

Abstract
Background:
Epigenetic drugs like sodium butyrate (NaB) show antidepressant-like effects in preclinical studies, but the exact molecular mechanisms of the antidepressant effects remain unknown. While research using NaB has mainly focused on its role as a histone deacetylase inhibitor (HDACi), there is also evidence that NaB affects DNA methylation.
Methods:
The purpose of this study was to examine NaB’s putative antidepressant-like efficacy in relation to DNA methylation changes in the prefrontal cortex of an established genetic rat model of depression (the Flinders Sensitive Line [FSL]) and its controls (the Flinders Resistant Line).
Results:
The FSL rats had lower levels of ten-eleven translocation methylcytosine dioxygenase 1 (TET1), which catalyzes the conversion of DNA methylation to hydroxymethylation. As indicated by the behavioral despair test, chronic administration of NaB had antidepressant-like effects in the FSL and was accompanied by increased levels of TET1. The TET1 upregulation was also associated with an increase of hydroxymethylation and a decrease of methylation in brain-derived neurotrophic factor (Bdnf), a gene associated with neurogenesis and synaptic plasticity. These epigenetic changes were associated with a corresponding BDNF overexpression.
Conclusions:
Our data support the antidepressant efficacy of HDACis and suggest that their epigenetic effects may also include DNA methylation changes that are mediated by demethylation-facilitating enzymes like TET1.
Keywords: depression, DNA methylation, epigenetics, TET1
Introduction
Depression is the leading cause of disability worldwide and has an average prevalence of approximately 20% (Kessler et al., 1994). Current pharmacotherapy for depression acts primarily on the monoamine neurotransmitter systems, modulating the synaptic availability of neurotransmitters like serotonin and noradrenaline (Connolly and Thase, 2012). However, only about 60–70 percent of depressed patients achieve full remission, indicating a need for development of alternative antidepressant drugs (Fava, 2003; Ressler and Mayberg, 2007). In the last decade, a number of studies have associated aberrations in epigenetic modifications, like DNA methylation and histone modifications, with psychiatric disorders, including depression (Tsankova et al., 2007). Drugs affecting the epigenome have thus gained increased attention with regard to their potential therapeutic efficacy (Insel, 2012; Peedicayil and Kumar, 2012). Histone deacetylase inhibitors (HDACis), for instance, have been extensively used in animal models and have shown antidepressant-like effects (Covington et al., 2009).
Sodium butyrate (NaB) is an established and widely-used HDACi (Candido et al., 1978; Sealy and Chalkley, 1978) that exerts antidepressant-like effects while simultaneously increasing the levels of brain-derived neurotrophic factor (BDNF; Schroeder et al., 2007). BDNF is a neurotrophin that promotes neurogenesis and neuroplasticity, and there is strong evidence for its involvement in the neurobiology of psychiatric disorders like depression (Angelucci et al., 2005). The Bdnf gene is highly conserved between humans and rodents, and has multiple epigenetically-controlled messenger RNA (mRNA) isoforms that are formed by different 5’ untranslated exons splicing to a common 3’ protein-coding exon (Aid et al., 2007; Boulle et al., 2012). One of the best-characterized Bdnf mRNA isoforms is the one driven by the promoter of exon 4 (Bdnf P4; Martinowich et al., 2003; Tsankova et al., 2006; Lubin et al., 2008). The expression of Bdnf P4 correlates well with the expression of total brain BDNF and is highly responsive to neuronal activity and antidepressant drug treatment (West et al., 2001; Dias et al., 2003; Dwivedi et al., 2006). Bdnf P4 was also shown to be regulated both by chromatin modifications through the actions of HDACs and by DNA methylation through the action of DNA methyltransferases (DNMTs; Martinowich et al., 2003).
While most studies using NaB have focused on its effects on histone acetylation, there are also findings showing that it affects DNA methylation (de Haan et al., 1986; Parker et al., 1986; Cosgrove and Cox, 1990; Biard et al., 1992; Boffa et al., 1994; Chiurazzi et al., 1999; Benjamin and Jost, 2001; Detich et al., 2003; Milutinovic et al., 2007; Sarkar et al., 2011; Gu et al., 2012; Shin et al., 2012). DNA methylation in vertebrates has been known to involve the DNMT-catalyzed addition of methyl groups to cytosine residues (5mC), but the mechanisms underlying DNA demethylation have been debated for a long time (Wu and Zhang, 2010). However, recently it was shown that hydroxylation of 5mC by ten-eleven translocation (TET) proteins leads to the formation of 5-hydroxymethylcytosine (5hmC), which can then mediate active DNA demethylation (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009; Guo et al., 2011). Interestingly, brain levels of tet methylcytosine dioxygenase 1 (TET1) were also found to correlate positively with Bdnf expression (Guo et al., 2011; Kaas et al., 2013).
In the present report, we hypothesized a mechanism of action for NaB as an enhancer of gene-specific demethylation in the brain, possibly mediated by NaB’s epigenetic effects on the transcription of DNMTs and/or TETs. For the purpose of this study, and to examine NaB’s antidepressant efficacy in parallel, we used a genetic rat model of depression (the Flinders Sensitive Line [FSL]) and its controls (the Flinders Resistant Line [FRL]). The FSL rats exhibit depression-like behavioral characteristics, including psychomotor retardation and sleep disturbances, as well as emotional memory impairments and dysfunctional regulation of glutamate transmission (Overstreet et al., 2005; Eriksson et al., 2012; Gomez-Galan et al., 2013). The FSL have also been successfully used to study both pharmacological and non-pharmacological interventions with epigenetic and antidepressant-like effects, including the use of acetylating agents to epigenetically modulate Grm2 (Nasca et al., 2013), selective serotonin reuptake inhibitors to epigenetically modulate p11 (Melas et al., 2012) and physical activity to epigenetically modulate Npy (Melas et al., 2013).
Methods
Animals, Drug Administration, and Behavioral Testing
Male FRL and FSL rats (3 months old) were obtained from the breeding stables at the Centre for Psychiatric Research, Aarhus University, Aarhus, Denmark. NaB, diluted in saline, was administered intraperitoneally (i.p.) to FRL (FRL-NaB; n = 7) and FSL (FSL-NaB; n = 7), in parallel with saline (vehicle [Veh]) i.p. injections (FRL-Veh; n = 7; FSL-Veh; n = 7). Both agents were administered chronically, twice a day for 23 days, with an injection volume of 1ml/kg body weight and a dosage of 0.4g NaB/kg of body weight. To assess antidepressant-like behavior, a behavioral despair test (the Porsolt forced swimming test [FST]) was used, which has an excellent predictive validity for antidepressant agents (Porsolt et al., 1977). FST experiments were performed as previously described (Fischer et al., 2012). In brief, FRL and FSL rats were placed in transparent cylinders (24cm diameter, 60cm height) filled with 40cm water at a temperature of 25±1 ˚C for 5min and the immobility time was scored by a person blind to the treatment conditions. The rats were sacrificed 24h after the FST and the brains were harvested and kept at -80 ˚C until shipment to the Karolinska Institutet for brain dissection according to the method of Glowinski and Iversen (1966). For the purpose of the molecular analyses, the PFC was separated in two parts: left and right. The left part was homogenized and divided into equal amounts for DNA and RNA extraction. The right part of the PFC was used for protein analyses. All experiments were approved by the Danish National Committee for Ethics in Animal Experimentation and the Ethical Committee for Protection of Animals at the Karolinska Institutet.
DNA/RNA Extraction and Reverse Transcription
DNA was extracted using the QIAamp DNA Mini Kit (Qiagen GmbH) and total RNA was isolated using the miRNeasy Mini Kit (Qiagen) following treatment with DNase I (Qiagen) to digest contaminating DNA. Complementary DNA (cDNA) was synthesized using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen; Life Technologies) according to the manufacturer’s protocol. In brief, equal amounts of RNA were random-hexamer primed at 25°C for 10min, followed by an incubation with SuperScript III RT at 50°C for 50min, and termination of the reaction at 85°C for 5min. DNA/complementary DNA was stored at -20oC and RNA at -80oC until further processing.
Gene Expression
Quantitative real-time polymerase chain reaction (qRT-PCR) was used to assess mRNA expression of target and reference genes. All qRT-PCR amplifications were performed in triplicate using Power SYBR Green (Applied Biosystems; Life Technologies) on an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems), with the following conditions: 95°C for 10min, followed by 40 repeats of 95°C for 15sec and 60°C for 1min, and a final dissociation stage to monitor amplification specificity. Target genes included Tet1, tet methylcytosine dioxygenase 2 (Tet2), tet methylcytosine dioxygenase 3 (Tet3), DNA (cytosine-5) methyltransferase 1 (Dnmt1), DNA (cytosine-5) methyltransferase 3 alpha (Dnmt3a), and brain-derived neurotrophic factor (Bdnf). Two reference genes were used for normalization purposes (glyceraldehyde-3-phosphate dehydrogenase [Gapdh] and cyclophilin A [Ppia]). Relative quantification of gene expression was calculated using the qBase software (version 1.3.4; Hellemans et al., 2007). The 5’-to-3’ primer sequences were: Tet1 forward (Fw): CCGGTCGCCAAGTGGGTGAT; Tet1 reverse (Rv): GGTCCACACGCTCACGAACCA; Tet2 Fw: TACCGTACAGCCACCCAAAC; Tet2 Rv: CGT GACT GGAACTGC TCACT; Tet3 Fw: GGACTTCTGTGCCCACGCCC; Tet3 Rv: TCAGGGTGCAGACCACAGTGC; Dnmt1 Fw: GGCCAGCCCCAT GAAACGCT; Dnmt1 Rv: GGG GCGTCCAGGTTGCTTCC; Dnmt3a Fw: TCCAACATGAGCCGCTTGGCG; Dnmt3a Rv: GGTGGC GGATGACTGGCACG; Bdnf P4 Fw: GCTG CCT TGATGTTTACTTTGA; Bdnf P4 Rv: GCAACCGAAGTATGAAATAACC; total Bdnf Fw: GGCCCAACGAAGAAAACCAT; total Bdnf Rv: AGCATCACCC GGGAAGTGT; Gapdh Fw: TCGGTGTGAACGGATTTGGCCG; Gapdh Rv: CCGTTGAACTTGCCGTGGGT; Ppia Fw: GGCTGATGGC GAGCCCTTGG; and Ppia Rv: CGTGTGAAGTCACCACCCTGGC. Dnmt3b mRNA levels were not assessed since we have previously found this gene not to be abundantly expressed (Ct values > 35) in the prefrontal cortex region of FSL/FRL animals (Melas et al., 2012).
Protein Expression
Genes showing a difference on the mRNA level were also tested for changes on the protein levels using a modified Western blot protocol, based on Lindfors et al. (2011). Briefly, following sample homogenization and centrifugation, lysates were boiled for 5min and protein concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Inc.) and equal amounts of protein (20 µg for TET1, 20 µg for BDNF, and 60 µg for DNMT1) were loaded on a NuPAGE Novex 4–12% Bis-Tris Gel (Invitrogen). The separated protein was transferred to Amersham Hybond ECL Nitrocellulose Membrane (GE Healthcare UK Limited) overnight at 4°C, and then blocked with 5% nonfat milk for 1h at room temperature. Immunoblotting was performed overnight at 4°C with a polyclonal rabbit anti-methylcytosine dioxygenase 1 (TET1) antibody (1:1 000 dilution; 09–872, Millipore), a monoclonal rabbit anti-BDNF antibody (1:1 000 dilution; ab108319, Abcam), or with a monoclonal rabbit anti- DNMT1 antibody (1:1 000 dilution; No.5119, Cell Signaling Technology) and, separately, with a mouse monoclonal anti-β-actin antibody (1:10 000; A5316, Sigma-Aldrich). After washing, the membrane for detecting TET1, BDNF, or DNMT1 was incubated with HRP-linked goat anti-rabbit secondary antibody (1:60 000 for TET1, 1:50 000 for BDNF, and 1:100 000 for DNMT1; Santa Cruz Biotechnology) and the membrane for detecting β-actin was incubated with HRP-linked goat anti-mouse secondary antibody (1:20 000; Santa Cruz Biotechnology) for 50min at room temperature. Finally, immunoreactive bands were visualized with the Amersham ECL Plus Western Blotting Detection System (GE Healthcare), exposed to Amersham Hyperfilm ECL (GE Healthcare), and optical densities were quantified using the NIH ImageJ software (1.47 version). TET1, BDNF, and DNMT1 expression levels were normalized to the expression of β-actin and the data are presented as relative quantifications.
DNA Hydroxymethylation and Methylation Analyses
Global levels of DNA hydroxymethylation (5hmC)—i.e., the average percent of genomic DNA cytosines that are hydroxymethylated at the 5th carbon—were assessed using the Quest 5-hmC DNA ELISA Kit (Zymo Research Corp.) according to the manufacturer’s protocol. All samples were processed in duplicate and control DNAs were used to generate a standard curve for quantitation of the 5hmC percentage. To measure gene-specific 5hmC levels at the Bdnf P4 promoter, the Hydroxymethyl Collector Kit (Active Motif Inc.) was used according to the manufacturer’s sonication protocol. Gene-specific levels of DNA methylation (5mC) at the Bdnf P4 promoter were determined using the MethylCollector Ultra kit (Active Motif) again according to the manufacturer’s sonication protocol. Samples and inputs from the 5hmC and 5mC assays were analyzed in triplicate with qRT-PCR, using Power SYBR Green (Applied Biosystems) on an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems) under the same conditions described in the gene expression section. Data are presented as % input, which is calculated as 100 x 2-ΔCt, where ΔCt = the average Ct value of the input triplicate minus the average Ct value of the sample triplicate. The 5’-to-3’ primer sequences capturing part of the Bdnf P4 promoter (-67 nucleotides to +99; referenced from P4’s transcription start site) were: Bdnf P4 promoter Fw: TCGAGGCAGAGGAGGTATCA and Bdnf P4 promoter Rv: TCTA GGCAGA GAC TGGGAGA.
Statistical Analyses
Data in bar graphs are presented as mean values ± 1 standard error of the mean (SEM). The Shapiro-Wilk and Levene’s tests were used for assessing the normality of the data and the homogeneity of the variance, respectively. Group differences were analyzed using one-way ANOVA and significant effects were further investigated with the Fisher’s least significant difference (LSD) post-hoc analysis for multiple comparisons. The Pearson’s R correlation test was used to assess the correlation between levels of 5hmC and 5mC. Differences were regarded as statistically significant when p < 0.05. The number of animals used in each experiment, in addition to potential outliers, is denoted in the figure legends. Outliers were excluded from the statistical analyses. All analyses were performed using IBM SPSS Statistics 22 (IBM Corporation).
Results
Sodium Butyrate Exerts Antidepressant-Like Effects
As a first step in our study we assessed behavioral despair in FSL/FRL, both with Veh and NaB treatment, using the forced swimming test. In line with previous results, FSL-Veh rats showed increased immobility compared to FRL-Veh (ANOVA, p = 0.0022; FSL-Veh vs. FRL-Veh, Fisher’s LSD, post-hoc p = 0.0014; Figure 1). Significantly, chronic NaB-treatment rescued the FSL depression-like phenotype; there was a pronounced decrease in immobility time, which reached the levels similar to those of both Veh and NaB-treated FRL (Figure 1).
Figure 1.

The behavioral despair test (Porsolt forced swimming test) was used in our study to evaluate the antidepressant-like effects of NaB. In line with the use of FSL as a genetic model of depression-like states, Veh-treated FSL animals showed significantly higher immobility compared to the FRL-Veh. Treatment with NaB significantly decreased the immobility time in the FSL but had no effects in the FRL strain. Data are presented as means ± SEM. n = 7 animals per group. **p < 0.01.
Aberrant TET1 Levels in the Prefrontal Cortex of FSL
Next, in order to address the hypothesis that NaB’s antidepressant-like effects may be related to changes in the expression of enzymes regulating DNA methylation in the prefrontal cortex, we measured mRNA levels of candidate genes involved in DNA hydroxymethylation and methylation (Tet1, Tet2, Tet3, Dnmt1, and Dnmt3a). Among all the candidate genes tested, only Tet1 showed a difference between vehicle-treated FRL and FSL. More specifically, TET1 mRNA levels were decreased in FSL-Veh compared to FRL-Veh (ANOVA, p = 0.0038; FSL-Veh vs. FRL-Veh, Fisher’s LSD, post-hoc p = 0.039; Figure 2A). NaB treatment, which was previously shown to exert antidepressant-like effects, was associated with an upregulation of Tet1 mRNA in the FSL-NaB group (FSL-Veh vs. FSL-NaB, Fisher’s LSD, post-hoc p = 0.0067; Figure 2A). Western blot experiments confirmed that the Tet1 group-differences observed on the mRNA levels were also present on the protein levels (ANOVA, p = 0.0029; FSL-Veh vs. FRL-Veh, Fisher’s LSD, post-hoc p = 0.0012; FSL-Veh vs. FSL-NaB, Fisher’s LSD, post-hoc p < 0.001; Figure 2B). NaB treatment, though less significantly, was also associated with downregulated Dnmt1 mRNA levels in the FSL-NaB group (ANOVA, p = 0.039; FSL-Veh vs. FSL-NaB, Fisher’s LSD, post-hoc p = 0.014; Figure 2C). However, this difference did not reach significance when measuring the protein levels of DNMT1 (ANOVA, p = 0.84; FSL-Veh vs. FSL-NaB, Fisher’s LSD, post-hoc p = 0.46; Figure 2D).
Figure 2.

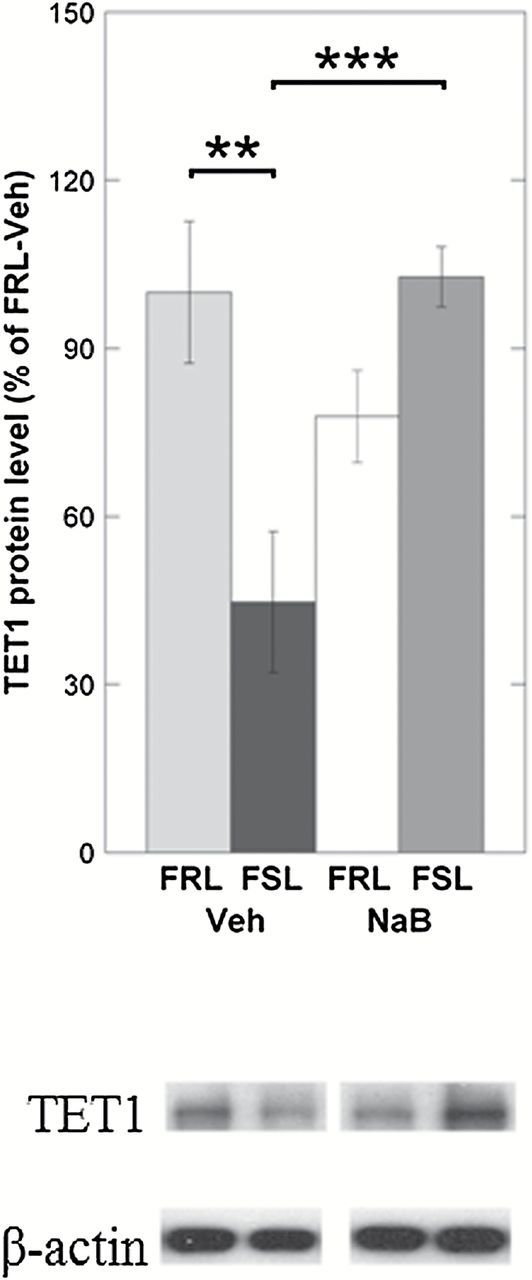

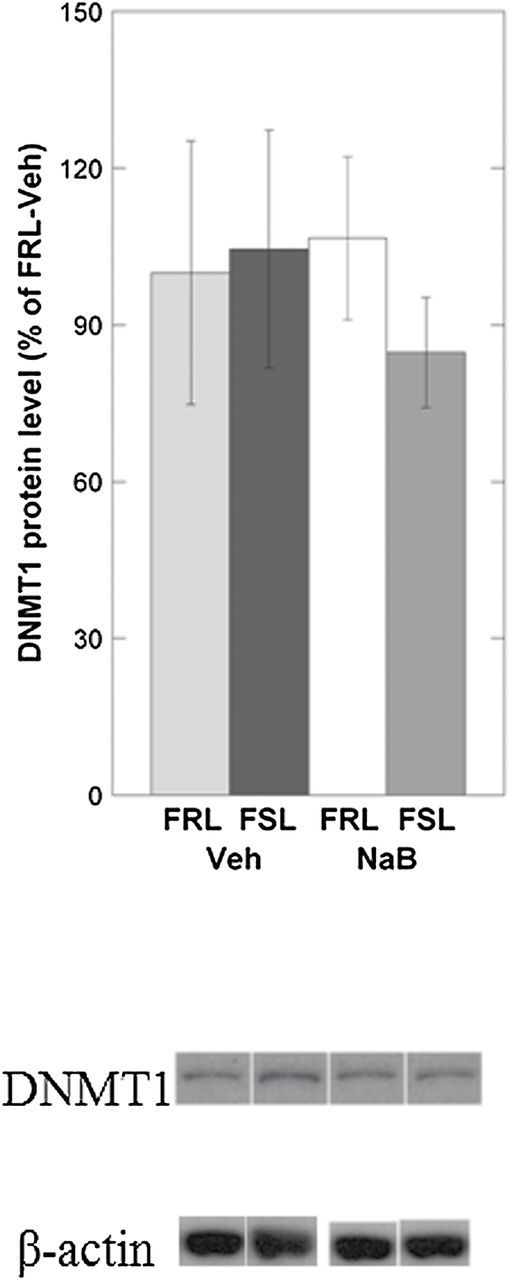
Gene expression levels of Tet and Dnmt mRNAs were measured in the prefrontal cortex of Veh- and NaB-treated FSL and FRL animals using qRT-PCR. Differences found on the mRNA levels were also tested on the protein levels using the Western blot. (A) Only Tet1 mRNA was differentially expressed between FRL-Veh and FSL-Veh, with reduced levels in the FSL. NaB treatment significantly increased Tet1 mRNA expression in the FSL-NaB group. (B) TET1 protein levels were also reduced in FSL-Veh and were “rescued” in the FSL-NaB animals. (C) Even though Dnmt1 mRNA levels were reduced in the FSL-NaB group, (D) protein measurements of DNMT1 did not reach a statistically significant level of difference. Gene expression data are presented as relative quantifications (R.Q.); two reference genes (Gapdh and Ppia) were used for normalization. Protein data are presented as % of FRL-Veh. Lower panels in (B) and (D) show representative Western blot images of TET1 and DNMT1 from the same gel, respectively, with β-actin as loading control. For each panel (B) and (D), the Western blot images were from the same gel. Data are presented as group means ± SEM. For all figures: n = 5–7 animals per group; n = 1 FSL-Veh outlier excluded. (A) n = 1 FSL-NaB outlier excluded. *p < 0.05, **p < 0.01, ***p < 0.001.
TET1 Levels Correlate Positively with DNA Hydroxymethylation Levels at BDNF
The aberrant levels of TET1 in the prefrontal cortex of FSL-Veh suggested putative changes in DNA hydroxymethylation. However, the assessment of global (genome-wide) 5hmC levels revealed no differences between groups (ANOVA, p = 0.12; Figure 3A). This led us to test the hypothesis that TET1 may affect specific genes. Among the list of candidate genes, we decided to study Bdnf since it is both associated with depression’s pathophysiology and is affected by brain levels of TET1 (Guo et al., 2011; Kaas et al., 2013). In line with our data showing a TET1 downregulation in FSL-Veh, we found a significant reduction in 5hmC levels at the Bdnf P4 locus in this group (ANOVA, p = 0.039; FSL-Veh vs. FRL-Veh, Fisher’s LSD, post hoc p = 0.032; Figure 3B). Interestingly, we also found that the NaB-dependent TET1 overexpression in FSL was associated with higher 5hmC levels at Bdnf P4 (FSL-Veh vs. FSL-NaB, Fisher’s LSD, post-hoc p = 0.0059; Figure 3B). Next, since hydroxymethylation is considered a mediator of active demethylation in the adult brain (Guo et al., 2011), we also examined the levels of 5mC at the same Bdnf locus. In accord with the decreased 5hmC levels in FSL-Veh, the same group was hypermethylated at Bdnf P4 compared to FRL-Veh (ANOVA, p = 0.023; FSL-Veh vs. FRL-Veh, Fisher’s LSD, post-hoc p = 0.025; Figure 3B). Additionally, the NaB-dependent increase in 5hmC levels in Bdnf P4 of FSL was associated with DNA hypomethylation at the same locus (FSL-Veh vs. FSL-NaB, Fisher’s LSD, post-hoc p = 0.0035; Figure 3B). Finally, this inverse relationship between 5hmC and 5mC levels at the Bdnf P4 promoter was supported by a correlation analysis (p = 0.045, correlation coefficient = -0.48, Figure 3C).
Figure 3.
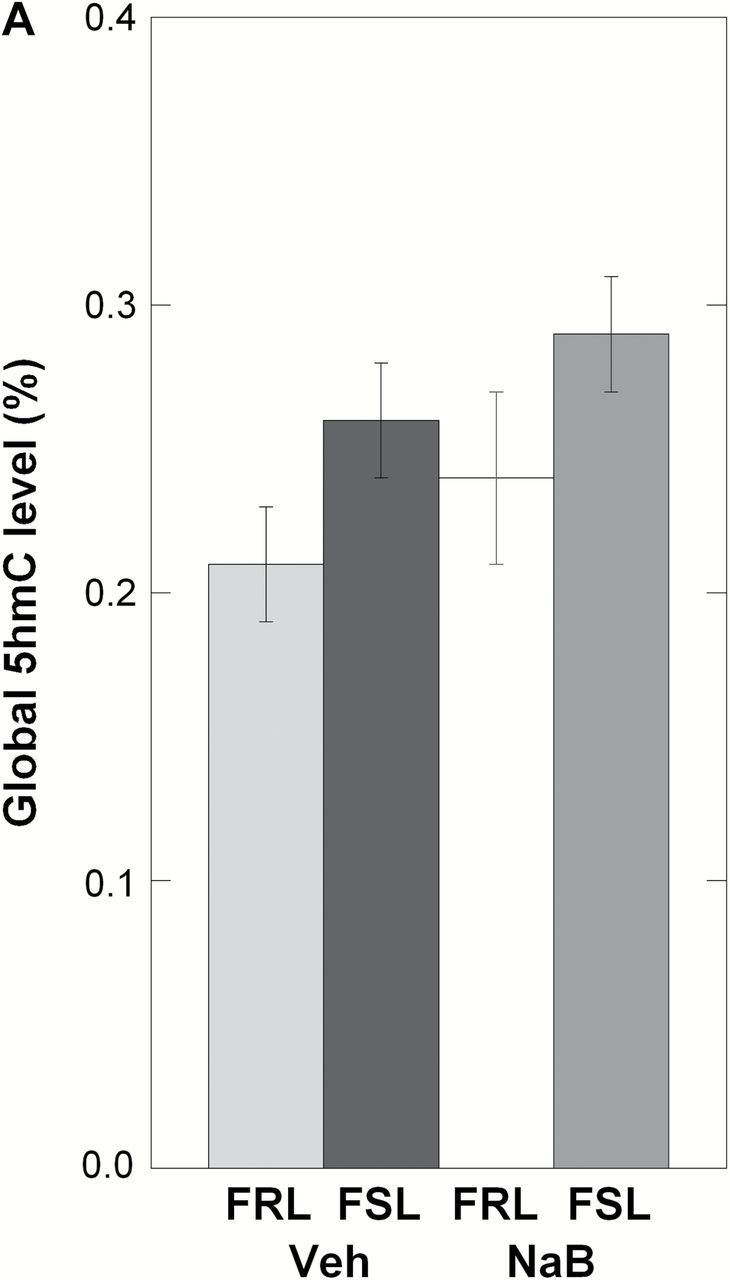


DNA hydroxymethylation (5hmC) and methylation (5mC) analyses were performed in the prefrontal cortex of Veh- and NaB-treated FRL and FSL animals. (A) Global 5hmC level was measured using ELISA and showed no significant changes between any of the groups. Data are presented as the average percent of the genomic DNA cytosines that are hydroxymethylated at the 5th carbon. (B) Region-specific 5hmC and 5mC levels were measured at the Bdnf promoter 4 (P4) region, spanning the transcription start site, and data are presented as relative values. FSL-Veh animals had a significant reduction in 5hmC levels at the Bdnf P4 region, and NaB treatment was associated with increased 5hmC levels in the FSL-NaB group. In contrast, 5mC levels at the same locus showed an inverse pattern compared to 5hmC levels, with hypermethylation in FSL-Veh but hypomethylation in the FSL-NaB group. (C) Pearson’s correlation analysis supported an inverse relationship between 5hmC and 5mC level at the Bdnf P4 locus (rho = -0.48; p = 0.045). Data in A and B are presented as group means ± SEM. n = 4–6 animals per group; n = 1 FSL-Veh outlier excluded. *p < 0.05, **p < 0.01.
DNA Hydroxymethylation Is Associated with BDNF Expression
TET1-driven DNA hydroxymethylation is emerging as a marker of gene activation (Guo et al., 2011). In order to examine this relationship, we measured mRNA levels of the Bdnf P4 isoform. Indeed, FSL-Veh rats that were found to have lower TET1 and 5hmC levels had downregulated levels of the Bdnf P4 transcript in the prefrontal cortex (ANOVA, p = 0.0064; FSL-Veh vs. FRL-Veh, Fisher’s LSD, post-hoc p = 0.0012; Figure 4A). In addition, NaB-dependent increase in TET1 and 5hmC levels in the FSL group was associated with a Bdnf P4 overexpression (FSL-Veh vs. FSL-NaB, Fisher’s LSD, post hoc p = 0.014; Figure 4A). Since previous studies (West et al., 2001; Dias et al., 2003; Dwivedi et al., 2006) have shown a positive correlation between P4-driven Bdnf mRNA levels and the expression of total Bdnf mRNA levels, we also wanted to test this finding in our model. In agreement with the latter studies, measurements of total Bdnf levels in our samples revealed similar expression patterns as with BDNF P4 (ANOVA, p < 0.001; FSL-Veh vs. FRL-Veh, Fisher’s LSD, post hoc p = 0.045; FSL-Veh vs. FSL-NaB, Fisher’s LSD, post hoc p = 0.014; Figure 4A). Finally, the differences of total BDNF mRNA were confirmed on the protein levels (ANOVA, p = 0.021; FSL-Veh vs. FRL-Veh, Fisher’s LSD, post-hoc p = 0.017; FSL-Veh vs. FSL-NaB, Fisher’s LSD, post-hoc p = 0.012; Figure 4B). These data suggest that the P4 promoter of Bdnf contributes substantially to the levels of total Bdnf in the prefrontal cortex.
Figure 4.

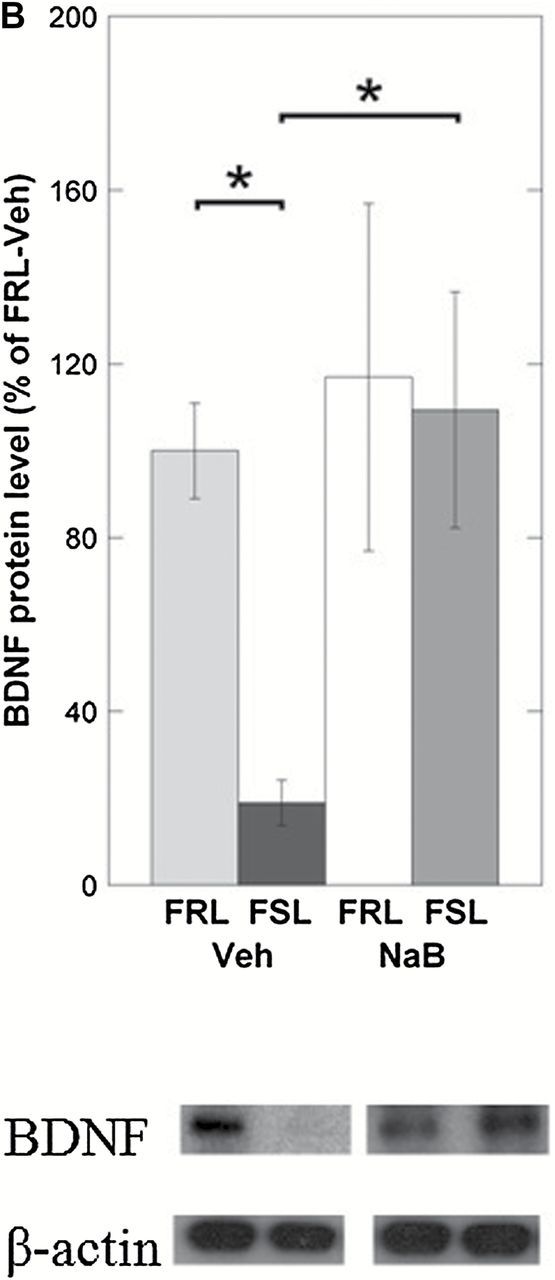
The expression of Bdnf P4 and total Bdnf mRNA levels were measured in the prefrontal cortex of Veh- and NaB-treated FRL and FSL animals using qRT-PCR; BDNF protein levels were tested using the Western blot. (A) While Bdnf P4 was downregulated in the FSL-Veh, NaB treatment increased Bdnf P4 levels in the FSL-NaB group. (B) This expression pattern was similar to the 5hmC level pattern at the Bdnf P4 locus. Total Bdnf exhibited a similar expression pattern as the Bdnf P4–driven expression. BDNF protein levels were also reduced in FSL-Veh and were “rescued” in the FSL-NaB animals. Gene expression data are presented as relative quantifications (R.Q.); two reference genes (Gapdh and Ppia) were used for normalization. Protein data are presented as % of FRL-Veh. The lower panel in (B) shows representative Western blot images of BDNF from the same gel with β-actin as loading control. All data represent group means ± SEM. n = 5–7 animals per group; n = 1 FRL-NaB outlier excluded in mRNA analysis. *p < 0.05, **p < 0.01.
Discussion
General Points
The epigenetic regulation of the genome refers to a number of mechanisms and modifications that occur at the level of DNA, RNA, histones, and chromatin remodeling complexes. Among these epigenetic modifications, histone acetylation and DNA methylation are the most extensively studied processes that have also been associated with psychopathology (Tsankova et al., 2007). Histone acetylation occurs at lysine residues on their amino-terminal tails and, when acetylated, the positive charge of lysine is eliminated. This decreases the histones’ affinity to the negatively-charged DNA, which is thought to lead to a chromatin loosening that gives access to regulatory proteins that increase transcriptional activity (Struhl, 1998). Histone deacetylases are enzymes that can remove these histone acetyl-groups, allowing the DNA to wrap the histones more tightly and thus repress transcription. Of note, there are already drugs (e.g., SAHA) that act by inhibiting histone deacetylation and are used as therapeutic agents against cancer. However, during recent years, HDACis have also received increased attention due to their putative efficacy in the therapy of psychiatric disorders (Grayson et al., 2010; Menke et al., 2012). NaB belongs to the aforementioned class of epigenetic agents, with an established role in the inhibition of most class I and II histone deacetylases (Candido et al., 1978; Sealy and Chalkley, 1978). While the majority of studies have utilized NaB for its HDACi properties and examined downstream acetylation changes, reports have also indicated an additional role for NaB in affecting DNA methylation (de Haan et al., 1986; Parker et al., 1986; Cosgrove and Cox, 1990; Biard et al., 1992; Boffa et al., 1994; Chiurazzi et al., 1999; Benjamin and Jost, 2001; Sarkar et al., 2011; Shin et al., 2012). NaB was also found to affect DNA methylation in a cell type–specific manner. Specifically, while NaB incubation of HeLa cells (cells with a cervical origin) resulted in DNA hypermethylation, incubation of glioblastoma cells (cells with a brain origin) resulted in DNA hypomethylation (Cosgrove and Cox, 1990). In line with the NaB demethylating data, studies using the HDACi valproic acid (VPA) also showed decreased methylation of tumor suppressor genes in neuroblastoma (Gu et al., 2012) and of other specific genes in HEK 293 cells (Detich et al., 2003; Milutinovic et al., 2007). Importantly, the VPA demethylating activity was shown to be DNA replication-independent (Detich et al., 2003), which is relevant for studies using postmitotic tissue like the prefrontal cortex. DNA methylation surrounding transcriptional start sites has generally been coupled to gene silencing (Brenet et al., 2011), and aberrant DNA methylation changes have been associated with psychiatric disorders, including depression (Klengel et al., 2014). DNA methylation, once thought to be a rather permanent modification of cytosine residues, was recently found to be reversible through the action of TET-family enzymes. TETs can oxidize methylated cytosines to produce hydroxymethylated residues and, through this process, 5mC can either be passively reverted to unmethylated cytosine through DNA replication or it can be actively depleted through additional oxidation and thymine DNA glycosylase-mediated base excision repair (Kohli and Zhang, 2013). The latter mechanism has important implications for studies of postmitotic tissues where cell division and DNA replication have ceased; as is often the case for neurons. In addition, 5hmC has been found to be most abundant in the brain, where it is particularly enriched in active genes, indicating a crucial role for this DNA modification in neuronal gene expression and memory formation (Mellen et al., 2012; Kaas et al., 2013).
Sodium Butyrate’s Antidepressant-Like Effects are Associated with Increased Levels of TET1 in the Prefrontal Cortex
We hypothesized that part of NaB’s antidepressant-like properties may involve changes on the DNA methylation level. We chose to work with the FSL rat line, which is an established genetic model of depression that has been successfully used for the past 25 years in depression-like and antidepressant-related studies (Overstreet et al., 2005; Overstreet and Wegener, 2013). The first key finding was that NaB exerted a clear antidepressant-like effect in the FSL, in accord with previous behavioral data from NaB studies in mice (Schroeder et al., 2007). Next, we examined whether NaB affected the expression of key enzymes regulating DNA methylation (Dnmt1 and Dnmt3a) and hydroxymethylation (Tet1, Tet2, and Tet3) in the prefrontal cortex, a critical region implicated in the pathophysiology of depression (Serafini, 2012). We found that TET1 was the only enzyme that differed between vehicle-treated FSL and controls, and that it was also affected by NaB treatment. Specifically, FSL-Veh had decreased Tet1 mRNA levels and, following NaB treatment, these levels were restored to normal, a finding that was also confirmed on the protein level. The treatment-related TET1 upregulation may be the consequence of a NaB-mediated increase in histone acetylation at Tet1, a finding that warrants future investigation. On the other hand, the TET1 difference between FSL-Veh and controls may be related to nucleotide variations, given that the FSL is a genetic model. In line with this assumption, we recently found that the FSL harbors a functional nucleotide polymorphism in the promoter of the neuropeptide Y (Npy) gene, which affects Npy’s transcriptional activity (Melas et al., 2013). However, one limitation that needs to be acknowledged at this point is related to the pharmacological intervention. Specifically, the chronically i.p.-administered injections could represent a chronic stress paradigm that needs to be taken into consideration when interpreting the present report’s results.
TET1 Upregulation is Associated with Increased Hydroxymethylation and Gene-Expression Levels of Bdnf
The second part of our molecular analyses focused on a specific gene, the Bdnf, which has been repeatedly associated with depression (Angelucci et al., 2005). We found that the decreased levels of TET1 in FSL-Veh were associated with decreased levels both of 5hmC in the Bdnf P4 promoter and of Bdnf P4 mRNA levels. Interestingly, the NaB-associated upregulation of TET1 led to an increase in both 5hmC and Bdnf mRNA levels. These findings are in line with two recent studies showing that TET1 overexpression in the mouse hippocampus also leads to increased Bdnf expression (Guo et al., 2011; Kaas et al., 2013). Next, we also analyzed the 5mC levels at the same Bdnf locus and convincingly found a reversed pattern. More specifically, increased levels of 5mC were found in the FSL-Veh group, which had reduced TET1 and 5hmC levels, whereas lower levels of 5mC were present in the NaB-treated FSL group, which had higher TET1 and 5hmC levels. The latter group also had decreased Dnmt1 mRNA levels, but we were not able to replicate this finding on the protein level. Nonetheless, it is worth mentioning that BDNF P4 was shown to be regulated by DNMT1 in cortical mouse neurons (Martinowich et al., 2003) and treatment with DNMT inhibitors was found to increase Bdnf levels in the rat hippocampus (Sales et al., 2011). Taken together, our analyses of modified cytosines at the Bdnf locus provide evidence that HDACi-associated upregulation of TET1 in the prefrontal cortex leads to increased 5mC to 5hmC conversion, which may reflect active DNA demethylation. However, it is noteworthy that the changes in TET1 levels were not associated with changes in global 5hmC levels. TET1 has been found to be present in both the nucleus and the soma of neurons, and also in the soma of astrocytes (Kaas et al., 2013). This could imply that the global effect of TET1 on the nuclear 5hmC levels can be buffered”by the TET1 localization in regions other than the nucleus. Thus, the ability of other studies to detect global 5hmC changes may be due to the employment of more drastic (e.g., viral-mediated) methodologies for TET1 overexpression (Guo et al., 2011; Kaas et al., 2013). Finally, one should also keep in mind that while only two, and not six, comparisons were of main interest (FSL-Veh vs FRL-Veh and FSL-Veh vs FSL-NaB), Fisher’s LSD was selected as the post-hoc test; this test does not provide an adequate correction for multiple comparisons. Also, the present data derive from the PFC of male rats only and, thus, both regional and gender differences need to be addressed in the future. For example, studies using female FSL have showed that certain selective serotonin reuptake inhibitors (e.g., escitalopram) lead to decreased expression of BDNF in the hippocampus (Hansson et al., 2011).
Clinical Relevance
Our preclinical study adds to the growing evidence suggesting that epigenetic-acting agents such as HDAC inhibitors (e.g., NaB, SAHA, and MS275) and DNMT inhibitors (e.g., 5-AzaC) have potential antidepressant properties (Schroeder et al., 2007; Covington et al., 2009; Zhu et al., 2009; Sales et al., 2011; Uchida et al., 2011; Yamawaki et al., 2012). The HDAC-dependent regulation of Bdnf P4 has repeatedly been supported by studies using HDACis, like NaB and VPA, both of which exert a positive effect on BDNF P4 transcription (Tsankova et al., 2006; Bredy et al., 2007). Recently, another preclinical study showed that treatment with a different HDACi (CI-994) ameliorated anxiety-related phenotypes (Graff et al., 2014). Anxiety disorders show high comorbidity with depression and have also been repeatedly associated with reduced levels of BDNF (Suliman et al., 2013). In addition, both depression and anxiety often co-occur with substance use disorders. This high co-occurrence has often been explained in terms of self-medication, with the patient using drugs in an attempt to reduce anxiety (Robinson et al., 2009). Interestingly, drugs like alcohol, cocaine, and nicotine have all been associated with a decrease in HDAC activity (Kumar et al., 2005; Pandey et al., 2008; Levine et al., 2011). However, one should keep in mind that there are four major classes of HDACs with different members in each class, and some members (class II) have the ability to shuttle between the nucleus and the cytoplasm. In addition, each member shows variability in cell-type distribution in the brain (Takase et al., 2013). HDACis, like NaB, act by inhibiting a number of HDACs non-specifically. Therefore, it is very unlikely that the therapeutic-like effects of current HDACis lie within the regulation of single genes like Tet1 and Bdnf; rather, they most probably depend on the varying contributions of HDAC members present within a specific cell population. Thus, there is not only a need for elucidating the role that each HDAC plays in psychopathology but also a need for the development of more specific inhibitors
Concluding Remarks
In the present study we found depression-like states to be associated both with changes in levels of the TET1 demethylation-facilitating enzyme and with concomitant alterations in hydroxymethylation of Bdnf. The molecular aberrations in these genes were found to be reversible with the use of an epigenetic targeting drug. These preclinical data obviously represent an oversimplified picture of the underpinnings of major depressive disorder in humans, and may perhaps relate to certain endophenotypes of the disorder. It has been recognized that most neuropsychiatric disorders, including depression, are not the result of mutations in a single gene but represent the outcome of molecular disturbances with low effect sizes in multiple genetic loci that are part of distinct regulatory pathways (Jia et al., 2011). This also explains why the testing of single-target agents has mostly proven ineffective in psychopharmacological trials. Therefore, the trend has shifted towards the development of multi-target drugs for treating complex disorders (Lu et al., 2012). The latter concept in drug design is also supported by findings showing that many genes are part of co-expression networks, meaning that their expression covaries (Oldham et al., 2008). A recent study in substance use disorders identified epigenetic modifications as being determinant factors of gene co-expression associated with alcoholism (Ponomarev et al., 2012). This implies that certain epigenetic marks may regulate a number of disorder-associated genes in a similar manner. Thus, pinpointing these aberrant epigenetic mechanisms and their target genes may lead to the discovery of novel multi-target epigenetic therapeutics with increased antidepressant efficacies.
Statement of Interest
None.
Acknowledgments
This work was supported by the Karolinska Institutet’s Faculty Funds, the Swedish Research Council (grant numbers 2010–3631 CL, 10414 AAM), the Fredrik and Ingrid Thurings Foundation, the regional agreement on medical training and clinical research (ALF) between the Stockholm County Council and Karolinska Institutet (CL), the Danish Medical Research Council, and the Lundbeck foundation. These funding sources had no involvement in the study design, analysis, and interpretation of the data, in the writing of the manuscript, or in the decision to submit the report for publication. We thank Dr Ida Nilsson for helping in the experimental procedures.
References
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. (2007). Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res 85 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Brene S, Mathe AA. (2005). BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry 10:345–352. [DOI] [PubMed] [Google Scholar]
- Benjamin D, Jost JP. (2001). Reversal of methylation-mediated repression with short-chain fatty acids: evidence for an additional mechanism to histone deacetylation. Nucleic Acids Res 29:3603–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biard DS, Maratrat M, Thybaud V, Melcion C, Sarasin A. (1992). Flow cytometric detection of drugs altering the DNA methylation pattern. Cancer Res 52:5213–5218. [PubMed] [Google Scholar]
- Boffa LC, Mariani MR, Parker MI. (1994). Selective hypermethylation of transcribed nucleosomal DNA by sodium butyrate. Exp Cell Res 211:420–423. [DOI] [PubMed] [Google Scholar]
- Boulle F, van den Hove DL, Jakob SB, Rutten BP, Hamon M, van Os J, Lesch KP, Lanfumey L, Steinbusch HW, Kenis G. (2012). Epigenetic regulation of the BDNF gene: implications for psychiatric disorders. Mol Psychiatry 17:584–596. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. (2007). Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem 14:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, Scandura JM. (2011). DNA methylation of the first exon is tightly linked to transcriptional silencing. PLOS ONE 6:e14524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido EP, Reeves R, Davie JR. (1978). Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14:105–113. [DOI] [PubMed] [Google Scholar]
- Chiurazzi P, Pomponi MG, Pietrobono R, Bakker CE, Neri G, Oostra BA. (1999). Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum Mol Gen 8:2317–2323. [DOI] [PubMed] [Google Scholar]
- Connolly KR, Thase ME. (2012). Emerging drugs for major depressive disorder. EOMD 17:105–126. [DOI] [PubMed] [Google Scholar]
- Cosgrove DE, Cox GS. (1990). Effects of sodium butyrate and 5-azacytidine on DNA methylation in human tumor cell lines: variable response to drug treatment and withdrawal. Biochim Biophys Acta 1087:80–86. [DOI] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. (2009). Antidepressant actions of histone deacetylase inhibitors. J Neurosci 29:11451–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan JB, Gevers W, Parker MI. (1986). Effects of sodium butyrate on the synthesis and methylation of DNA in normal cells and their transformed counterparts. Cancer Res 46:713–716. [PubMed] [Google Scholar]
- Detich N, Bovenzi V, Szyf M. (2003). Valproate induces replication-independent active DNA demethylation. J Biol Chem 278:27586–27592. [DOI] [PubMed] [Google Scholar]
- Dias BG, Banerjee SB, Duman RS, Vaidya VA. (2003). Differential regulation of brain derived neurotrophic factor transcripts by antidepressant treatments in the adult rat brain. Neuropharmacology 45:553–563. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Pandey GN. (2006). Antidepressants reverse corticosterone-mediated decrease in brain-derived neurotrophic factor expression: differential regulation of specific exons by antidepressants and corticosterone. Neuroscience 139:1017–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson TM, Delagrange P, Spedding M, Popoli M, Mathe AA, Ogren SO, Svenningsson P. (2012). Emotional memory impairments in a genetic rat model of depression: involvement of 5-HT/MEK/Arc signaling in restoration. Mol Psychiatry 17:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava M. (2003). Diagnosis and definition of treatment-resistant depression. Biol Psychiatry 53:649–659. [DOI] [PubMed] [Google Scholar]
- Fischer CW, Liebenberg N, Elfving B, Lund S, Wegener G. (2012). Isolation-induced behavioural changes in a genetic animal model of depression. Behav Brain Res 230:85–91. [DOI] [PubMed] [Google Scholar]
- Glowinski J, Iversen LL. (1966). Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J Neurochem 13:655–669. [DOI] [PubMed] [Google Scholar]
- Gomez-Galan M, De Bundel D, Van Eeckhaut A, Smolders I, Lindskog M. (2013). Dysfunctional astrocytic regulation of glutamate transmission in a rat model of depression. Mol Psychiatry 18:582–594. [DOI] [PubMed] [Google Scholar]
- Graff J, Joseph NF, Horn ME, Samiei A, Meng J, Seo J, Rei D, Bero AW, Phan TX, Wagner F, Holson E, Xu J, Sun J, Neve RL, Mach RH, Haggarty SJ, Tsai LH. (2014). Epigenetic Priming of Memory Updating during Reconsolidation to Attenuate Remote Fear Memories. Cell 156:261–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson DR, Kundakovic M, Sharma RP. (2010). Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol 77:126–135. [DOI] [PubMed] [Google Scholar]
- Gu S, Tian Y, Chlenski A, Salwen HR, Lu Z, Raj JU, Yang Q. (2012). Valproic acid shows a potent antitumor effect with alteration of DNA methylation in neuroblastoma. Anticancer Drugs 23:1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson AC, Rimondini R, Heilig M, Mathe AA, Sommer WH. (2011). Dissociation of antidepressant-like activity of escitalopram and nortriptyline on behaviour and hippocampal BDNF expression in female rats. J Psychopharmacol 25:1378–1387. [DOI] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. (2007). qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. (2012). Next-generation treatments for mental disorders. Sci Transl Med 4:155–119. [DOI] [PubMed] [Google Scholar]
- Jia P, Kao CF, Kuo PH, Zhao Z. (2011). A comprehensive network and pathway analysis of candidate genes in major depressive disorder. BMC Syst Biol 5(Suppl 3):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, King JR, Song H, Sweatt JD. (2013). TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 79:1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S, Wittchen HU, Kendler KS. (1994). Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry 51:8–19. [DOI] [PubMed] [Google Scholar]
- Klengel T, Pape J, Binder EB, Mehta D. (2014). The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology Jan 19. [DOI] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. (2009). The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 324:929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. (2005). Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48:303–314. [DOI] [PubMed] [Google Scholar]
- Levine A, Huang Y, Drisaldi B, Griffin EA, Jr., Pollak DD, Xu S, Yin D, Schaffran C, Kandel DB, Kandel ER. (2011). Molecular mechanism for a gateway drug: epigenetic changes initiated by nicotine prime gene expression by cocaine. Sci Transl Med 3:107–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindfors C, Nilsson IA, Garcia-Roves PM, Zuberi AR, Karimi M, Donahue LR, Roopenian DC, Mulder J, Uhlen M, Ekstrom TJ, Davisson MT, Hokfelt TG, Schalling M, Johansen JE. (2011). Hypothalamic mitochondrial dysfunction associated with anorexia in the anx/anx mouse. Proc Natl Acad Sci USA 108:18108–18113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JJ, Pan W, Hu YJ, Wang YT. (2012). Multi-target drugs: the trend of drug research and development. PLOS ONE 7:e40262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. (2008). Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci 28:10576–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302:890–893. [DOI] [PubMed] [Google Scholar]
- Melas PA, Rogdaki M, Lennartsson A, Bjork K, Qi H, Witasp A, Werme M, Wegener G, Mathe AA, Svenningsson P, Lavebratt C. (2012). Antidepressant treatment is associated with epigenetic alterations in the promoter of P11 in a genetic model of depression. Int J Neuropsychop 15:669–679. [DOI] [PubMed] [Google Scholar]
- Melas PA, Lennartsson A, Vakifahmetoglu-Norberg H, Wei Y, Aberg E, Werme M, Rogdaki M, Mannervik M, Wegener G, Brene S, Mathe AA, Lavebratt C. (2013). Allele-specific programming of Npy and epigenetic effects of physical activity in a genetic model of depression. Transl Psychiatry 3:e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151:1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A, Klengel T, Binder EB. (2012). Epigenetics, depression and antidepressant treatment. Curr Pharm Des 18:5879–5889. [DOI] [PubMed] [Google Scholar]
- Milutinovic S, D’Alessio AC, Detich N, Szyf M. (2007). Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis 28:560–571. [DOI] [PubMed] [Google Scholar]
- Nasca C, Xenos D, Barone Y, Caruso A, Scaccianoce S, Matrisciano F, Battaglia G, Mathe AA, Pittaluga A, Lionetto L, Simmaco M, Nicoletti F. (2013). L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA 110:4804–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. (2008). Functional organization of the transcriptome in human brain. Nat Neurosci 11:1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Wegener G. (2013). The flinders sensitive line rat model of depression--25 years and still producing. Pharmacol Rev 65:143–155. [DOI] [PubMed] [Google Scholar]
- Overstreet DH, Friedman E, Mathe AA, Yadid G. (2005). The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci Biobehav Rev 29:739–759. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. (2008). Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci 28:3729–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MI, de Haan JB, Gevers W. (1986). DNA hypermethylation in sodium butyrate-treated WI-38 fibroblasts. J Biol Chem 261:2786–2790. [PubMed] [Google Scholar]
- Peedicayil J, Kumar A. (2012). Time for clinical trials of epigenetic drugs in psychiatric disorders? Br J Clin Pharmacol 73:309–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. (2012). Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci 32:1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M. (1977). Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther 229:327–336. [PubMed] [Google Scholar]
- Ressler KJ, Mayberg HS. (2007). Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat Neurosci 10:1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Sareen J, Cox BJ, Bolton J. (2009). Self-medication of anxiety disorders with alcohol and drugs: Results from a nationally representative sample. J Anxiety Disord 23:38–45. [DOI] [PubMed] [Google Scholar]
- Sales AJ, Biojone C, Terceti MS, Guimaraes FS, Gomes MV, Joca SR. (2011). Antidepressant-like effect induced by systemic and intra-hippocampal administration of DNA methylation inhibitors. Br J Pharmacol 164:1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Abujamra AL, Loew JE, Forman LW, Perrine SP, Faller DV. (2011). Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res 31:2723–2732. [PubMed] [Google Scholar]
- Schroeder FA, Lin CL, Crusio WE, Akbarian S. (2007). Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry 62:55–64. [DOI] [PubMed] [Google Scholar]
- Sealy L, Chalkley R. (1978). The effect of sodium butyrate on histone modification. Cell 14:115–121. [DOI] [PubMed] [Google Scholar]
- Serafini G. (2012). Neuroplasticity and major depression, the role of modern antidepressant drugs. World J Psychiatry 2:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Kim JH, Lee YS, Lee YC. (2012). Change in gene expression profiles of secreted frizzled-related proteins (SFRPs) by sodium butyrate in gastric cancers: induction of promoter demethylation and histone modification causing inhibition of Wnt signaling. Int J Oncol 40:1533–1542. [DOI] [PubMed] [Google Scholar]
- Struhl K. (1998). Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 12:599–606. [DOI] [PubMed] [Google Scholar]
- Suliman S, Hemmings SM, Seedat S. (2013). Brain-Derived Neurotrophic Factor (BDNF) protein levels in anxiety disorders: systematic review and meta-regression analysis. Front Integr Neurosci 7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takase K, Oda S, Kuroda M, Funato H. (2013). Monoaminergic and neuropeptidergic neurons have distinct expression profiles of histone deacetylases. PLOS ONE 8:e58473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 9:519–525. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. (2007). Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8:355–367. [DOI] [PubMed] [Google Scholar]
- Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, Suzuki T, Miyata N, Watanabe Y. (2011). Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69:359–372. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. (2001). Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98:11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Zhang Y. (2010). Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 11:607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamawaki Y, Fuchikami M, Morinobu S, Segawa M, Matsumoto T, Yamawaki S. (2012). Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus. World J Biol Psychiatry 13:458–467. [DOI] [PubMed] [Google Scholar]
- Zhu H, Huang Q, Xu H, Niu L, Zhou JN. (2009). Antidepressant-like effects of sodium butyrate in combination with estrogen in rat forced swimming test: involvement of 5-HT(1A) receptors. Behav Brain Res 196:200–206. [DOI] [PubMed] [Google Scholar]