosphorylation of CAMK sites restores synapsin binding to synaptic vesicles and tethering to the cytoskeleton in the reserve pool. In contrast, there is an inverse relationship between Ca++ influx and synapsin phosphorylation at Ser62/67 by extracellular signal-regulated kinase mitogen-activated protein (ERK MAP) kinase. High-frequency stimulation promotes transient ERK-mediated phosphorylation of synapsin at Ser62/67 sites that inhibits posttetanic potentiation and is terminated by PP2B (calcineurin)-dependent dephosphorylation (Jovanovic et al., 2001; Vara et al., 2009). However, low-frequency stimulation can promote phosphorylation of synapsin Ser62/67 (and Ser 9), facilitating dissociation of synaptic vesicles from the actin cytoskeleton (Chi et al., 2003). In addition, BDNF stimulates ERK/MAPK-mediated synapsin I Ser62/67 phosphorylation that augments glutamate release in cortical synaptosomes in a calcium-independent manner (Jovanovic et al., 2000).
Based on this background, we first evaluated whether cocaine, in the presence or absence of intra-dmPFC BDNF infusion at the end of SA, affects the phosphorylation of synapsins at the 3 major phosphorylation sites during early withdrawal (2 or 22 hours after the end of the last session of cocaine SA). These are the time points at which a normalizing effect of BDNF on cocaine SA-induced changes in phospho-ERK has been detected in both PFC and NAc (Berglind et al., 2007; Whitfield et al., 2011). Further, after determining that phosphorylation was altered by cocaine SA, we explored the possible association of PP2A with synapsin after cocaine SA and/or intra-dmPFC BDNF infusion.
Methods
Animals
Adult male Sprague-Dawley rats (Charles River, MA) weighing 301 to 325g at the beginning of each experiment were housed individually in a temperature- and humidity-controlled vivarium on a reversed 12-h–light/–dark cycle (lights off at 7:00 am). Animals were given 20 to 25g of standard rat chow (Harlan) with ad libitum access to water daily. Experimental procedures were performed during the dark cycle. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina and complied with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication no. 80-23, revised 1996). All efforts were made to minimize animal suffering and reduce the number of animals used.
Surgery
Animals were given at least a 5-day acclimation period. On the day of surgery, rats were anesthetized with a mixture of ketamine (66mg/kg, intraperitoneally; Vedco Inc) and xylazine (1.33mg/kg, intraperitoneally; Lloyd Laboratories), respectively, followed by equithesin (0.5mL/kg) and ketorolac (2mg/kg; Sigma-Aldrich). A silastic catheter was implanted into the right jugular vein with the distal end threaded subcutaneously and exiting on the back to connect to an infusion harness (Instech Solomon) for intravenous infusions. Immediately after catheterization, rats were implanted with 28-gauge bilateral stainless-steel guide cannulae (Plastics One) aimed 1mm above the dmPFC infusion site. Coordinates used in the stereotaxic procedures were +3.0mm anterior-posterior (AP), ±0.6 mm mediolateral, and −1.6mm dorsoventral from the dural surface (Paxinos and Watson, 2005). Guide cannulae were secured to the skull with dental cement and steel screws. Two 10-mm stylets (Plastics One) were placed into the guide cannulae following surgery to prevent blockage throughout the experiment. After surgeries, daily intravenous infusions of 0.1mL of the antibiotics, Cefazolin (10mg/mL; Schein Pharmaceuticals) or Timentin (2.4mg/mL), and 0.1mL of heparinized saline (100U/mL; Elkins-Sinn) were administered during surgical recovery and throughout SA to maintain catheter patency. Catheter patency was verified as needed with an intravenous infusion of 0.1mL of methohexital sodium (10mg/mL; Eli Lilly), which produces a rapid loss of muscle tone.
Cocaine SA and Intracranial Infusion
After 5 days of recovery, rats were food restricted to 90% of their body weight and subjected to 2-hour daily cocaine or yoked-saline SA sessions under a FR1 reinforcement schedule in standard operant conditioning chambers (MED Associates) for 14 days after they reached 10 infusions per session for 3 consecutive days. Each chamber was equipped with 2 retractable levers (7cm above floor) and a circular stimulus light above each lever. Tygon infusion lines within the spring leash were mounted to a balanced metal arm and attached to a liquid swivel (Instech Solomon). Before each session, the catheter was secured to the infusion line. Presses on the active lever resulted in a 2-second cocaine hydrochloride (0.2mg/50 µL/infusion; NIDA) infusion via a computer-controlled pump. Cocaine infusions were paired with a 5-second illumination of a white stimulus light above the active lever and a tone (2kHz, 15 dB above ambient noise) followed by a 20-second timeout period, during which additional responses on the active lever resulted in no programmed consequences. Responses on the inactive lever were recorded but had no consequence. For each session, rats had to self-administer at least 10 infusions to fulfill the maintenance criterion. Immediately after the end of the last SA session, BDNF (0.75 µg/side; R&D Systems) or its vehicle, phosphate buffered saline (pH=7.4), was infused into the dmPFC. Each bilateral infusion was administered through an injector (33 gauge; Plastics One) inserted into the guide cannula so that 1mm of the injector length extended beyond the guide cannula. After a 2-minute infusion (0.25 μL/min), the injectors remained in the cannulae for 1 minute to allow complete diffusion. Rats were then directly placed into their home cages.
Immunoblotting
Two or 22 hours after infusions, rats were rapidly decapitated and brains were removed and placed in a brain matrix (Braintree Scientific). Two-mm coronal slices at AP 2.7 to 0.7 rostral to Bregma were cut, and the NAc was bilaterally punched and frozen at −80oC. Coronal slices were also cut through the PFC between AP 2.7 and 3.7mm rostral to Bregma to visually identify cannula placements in the dmPFC (all placements were verified to be in anterior cingulate/prelimbic cortex) before freezing and immunoblotting of other proteins (Sun et al., 2013). Protein from NAc samples was extracted by sonication in ice-cold RIPA lysis buffer and centrifuged at 10000 × g for 20 minutes at 4°C. The resulting supernatants were collected and subjected to protein analysis with the Micro-Bicinchoninic Acid assay kit (Pierce, Rockford, IL). Equal amounts of protein extracts were run on SDS-PAGE gels and then transferred to PVDF membranes. Membranes were then blocked with 5% nonfat dry milk in Tris-buffered saline with Tween-20 (TBST; pH=7.6) for 1 hour at room temperature and incubated with primary antibody against p-synapsin Ser9 recognizing all synapsin isoforms at the Ser9 site (1:2000; Cell Signaling), p-synapsin I Ser62/67 (1:1000), p-synapsin I Ser603 (1:3000; Millipore), or p-PP2Ac Tyr307 (1:5000; Abcam) overnight at 4°C. After 3 washes with TBST, membranes were incubated with their appropriate secondary antibodies for 1 hour at room temperature followed by 3 more washes with TBST. Antibody binding was detected by using an enhanced chemiluminescence kit (ECL Plus; GE Healthcare Bio-Sciences). Membranes were then stripped and reprobed for total protein by antisera against synapsin (1:2000; Cell Signaling) or PP2Ac (1:5000; Millipore) for quantitation. Equal loading proteins were further confirmed by probing with anti-calnexin antiserum (1:10000; Enzo Life Sciences). All Western-blot analyses were performed a minimum of twice and the data were averaged. The integrated density value of each phosphoprotein (phospho-synapsin Ia, Ib, and IIa Ser9 measured together) and total protein band were measured using ImageJ software (NIH).
Co-Immunoprecipitation
The procedure of co-immunoprecipitation was previously described (Sun et al., 2009). Briefly, equal amounts of protein extracts (250 µg) were pooled from 2 to 3 rats per group and incubated in RIPA lysis buffer containing antibody against total synapin (9 µg; Cell Signaling) or PP2Ac (8 µg; Millipore) overnight, followed by the addition of protein A/G agarose beads (30 µL; Santa Cruz Technologies) for 4 hours at 4°C. Beads were then washed 4 times in the RIPA lysis buffer. After final centrifugation, Lammeli buffer containing 1% β-mercaptoethanol was added, boiled for 5 minutes, loaded into gels, and analyzed by immunoblotting procedures as described above. Two percent of protein extract was used as input to normalize the protein band intensity.
Statistical Analysis
Two standard deviations from the mean were used to exclude potential outliers of each group. Independent t tests were used for the SA behavioral measurements. For biochemical data, 2-way analysis of variances (ANOVAs) [treatment (saline vs cocaine) × infusion (vehicle vs BDNF)] were used followed by Student–Newman–Keuls (SNK) multiple comparison tests when a significant interaction or main effect was found after ANOVA. Because of the small sample size (3–5 samples/group), Fisher’s Least Significant Difference test was used to analyze the co-immunoprecipitation data after ANOVA. The criterion for statistically significant differences was set at P<.05.
Results
During the last 3 SA sessions, cocaine self-administering rats exhibited significantly lower inactive lever pressing and higher active lever pressing than yoked-saline controls, indicating the acquisition of cocaine-associated reinforcing effects (Table 1; 2 hours: inactive lever: t29=−3.31, P<.01; active lever: t29=10.53, P<.001; 22 hours: inactive lever: t23=−4.10, P<.001; active lever: t23=7.66, P<.001). Before receiving intra-dmPFC vehicle or BDNF infusion, rats with the cocaine SA history pressed a similar amount on the active levers and received a similar number of cocaine infusions.
Table 1.
Average Number of Lever Presses and Cocaine Infusions during the Last 3 Sessions of SA
| Group | Treatment | Inactive Lever | Active Lever | Cocaine Infusions |
|---|---|---|---|---|
| 2 h | Yoked-saline | 4.19±1.40 | 5.50±1.23 | — |
| Cocaine SA | 0.40±0.25* | 35.31±2.10* | 27.49±1.35 | |
| 22 h | Yoked-saline | 3.81±0.88 | 6.83±1.45 | — |
| Cocaine SA | 0.26±0.18* | 35.13±3.69* | 28.92±3.69 |
Abbreviation: SA, self-administration.
Data represent mean ± SEM of each group and sacrificed 2 or 22h after the end of the last SA session (2 h: yoked-saline [N=12], cocaine SA [N=19]; 22 h: yoked-saline [N=12], cocaine SA [N=13]). * P <.05 vs respective yoked-saline controls, independent t test.
Two hours after intra-dmPFC infusions, there was a significant treatment × infusion interaction for p-synapsin Ser9 in the NAc (F 1,24=5.20, P<.05). A SNK test revealed greater p-synapsin Ser9 protein expression in the cocaine-/vehicle-treated group than in the saline-/vehicle- and cocaine-/BDNF-treated groups (P<.05) (Figure 1a). Similarly, for the protein expression of p-synapsin Ser62/67, there was a trend toward a main effect of treatment (F 1,27=3.68, P=.07) with a significant treatment × infusion interaction (F 1,27=7.16, P<.05). The cocaine-/vehicle-treated group alone had higher p-synapsin Ser62/67 in the NAc than in other groups (P<.05) (Figure 1b). Neither cocaine nor BDNF altered p-synapsin Ser603 protein expression. Twenty-two hours after infusion, there was also a trend toward a main effect of infusion and a significant treatment × infusion interaction for p-synapsin Ser9 (F 1,21=3.49, P=.08 and F 1,21=5.01, P<.05, respectively) (Figure 2a). The SNK multiple comparison revealed that the protein expression of p-synapsin Ser9 was significantly greater in the cocaine/vehicle group than in other groups (P<.05). The protein levels of p-synapsin Ser62/67 and Ser603 were not changed 22 hours after infusion (Figure 2b and c). Further, there was no alteration in total synapsin expression at either time point examined.
Figure 1.
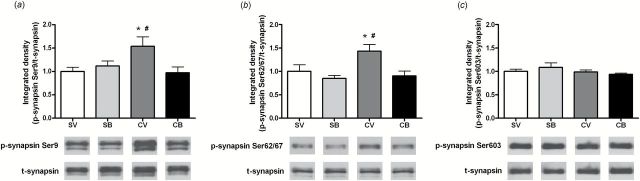
Effects of intra-dorsomedial (dm) prefrontal cortex (PFC) brain-derived neurotrophic factor (BDNF) infusion on p-synapsin protein expression in the nucleus accumbens (NAc) after 2 hours of withdrawal from cocaine self-administration (SA). (a) Top: Quantitative analysis of p-synapsin Ser9 protein levels in the NAc. Bottom: Representative immunoblots for p-synapsin Ser9 and t-synapsin from each group (SV: N=6; SB: N=6; CV: N=8; CB: N=8). (b) Top: Quantitative analysis of p-synapsin Ser62/67 protein levels in the NAc. Bottom: Representative subsections of an immunoblot for p-synapsin Ser62/67 and t-synapsin from each group (SV: N=6; SB: N=6; CV: N=9; CB: N=10). (c) Top: Quantitative analysis of p-synapsin Ser603 protein levels in the NAc. Bottom: Representative immunoblots illustrating bands of p-synapsin Ser603 and t-synapsin from each group (SV: N=6; SB: N=6; CV: N=9; CB: N=10). Bar graphs show the mean±SEM of each group. * P<.05 vs SV; # P<.05 vs CB, 2-way analysis of variance (ANOVA) followed by Student–Newman–Keuls (SNK) multiple comparisons. B, BDNF; C, cocaine; S, saline; V, vehicle.
Figure 2.
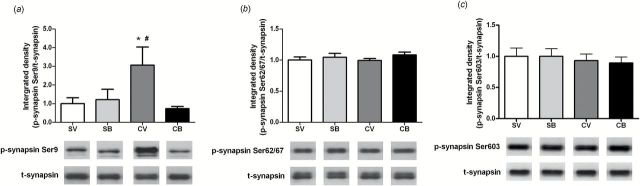
Effects of intra-dorsomedial (dm) prefrontal cortex (PFC) brain-derived neurotrophic factor (BDNF) infusion on p-synapsin protein expression in the nucleus accumbens (NAc) after 22 hours of withdrawal from cocaine self-administration (SA). (a) Top: Quantitative analysis of p-synapsin Ser9 protein levels in the NAc. Bottom: Representative immunoblots for p-synapsin Ser9 and t-synapsin from each group (SV: N=6; SB: N=6; CV: N=6; CB: N=7). (b) Top: Quantitative analysis of p-synapsin Ser62/67 protein levels in the NAc. Bottom: Representative subsections of an immunoblot for p-synapsin Ser62/67 and t-synapsin from each group (SV: N=6; SB: N=6; CV: N=6; CB: N=7). (c) Top: Quantitative analysis of p-synapsin Ser603 protein levels in the NAc. Bottom: Representative immunoblots illustrating bands of p-synapsin Ser603 and t-synapsin from each group (SV: N=6; SB: N=6; CV: N=6; CB: N=7). Bar graphs show the mean±SEM of each group. * P<.05 vs SV; # P<.05 vs CB, 2-way analysis of variance (ANOVA) followed by Student–Newman–Keuls (SNK) multiple comparison. B, BDNF; C, cocaine; S, saline; V, vehicle.
PP2Ac is the protein phosphatase that dephosphorylates p-synapsin Ser9 (Jovanovic et al., 2001). We further evaluated p-PP2Ac Tyr307 (associated with inactivation of PP2A) protein levels due to the augmentation of p-synapsin Ser9 in the NAc 2 and 22 hours after the end of cocaine SA. Two hours after infusion, 2-way ANOVA indicated that there was a significant main effect of treatment (F 1,26=10.68, P<.01) and a treatment × infusion interaction (F 1,26=10.03, P<.01). SNK multiple comparisons showed a higher p-PP2Ac Tyr307 protein induction in the cocaine/vehicle group than in other groups (P<.01) (Figure 3a). Similarly, 22 hours after infusion, there was a significant main effect of treatment t (F 1,26=9.60, P<.05) with a treatment × infusion interaction (F 1,26=5.28, P<.05). Further analysis indicated that the protein expression of p-PP2Ac in the cocaine-/vehicle-treated group was significantly increased compared with other groups (P<.01) (Figure 3b). The total PP2Ac protein levels were not altered at either time point examined.
Figure 3.
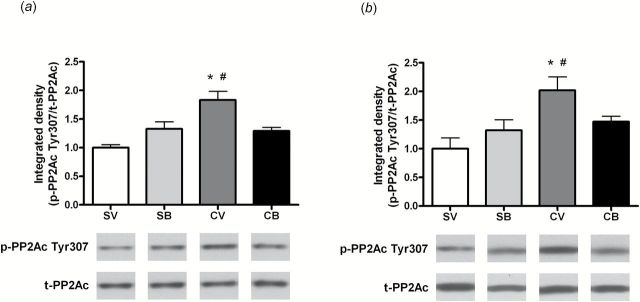
Intra-dorsomedial (dm) prefrontal cortex (PFC) brain-derived neurotrophic factor (BDNF) infusion suppresses p-PP2Ac Tyr307 protein induction in the nucleus accumbens (NAc) after cocaine self-administration (SA). (a) Top: Quantitative analysis of p-PP2Ac Tyr307 protein levels in the NAc 2 hours after the end of cocaine SA. Bottom: Representative immunoblots for p-PP2Ac Tyr307 and t-PP2Ac from each group (SV: N=6; SB: N=6; CV: N=8; CB: N=10). (b) Top: Statistical analysis of p-PP2Ac Tyr307 protein levels in the NAc after 22 hours of withdrawal from cocaine SA. Bottom: Representative immunoblots illustrating bands of p-PP2Ac Tyr307 and t-PP2Ac from each group (SV: N=6; SB: N=6; CV: N=7; CB: N=6). Histograms show the mean±SEM of each group. * P<.05 vs SV; # P<.05 vs CB, 2-way analysis of variance (ANOVA) followed by Student–Newman–Keuls (SNK) multiple comparison. B, BDNF; C, cocaine; S, saline; V, vehicle.
Given that the modulatory effect of PP2Ac on synapsin may be through a protein-protein interaction, we further evaluated the possibility of an alteration in a PP2Ac:synapsin association after cocaine SA with or without BDNF infusion. First, an interaction between PP2Ac and synapsin was detected in the NAc of drug naïve rats (Figure 4a left). This protein-protein interaction was further confirmed using a reciprocal co-immunoprecipitation approach: immunoprecipitation using PP2Ac antibody followed by probing with the synapsin antibody (Figure 4a right). In the 2-hour time point samples, a 2-way ANOVA revealed a significant treatment × infusion interaction (F 1,11=5.23, P<.05). Multiple comparison tests indicated that the PP2Ac:synapsin protein complex was significantly lower in the cocaine-/vehicle-treated group than in saline-/vehicle- or cocaine-/BDNF-treated groups (Fisher’s Least Significant Difference test; p<.05) (Figure 4b). No alteration of total PP2Ac as input was detected.
Figure 4.
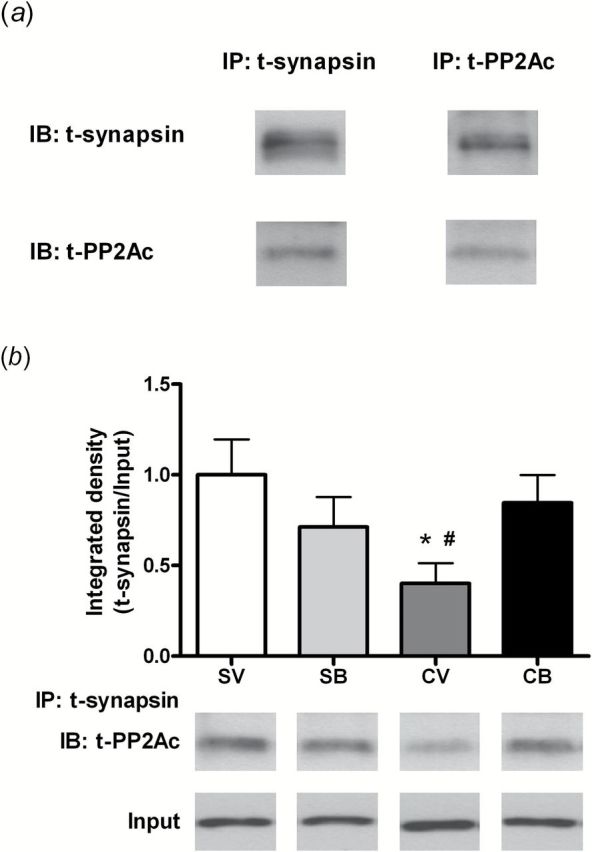
Intra-dorsomedial (dm) prefrontal cortex (PFC) brain-derived neurotrophic factor (BDNF) infusion normalizes the reduction of PP2A:synapsin I binding in the nucleus accumbens (NAc) after cocaine self-administration (SA). (a) A control experiment shows images of specificity for PP2A and synapsin binding in the NAc from drug naïve rats. Left: Representative images of NAc homogenates immunoprecipitated by t-synapsin followed by probing with t-synapsin and t-PP2Ac antibodies. Right: Immunoblots illustrating the bands of t-synapsin I and t-PP2Ac after t-PP2Ac antibody immunoprecipitation. (b) Top: Quantitative analysis of PP2Ac:synapsin protein binding levels in the NAc 2 hours after the end of cocaine SA. Bottom: Representative bands of an immunoblot of t-PP2Ac and its input protein levels after immunoprecipitation by t-synapsin antibody from each group (SV: N=3; SB: N=3; CV: N=4; CB: N=5). Histograms show the mean±SEM of each group. * P<.05 vs SV; # P<.05 vs CB, 2-way analysis of variance (ANOVA) followed by Fisher’s Least Significant Difference (LSD) multiple comparison. B, BDNF; C, cocaine; CNXN, calnexin; IB, immunoblotting; IP, immunoprecipitation; S, saline; V, vehicle.
Discussion
In the present study, we demonstrated that p-synapsin Ser9 was increased in the NAc after 2 and 22 hours of withdrawal from cocaine SA. Elevation of p-synapsin Ser62/67 was also observed in cocaine-treated rats, but only at the 2-hour time point. Phosphorylation of PP2Ac was significantly elevated (indicating inactivation) after cocaine SA at the 2 time points examined. This elevation and that of p-synapsin at both the Ser9 and Ser62/67 sites was normalized by intra-dmPFC BDNF infusion. Further, co-immunoprecipitation revealed a protein-protein interaction between synapsin and PP2Ac in presynaptic terminals that cocaine reduced and intra-dmPFC BDNF infusion reversed.
These data indicate that during early withdrawal from cocaine SA, p-synapsin Ser9 is elevated due to a dissociation of PP2Ac from synapsin and a decrease in PP2A activity in presynaptic terminals in the NAc. Intra-dmPFC BDNF reactivates PP2A, resulting in enhanced binding of PP2A to, and dephosphorylation of, p-synapsin Ser9 in NAc terminals. Although these terminals likely originate from PFC neurons, indirect effects via the amygdala, for example, cannot be ruled out. Since PP2A also mediates the deactivation of CaMKI (DeRemer et al., 1992) and CAMKI is localized in the neuropil (Picciotto et al., 1995), future studies can focus on whether elevation of p-synapsin Ser9 is associated with a reduction of PP2A’s inhibitory effect on CaMKI in PFC-NAc terminals. Elevated p-synapsin Ser9 in NAc immediately after the last cocaine SA session using a different regimen (4-hour daily sessions) has been previously detected (Edwards et al., 2007), suggesting that changes in p-synapsin Ser9 expression are common time-dependent monitors of presynaptic plasticity in NAc in response to cocaine SA.
In contrast to the elevated p-synapsin Ser9 after 2 and 22 hours of withdrawal from cocaine, an increase in ERK-mediated p-synapsin Ser62/67 was observed only at the 2-hour time point. The phosphorylation of ERK-mediated p-synapsin in response to low activity at 2 hours may be reversed as neuronal activity is restored and more Ca2+ influx triggers PP2B activation with time (22 hours). A similar upregulation of p-synapsin Ser9 and downregulation of ERK-activated p-synapsin Ser62/67 has been documented after 4-aminopyridine induced Ca2+ influx (Jovanovic et al., 2001). Moreover, a decrease in phosphorylation of ERK and CREB in dmPFC was observed at 2 hours, but not after 22 hours, of abstinence from cocaine SA (Whitfield et al., 2011), suggesting a gradual recovery of neuronal activity in the PFC as the phase of withdrawal changes. Therefore, the dynamic alteration of p-synapsin 62/67 in the NAc may represent a sensitive biomarker of PFC neuronal activity changes. Because CaMK- and ERK-mediated phosphorylation of synapsin play different roles in modulating the tethering of synaptic vehicles to the actin cytoskeleton (Huttner et al., 1983; Sihra et al., 1989; Jovanovic et al., 1996; Hosaka et al., 1999), differential regulation of their phosphorylation states in the NAc is likely to have a functionally significant impact on synaptic transmission after cocaine SA ends.
The differential phosphorylation of synapsin sites by cocaine in this study may reflect compensatory responses and be related to other findings that basal cortical hypoactivity and sensitized responses to evoked activity occur during early withdrawal from cocaine. First, dephosphorylation of ERK and CREB in the dmPFC (Whitfield et al., 2011) is accompanied by the attenuated phosphorylation of striatal-enriched protein tyrosine phosphatase and its targets, GluN1/2B, in the dmPFC after 2 hours of abstinence from cocaine SA (Sun et al., 2013). Increased phosphatase activity and decreased glutamatergic transmission may indicate synaptic depression in the PFC as a critical part of the withdrawal cascade. Second, basal extracellular glutamate levels in the NAc are similarly reduced after 1, 21, or 60 days of cocaine SA abstinence (Lutgen et al., 2014; Wydra et al., 2013), but a specific enhancement of evoked release probability at PFC-NAc synapses occurs after 1 day of abstinence (Suska et al., 2013) and excessive evoked glutamate release occurs in response to cocaine prime-induced reinstatement after extinction (McFarland et al., 2003). Third, basal electrophysiological activity in the PFC of rats is suppressed, but stimulus-evoked burst duration and firing rate are enhanced within 24 hours after the end of cocaine SA (Sun and Rebec, 2006). Fourth, downregulation of immediate early gene and Bdnf mRNA occurs within 22 hours of cocaine withdrawal, but robust upregulation occurs in response to cue-induced relapse to drug-seeking (Hearing et al., 2008; McGinty et al., 2010). Finally, basal hypoactivity punctuated by hyper-responsiveness of PFC to cues associated with cocaine are critical disturbances in abstinent human cocaine addicts that are associated with craving and relapse (Childress et al., 1999; Goldstein and Volkow, 2011). Thus, it is possible that by repeatedly driving synaptic activity and excessive glutamate release in NAc (Miguens et al., 2008), cocaine SA causes hyper-phosphorylation of synapsins and depletion of the reserve SV pool along with increased evoked release probability that extends into withdrawal.
The mechanism underlying the ability of a BDNF infusion into dmPFC to normalize synapsin and PP2Ac phosphorylation in the NAc during early withdrawal is likely related to BDNF’s ability to increase synaptic activity. Emerging evidence indicates that inhibition of synaptic activity in the PFC blocks the ability of intra-PFC BDNF to suppress cocaine-seeking (Go and McGinty, 2013). In addition, BDNF has been demonstrated to increase excitatory glutamatergic neuronal activity (Haubensak et al., 1998) as well as PP2A activity in cortical neurons (Jovanovic et al., 2004). Sustained depolarization also increases PP2A activity in midbrain dopamine neurons (Padmanabhan and Prasad, 2009). Thus, it is possible that intra-dmPFC BDNF infusion activates PP2A/PP2B and normalizes p-synapsin Ser9 and Ser62/67 levels and restores the cocaine-depleted reserve pool of synaptic vesicles in NAc terminals as a function of restoring PFC neuronal activity during early withdrawal. Future studies will attempt to extend and confirm this hypothesis by establishing whether cocaine SA depletes, and BDNF restores, the reserve pool of SVs in the NAc and the possible underlying mechanisms.
Surprisingly, in the NAc of yoked-saline animals, we did not observe any p-synapsin changes after intra-dmPFC BDNF infusion, which is in contrast to a previous study demonstrating the elevation of p-synapsin in the synaptosomal fraction induced by local BDNF application (Jovanovic et al., 2010). Because total homogenates of NAc were used and BDNF was infused in the dmPFC in the present study, the underlying mechanism of BDNF effects on the local axon terminals in the NAc could not be examined in detail, which may partially explain the discrepancy. Further experiments using synapsomal and/or presynaptic fractions are needed to elucidate BDNF’s effects on p-synapsin and its upstream activators as well as other possible alternative intracellular mechanisms.
In conclusion, we demonstrated both p-synapsin Ser9 and Ser62/67 in the NAc were induced during early withdrawal from cocaine SA. This hyper-phosphorylation of synapsins may represent a molecular disturbance of the reserve pool of synaptic vesicles that contributes to synaptic depression and alters release probability in the PFC-NAc pathway during withdrawal from cocaine SA. However, the possible contribution of PFC-amygdala-NAc input on synapsin phosphorylation cannot be excluded, since BDNF was elevated in the amygdala after intra-dmPFC BDNF infusion (McGinty et al., 2010). Nevertheless, together with our previous findings (Berglind et al., 2007, 2009; Whitfield et al., 2011), the ability of intra-dmPFC BDNF infusion to rescue p-synapsin in the NAc and related signaling cascades in the PFC suggests that the restoration of PFC function during early withdrawal may abrogate maladaptive responses that disturb the PFC output to the NAc. Therefore, the BDNF/TrkB system in the PFC-NAc pathway remains a potential therapeutic target for early intervention in cocaine addiction.
Statement of Interest
None.
Acknowledgments
We thank Phong Do, Andrew Nowak, and John Yang for excellent technical assistance. This research was supported by P50 DA015369, T32 DA007288, and the MUSC Neuroscience Institute.
References
- Baldelli P, Fassio A, Valtorta F, Benfenati F. (2007). Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosci 27:13520–13531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Bahler M, Jahn R, Greengard P. (1989). Interactions of synapsin I with small synaptic vesicles: distinct sites in synapsin I bind to vesicle phospholipids and vesicle proteins. J Cell Biol 108:1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Neyroz P, Bahler M, Masotti L, Greengard P. (1990). Time-resolved fluorescence study of the neuron-specific phosphoprotein synapsin I. Evidence for phosphorylation-dependent conformational changes. J Biol Chem 265:12584–12595. [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. (2007). A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci 26:757–766. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, Whitfield TW, Jr, LaLumiere RT, Kalivas PW, McGinty JF. (2009). A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci 29:3715–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogen IL, Boulland JL, Mariussen E, Wright MS, Fonnum F, Kao HT, Walaas SI. (2006). Absence of synapsin I and II is accompanied by decreases in vesicular transport of specific neurotransmitters. J Neurochem 96:1458–1466. [DOI] [PubMed] [Google Scholar]
- Bogen IL, Haug KH, Roberg B, Fonnum F, Walaas SI. (2009). The importance of synapsin I and II for neurotransmitter levels and vesicular storage in cholinergic, glutamatergic and GABAergic nerve terminals. Neurochem Int 55:13–21. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. (2001). Synapsin dispersion and reclustering during synaptic activity. Nature Neurosci 4:1187–1193. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. (2003). Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38:69–78. [DOI] [PubMed] [Google Scholar]
- Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. (1999). Limbic activation during cue-induced cocaine craving. Am J Psychiatr 156:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRemer MF, Saeli RJ, Edelman AM. (1992). Ca(2+)-calmodulin-dependent protein kinases Ia and Ib from rat brain I. Identification, purification, and structural comparisons. J Biol Chem 267:13460–13465. [PubMed] [Google Scholar]
- Edwards S, Graham DL, Bachtell RK, Self DW. (2007). Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur J Neurosci 25:2201–2213. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr., Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. (2004). Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304:1815–1819. [DOI] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, Augustine GJ. (2004a) Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci 24:3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. (2004b) Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci 24:11368–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Cheng Q, Greengard P, Augustine GJ. (2008). Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci 28:10835–10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go BS, McGinty JF. (2013). Synaptic activity mediates ability of BDNF-TrkB signaling to suppress cocaine-seeking in rats. Soc Neurosci NeurOnline Planner 82015. [Google Scholar]
- Goldstein RZ, Volkow ND. (2011). Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci 12:652–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Narz F, Heumann R, Lessmann V. (1998). BDNF-GFP containing secretory granules are localized in the vicinity of synaptic junctions of cultured cortical neurons. J Cell Sci 111 (Pt 11):1483–1493. [DOI] [PubMed] [Google Scholar]
- Hearing MC, Miller SW, See RE, McGinty JF. Relapse to cocaine seeking increases activity-regulated gene expression differentially in the prefrontal cortex of abstinent rats. Psychopharmacology 198:77–91, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. (1999). Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci 354:269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Benfenati F, Doussau F, Nairn AC, Czernik AJ, Augustine GJ, Greengard P. (2005). Structural domains involved in the regulation of transmitter release by synapsins. J Neurosci 25:2658–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M, Hammer RE, Sudhof TC. (1999). A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 24:377–387. [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P. (1983). Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96:1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. (1996). Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci U S A 93:3679–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. (2000). Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci 3:323–329. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr, Greengard P, Czernik AJ. (2001). Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci 21:7944–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. (2004). Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J Neurosci 24:522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile BM, Guillot TS, Venton BJ, Wetsel WC, Augustine GJ, Wightman RM. (2010). Synapsins differentially control dopamine and serotonin release. J Neurosci 30:9762–9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Caine SB, Parsons L, Markou A, Weiss F. (1997). Opponent process model and psychostimulant addiction. Pharmacol Biochem Behav 57:513–521. [DOI] [PubMed] [Google Scholar]
- Lutgen V, Kong L, Kau KS, Madayag A, Mantsch JR, Baker DA. (2014). Time course of cocaine-induced behavioral and neurochemical plasticity. Addict Biol 19:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. (2003). Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23:3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr, Berglind WJ. (2010). Brain-derived neurotrophic factor and cocaine addiction. Brain Res 1314:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguens M, Del Olmo N, Higuera-Matas A, Torres I, Garcia-Lecumberri G, Ambrosio E. (2008). Glutamate and aspartate levels in the nucleus accumbens during cocaine self-administration and extinction: a time course microdialysis study. Psychopharmacol 196:303–313. [DOI] [PubMed] [Google Scholar]
- Padmanabhan S, Prasad BM. (2009). Sustained depolarization decreases calcium/calmodulin-dependent protein kinase II activity and gene expression in dopamine neurons. Neuroscience 163:277–285. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. (2005). The rat brain in stereotaxic coordinates. Burlington, MA: Elsevier Academic. [Google Scholar]
- Picciotto MR, Zoli M, Bertuzzi G, Nairn AC. (1995). Immunochemical localization of calcium/calmodulin-dependent protein kinase I. Synapse 20:75–84. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Sudhof TC. (1995). Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375:488–493. [DOI] [PubMed] [Google Scholar]
- Sihra TS, Wang JK, Gorelick FS, Greengard P. (1989). Translocation of synapsin I in response to depolarization of isolated nerve terminals. Proc Natl Acad Sci U S A 86:8108–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Rebec GV. (2006). Repeated cocaine self-administration alters processing of cocaine-related information in rat prefrontal cortex. J Neurosci 26:8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL, Zhou L, Quinones-Jenab V, Jenab S. (2009). Cocaine effects on dopamine and NMDA receptors interactions in the striatum of Fischer rats. Brain Res Bull 80:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL, Zelek-Molik A, McGinty JF. (2013). Short and long access to cocaine self-administration activates tyrosine phosphatase STEP and attenuates GluN expression but differentially regulates GluA expression in the prefrontal cortex. Psychopharmacology (Berl) 229:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suska A, Lee BR, Huang YH, Dong Y, Schluter OM. (2013). Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proc Natl Acad Sci USA 110:713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller L. (2001). BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zone of hippocampal excitatory synapses. J Neurosci 21:4249–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara H, Onofri F, Benfenati F, Sassoe-Pognetto M, Giustetto M. (2009). ERK activation in axonal varicosities modulates presynaptic plasticity in the CA3 region of the hippocampus through synapsin I. Proc NAtl Acad Sci USA 106:9872–9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venton BJ, Seipel AT, Phillips PE, Wetsel WC, Gitler D, Greengard P, Augustine GJ, Wightman RM. (2006). Cocaine increases dopamine release by mobilization of a synapsin-dependent reserve pool. J Neurosci 26:3206–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, McGinty JF. (1997). Glutamate-dopamine interactions mediate the effects of psychostimulant drugs. Addict Biol 4:141–150. [DOI] [PubMed] [Google Scholar]
- White FJ, Kalivas PW. (2001). Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend 51:141–153. [DOI] [PubMed] [Google Scholar]
- Whitfield TW, Jr, Shi X, Sun WL, McGinty JF. (2011). The suppressive effect of an intra-prefrontal cortical infusion of BDNF on cocaine-seeking is Trk receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J Neurosci 31:834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wydra K, Golembiowska K, Zaniewska M, Kaminska K, Ferraro L, Fuxe K, Filip M. (2013). Accumbal and pallidal dopamine, glutamate and GABA overflow during cocaine self-administration and its extinction in rats. Addict Biol 18:307–324. [DOI] [PubMed] [Google Scholar]