

Abstract
Background:
Repeated alcohol exposure is known to increase subsequent ethanol consumption in mice. However, the underlying mechanisms have not been fully elucidated. One postulated mechanism involves epigenetic modifications, including histone modifications and DNA methylation of relevant genes such as NR2B or BDNF.
Methods:
To investigate the role of epigenetic mechanisms in the development of alcohol drinking behavior, an established chronic intermittent ethanol exposure reinforced ethanol drinking mouse model with vapor inhalation over two 9-day treatment regimens was used. The DNA methyltransferase inhibitor, 5-azacytidine or the histone deacetylase inhibitor, Trichostatin A was administered (intraperitoneally) to C57BL/6 mice 30min before daily exposure to chronic intermittent ethanol. Changes in ethanol consumption were measured using the 2-bottle choice test.
Results:
The results indicated that systemic administration of Trichostatin A (2.5 µg/g) facilitated chronic intermittent ethanol-induced ethanol drinking, but systemic administration of 5-azacytidine (2 µg/g) did not cause the same effect. However, when 5-azacytidine was administered by intracerebroventricular injection, it facilitated chronic intermittent ethanol-induced ethanol drinking. Furthermore, the increased drinking caused by chronic intermittent ethanol was prevented by injection of a methyl donor, S-adenosyl-L-methionine. To provide evidence that chronic intermittent ethanol- or Trichostatin A-induced DNA demethylation and histone modifications of the NR2B promoter may underlie the altered ethanol consumption, we examined epigenetic modifications and NR2B expression in the prefrontal cortex of these mice. Chronic intermittent ethanol or Trichostatin A decreased DNA methylation and increased histone acetylation in the NR2B gene promoter, as well as mRNA levels of NR2B in these mice.
Conclusions:
Taken together, these results indicate that epigenetic modifications are involved in regulating ethanol drinking behavior, partially through altering NR2B expression.
Keywords: alcohol drinking, epigenetics, mice, Trichostatin-A, 5-azacytidine, SAM, NR2B
Introduction
Continuous consumption of excessive ethanol can lead to the development of dependence and addiction. The neuroadaptation of the brain due to repeated alcohol exposure may serve as the neural basis responsible for alcohol craving and drinking during withdrawal. However, the underlying mechanisms are not fully elucidated. Recent studies of the mechanisms of learning and memory indicate that active regulation of gene expression is necessary for experience-triggered lasting functional plasticity (Zovkic and Sweatt, 2012), which also supports mechanisms of long-lasting addictive behaviors. The role of epigenetic mechanisms is involved in various types of neuronal plasticity and behavioral disorders, such as long-term behavior in maternal caring (Weaver et al., 2004; Meaney and Szyf, 2005), memory formation and maintenance (Levenson and Sweatt, 2005; Lubin et al., 2008; Roth and Sweatt, 2009; Day and Sweatt, 2010; Johnson et al., 2012), as well as the development of addictive behavior (Marutha Ravindran and Ticku, 2004; Pandey et al., 2008; Malvaez et al., 2009; Maze et al., 2010; Wong et al., 2011; Moonat et al., 2013; Warnault et al., 2013). It has been reported that ethanol exposure produces epigenetic modifications, including histone acetylation in amygdala (Pandey et al., 2008; Moonat et al., 2013), mesolimbic dopaminergic system, prefrontal cortex, nucleus accumbens, and striatum (Pascual et al., 2009; Warnault et al., 2013) in both adult and adolescent rats. DNA methylation has also been proposed to play a role in alcohol dependence based on studies in alcohol-dependent patients (Bonsch et al., 2005; Bleich et al., 2006). In our laboratory, we found significant DNA demethylation and changes in modifications of histone H3K9 in the NR2B gene promoter following chronic intermittent ethanol (CIE) administration using either primary neuronal cultures or mice in vivo. These epigenetic modifications were shown to regulate NR2B transcription in primary neuronal cultures (Marutha Ravindran and Ticku, 2005; Qiang et al., 2010 2011). The results are consistent with our earlier finding that NR2B gene expression was upregulated following CIE exposure (Hu et al., 1996; Sheela Rani and Ticku, 2006; Qiang et al., 2007). Moreover, the CIE-induced DNA demethylation and NR2B upregulation were efficiently reversed by co-treatment with a methyl donor, S-adenosyl-L-methionine (SAM), which confirmed that DNA and histone methylation underlie the change of NR2B expression.
The N-methyl-D-aspartate (NMDA) receptor is a well-established target of ethanol in the brain and has been implicated in ethanol-associated phenotypes such as tolerance, dependence, withdrawal, craving, and relapse (Hoffman et al., 1990; Trujillo and Akil, 1995; Krystal et al., 2003; Ron and Wang, 2008). One key neuroadaptative process induced by CIE is the upregulation of the R2B subunit (NR2B) of the NMDA receptor. Recent studies using a forebrain conditional NR2B knockout in adult mice revealed behavioral alterations in response to acute ethanol treatment (Badanich et al., 2011) and electrophysiological alterations following CIE exposure (Wills et al., 2012). In the present study, we explored the role of epigenetic modifications in regulating ethanol consumption using an established CIE exposure-reinforced ethanol-drinking mouse model during withdrawal (Becker and Lopez, 2004; Lopez and Becker, 2005; Finn et al., 2007) combined with the utilization of epigenetic enzyme inhibitors 5-azacytidine (5’AZA), a DNA methyltransferase inhibitor, and Trichostatin A (TSA), a histone deacetylase (HDAC) inhibitor (class I and II). These compounds increase gene expression by inhibiting the repressive enzymes in repressor complexes, which mediates chromatin remodeling. The results show that TSA and 5’AZA facilitated CIE-induced ethanol drinking; CIE or TSA decreased DNA methylation and increased histone acetylation in the NR2B gene promoter of these mice.
Animals and methods
Animals
Male C57BL/6J mice 8 weeks old were purchased from Jackson Laboratory (Bar Harbor, ME) and given a minimum 14-day acclimation period of a reversed light/dark cycle (lights on 9:00 pm–9:00 am) prior to any experiment. Mice were maintained under a controlled ambient temperature (21–23ºC) and humidity (40–55%) and were provided free access to water and chow. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio and conducted in accordance with National Institutes of Health guidelines for Institutional Animal Care.
Stereotaxic Surgery and Microinjection
Mice were anesthetized with pentobarbital (90 mg⁄kg, intraperitoneally [i.p.]) and stereotaxically implanted with a 26-gauge stainless-steel guide cannula (Plastics One, Inc., Roanoke, VA) into the lateral ventricles bilaterally. The coordinates for the target areas were −0.5mm posterior to bregma, ±1.0mm lateral to midline, and −2.8mm below the surface of the skull. The guide cannula (C315GS-5/SPC, 26-gauge) was secured using dental cement to affix to the skull. A stainless-steel dummy cannula (C315DCS-5/SPC), extending 0.5mm beyond the guide, was used to seal the guide cannula when not in use. After surgery, all animals were housed individually and allowed to recover for 1 week postcannulation. During this period, they were handled daily to minimize stress derived from the microinjection procedure. All intra-lateral ventricular microinjections were performed on conscious, unrestrained, freely moving mice in cages. Two microliters (1 µL per side) of the solution or vehicle (0.9% saline) was administered by bilateral intracerebroventricular injection (icv) for 2 minutes operated by an infusion pump. The injector was inserted and extended 1mm beyond the tip of the guide cannula. The injection cannulas were left in place for an additional 2 minutes to allow for drug diffusion and prevent backflow.
Two-Bottle Choice Test
The general experimental design is depicted in Figure 1. A voluntary 2-bottle choice drinking test was performed as previously described (Becker and Lopez, 2004; Yang et al., 2008) with minor modifications. Mice were housed individually and trained to voluntarily drink ethanol in a 2-hour limited access (in the dark) regimen. Mice were initially trained to drink ethanol until stable intake was achieved. They were allowed equal access to 2 drinking bottles containing water or different increasing concentrations of ethanol as follows: 3% ethanol for 2 days, 6% ethanol for 2 days, 12% ethanol for 5 days. Ethanol and water consumption were immediately recorded as baseline consumption after a 2-hour limited access to 2 bottles during the 5 days of 12% ethanol intake. Mice were then separated into chronic ethanol and control groups. After each treatment of ethanol vapor inhalation exposure, mice were again evaluated for ethanol drinking by allowing 2-hour limited access to 2 bottles containing a 12% (vol/vol) alcohol solution and water for 5 consecutive days. The bottle positions were switched every day. Body weight and food consumption were measured on the first day and the last day of each 2-bottle test period (5 days) in the experiments. The ethanol consumption (g/kg) was calculated by using an average value of 5-day body weight values, which were recorded at the same time when ethanol was consumed. Data are presented as ethanol preference defined as the volume of ethanol consumption divided by total fluid volume consumed (ethanol + water)×100. The volume of ethanol intake was converted to a value in g/kg/2h and expressed as a mean±SEM (Yang et al., 2008).
Figure 1.

General experimental design. In all experiments, mice were initially trained to drink ethanol voluntarily in a 2-hour limited-access paradigm for 5 consecutive days (baseline drinking). The treatments are depicted in 2 ways. (A) Two exposures to chronic intermittent ethanol (CIE). Once stable daily baseline intake was established, mice were subjected to 2 treatments (9 days each) of CIE or room air exposure. There was a 2-week period of rest between treatment 1 and treatment 2. On the day before and after each CIE treatment (rest), no manipulation was performed on the mice. From the second day after each CIE treatment, mice were given the 2-bottle choice test for 5 days. (B) Mice were given Trichostatin A (TSA), 5-azacytidine (5’AZA), S-adenosyl-L-methionine (SAM), or saline 30 minutes before being placed into the vapor chamber cages daily during the 9-day exposure to CIE. All mice were then given the 2-bottle choice test for 5 consecutive days after vapor chamber exposure.
CIE Exposure
This model was previously described (Lopez and Becker, 2005) with some modifications, for example, pyrazole is not used to stabilize blood ethanol concentrations (BECs). Mice were randomly divided into 2 groups. The mice in the ethanol group were placed into standard plastic box cages with ethanol vapor (La Jolla Alcohol Research, CA), whereas those in the control group were placed in identical chambers receiving room air. Animals were exposed to ethanol (or air) vapor for 1 or 2 treatments (Figure 1) where each treatment included 9 cycles of 10 hours of vapor exposure followed by 14-hour withdrawal periods. Ethanol vapor was delivered to the inhalation chambers in the dark (between 9:30 am and 7:30 pm) at flow rates between 50 and 60mL/h to inhalation chambers that each housed 5 mice (Kang et al., 2004). Ethanol vapor was adjusted to yield actual BEC measurements of 150 to 250mg/dL (average: 231mg/dL) in the animals exposed to ethanol vapor. Water and food were available ad libitum throughout the period of ethanol vapor exposure.
5’AZA, TSA, and SAM Preparation
TSA (Sigma, T8552) was dissolved in dimethylsulfoxide (stock at 10mg/mL) and then freshly diluted (1:5 dilution) with saline before use. 5’AZA (Sigma, A2385) was dissolved in H2O (stock, 10mM). SAM was purchased from Sigma (A7007) and dissolved in H2O.
BEC Measurements
Tail blood samples were collected on days 4 and 7 during the 9-day CIE period. Tail blood samples were collected in capillary tubes containing evaporated heparin. The plasma was separated by centrifuging blood samples and then injected into an oxygen-rate Analox AM1 analyzer (Analox Instruments, Lunenburg, MA) for blood alcohol determination. Chamber ethanol concentrations were monitored every 2 hours by collecting air samples (2mL) through a port in the inhalation chamber wall.
Pyrosequencing
The pyrosequencing analysis was described in our previous study (Qiang et al., 2010). Briefly, genomic DNA (1 µg) isolated from prefrontal cortex using Blood and Cell Culture DNA kit (Qiagen) was bisulfate treated using the Zymo DNA Methylation kit (Zymo Research). Bisulfate-treated DNA was eluted in 10 µL volume with 1 µL used for each polymerase chain reaction (PCR). The PCR was performed with primers biotinylated to convert the PCR product to single-stranded DNA templates. The PCR products (10 µL) were sequenced by pyrosequencing PSQ96 HS System (Biotage, Kungsgatan, Sweden) following the manufacturer’s instructions. The methylation status of each locus was individually analyzed as a T/C single-nucleotide polymorphism using QCpG software.
Chromatin Immunoprecipitation (ChIP) Assay and Quantitative PCR (qChIP PCR)
ChIP analyses were performed as previously described (Qiang et al., 2011). Briefly, the brain tissue was chopped into small pieces using 2 razor blades. The chromatin was cross-linked with 1% formaldehyde for 10 minutes at room temperature. After sonication in lysis buffer, an aliquot of the chromatin was reversely cross-linked, and the efficiency of sonication was verified on agarose gel with an average length of about 600bp. An aliquot of the chromatin was reverse cross-linked and quantitated by optical density. Precleared chromatin with protein A/G beads was incubated with 2 µg of anti-acetylated histone H3 at Lys9 (ac-H3K9) antibody or IgG (negative control, Santa Cruz Biotechnology, sc-324) and rotated at 4°C overnight. Immunoprecipitation was carried out with protein A/G beads. The immune complexes were eluted by elution buffer (1% sodium dodecyl sulfate, 0.1M NaHCO3). The cross-link was reversed and the DNA was purified using a PCR purification column. For qPCR quantitation, we designed the primers to amplify GAPDH promoter (F: 5’-ccaaagacagaagccaggag-3’; R: 5’-catcgaacctctccccatta-3’) as the internal control. The levels of DNA associated with acetylated histone H3K9 (Upstate, 07-352) at the promoter of the gapdh gene was measured and no difference between CIE-treatments and control group (data not shown).
Statistical Analyses
Results are expressed as means ± SEM. Ethanol preference and intake across daily test sessions were analyzed by 2-way analysis of variance (ANOVA) with repeated measures for the effects of treatments. Whenever a significant main effect or interaction was indicated in the ANOVA, posthoc analyses were conducted using Newman-Keuls tests for individual comparisons.
Results
Baseline Drinking
Before exposure to CIE, all mice were measured for a baseline drinking of ethanol voluntarily during a 2-hour limited access regimen and drinking (in the dark) was then recorded for 5 days. As expected, during the baseline periods, no significant differences were observed in either the level of ethanol preference or ethanol intake in the groups that subsequently were or were not exposed to the CIE condition. These baseline data are shown in Figures 2 to 5. The BEC during the 5-day baseline drinking was not significantly increased compared with nondrinking mice (data not shown); therefore, it was not considered as a part of CIE exposure.
Figure 2.
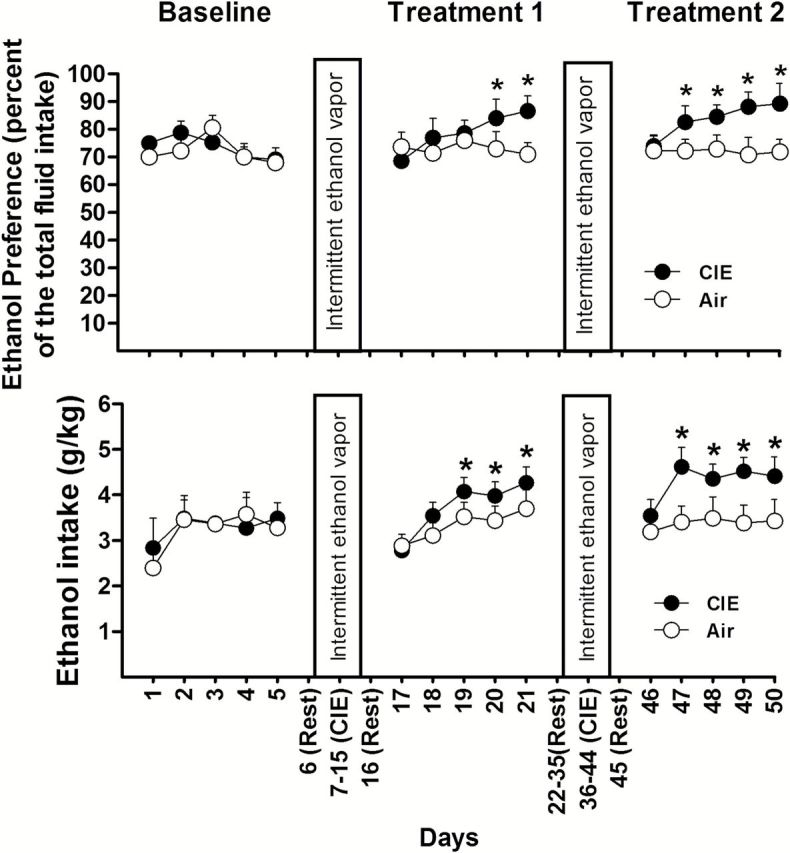
Ethanol vapor exposure significantly increases ethanol preference and intake after each section of intermittent ethanol exposure. Mice were exposed to 2 treatments of chronic intermittent ethanol (CIE) or air. Ethanol preference or ethanol intake was compared between the CIE (n = 16) and air groups (n = 12). Values represent mean ± SEM. *P<.05 vs their corresponding control (posthoc Newman-Keuls test).
5’AZA and TSA Facilitate an Increase in Ethanol Consumption after CIE Exposure and Withdrawal
After stable baseline drinking was established, mice were randomly divided into 2 groups: CIE or control (air). After a 1-day rest period following each CIE exposure, mice were subjected to another 5-day period for ethanol drinking using the 2-bottle choice test. As shown in Figure 2, ethanol consumption of mice in the air group did not differ significantly in their ethanol preference or intake compared with that measured during the baseline period (P>.05). By contrast, the mice exposed to CIE exhibited an increase in both the preference and intake of ethanol during the subsequent daily 2-bottle choice test periods. Two-way ANOVA with repeated measures indicated a significant effect of CIE treatment on ethanol preference [F(1,26)=6.343; P=.013] and intake [F(1,26)=6.223; P=.014] in the CIE group after treatment 1; ethanol preference [F(1,26)=23.762, P<.01] and intake [F(1,26)=24.515, P<.01] after treatment 2 compared with that in the air group. In addition, posthoc tests showed significant effects on days 3 or 4 after treatment 1 and as early as day 2 after treatment 2 (Figure 2). Such results are consistent with previous findings (Becker and Lopez, 2004; Lopez and Becker, 2005; Finn et al., 2007).
To investigate whether epigenetic modifications play a role in the ethanol drinking in this mouse model, 5’AZA and TSA were used. The mice were divided into the following 4 groups: (1) saline+CIE; (2) TSA+CIE; (3) 5’AZA+CIE; and (4) saline+air. 5’AZA and TSA were administered daily to mice 30 minutes before the CIE exposure (Figure 1B). Body weight was measured each day before injection to determine the amount of drug to administer to an individual mouse. At 24 hours after the last cycle of CIE exposure, the 2-bottle choice test was carried out for 5 days. In confirmation of the previous experiment, CIE increased both ethanol preference and intake after a single treatment of CIE exposure (Figure 3). The mice treated with TSA showed a greater increase in ethanol preference and intake than those treated with saline, whereas this increase did not occur in the mice receiving TSA only without CIE (supplementary Figure S1). Two-way ANOVA with repeated measures indicated a significant effect of the TSA treatment on ethanol preference [F(3,40)=6.443, P=.013] and ethanol intake [F(3,40)=8.462, P<.001]. As shown in Figure 3, posthoc tests indicated significant differences between the saline+CIE and air groups as well as between the TSA+CIE and saline+CIE groups. Although the effects were not significant on the first day of the 2-bottle choice test, the overall effects for the entire experimental period indicate that TSA facilitates CIE-induced voluntary ethanol consumption, suggesting that histone modifications mediate the effect on CIE-induced ethanol drinking. In contrast, i.p. injection of 5’AZA had no significant effect on ethanol preference or intake compared with CIE alone.
Figure 3.
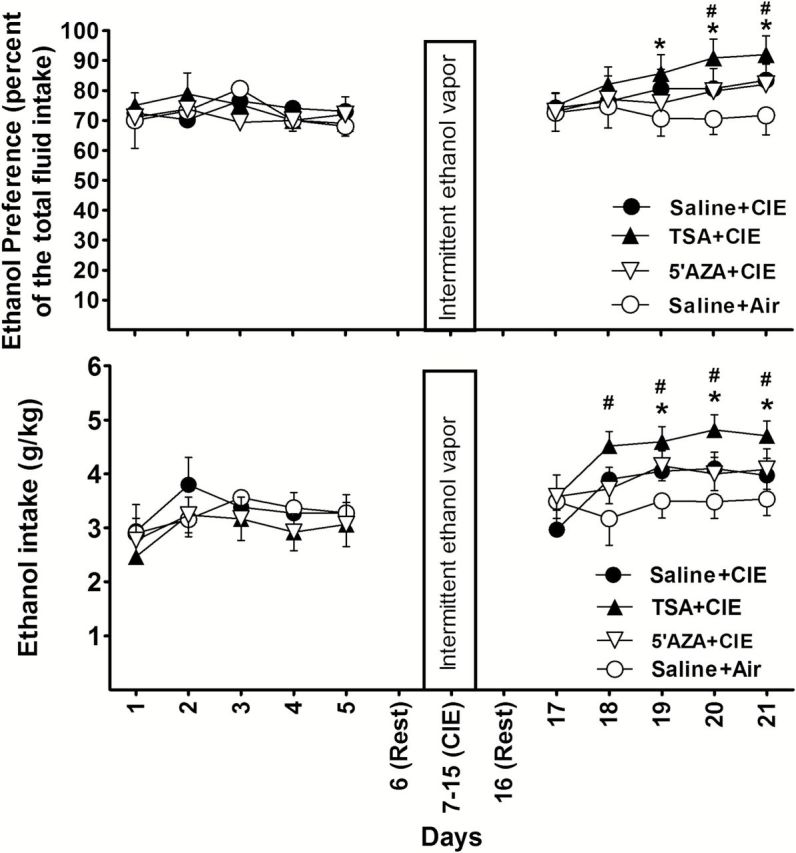
The effects of Trichostatin A (TSA) or 5-azacytidin (5’AZA) injection (intraperitoneally [i.p.]) on chronic intermittent ethanol (CIE)-induced ethanol consumption during withdrawal. The daily changes of ethanol preference or intake before and after CIE are shown. Mice (TSA+CIE; n=12) received daily TSA injections (2.5 µg/g) or (5’AZA+CIE; n=12) daily 5’AZA (2 µg/g) or saline injection (saline+CIE; n=11) 30 minutes before exposure to CIE. Air mice (n=10) were handled in the same way as previously described. Values represent mean±SEM. *P<.05 indicates a significant difference between saline+CIE and saline+air groups; #P<.05 indicates a significant difference between TSA+CIE and saline+CIE groups (posthoc Newman-Keuls test).
To determine if the lack of effect of 5’AZA was due to its not adequately penetrating the blood-brain barrier (BBB) to reach a sufficient concentration in brain, we delivered 5’AZA and TSA by icv. In this procedure, baseline drinking was carried out before cannulas were implanted into the lateral ventricles of mice in order to shorten the duration that the mice had to carry cannulas during treatment and testing. One week after surgery, mice were divided into 3 groups receiving bilateral injections of the following: (1) TSA (1 µg/µL; 1 µL per side); (2) 5’AZA (10 µg/µL; 1 µL per side); or (3) saline (1 µL per side). The mice in groups 1 and 2 received a daily CIE exposure 30 minutes after the injection. Eight mice in group 3 received daily CIE exposure and the other 7 were exposed to air. The 2-bottle choice test was conducted after a 24-hour rest period. Two-way ANOVA with repeated measures revealed a significant main effect of treatment on ethanol preference [F(3,31)=7.29; P=.01] and ethanol intake [F(3,31) = 21.01; P < .01]. As shown in Figure 4, post-hoc tests indicated a significant increase of ethanol intake in all mice of the three CIE-exposed groups compared to air control; and a significant increase of ethanol intake in the mice receiving TSA+CIE or 5’AZA+CIE compared to those receiving Saline+CIE. The TSA+CIE group showed a similar pattern to that in those mice receiving TSA by systematic injection. However, when given icv, 5’AZA produced a greater increase in ethanol consumption in the mice compared with those given saline, with the increases beginning on days 4 to 6 after CIE exposure.
Figure 4.
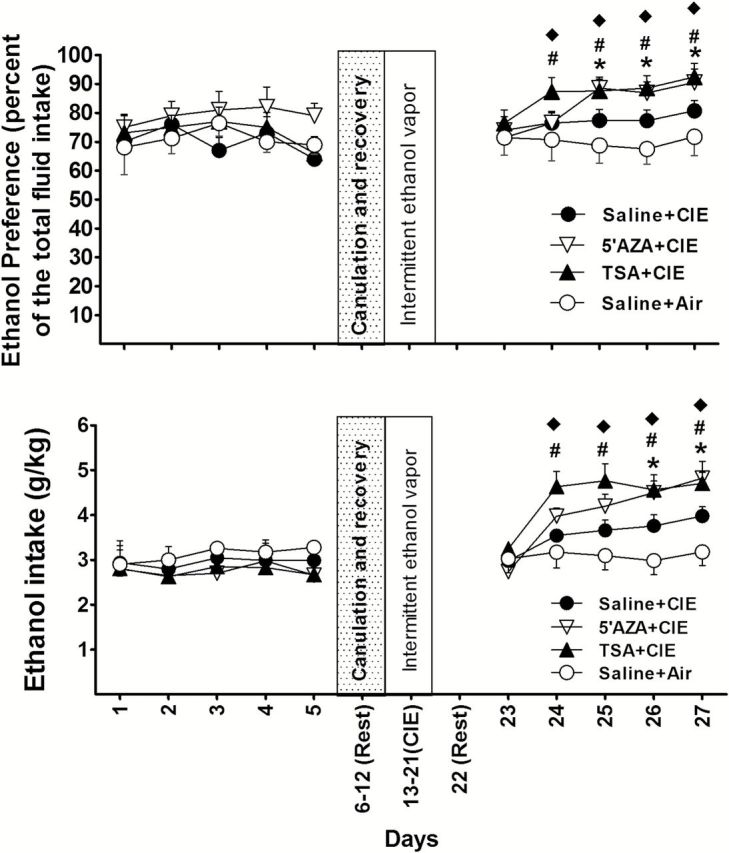
The effect of intracerebroventricular injection (icv) of 5-azacytidine (5’AZA) or Trichostatin A (TSA) on chronic intermittent ethanol (CIE)-induced ethanol consumption during withdrawal. The daily changes of ethanol preference or intake before and after exposure to CIE are shown. Mice received TSA (TSA+CIE, n=9) or 5’AZA (5’AZA+CIE, n=8) by daily icvs or saline (saline+CIE; n=10) injections 30 minutes before daily exposure to CIE. Mice (n=8) that received saline+air were used as control. Values represent mean±SEM. *P<.05 indicates a significant difference between the 5’AZA+CIE and saline+CIE groups; #P<.05 indicates a significant difference between the TSA+CIE and saline+CIE groups; ♦P<.05 indicates a significant difference between the saline+air and other groups (posthoc Newman-Keuls test).
A separate experiment was then carried out to determine whether a similar effect would occur when mice were given the opportunity to drink a sucrose solution rather than ethanol using the same regimen. As shown in supplementary Figure S2, CIE exposure did not significantly change sucrose intake (P>.05) in animals with or without pretreatments with TSA, 5’AZA, or SAM.
Effect of SAM Treatment on the Prevention of CIE-Induced Increases in Ethanol Consumption
To provide more direct evidence that DNA methylation and histone methylation contribute to the CIE-induced increase in ethanol consumption, we administered a substrate of DNA methyltransferases and histone methylases, SAM, to mice 30 minutes prior to daily ethanol exposure during 9-day CIE vapor exposure. Mice were randomly divided into the following 3 groups: (1) saline+CIE; (2) SAM treatment with 100mg/kg, i.p. (SAMLo); or (3) SAM treatment with 300mg/kg, i.p. (SAMHi). Two-way ANOVA with repeated measures indicated that SAM treatment had a significant main effect on ethanol preference [F(3,33)=17.3; P<.001] and intake [F(3,33)=16.9; P<.001]. Saline+CIE-exposed mice showed an increase compared with saline+air; the mice receiving 300mg/kg, but not 100mg/kg SAM, showed a significant reduction in both ethanol preference and intake during withdrawal compared with the values of saline+CIE (Figure 5).
Figure 5.

The effect of S-adenosyl-L-methionine (SAM) injection (intraperitoneally [i.p.]) on chronic intermittent ethanol (CIE)-induced ethanol drinking during withdrawal. The daily changes of ethanol preference or intake before and after exposure to CIE are shown. Mice were given an injection of either 100mg/kg (SAMLo, n=10) or 300mg/kg (SAMHi, n=10) of SAM or saline (n=9) injections 30 minutes before daily exposure to CIE. Mice (n=8) that received saline+air were used as control. Values represent mean ± SEM. *P<.05 compared with saline+CIE; ♦ P<.05 indicates a significant difference between the saline+air and saline+CIE (posthoc Newman-Keuls test).
CIE Induced DNA Demethylation and Increased Histone H3K9 Acetylation of the NR2B Promoter in the Prefrontal Cortex of Mice
We next assessed potential epigenetic modifications in the NR2B promoter after CIE treatment. Five days following the last day of CIE exposure, 8 mice from each treatment group were sacrificed, and the tissue from prefrontal cortex was collected to examine whether changes had occurred in DNA methylation and histone acetylation in the NR2B promoter of these mice. As expected, bisulfite pyrosequencing analysis indicated that 18 CpG sites in the regions b, c, and g of the NR2B promoter previously described (Qiang et al., 2010) were demethylated in CIE-exposed mice compared with control mice (Figure 6A). In addition, a qChIP assay was carried out with the tissue from prefrontal cortex of these mice using antibodies specific to ac-H3K9. The qPCR results showed that ac-H3K9 levels in regions b, c, and g of the NR2B promoter were increased in mice exposed to CIE compared with the values in those mice receiving room air (Figure 6B). These altered modifications by CIE are consistent with the corresponding upregulation of the NR2B gene (as shown in Figure 8).
Figure 6.
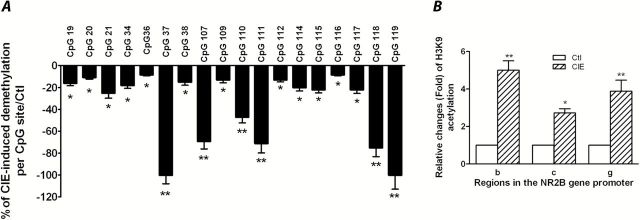
Chronic intermittent ethanol (CIE) induced DNA demethylation and histone H3K9 acetylation in the NR2B promoter in the prefrontal cortex of the mice. After 5 days withdrawal from CIE exposure, 8 mice from each group were sacrificed and the tissues of prefrontal cortex were collected. (A) Genomic DNA isolated from these mice was subjected to bisulfite pyrosequencing analysis. The values in negative are defined in this study to represent demethylation. Results are presented as the mean percentage of demethylation level in the CIE group vs control group±SEM; *P<.05; **, P<.01 compared with their corresponding controls. (B) Eight mice from each group were sacrificed and prefrontal cortexes were collected. Quantitative chromatin immunoprecipitation (qChIP) assays were performed using these prefrontal cortical tissues to examine the acetylation levels of histone H3K9 in the NR2B promoter in the regions b, c, and g of the NR2B (Qiang et al., 2010). Values are presented as average fold changes over their corresponding controls (control=1)±SEM (n=8). *P<.05, **P<.01 compared with their corresponding controls (2-way analysis of variance [ANOVA] followed by posthoc Newman-Keuls test).
Figure 8.

Chronic intermittent ethanol (CIE) and Trichostatin A (TSA) increase NR2B gene transcription. Eight mice in each treatment group of CIE, air, or TSA were sacrificed, and total RNA was isolated from the prefrontal cortex using the mirVanaTM miRNA isolation kit (Ambion, AM1561). Two-step quantitative PCR (qPCR) was performed as previously described (Qiang et al., 2011). Results are presented as the mean ratio to control ± SEM; **P<.01 compared with control levels.
TSA-Induced Increase in Histone H3K9 Acetylation and DNA Demethylation of the NR2B Promoter in the Prefrontal Cortex of Mice
To confirm that TSA induces histone acetylation and DNA demethylation of the NR2B promoter, 8 mice from each treatment group of TSA or saline were sacrificed and the tissue from prefrontal cortex was collected. The qChIP assays were then performed with an antibody specific to ac-H3K9. There was a robust increase in ac-H3K9 levels in regions b, c, and g of the NR2B promoter in TSA-treated mice compared with those in saline-treated mice (Figure 7A). In addition, genomic DNA was also assayed by bisulfite pyrosequencing analysis. TSA-treated mice had a decrease in DNA methylation in the NR2B promoter (Figure 7B), which is similar as that observed in the mice exposed to CIE (Figure 6A). The altered modifications by TSA are consistent with the corresponding results of upregulation of the NR2B gene observed in the same tissue (Figure 8).
Figure 7.
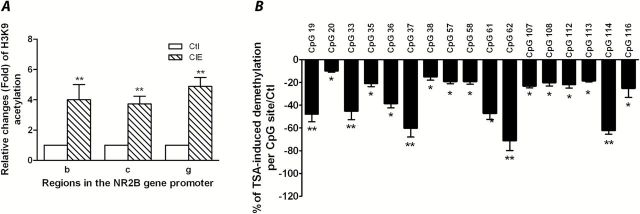
Trichostatin A (TSA) induced histone H3K9 acetylation and DNA demethylation in the NR2B promoter in the prefrontal cortex of mice. Mice received an injection of either TSA (2.5 µg/g, intraperitoneally [i.p.]) or saline for 9 days. (A) Quantitative chromatin immunoprecipitation assays were performed to examine the acetylation levels in regions b, c, and g in the NR2B promoter (Qiang et al., 2010). Values are presented as average fold changes over their corresponding controls (control=1)±SEM (n=8). **p<.01 (2-way analysis of variance [ANOVA] followed by Newman-Keuls posthoc comparison). (B) Genomic DNA isolated from prefrontal cortex in the control and TSA-treated mice was subjected to bisulfite pyrosequencing analysis. Results are presented as the mean percentage of demethylation levels in TSA group vs control group±SEM; *P<.05; **P<.01 compared with their corresponding controls.
Discussion
Epigenetic Regulatory Mechanisms Are Involved in Alcohol-Reinforced Drinking
Epigenetic modifications control gene transcription over time by dynamically responding to environmental stimuli and marking these changes into long-lasting code in chromatin. This may offer a possible explanation of how individual behavior is established and maintained. Repeated CIE exposure has been reported to increase subsequent ethanol consumption (Becker and Lopez, 2004; Lopez and Becker, 2005; Finn et al., 2007), which provides a good model for studying the mechanisms of ethanol-reinforced drinking. To address the underlying mechanisms of this phenomenon, we used the epigenetic enzyme inhibitors TSA or 5’AZA to change the status of histone modifications and DNA methylation in the brain, respectively. Administration of these drugs during CIE exposure reduced DNA methylation and increased histone acetylation in the NR2B promoter, and promoted CIE-induced ethanol drinking in the withdrawal period. By contrast, administration of these drugs had no effect on sucrose consumption, suggesting that epigenetic regulation has a selective effect on regulating ethanol-reinforced drinking by altering DNA methylation or histone modifications rather than just a general increase in the consumption of fluids. The effect of TSA to increase ethanol consumption has recently been reported in C57BL/6 mice using a 2-bottle choice test 4 weeks postadministration, but not after acute administration (Wolstenholme et al., 2011). In agreement with this result, we found that when mice were administered these drugs only, instead of exposing them to the CIE, neither 5’AZA nor TSA increased ethanol drinking within the 1-week postadministration period used in this study. However, when the drugs were administered in combination with CIE exposure, they promoted ethanol drinking.
However, in disagreement with the findings of this study, recent published studies showed an opposite alteration, namely that systemic administration of 5-AzaC and HDAC inhibitors reduced binge-like alcohol drinking in mice (Warnault et al., 2013). Also, HDAC2 knockdown in the CeA of rats attenuated anxiety-like behaviors and voluntary alcohol drinking (Moonat et al., 2013). These differences in results may be because of the different procedures with which the experiments were carried out. For example, use of 5’AZA or HDAC inhibitors in different stages, that is, during or after the formation of increased ethanol drinking. In our study, the inhibitors were given 30 minutes prior to CIE exposure on each day for 9 days. In the study by Warnault et al. (2013), the inhibitors were given after the mice had been exposed to intermittent voluntary ethanol drinking for 8 weeks. Similarily, Moonat et al. (2013) knocked down HDAC2 with siRNA after rats were allowed to voluntarily drink ethanol for 6 days. Also, in our experiments, drinking behavor was measured 5 days post-CIE exposure and the inhibitors were not given during this time. By contrast, in the studies of Moonat et al. (2013) and Warnault et al. (2013), inhibition of HDAC was ongoing during the test sesions. Finally, our mice were forced to inhale ethanol during the 10 hours in the chamber, whereas ethanol exposure/consumption was volutary in the other 2 studies.
In this study, we showed that TSA and 5’AZA have a similar effect on regulating ethanol consumption although they activate gene transcription through different mechanisms. It was reported earlier that histone modifications and DNA methylation have extensive and complex interactions in neurons (Kondo et al., 2003), and a mutual regulatory relationship has previously been shown. When DNA methyltransferases were inhibited by 5’AZA, promoter demethylation occurred, which also resulted in increased acetylation and decreased histone methylation (Meldi et al., 2012); by contrast, the HDAC inhibitor, TSA, produced “demethylation” of the 5’ CpG and 3’ CpG dinucleotides of the glucocorticoid receptor gene promoter in the offspring of low- and high-licking/grooming and of arched-back nursing mothers (Weaver et al., 2004). In the present study, the application of TSA caused a significant increase in histone H3K9 acetylation as well as the DNA demethylation of the NR2B promoter, which agrees with a previous report that CIE-induced ethanol drinking may be controlled by the mechanisms involving a crosstalk between the 2 epigenetic modifications.
NR2B Is a Potential Target of Epigenetic Modifications Involved in Alcohol-Drinking Behavior
In mice, repeated CIE/withdrawal experiences have been shown to increase subsequent ethanol consumption (Lopez and Becker, 2005; Finn et al., 2007). However, the mechanisms underlying the motivation to drink ethanol during withdrawal are not fully understood. Our previous studies showed that CIE caused DNA demethylation and increased histone acetylation in the promoter region of the NR2B gene (Qiang et al., 2010 2011). Using cortical neuronal cultures, we also found that treatment of cells with 5’AZA or TSA for 3 to 5 days induced long-lasting upregulation of the NR2B gene, which is similar to the effect we have seen following CIE treatment. These results may explain the CIE-induced upregulation of NR2B gene transcription through epigenetic mechanisms. Since NR2B is a major target of ethanol-induced epigenetic regulation, we hypothesized that these epigenetic events may be involved in the formation of ethanol-drinking phenotype, that is, the CIE-induced ethanol drinking could be mimicked and reinforced by 5’AZA or TSA treatment during CIE reinforcement. Here, we found that administration of these inhibitors altered ethanol consumption in the same way that CIE does, enhancing the likelihood that CIE-induced epigenetic effects are linked to its behavioral effects. In addition, these results are consistent with that from a recent network analysis supporting the correlation between alcohol consumption and the expression level of the NR2B gene; C57BL/6 mice consuming the least amount of ethanol exhibited the lowest expression of the NR2B gene in the prefrontal cortex and vice versa (Wolstenholme et al., 2011).
The Potential Role of Sam in the Prevention of Alcohol Addiction
SAM is a substrate for DNA methyltransferases and histone methylases that has been previously used to rescue hypomethylation by providing methyl donors for DNA methylation (Detich et al., 2003). In our previous in vitro study, the application of SAM blocked ethanol-induced DNA demethylation and the upregulation of NR2B gene expression (Qiang et al., 2010 2011). To provide more direct evidence that DNA and/or histone methylation contribute to the CIE-induced increase in ethanol consumption, we administered SAM to mice before daily CIE exposure. As expected, SAM reversed the CIE-induced increase in ethanol drinking. SAM has received increased attention recently for its ability to protect the liver from alcohol toxicity together with folic acid and vitamin A (Purohit et al., 2007; Moghe et al., 2011). SAM may stabilize methylation in the brain as well, thus protecting the brain from CIE-induced demethylation. Chronic ethanol consumption can influence methionine synthesis by decreasing methionine synthase activity (Villanueva and Halsted, 2004; Kharbanda et al., 2005), which is involved in producing SAM. Studies in humans have also reported improved cognitive and psychiatric symptoms in Alzheimer’s disease patients because of partial reversal of DNA hypomethylation through supplementation with foliate, SAM, and vitamin B6 (Narayan and Dragunow, 2010). Together, these results suggest the possibility that epigenetic intervention and therapeutic strategies may be effective in alcoholics.
5’AZA has also been used as a DNA hypomethylation agent for improving cognition and regulating learning and memory as a research tool (for review, see Weeber et al., 2002). However, whether this compound can cross the BBB or it is safe at a specific dose remains controversial in the literature, with some studies indicating that it can cross the BBB (Chabot et al., 1983) and others showing it cannot (Simonini et al., 2006) or being inconclusive (Jaenisch et al., 1985). Previous studies have delivered 5’AZA to brain by icv administration (Endres et al., 2000) or through an intra-brain injection (Miller and Sweatt, 2007). In the present study, we observed that 5’AZA enhanced ethanol consumption only when it was delivered by icv injection, but not systematically administered. Therefore, it is possible that 5’AZA either does not cross the BBB or does so poorly so as to not reach a concentration in certain brain regions that is sufficient for affecting ethanol-drinking behavior. Our results underscore that results based on systemic administration of 5’AZA should be interpreted cautiously.
Supplementary Material
Acknowledgments
The authors specially thank Dr. Alan Frazer for the helpful discussion of the results and critically reading the manuscript. This research is supported by NIAAA AA017362 (Alan Frazer), ABMRF/The Foundation for Alcohol Research (Mei Qiang), and NIAAA AA021475 (Mei Qiang).
Interest statement: None.
References
- Badanich KA, Doremus-Fitzwater TL, Mulholland PJ, Randall PK, Delpire E, Becker HC. (2011). NR2B-deficient mice are more sensitive to the locomotor stimulant and depressant effects of ethanol. Genes Brain Behav 10:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HC, Lopez MF. (2004). Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res 28:1829–1838. [DOI] [PubMed] [Google Scholar]
- Bleich S, Lenz B, Ziegenbein M, Beutler S, Frieling H, Kornhuber J, Bonsch D. (2006). Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcohol Clin Exp Res 30:587–591. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Lenz B, Kornhuber J, Bleich S. (2005). DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport 16:167–170. [DOI] [PubMed] [Google Scholar]
- Chabot GG, Rivard GE, Momparler RL. (1983). Plasma and cerebrospinal fluid pharmacokinetics of 5-Aza-2’-deoxycytidine in rabbits and dogs. Cancer Res 43:592–597. [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. (2010). DNA methylation and memory formation. Nat Neurosci 13:1319–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detich N, Hamm S, Just G, Knox JD, Szyf M. (2003). The methyl donor S-Adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S-Adenosylmethionine. J Biol Chem 278:20812–20820. [DOI] [PubMed] [Google Scholar]
- Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, Dirnagl U. (2000). DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci 20:3175–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn DA, Snelling C, Fretwell AM, Tanchuck MA, Underwood L, Cole M, Crabbe JC, Roberts AJ. (2007). Increased drinking during withdrawal from intermittent ethanol exposure is blocked by the CRF receptor antagonist D-Phe-CRF(12–41). Alcohol Clin Exp Res 31:939–949. [DOI] [PubMed] [Google Scholar]
- Hoffman PL, Rabe CS, Grant KA, Valverius P, Hudspith M, Tabakoff B. (1990). Ethanol and the NMDA receptor. Alcohol 7:229–231. [DOI] [PubMed] [Google Scholar]
- Hu XJ, Follesa P, Ticku MK. (1996). Chronic ethanol treatment produces a selective upregulation of the NMDA receptor subunit gene expression in mammalian cultured cortical neurons. Brain Res Mol Brain Res 36:211–218. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Schnieke A, Harbers K. (1985). Treatment of mice with 5-azacytidine efficiently activates silent retroviral genomes in different tissues. Proc Natl Acad Sci U S A 82:1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LR, McGuire J, Lazarus R, Palmer AA. (2012). Pavlovian fear memory circuits and phenotype models of PTSD. Neuropharmacology 62:638–646. [DOI] [PubMed] [Google Scholar]
- Kang SS, Cole M, Lee S, Rivier C. (2004). Development of individual alcohol inhalation chambers for mice: validation in a model of prenatal alcohol. Alcohol Clin Exp Res 28:1549–1556. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, Rogers DD, 2nd, Mailliard ME, Siford GL, Barak AJ, Beckenhauer HC, Sorrell MF, Tuma DJ. (2005). A comparison of the effects of betaine and S-adenosylmethionine on ethanol-induced changes in methionine metabolism and steatosis in rat hepatocytes. J Nutr 135:519–524. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Issa JP. (2003). Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol Cell Biol 23:206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Mason G, Trevisan L, D’Souza DC. (2003). N-methyl-D-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther 99:79–94. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. (2005). Epigenetic mechanisms in memory formation. Nat Rev Neurosci 6:108–118. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Becker HC. (2005). Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology (Berl) 181:688–696. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. (2008). Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci 28:10576–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, Barrett RM, Wood MA, Sanchis-Segura C. (2009). Epigenetic mechanisms underlying extinction of memory and drug-seeking behavior. Mamm Genome 20:612–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marutha Ravindran CR, Ticku MK. (2004). Changes in methylation pattern of NMDA receptor NR2B gene in cortical neurons after chronic ethanol treatment in mice. Brain Res Mol Brain Res 121:19–27. [DOI] [PubMed] [Google Scholar]
- Marutha Ravindran CR, Ticku MK. (2005). Role of CpG islands in the up-regulation of NMDA receptor NR2B gene expression following chronic ethanol treatment of cultured cortical neurons of mice. Neurochem Int 46:313–327. [DOI] [PubMed] [Google Scholar]
- Maze I, Covington HE, 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. (2010). Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327:213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. (2005). Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci 28:456–463. [DOI] [PubMed] [Google Scholar]
- Meldi KM, Gaconnet GA, Mayo KE. (2012). DNA methylation and histone modifications are associated with repression of the inhibin alpha promoter in the rat corpus luteum. Endocrinology 153:4905–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. (2007). Covalent modification of DNA regulates memory formation. Neuron 53:857–869. [DOI] [PubMed] [Google Scholar]
- Moghe A, Joshi-Barve S, Ghare S, Gobejishvili L, Kirpich I, McClain CJ, Barve S. (2011). Histone modifications and alcohol-induced liver disease: are altered nutrients the missing link? World J Gastroenterol 17:2465–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC. (2013). Aberrant histone deacetylase2-mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biol Psychiatry 73:763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan P, Dragunow M. (2010). Pharmacology of epigenetics in brain disorders. Br J Pharmacol 159:285–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. (2008). Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci 28:3729–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Boix J, Felipo V, Guerri C. (2009). Repeated alcohol administration during adolescence causes changes in the mesolimbic dopaminergic and glutamatergic systems and promotes alcohol intake in the adult rat. J Neurochem 108:920–931. [DOI] [PubMed] [Google Scholar]
- Purohit V, Abdelmalek MF, Barve S, Benevenga NJ, Halsted CH, Kaplowitz N, Kharbanda KK, Liu QY, Lu SC, McClain CJ, Swanson C, Zakhari S. (2007). Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: summary of a symposium. Am J Clin Nutr 86:14–24. [DOI] [PubMed] [Google Scholar]
- Qiang M, Denny AD, Ticku MK. (2007). Chronic intermittent ethanol treatment selectively alters NMDA receptor subunits surface expression in cultured cortical neurons. Mol Pharmacol 72:95–102. [DOI] [PubMed] [Google Scholar]
- Qiang M, Denny A, Chen J, Ticku MK, Yan B, Henderson G. (2010). The site specific demethylation in the 5’-regulatory area of NMDA receptor 2B subunit gene associated with CIE-induced up-regulation of transcription. PLoS One 5:e8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang M, Denny A, Lieu M, Carreon S, Li J. (2011). Histone H3K9 modifications are a local chromatin event involved in ethanol-induced neuroadaptation of the NR2B gene. Epigenetics 6:1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Wang J. (2008). NMDA receptors and alcohol addiction. In: Biology of the NMDA receptor (Van Dongen AM, ed). Boca Raton: Taylor and Francis/CRC Press. [PubMed] [Google Scholar]
- Roth TL, Sweatt JD. (2009). Regulation of chromatin structure in memory formation. Curr Opin Neurobiol 19:336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheela Rani CS, Ticku MK. (2006). Comparison of chronic ethanol and chronic intermittent ethanol treatments on the expression of GABA(A) and NMDA receptor subunits. Alcohol 38:89–97. [DOI] [PubMed] [Google Scholar]
- Simonini MV, Camargo LM, Dong E, Maloku E, Veldic M, Costa E, Guidotti A. (2006). The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc Natl Acad Sci U S A 103:1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo KA, Akil H. (1995). Excitatory amino acids and drugs of abuse: a role for N-methyl-D-aspartate receptors in drug tolerance, sensitization and physical dependence. Drug Alcohol Depend 38:139–154. [DOI] [PubMed] [Google Scholar]
- Villanueva JA, Halsted CH. (2004). Hepatic transmethylation reactions in micropigs with alcoholic liver disease. Hepatology 39:1303–1310. [DOI] [PubMed] [Google Scholar]
- Warnault V, Darcq E, Levine A, Barak S, Ron D. (2013). Chromatin remodeling--a novel strategy to control excessive alcohol drinking. Transl Psychiatry 3:e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. (2004). Epigenetic programming by maternal behavior. Nat Neurosci 7:847–854. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, Herz J. (2002). Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem 277:39944–39952. [DOI] [PubMed] [Google Scholar]
- Wills TA, Klug JR, Silberman Y, Baucum AJ, Weitlauf C, Colbran RJ, Delpire E, Winder DG. (2012). GluN2B subunit deletion reveals key role in acute and chronic ethanol sensitivity of glutamate synapses in bed nucleus of the stria terminalis. Proc Natl Acad Sci U S A 109:E278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstenholme JT, Warner JA, Capparuccini MI, Archer KJ, Shelton KL, Miles MF. (2011). Genomic analysis of individual differences in ethanol drinking: evidence for non-genetic factors in C57BL/6 mice. PLoS One 6:e21100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CC, Mill J, Fernandes C. (2011). Drugs and addiction: an introduction to epigenetics. Addiction 106:480–489. [DOI] [PubMed] [Google Scholar]
- Yang X, Wang S, Rice KC, Munro CA, Wand GS. (2008). Restraint stress and ethanol consumption in two mouse strains. Alcohol Clin Exp Res 32:840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Sweatt JD. (2013). Epigenetic mechanisms in learned fear: implications for PTSD. Neuropsychopharmacology 38:77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.