

Abstract
Background:
Adolescent exposure to cannabinoids in vulnerable individuals is proposed to be a risk factor for psychiatric conditions later in life, particularly schizophrenia. Evidence from studies in animals has indicated that a combination of repeated pubertal cannabinoid administration with either neonatal prefrontocortical lesion, isolation rearing, or chronic NMDA receptor antagonism administration induces enhanced schizophrenia-like behavioral disruptions. The effects of adolescent exposure to CB1 receptor agonists, however, have not been tested in a developmental disruption model of schizophrenia.
Methods:
This was tested in the methylazoxymethanol (MAM) model, in which repeated treatment with the synthetic cannabinoid agonist WIN 55,212-2 (WIN; 1.2mg/kg) was extended over 25 days throughout puberty (postnatal days 40–65) in control and MAM rats. The rats received 20 injections, which were delivered irregularly to mimic the human condition. Adult rats were tested for attentional set-shifting task and locomotor response to amphetamine, which was compared with in vivo recording from ventral tegmental area (VTA) dopamine (DA) neurons.
Results:
MAM-treated rats showed impairment in the attentional set-shifting task, augmented locomotor response to amphetamine administration, and an increased number of spontaneously active DA neurons in the VTA. Interestingly, pubertal WIN treatment in normal animals induced similar changes at adulthood as those observed in MAM-treated rats, supporting the notion that adolescence exposure to cannabinoids may represent a risk factor for developing schizophrenia-like signs at adulthood. However, contrary to expectations, pubertal WIN administration did not exacerbate the behavioral and electrophysiological changes in MAM-treated rats beyond that observed in WIN-treated saline rats (Sal). Indeed, WIN treatment actually attenuated the locomotor response to amphetamine in MAM rats without impacting DA neuron activity states.
Conclusions:
Taken together, the present results indicate that the impact of cannabinoids during puberty/adolescence on schizophrenia models is more complex than may be predicted.
Keywords: adolescence, cannabinoid, dopamine, MAM treatment, schizophrenia.
Introduction
Early insults during brain development have been associated with increased risk of schizophrenia (Harrison and Weinberger, 2005; Rapoport et al., 2005). Evidence for a neurodevelopmental disruption is largely based on follow-back, cohort, and population studies in which the pre-morbid history is associated with the presence of subtle prenatal perturbations that may interact with genetic predisposition to result in the schizophrenia phenotype (van Os et al., 2010). This finding is central to the development of animal models which utilize perinatal insults to produce a behavioral phenotype as adult.
One model that has substantial face validity utilizes the administration of the DNA methylating agent methylazoxymethanol acetate (MAM) to pregnant dams on gestational day (GD) 17 (Moore et al., 2006). This model utilizes a non-selective developmental disruption with no selective genetic manipulation or loss of a specific brain structure, and has findings consistent with those seen in schizophrenia patients. Furthermore, the deficits observed in this model parallel those observed in schizophrenia patients, including anatomical changes (Moore et al., 2006), behavioral deficits (Talamini et al., 2000; Flagstad et al., 2004; Moore et al., 2006), and disruption of rhythmic activity in the frontal cortex (Goto and Grace, 2006). In addition, the MAM model also shows pharmacological validity, with typical and atypical antipsychotics being able to reverse MAM-induced behavioral and electrophysiological changes (Pen et al., 2010; Valenti et al., 2011; Belujon et al., 2013) at a time course consistent with schizophrenia in humans (Agid et al., 2003; Valenti et al., 2011).
In addition to the perinatal phase, adolescence is also a period extremely vulnerable to disruption by environmental influence. This period is characterized by cognitive, emotional, and social maturation. Moreover, other important changes observed in adolescence are risk taking and novelty seeking (Kelley et al., 2004). Besides their adaptive benefits, these behaviors also render adolescents more vulnerable to pathology. For example, epidemiological studies showed an increased risk for drug abuse during adolescence (Fried et al., 2001; Martin et al., 2002). Among these drugs, epidemiological data indicate a causal association between early cannabis abuse and development of psychiatric conditions later in life, including schizophrenia (Arseneault et al., 2004; Degenhardt and Hall, 2006; Fergusson et al., 2006; van Laar et al., 2007). Adolescents initiate cannabis use before consuming other illicit drugs (Cleveland and Wiebe, 2008) and, although the majority of people who experience this drug during adolescence do not develop a psychiatric condition later in life (Gregg et al., 2007; Dekker et al., 2009; Kolliakou et al., 2011), genetic and environmental factors, such as childhood trauma or psychosocial stress, may predispose them to be particularly vulnerable to the effects of cannabis (Caspi et al., 2005; Arseneault et al., 2011; Kuepper et al., 2011). Therefore, adolescent exposure to cannabinoids (that is, CB1 receptor agonists) in vulnerable individuals is proposed to act as a risk factor for inducing behavioral disturbances (Casadio et al., 2011). This conclusion seems to be more credible when the two-hit hypothesis of schizophrenia is taken into account. In this hypothesis, genetic or environmental factors disrupt early central nervous system development, producing vulnerability to a “second hit” that then may lead to the onset of schizophrenia symptoms. In fact, a combination of a neonatal prefrontocortical lesion with repeated pubertal cannabinoid (CB1/2 receptor agonist WIN55,212-2) administration leads to greater impairments in social behavior (Schneider and Koch, 2005) and object recognition memory (Schneider and Koch, 2007), suggesting that pubertal cannabinoid administration in vulnerable individuals might induce enhanced behavioral disturbances. Furthermore, pubertal exposure to Δ9-tetrahydrocannabinol (THC), the major psychotomimetic compound present in cannabis, worsened disruption of prepulse inhibition induced by isolation rearing (Malone and Taylor, 2006) and impairment in the object recognition test, induced by chronic administration of phencyclidine (Vigano et al., 2009), an NMDA receptor antagonist. The effects of pubertal exposure to CB1 receptor agonists, however, have not been tested in neurodevelopmental disruption models of schizophrenia which, due to their delayed onset, would be expected to have greater interaction with adolescent cannabis use.
MAM-treated animals show increased dopamine (DA) neuron population activity in the ventral tegmental area (VTA) that correlates with the enhanced locomotor response to amphetamine (Lodge and Grace, 2007), indicating an enhanced activity in the mesolimbic DA system. Cannabis, like most drugs of abuse, causes an increase in extracellular DA levels (Gardner, 2005). In addition, repeated use of cannabis in adolescence could lead to sensitization of the mesolimbic DA system, a fact that would help to explain why cannabis use during adolescence may facilitate the development of schizophrenia (Stefanis et al., 2004). Moreover, altered DA function is proposed to make schizophrenia patients more vulnerable to the effects of CB1 receptor agonists (Abi-Dargham, 2004).
Based on this evidence, the effects of pubertal exposure to the CB1/2 receptor agonist WIN55,212-2 (WIN) were tested on the behavioral changes and VTA DA neuronal activity observed in MAM-treated rats.
Material and Methods
Animals and MAM Treatment
All experiments were conducted according to the guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Pregnant female Sprague–Dawley rats were obtained from Hilltop Lab Animals on GD 15 and individually housed in ventilated plastic breeding tubs. MAM (20mg/kg, i.p.; Midwest Research Institute) was administered on GD 17. Control dams received injections of saline (1mL/kg, i.p.). Male pups were weaned on postnatal day (PD) 21 and housed in groups of two to three with littermates approximately 3–4 months of age, at which time they were used for behavioral and electrophysiological experiments. All experiments were performed on multiple litters of MAM- and saline-treated rats.
Experimental Design
Pregnant rats were administered MAM or saline on GD 17. Male offspring of both groups were then administered either the CB1/2 receptor agonist WIN55,212-2 (Sigma-Aldrich) or vehicle (Veh). WIN was emulsified in 0.5% Tween 80 and then diluted in saline (0.9%). The drug was administered intraperitoneally at a dose of 1.2mg/kg in a volume of 1mL/kg. The treatment with either the synthetic cannabinoid WIN or Veh lasted 25 days, from PD40 to PD65. This period corresponds to the pubertal phase in male rats. It should be noted that puberty and adolescence are overlapping time periods with puberty being a part of adolescence (Schneider, 2013).
During the treatment period the rats received either one or two injections daily or no injection at all (10 times one injection, 5 times two injections, and 10 times no injection per day, for a total of 20 injections). This protocol was chosen in order to mimic the irregular consumption practice in humans.
As adults, the animals were submitted to the behavioral (PD85–PD100) and electrophysiological tests (PD100–PD125; Figure 1). The WIN dose and the experimental design were based on previous studies (Schneider and Koch, 2002, 2003 2005, 2007; Schneider et al., 2005; Schneider et al., 2008; Du and Grace, 2013; Zimmerman et al., 2013). All experiments were performed with investigators blinded to treatment.
Figure 1.
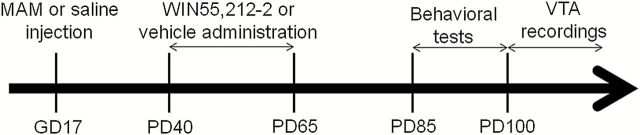
Experimental design. Pregnant rats were administered MAM or saline on GD 17. Rats from each group received either i.p. administration of the CB1/2 receptor agonist WIN55,212-2 (1.2mg/kg) or Veh during PD40 to PD65. These rats were used for behavioral (attentional set-shifting task and amphetamine-induced hyperlocomotion) and electrophysiology tests as adults (PD > 85).
The rats showed a normal increase in body weight (measured every other day from PD40 to PD80) that was independent of prenatal treatment (MAM or saline) or pubertal drug administration (WIN or Veh; data not shown).
Attentional Set-Shifting Task
Both five days before the attentional set-shifting task and during testing, rats were food restricted to approximately 85% of their body weight. The attentional set-shifting task, designed to evaluate extra-dimensional shift as a rodent analog of the Wisconsin Card Sort Test (Tait et al., 2014), was conducted in a white test box (L70 x W40 x H30cm3) in which a wood panel was used to divide one-third of the box length into two equal sections, forming the choice chambers to which access could be blocked via removable doors. During behavioral testing, one ceramic bowl (diameter of 8cm and depth of 4cm) was placed in each choice chamber. Food rewards were one-third pieces of Honey Nut Cheerios (General Mills), which were placed in one bowl per trial and covered with digging media. The media varied by odor and/or texture to provide two different stimulus dimensions to guide choice behavior. Testing was performed according to a modified version of the protocol previously described (Birrell and Brown, 2000; Gastambide et al., 2012). Rats were habituated to the testing box and then initially trained to dig in bowls filled with cage bedding to retrieve food rewards. Once habituated, rats were trained on two simple discriminations (SDs): one based on odor (mustard vs. celery) and one based on texture (shredded paper vs. styrofoam). SD order and reinforced stimuli were pseudo-randomly chosen per rat, but counterbalanced across the rat groups. These odor and texture stimuli were not used again in later phases of the experiment. The purpose of this preliminary phase was to acquaint rats with the basic discrimination learning process, as well as to encourage attention to the two different dimensions of the digging media that could be relevant for subsequent stages of discrimination learning. The following day, rats were given a series of seven discriminations (Table 1): a simple discrimination (SD); a compound discrimination (CD) in which digging media differed according to both odor and texture, but with correct and incorrect exemplars remaining similar to the preceding SD; a reversal (Rev1) in which the reward contingency of the CD exemplars is reversed; an intra-dimensional shift (IDS) in which a novel discrimination is learned with new stimuli, the new correct exemplar being of the same dimension as before; a second reversal (Rev2); and an extra-dimensional shift (EDS), in which another discrimination with new stimuli is learned, but in this case the correct exemplar is now from the other previously irrelevant dimension; and finally a third reversal. For each discrimination stage, testing continued until rats reached a criterion level of six correct consecutive trials. The procedure was the same for each stage: a trial was initiated by raising the removable doors to give rats access to the two digging bowls, only one of which was baited. The first four trials of each discrimination stage were deemed discovery trials, where rats were permitted to dig in both bowls if they chose the incorrect bowl first. An error was recorded if rats dug first in the unbaited bowl. On subsequent trials, if rats started to dig in the unbaited bowl, an error was recorded and the trial was terminated. If rats did not dig at all in either bowl within 3min, the trial was aborted, recorded as an omission, and reinitiated. The number of errors made to reach criterion was recorded per rat for each stage of the test.
Table 1.
Example of Order of Exemplar Exposure in the Attentional Set-Shifting Task.
| Discrimination | Odor Pair | Medium Pair |
|---|---|---|
| Simple discrimination | cumin/cinnamon | bedding |
| Compound discrimination | cumin/cinnamon | cedar shavings/boxo |
| Reversal 1 | cumin/cinnamon | cedar shavings/boxo |
| Intra-dimensional shift | thyme/cloves | fine shavings/paperchips |
| Reversal 2 | thyme/cloves | fine shavings/paperchips |
| Extra-dimensional shift | oregano/paprika | Cat litter/ground cat litter |
| Reversal 3 | oregano/paprika | Cat litter/ground cat litter |
The table shows a possible order of exposure to the exemplar, where the rat must shift its attention from odor to digging medium at the extra-dimensional shift acquisition. The rewarded exemplar is indicated by the underlined words. Presented with either of the two exemplars from the irrelevant dimension, so that during each trial within a discrimination, all four exemplars are present. Rats were counterbalanced so that 50% underwent odor to medium shift.
Locomotor Response to Amphetamine
Adult rats were tested in an open-field chamber (Coulbourn Instruments) in which locomotor activity was determined by beam breaks and recorded with TruScan software (Coulbourn Instruments). All experiments were conducted at the same time each day. Spontaneous activity was recorded for 30min. After that, rats were injected with D-amphetamine sulfate (0.5mg/kg, i.p.; Sigma-Aldrich) and their locomotor activity was recorded for another 90min.
In Vivo Recording from VTA DA Neurons
Rats were anesthetized with chloral hydrate and mounted on a stereotaxic frame (Kopf). The body temperature was maintained at 37ºC using a thermostatically-controlled feedback heating pad (Fintronics). A burr hole was drilled in the skull overlying the right VTA. Extracellular recording microelectrodes were pulled from Omegadot 2.0mm glass tubing on a Narishige P-5 vertical electrode puller, the tip broken back under microscopic control, and filled with 2M NaCl containing 2% Pontamine Sky Blue dye. The impedance of the electrodes tested in situ ranged from 6 to 15 MΩ. The stereotaxic coordinates for the VTA were 5.3mm posterior from bregma, 0.6mm lateral to the midline, and 6.5–9.0mm ventral from the brain surface. Single-unit activity was filtered using a highpass filter at 30 Hz and lowpass at 10kHz. All data analysis was performed using custom software (Neuroscope). Only neuronal activity with a signal-to-noise ratio greater than 3:1 and at least 1–3min of stable spontaneous activity was used. Six to nine vertical tracks, separated by 200mm, were sampled in a predetermined pattern within the VTA of each rat. DA neurons were identified according to well-established electrophysiological features (Grace and Bunney, 1983; Ungless and Grace, 2012), which included the following criteria: (1) location; (2) an action potential duration > 2.2ms with variable waveform within a train; (3) slow firing rate (1–10 Hz); and (4) irregular and burst firing patterns, with the start of burst characterized by inter-spike interval < 80ms, and the end of burst characterized by inter-spike interval > 160ms. The activity of each identified DA neuron was recorded for 1–3min. Three parameters of the DA neuron activity were analyzed: (1) the number of spontaneously active DA neurons per electrode track; (2) average firing rate; and (3) the percentage of spikes that occurred in bursts. At the end of recordings, the recording sites were marked via electrophoretic ejection of Pontamine Sky Blue dye from the tip of the electrode (20 µA constant negative current, 30min). Rats were euthanized by an overdose of anesthetic; the brains were removed, fixed for at least 48h in 8% paraformaldehyde, cryoprotected in 25% sucrose, and sectioned for histological confirmation of the electrode sites.
Statistical Analysis
The attentional set-shifting task was analyzed using repeated-measures 3-way ANOVA with condition (prenatal treatment, MAM, or Sal) and pubertal treatment (Veh or WIN) as the main independent factors, and discrimination type as a repeated measurement followed by Bonferroni post hoc tests. Locomotor activity was analyzed by TruScan software and compared using repeated-measures 3-way ANOVA with condition and treatment as the main independent factors, and time as a repeated measurement. As the 3-way ANOVA indicated a significant effect of condition to treatment interaction but not to condition or treatment, we performed a 2-way ANOVA with group (Sal or MAM + Veh or WIN) as the main independent factor and time as a repeated measurement followed by Bonferroni post hoc tests. Electrophysiological analysis of DA neuron activity was analyzed using 2-way ANOVA with condition and treatment as the two factors followed by Bonferroni post hoc tests. All data are represented as the mean ± SEM. Results of statistical tests with p < 0.05 were considered significant.
Results
Effects of Pubertal WIN Exposure on the Behavioral Flexibility in the Attentional Set-Shifting Task in Normal and MAM-Treated Rats Tested as Adults
During the habituation and training sessions, all rats learned to dig in bowls to retrieve the food reward and perform the SDs. During the test session, Sal:WIN, MAM:Veh, and MAM:WIN rats required significantly more trials to reach criterion than the Sal:Veh group. The repeated-measures 3-way ANOVA indicated significant effects of condition (F1,29 = 18.3, p < 0.0001), treatment (F1,29 = 8.9, p < 0.01), and discrimination type (F6,174 = 68.7, p < 0.0001). There were also significant condition versus treatment (F1,29 = 5.4, p < 0.05), condition versus discrimination type (F6,174 = 3.1, p < 0.05), and treatment versus discrimination type interactions (F6,174 = 2.5, p < 0.05), but no effects for interaction among condition versus treatment versus discrimination type (F6,174 = 1.7, p > 0.05).
All rats required more trials to learn the reversals than they required for either initial acquisition (SD and CD stages) or the IDS (discrimination type: F6,174 = 68.7, p < 0.0001). Moreover, Sal:Veh animals made significantly more errors to reach the criterion of six consecutive correct trials in the EDS than in the IDS (p < 0.05), demonstrating that they had formed an attentional set towards the relevant dimension before the EDS stage (Birrell and Brown, 2000; Gastambide et al., 2012). Concerning the omitted trials, although no significant difference was observed, the animals were most likely to stop digging at Rev1 (Sal:Veh, -2.9±0.7; Sal:WIN, -3.6±0.6; MAM:Veh, -2.7±0.4; MAM:WIN, -3.2±0.5), while fewer stopped digging at the IDS or Rev2, and very few stopped digging after that.
Planned comparison analyses were conducted which compared groups during each stage of discrimination. MAM:Veh rats (n = 8) required a significantly greater number of trials to reach criterion during the three reversal discriminations and the IDS discrimination compared to Sal:Veh rats (n = 8; p < 0.05, Bonferroni post hoc test; Figure 2). A similar pattern was observed during two of the three reversal discriminations (Rev1 and Rev2) and IDS discrimination, with MAM:WIN animals (n = 9) requiring more trials to reach criterion compared to Sal:Veh rats (p < 0.05, Bonferroni post hoc test; Figure 2). Moreover, pubertal WIN treatment in normal rats (Sal:WIN rats, n = 8) required a significantly greater number of trials to reach criterion during the Rev1 and Rev2 (p < 0.05, Bonferroni post hoc test; Figure 2). No differences were observed among Sal:WIN, MAM:Veh, and MAM:WIN during any discriminations.
Figure 2.
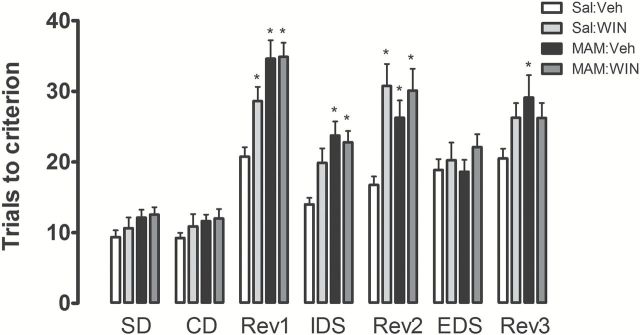
Sal:WIN, MAM:Veh, and MAM:WIN-treated rats exhibited deficits in the attentional set shifting task (n = 8−9/group). Graph bars represent the mean ± SEM of the number of errors made to reach the criterion of six correct consecutive trials in each test discrimination. *p < 0.05 vs. Sal:Veh rats; repeated measures of 3-way ANOVA followed by Bonferroni post hoc test. SD: simple discrimination; CD: compound discrimination; IDS: intra-dimensional shift; EDS: extra-dimensional shift; Rev: discriminations requiring a reversal learning.
Effects of Pubertal WIN Exposure on Amphetamine-Induced Hyperlocomotion in Normal and MAM-Treated Rats Tested as Adults
Both Sal:WIN and MAM:Veh rats exhibited greater locomotor response to amphetamine compared to Sal:Veh rats. However, the MAM:WIN rats showed significantly less amphetamine-induced locomotor activity than either Sal:Win or MAM:Sal rats and were not significantly different from controls. The repeated-measures 3-way ANOVA indicated no significant effects of condition (F1,29 = 1.3, p > 0.05) or treatment (F1,29 = 0.22, p > 0.05); however, there was a significant effect of time (F23,667 = 52.4, p < 0.001) and an interaction between condition and treatment (F1,29 = 6.5, p < 0.05). There were also significant condition versus time (F23,667 = 2.1, p < 0.05), treatment versus time (F23,667 = 1.9, p < 0.05), and condition versus treatment versus time interactions (F23,667 = 2.4, p < 0.01). Although the 3-way ANOVA did not indicate any effect of condition and treatment, the 2-way ANOVA showed significant effects of group (Sal or MAM + Veh or WIN; F3,667 = 3.5, p < 0.05), time (F23,667 = 53.12, p < 0.001), and interaction between group and time (F69,667 = 2.2, p < 0.001).
Consistent with previous studies showing that rats treated with MAM on GD17 exhibited an enhanced locomotor response to amphetamine (Flagstad et al., 2004; Moore et al., 2006), MAM:Veh rats (n = 8) showed significantly higher levels of locomotor activity in response to amphetamine administration (0.5mg/kg, i.p.) compared to controls (Sal:Veh, n = 8; p < 0.05 at 5, 10, and 15min after amphetamine, Bonferroni post hoc test; Figure 3). Likewise, pubertal WIN treatment in normal rats (Sal:WIN, n = 8) produced a significant enhancement in amphetamine-stimulated locomotion compared to Sal:Veh rats (p<0.05 at 5, 10, 20, and 40min after amphetamine, Bonferroni post hoc test; Figure 3). Surprisingly, WIN treatment in MAM rats (MAM:WIN, n = 9) induced a significantly lower level of amphetamine-stimulated locomotion compared to MAM:Veh rats (p < 0.05 at 10 and 15min after amphetamine, Bonferroni post hoc test; Figure 3), and was not significantly different from Sal:Veh rats (p > 0.05, Bonferroni post hoc test). The locomotor activity before amphetamine administration did not differ significantly among all four groups (p > 0.05, Bonferroni post hoc test; Figure 3).
Figure 3.
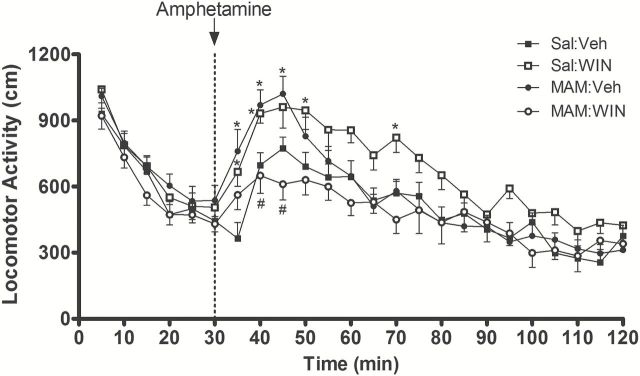
Pubertal WIN exposure produces opposite effects on MAM and normal rats. MAM:Veh rats (n = 8) showed significantly higher amphetamine-induced locomotion compared to Sal:Veh rats (n = 8). Normal rats treated with WIN (1.2mg/kg, i.p., PD40–PD65; Sal:WIN, n = 8) also showed a significantly higher amphetamine-induced locomotion compared to Sal:Veh rats. In contrast, MAM:WIN (n = 9) showed an attenuation of the aberrant enhancement of the locomotor response to amphetamine that was observed in MAM-treated rats. Sal:Veh and MAM:WIN rats were not significantly different. Locomotor activity was calculated within each bin (bin width = 5min). D-amphetamine (0.5mg/kg, i.p.) injection is indicated by the dashed line. Spontaneous activity before amphetamine injection was not significantly different among all four groups. *p < 0.05 vs. Sal:Veh rats; #p < 0.05 vs. MAM:Veh; repeated measures of 2-way ANOVA followed by Bonferroni post hoc test (see Statistical analysis).
Based on the opposite effects induced by the chronic pubertal treatment with WIN in normal and MAM-treated rats, we tested the effects of a WIN administration given once on PD65 to test whether repeated administration would be required to induce altered locomotor responses (Supplementary Figure 1). Indeed, no effect in the amphetamine-stimulated locomotion induced by the single WIN injection was observed in saline- or MAM-treated rats tested on PD85–PD90 (Supplementary Figure 2).
Effects of Pubertal WIN Exposure on VTA DA Neuron Activity in Normal and MAM-Treated Rats Tested as Adults
Both MAM groups as well as the Sal:WIN group demonstrated significant increases in the number of DA neurons firing spontaneously compared to Sal:Veh rats. The number of spontaneously active DA neurons was significantly affected by prenatal MAM (condition: F1,23 = 4.4, p < 0.05) and pubertal WIN administration (treatment: F1,23 = 12.9, p < 0.001), but no interaction was observed (F1,23 = 2.0, p > 0.05; 2-way ANOVA). Consistent with what has been reported previously (Lodge and Grace, 2007, 2009), recordings from MAM:Veh rats (n = 7 rats, 78 neurons) showed a significantly greater number of spontaneously active DA neurons per electrode track (1.6±0.1 cells/track, p < 0.05, Bonferroni post hoc test) compared to Sal:Veh rats (n = 6 rats, 46 neurons, 0.99±0.2 cells/track; Figure 4A).
Figure 4.
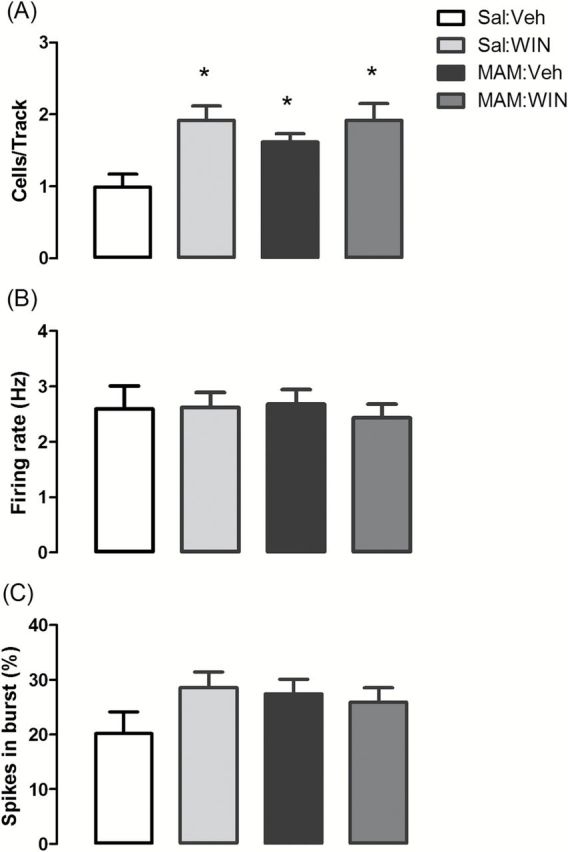
MAM rats and WIN-treated normal rats showed increases in the number of DA neurons firing spontaneously. (A) Pubertal WIN treatment (1.2mg/kg, i.p.; PD40–PD65; Sal:WIN, n = 7, 92 neurons) increased the number of spontaneously active DA neurons (presented as cells/track) compared to control group (Sal:Veh, n = 6 rats, 46 neurons). MAM:Veh (n = 7, 78 neurons) and MAM:WIN rats (n = 7, 90 neurons) also had a significantly higher number of DA neurons firing per electrode track compared to Sal:Veh, but WIN treatment did not significantly alter the number of DA neurons firing in MAM-treated rats compared to MAM:Veh. (B) Average firing rate and (C) percentage of spikes fired in bursts were not significantly different. *p < 0.05 vs. Sal:Veh rats, 2-way ANOVA followed by Bonferroni post hoc test.
Compared to Sal:Veh rats, Sal:WIN rats (n = 7, 92 neurons) showed a significantly greater number of spontaneously active DA neurons (1.9±0.2 cells/track, p < 0.05, Bonferroni post hoc test). These changes required repeated WIN exposure, given that no effect was observed after a single WIN injection on PD65 (Supplementary Figure 3).
Similar to the MAM:Veh rats, the numbers of DA neurons firing in the MAM:WIN (n = 7, 90 neurons; 1.9±0.3 cells/track) was greater than the Sal:Veh rats (p < 0.05, Bonferroni post hoc test). Importantly, despite differences in amphetamine-induced locomotion, no difference was observed between MAM:Veh and MAM:WIN rats with respect to DA neuron activity (p > 0.05, Bonferroni post hoc test; Figure 4A). The firing rate and percentage of spikes in bursts did not differ significantly across all four groups (p > 0.05 by two-way ANOVA; Figure 4B and 4C).
Discussion
Consistent with previous studies (Featherstone et al., 2007; Lodge and Grace, 2007; Gastambide et al., 2012), MAM-treated rats showed an impairment in the attentional set-shifting task, augmented locomotor response to amphetamine administration, and an increased number of spontaneously active DA neurons in the VTA. Interestingly, pubertal treatment with the CB1/2 receptor agonist WIN from PD40 through PD65 in normal animals induced similar changes at adulthood as those observed in MAM-treated rats. Moreover, WIN treatment did not exacerbate the changes in MAM-treated rats. Instead, WIN treatment actually prevented the increase in amphetamine-induced locomotion in the MAM rats without altering the increase in DA neuron activity.
The results of the present study are consistent with recent findings showing that repeated pubertal cannabinoid treatment induces lasting behavioral changes in adulthood, including sensorimotor gating impairment, abnormal social behavior, and anhedonia (Schneider and Koch, 2003, 2005, 2007; Schneider et al., 2008). Moreover, rodents chronically treated during different periods of adolescence with other CB1 receptor agonists (CP55,940 and THC) and tested as adults also exhibited deficits in sensorimotor gating, object recognition, and spatial working memory (O’Shea et al., 2004; Quinn et al., 2008; Rubino, Realini, Braida, Alberio, et al., 2009; Rubino, Realini, Braida, Guidi, et al., 2009; Gleason et al., 2012). The current study is the first time that the long-term effects of cannabinoid exposure during adolescence have been evaluated in the attentional set-shifting task.
In the set-shifting task, rats are required to solve a series of discriminations by attending to a particular perceptual dimension of a multidimensional stimulus. A critical discrimination occurs when rats are required to shift to an alternate perceptual dimension after having acquired an attentional set to the previous dimension (Tait et al., 2013). The neural substrates of set shifting and reversal learning are reasonably well-defined. Lesions of the monkey lateral prefrontal cortex (Dias et al., 1997) and the equivalent prelimbic and infralimbic regions of the rat medial prefrontal cortex (Birrell and Brown, 2000) disrupt attentional set-shifting ability, whereas lesions of the orbitofrontal cortex selectively impair reversal learning in both species (Dias et al., 1997; McAlonan and Brown, 2003). Impaired reversal learning has recently been highlighted to occur reliably in schizophrenia patients. Indeed, Leeson et al. (2009) found in a large group of first-episode schizophrenia patients that although they were impaired on set shifting, they also exhibited small but consistent deficits in reversal learning. Moreover, chronic cannabis use in adolescence appears to be associated with overall less efficient executive function and attention (Abdullaev et al., 2010).
Consistent with previous studies (Featherstone et al., 2007; Gastambide et al., 2012), MAM rats exhibited a variety of cognitive impairments, including reversal learning and attentional set shifting, as defined by requiring a greater number of trials than controls to successfully learn to shift cognitive set between stimuli belonging to the same perceptual dimension (IDS), and having difficulties in learning to reverse a previously acquired discrimination. Interestingly, the pubertal WIN treatment in normal rats also resulted in a significantly greater number of trials to reach criterion during two of the three reversal learning discrimination trials. Thus, the deficits observed in reversal learning, together with the preservation of ability to shift strategy, indicated that MAM-treated rats and the pubertal WIN exposure in normal animals induced an increased rigidity in the processes required to update responses based on affective associations between stimuli and reward presentation, but did not affect ability for higher order attentional flexibility (EDS). This suggests that the deficits in the reversal learning and IDS were not due to a generalized performance or cognitive impairment. Although it has been suggested that adolescent cannabinoid exposure in vulnerable individuals might induce even more pronounced behavioral disturbances, and studies with animals have shown an enhanced cognitive impairment observed after the combination of pubertal exposure to cannabinoids with neonatal prefrontocortical lesions, social isolation, or chronic administration of phencyclidine (Schneider and Koch, 2005, 2007; Malone and Taylor, 2006; Vigano et al., 2009), no significant difference was observed between Veh- and WIN-treated MAM rats in the attentional set-shifting task.
Similar to MAM-treated rats, WIN administration during puberty induced an augmented locomotor response to amphetamine and an increased number of spontaneously active DA neurons in the VTA in normal animals as adults. Previous studies have shown that acute CB1 receptor activation increases mesolimbic DA activity (French, 1997; Tanda et al., 1997; Wu and French, 2000). However, this is the first study showing persistent long-term changes in mesolimbic DA activity induced by pubertal cannabinoid exposure.
Prenatal MAM administration and pubertal cannabinoid exposure have been shown to induce similar changes in GABA neurons of adult rats. Zamberletti et al. (2014) observed that adolescent THC exposure in mice reduced GAD67 expression in interneurons containing the calcium binding protein parvalbumin (PV) within the adult prefrontal cortex. Moreover, repeated CB1 receptor activation in adolescence elicited an enduring state of prefrontal cortex disinhibition due to a developmental impairment of prefrontal GABAergic transmission (Cass et al., 2014). Decreased GABAergic signaling is among the most robust postmortem pathological changes observed in schizophrenia (Reynolds et al., 2002; Lewis et al., 2005). Specifically, a decrease in GAD67 protein is observed postmortem throughout the cortex of schizophrenia patients (Hashimoto et al., 2003) that are largely restricted to the GABAergic PV-positive interneurons (Lewis et al., 2005). Interestingly, a decrease in PV-containing interneurons is also a consistent observation in a diverse variety of animal models of schizophrenia, including the MAM model (Penschuck et al., 2006; Lodge et al., 2009; Gill and Grace, 2014).
It has been suggested that the augmented DA neuron activity and hyper-responsivity to psychomotor stimulants observed in MAM-treated rats results from an increased activity within ventral regions of the hippocampus due to a loss of PV interneurons (Lodge et al., 2009). In addition to the ventral hippocampus, adult MAM-treated rats also display specific reductions in the number of PV-positive interneurons throughout the medial prefrontal cortex (Penschuck et al., 2006; Lodge and Grace, 2009) and the orbitofrontal cortex (Gastambide et al., 2012), the main brain structures involved in set-shifting and reversal learning, respectively (Birrell and Brown, 2000; McAlonan and Brown, 2003). Thus, based on the evidence indicating that cannabinoid exposure during adolescence may reduce the number of PV-positive interneurons (Zamberletti et al., 2014), changes in the PV expression could be involved in the dopaminergic dysfunction and in the impairment observed during the attentional set-shifting task in WIN-treated normal, similar to that observed with MAM-treated rats.
No additive or synergic effect, however, was found in MAM-treated rats that received WIN. Although this could reflect a ceiling effect, it is not possible to rule out the involvement of at least partially distinct (and parallel) mechanisms for the attentional set-shifting task impairment induced by these two treatments.
The most intriguing finding of our study is that, although WIN-treated MAM rats showed an enhanced VTA DA neuronal spontaneous activity that was similar to Veh-treated MAM rats, pubertal WIN exposure in MAM rats decreased amphetamine-induced hyperlocomotion. The reason for this attenuation is unclear, particularly given that increases in DA neuronal activity in the VTA, such as those observed after either MAM (Lodge and Grace, 2007) or WIN administration, along with other models (e.g., amphetamine sensitization [Lodge and Grace, 2012], temporal lobe epilepsy [Cifelli and Grace, 2012]) have consistently shown parallel changes between the number of DA neurons firing spontaneously and amphetamine-induced locomotor activity. This suggests that the pubertal exposure to WIN may have induced compensatory changes in MAM rats that are downstream from DA neuron activity. Thus, it is known that exogenous cannabinoids affect the function of the endocannabinoid system. Indeed, pubertal cannabinoid exposure can change the expression of components of the endocannabinoid system in brain structures related to motivation and motor control (Marco et al., 2007; Ceci et al., 2014). Therefore, plastic changes in the endocannabinoid system induced by repeated CB1 receptor agonist administration could lead to plastic changes in the CB1 receptor modulation of GABA- or glutamate-mediated neurotransmission (Wilson and Nicoll, 2002; Fernandez-Ruiz et al., 2010) in key brain structures related to hyperlocomotion, such as the nucleus accumbens, thereby compensating for the modifications in other brain areas (for example, the ventral subiculum) induced by MAM. Interestingly, Spano et al. (2013) observed that both WIN self-administration and passive WIN administration (i.v.) over 14 days attenuated hyperlocomotion in response to an acute phencyclidine (PCP) challenge in adult rats treated chronically with PCP, a model of schizophrenia based on the N-methyl-d-aspartate receptor hypofunction.
Alternately, a decrease in the DA transporter seen in the caudate nucleus of schizophrenia patients was not observed in patients who had used cannabis (Dean et al., 2003), leading the investigators to suggest that THC might reverse the decreases in DA transporter expression associated to schizophrenia.
In conclusion, these results are consistent with the notion that adolescent exposure to cannabinoids may represent a risk factor for developing schizophrenia-like signs at adulthood. However, contrary to our hypothesis that pubertal MAM-treated rats would be more susceptible to the cannabinoid exposure, WIN administration did not exacerbate the behavioral and electrophysiological changes in MAM-treated rats, and in fact prevented the augmentation of the locomotor response to amphetamine. While several epidemiological studies have clearly shown an association between cannabis use and susceptibility to schizophrenia (Arseneault et al., 2004; Casadio et al., 2011), it is not possible to evaluate if subgroups of patients that may develop schizophrenia later in life are protected by cannabis use. Therefore, it is clear that the relationship between cannabinoid exposure and susceptibility to disease states is more complex than may be predicted by correlative epidemiological studies.
Supplementary Material
For supplementary material accompanying this paper, visit http://www.ijnp.oxfordjournals.org/
Statement of Interest
Drs Gomes and Guimarães declare no conflict of interest. Dr Grace has received funds from Johnson & Johnson, Lundbeck, Pfizer, GSK, Merck, Takeda, Dainippon Sumitomo, Otsuka, Lilly, Roche, Asubio, and Abbott.
Supplementary Material
Acknowledgments
We thank Niki MacMurdo for her technical assistance. This work was supported by the Brazilian Federal Agency for Support and Evaluation of Graduate Education, CAPES/PDSE (10865/13–7, FVG) and USPHS MH57440 (AAG). Drs Gomes and Guimarães are, respectively, recipients of a FAPESP doctoral (2010/17343-0) and of a CNPq researcher fellowship.
References
- Abdullaev Y, Posner MI, Nunnally R, Dishion TJ. (2010). Functional MRI evidence for inefficient attentional control in adolescent chronic cannabis abuse. Behav Brain Res 215:45–57. [DOI] [PubMed] [Google Scholar]
- Abi-Dargham A. (2004). Do we still believe in the dopamine hypothesis? New data bring new evidence. Int J Neuropsychopharmacol 7(Suppl 1):S1–5. [DOI] [PubMed] [Google Scholar]
- Agid O, Kapur S, Arenovich T, Zipursky RB. (2003). Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry, 60:1228–1235. [DOI] [PubMed] [Google Scholar]
- Arseneault L, Cannon M, Fisher HL, Polanczyk G, Moffitt TE, Caspi A. (2011). Childhood trauma and children’s emerging psychotic symptoms: A genetically sensitive longitudinal cohort study. Am J Psychiatry 168:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arseneault L, Cannon M, Witton J, Murray RM. (2004). Causal association between cannabis and psychosis: examination of the evidence. Br J Psychiatry 184:110–117. [DOI] [PubMed] [Google Scholar]
- Belujon P, Patton MH, Grace AA. (2013). Disruption of prefrontal cortical–hippocampal balance in a developmental model of schizophrenia: reversal by sulpiride. Int J Neuropsychopharmacol 16:507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell JM, Brown VJ. (2000). Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci 20:4320–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadio P, Fernandes C, Murray RM, Di Forti M. (2011). Cannabis use in young people: the risk for schizophrenia. Neurosci Biobehav Rev 35:1779–1787. [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE, Cannon M, McClay J, Murray R, Harrington H, Taylor A, Arseneault L, Williams B, Braithwaite A, Poulton R, Craig IW. (2005). Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol Psychiatry 57:1117–1127. [DOI] [PubMed] [Google Scholar]
- Cass DK, Flores-Barrera E, Thomases DR, Vital WF, Caballero A, Tseng KY. (2014). CB1 cannabinoid receptor stimulation during adolescence impairs the maturation of GABA function in the adult rat prefrontal cortex. Mol Psychiatry 19:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceci C, Mela V, Macri S, Marco EM, Viveros MP, Laviola G. (2014). Prenatal corticosterone and adolescent URB597 administration modulate emotionality and CB1 receptor expression in mice. Psychopharmacology (Berl) 231:2131–2144. [DOI] [PubMed] [Google Scholar]
- Cifelli P, Grace AA. (2012). Pilocarpine-induced temporal lobe epilepsy in the rat is associated with increased dopamine neuron activity. Int J Neuropsychopharmacol 15:957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland HH, Wiebe RP. (2008). Understanding the association between adolescent marijuana use and later serious drug use: gateway effect or developmental trajectory? Dev Psychopathol 20:615–632. [DOI] [PubMed] [Google Scholar]
- Dean B, Bradbury R, Copolov DL. (2003). Cannabis-sensitive dopaminergic markers in postmortem central nervous system: changes in schizophrenia. Biol Psychiatry 53:585–592. [DOI] [PubMed] [Google Scholar]
- Degenhardt L, Hall W. (2006). Is cannabis use a contributory cause of psychosis? Can J Psychiatry 51:556–565. [DOI] [PubMed] [Google Scholar]
- Dekker N, Linszen DH, De Haan L. (2009). Reasons for cannabis use and effects of cannabis use as reported by patients with psychotic disorders. Psychopathology 42:350–360. [DOI] [PubMed] [Google Scholar]
- Dias R, Robbins TW, Roberts AC. (1997). Dissociable forms of inhibitory control within prefrontal cortex with an analog of the Wisconsin Card Sort Test: restriction to novel situations and independence from “on-line” processing. J Neurosci 17:9285–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Grace AA. (2013). Peripubertal Diazepam Administration Prevents the Emergence of Dopamine System Hyperresponsivity in the MAM Developmental Disruption Model of Schizophrenia. Neuropsychopharmacology 38:1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone RE, Rizos Z, Nobrega JN, Kapur S, Fletcher PJ. (2007). Gestational methylazoxymethanol acetate treatment impairs select cognitive functions: parallels to schizophrenia. Neuropsychopharmacology 32:483–492. [DOI] [PubMed] [Google Scholar]
- Fergusson DM, Poulton R, Smith PF, Boden JM. (2006). Cannabis and psychosis. BMJ 332:172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Ruiz J, Hernandez M, Ramos JA. (2010). Cannabinoid-dopamine interaction in the pathophysiology and treatment of CNS disorders. CNS Neurosci Ther 16:e72–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagstad P, Mork A, Glenthoj BY, van Beek J, Michael-Titus AT, Didriksen M. (2004). Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology 29:2052–2064. [DOI] [PubMed] [Google Scholar]
- French ED. (1997). delta9-Tetrahydrocannabinol excites rat VTA dopamine neurons through activation of cannabinoid CB1 but not opioid receptors. Neurosci Lett 226:159–162. [DOI] [PubMed] [Google Scholar]
- Fried PA, James DS, Watkinson B. (2001). Growth and pubertal milestones during adolescence in offspring prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol 23:431–436. [DOI] [PubMed] [Google Scholar]
- Gardner EL. (2005). Endocannabinoid signaling system and brain reward: emphasis on dopamine. Pharmacol Biochem Behav 81:263–284. [DOI] [PubMed] [Google Scholar]
- Gastambide F, Cotel MC, Gilmour G, O’Neill MJ, Robbins TW, Tricklebank MD. (2012). Selective remediation of reversal learning deficits in the neurodevelopmental MAM model of schizophrenia by a novel mGlu5 positive allosteric modulator. Neuropsychopharmacology 37:1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill KM, Grace AA. (2014). Corresponding decrease in neuronal markers signals progressive parvalbumin neuron loss in MAM schizophrenia model. Int J Neuropsychopharmacol 17:1609–1619–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason KA, Birnbaum SG, Shukla A, Ghose S. (2012). Susceptibility of the adolescent brain to cannabinoids: long-term hippocampal effects and relevance to schizophrenia. Transl Psychiatry 2:e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Grace AA. (2006). Alterations in medial prefrontal cortical activity and plasticity in rats with disruption of cortical development. Biol Psychiatry 60:1259–1267. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1983). Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience 10:301–315. [DOI] [PubMed] [Google Scholar]
- Gregg L, Barrowclough C, Haddock G. (2007). Reasons for increased substance use in psychosis. Clin Psychol Rev 27:494–510. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 10:40–68. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 23:6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley AE, Schochet T, Landry CF. (2004). Risk taking and novelty seeking in adolescence: introduction to part I. Ann NY Acad Sci 1021:27–32. [DOI] [PubMed] [Google Scholar]
- Kolliakou A, Joseph C, Ismail K, Atakan Z, Murray RM. (2011). Why do patients with psychosis use cannabis and are they ready to change their use? Int J Dev Neurosci 29:335–346. [DOI] [PubMed] [Google Scholar]
- Kuepper R, van Os J, Lieb R, Wittchen HU, Henquet C. (2011). Do cannabis and urbanicity co-participate in causing psychosis? Evidence from a 10-year follow-up cohort study. Psychol Med 41:2121–2129. [DOI] [PubMed] [Google Scholar]
- Leeson VC, Robbins TW, Matheson E, Hutton SB, Ron MA, Barnes TR, Joyce EM. (2009). Discrimination learning, reversal, and set-shifting in first-episode schizophrenia: stability over six years and specific associations with medication type and disorganization syndrome. Biol Psychiatry 66:586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pen GL, Jay TM, Krebs MO. (2011). Effect of antipsychotics on spontaneous hyperactivity and hypersensitivity to MK-801-induced hyperactivity in rats prenatally exposed to methylazoxymethanol. J Psychopharmacol 25:822–835. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. (2005). Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6:312–324. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Behrens MM, Grace AA. (2009). A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci 29:2344–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. (2007). Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci 27:11424–11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. (2009). Gestational methylazoxymethanol acetate administration: a developmental disruption model of schizophrenia. Behav Brain Res 204:306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. (2012). Divergent activation of ventromedial and ventrolateral dopamine systems in animal models of amphetamine sensitization and schizophrenia. Int J Neuropsychopharmacol 15:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone DT, Taylor DA. (2006). The effect of Delta9-tetrahydrocannabinol on sensorimotor gating in socially isolated rats. Behav Brain Res 166:101–109. [DOI] [PubMed] [Google Scholar]
- Marco EM, Granstrem O, Moreno E, Llorente R, Adriani W, Laviola G, Viveros MP. (2007). Subchronic nicotine exposure in adolescence induces long-term effects on hippocampal and striatal cannabinoid-CB1 and mu-opioid receptors in rats. Eur J Pharmacol 557:37–43. [DOI] [PubMed] [Google Scholar]
- Martin CA, Kelly TH, Rayens MK, Brogli BR, Brenzel A, Smith WJ, Omar HA. (2002). Sensation seeking, puberty, and nicotine, alcohol, and marijuana use in adolescence. J Am Acad Child Adolesc Psychiatry 41:1495–1502. [DOI] [PubMed] [Google Scholar]
- McAlonan K, Brown VJ. (2003). Orbital prefrontal cortex mediates reversal learning and not attentional set shifting in the rat. Behav Brain Res 146:97–103. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. (2006). A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry 60:253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea M, Singh ME, McGregor IS, Mallet PE. (2004). Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J Psychopharmacol 18:502–508. [DOI] [PubMed] [Google Scholar]
- Pen GL, Jay TM, Krebs MO. (2011) Effect of antipsychotics on spontaneous hyperactivity and hypersensitivity to MK-801-induced hyperactivity in rats prenatally exposed to methylazoxymethanol. J Psychopharmacol 25:822–835. [DOI] [PubMed] [Google Scholar]
- Penschuck S, Flagstad P, Didriksen M, Leist M, Michael-Titus AT. (2006). Decrease in parvalbumin-expressing neurons in the hippocampus and increased phencyclidine-induced locomotor activity in the rat methylazoxymethanol (MAM) model of schizophrenia. Eur J Neurosci 23:279–284. [DOI] [PubMed] [Google Scholar]
- Quinn HR, Matsumoto I, Callaghan PD, Long LE, Arnold JC, Gunasekaran N, Thompson MR, Dawson B, Mallet PE, Kashem MA, Matsuda-Matsumoto H, Iwazaki T, McGregor IS. (2008). Adolescent rats find repeated Delta(9)-THC less aversive than adult rats but display greater residual cognitive deficits and changes in hippocampal protein expression following exposure. Neuropsychopharmacology 33:1113–1126. [DOI] [PubMed] [Google Scholar]
- Rapoport JL, Addington AM, Frangou S, Psych MR. (2005). The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry 10:434–449. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Beasley CL, Zhang ZJ. (2002). Understanding the neurotransmitter pathology of schizophrenia: selective deficits of subtypes of cortical GABAergic neurons. J Neural Transm 109:881–889. [DOI] [PubMed] [Google Scholar]
- Rubino T, Realini N, Braida D, Alberio T, Capurro V, Vigano D, Guidali C, Sala M, Fasano M, Parolaro D. (2009). The depressive phenotype induced in adult female rats by adolescent exposure to THC is associated with cognitive impairment and altered neuroplasticity in the prefrontal cortex. Neurotox Res 15:291–302. [DOI] [PubMed] [Google Scholar]
- Rubino T, Realini N, Braida D, Guidi S, Capurro V, Vigano D, Guidali C, Pinter M, Sala M, Bartesaghi R, Parolaro D. (2009). Changes in hippocampal morphology and neuroplasticity induced by adolescent THC treatment are associated with cognitive impairment in adulthood. Hippocampus 19:763–772. [DOI] [PubMed] [Google Scholar]
- Schneider M. (2013). Adolescence as a vulnerable period to alter rodent behavior. Cell Tissue Res 354:99–106. [DOI] [PubMed] [Google Scholar]
- Schneider M, Drews E, Koch M. (2005). Behavioral effects in adult rats of chronic prepubertal treatment with the cannabinoid receptor agonist WIN 55,212-2. Behav Pharmacol 16:447–454. [DOI] [PubMed] [Google Scholar]
- Schneider M, Koch M. (2002). The cannabinoid agonist WIN 55,212-2 reduces sensorimotor gating and recognition memory in rats. Behav Pharmacol 13:29–37. [DOI] [PubMed] [Google Scholar]
- Schneider M, Koch M. (2003). Chronic pubertal, but not adult chronic cannabinoid treatment impairs sensorimotor gating, recognition memory, and the performance in a progressive ratio task in adult rats. Neuropsychopharmacology 28:1760–1769. [DOI] [PubMed] [Google Scholar]
- Schneider M, Koch M. (2005). Deficient social and play behavior in juvenile and adult rats after neonatal cortical lesion: effects of chronic pubertal cannabinoid treatment. Neuropsychopharmacology 30:944–957. [DOI] [PubMed] [Google Scholar]
- Schneider M, Koch M. (2007). The effect of chronic peripubertal cannabinoid treatment on deficient object recognition memory in rats after neonatal mPFC lesion. Eur Neuropsychopharmacol 17:180–186. [DOI] [PubMed] [Google Scholar]
- Schneider M, Schomig E, Leweke FM. (2008). Acute and chronic cannabinoid treatment differentially affects recognition memory and social behavior in pubertal and adult rats. Addict Biol 13:345–357. [DOI] [PubMed] [Google Scholar]
- Spano MS, Fattore L, Cadeddu F, Fratta W, Fadda P. (2013). Chronic cannabinoid exposure reduces phencyclidine-induced schizophrenia-like positive symptoms in adult rats. Psychopharmacoloy (Berl) 225:531–542. [DOI] [PubMed] [Google Scholar]
- Stefanis NC, Delespaul P, Henquet C, Bakoula C, Stefanis CN, Van Os J. (2004). Early adolescent cannabis exposure and positive and negative dimensions of psychosis. Addiction 99:1333–1341. [DOI] [PubMed] [Google Scholar]
- Tait DS, Chase EA, Brown VJ. (2014). Attentional set-shifting in rodents: a review of behavioural methods and pharmacological results. Curr Pharm Des 20:5046–5059. [DOI] [PubMed] [Google Scholar]
- Talamini LM, Ellenbroek B, Koch T, Korf J. (2000). Impaired sensory gating and attention in rats with developmental abnormalities of the mesocortex. Implications for schizophrenia. Ann NY Acad Sci 911:486–494. [DOI] [PubMed] [Google Scholar]
- Tanda G, Pontieri FE, Di Chiara G. (1997). Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 276:2048–2050. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Grace AA. (2012). Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci 35:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Laar M, van Dorsselaer S, Monshouwer K, de Graaf R. (2007). Does cannabis use predict the first incidence of mood and anxiety disorders in the adult population? Addiction 102:1251–1260. [DOI] [PubMed] [Google Scholar]
- van Os J, Kenis G, Rutten BP. (2010). The environment and schizophrenia. Nature 468:203–212. [DOI] [PubMed] [Google Scholar]
- Valenti O, Cifelli P, Gill KM, Grace AA. (2011). Antipsychotic drugs rapidly induce dopamine neuron depolarization block in a developmental rat model of schizophrenia. J Neurosci 31:12330–12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigano D, Guidali C, Petrosino S, Realini N, Rubino T, Di Marzo V, Parolaro D. (2009). Involvement of the endocannabinoid system in phencyclidine-induced cognitive deficits modelling schizophrenia. Int J Neuropsychopharmacol 12:599–614. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. (2002). Endocannabinoid signaling in the brain. Science 296:678–682. [DOI] [PubMed] [Google Scholar]
- Wu X, French ED. (2000). Effects of chronic delta9-tetrahydrocannabinol on rat midbrain dopamine neurons: an electrophysiological assessment. Neuropharmacology 39:391–398. [DOI] [PubMed] [Google Scholar]
- Zamberletti E, Beggiato S, Steardo L, Jr., Prini P, Antonelli T, Ferraro L, Rubino T, Parolaro D. (2014). Alterations of prefrontal cortex GABAergic transmission in the complex psychotic-like phenotype induced by adolescent delta-9-tetrahydrocannabinol exposure in rats. Neurobiol Dis 63:35–47. [DOI] [PubMed] [Google Scholar]
- Zimmerman EC, Bellaire M, Ewing SG, Grace AA. (2013). Abnormal stress responsivity in a rodent developmental disruption model of schizophrenia. Neuropsychopharmacology 38:2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}