

Abstract
We report an unusual association of a pattern dystrophy of the retinal pigment epithelium and homonymous hemianopia in a woman diagnosed with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes syndrome.
Background
The syndrome of mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) is a maternally inherited disorder due to mitochondrial DNA point mutation affecting transfer RNA. Ophthalmic features, including external ophthalmoplaegia, ptosis, pigmentary retinopathy, pattern dystrophy, myopia, nuclear cataract and optic atrophy, have been described in patients with adenine-guanine 3243 point mutation.1 2
Pigmentary retinopathy, characterised by symmetric areas of depigmentation involving predominantly the posterior pole and midperipheral retina, are the most common ophthalmological findings in these patients.
The aim of this paper is to report an unusual association between homonymous hemianopia and pattern dystrophy of the retinal pigment epithelium (RPE) in a patient with MELAS syndrome.
Case presentation
A 24-year-old woman presented with intense oppressive occipital headache, vomiting, sonophobia and photophobia, as well as repeated motor focal seizures with oculocephalic deviation. She was admitted to the neurology unit, where she had the appropriate ancillary tests. Relevant personal history included migraine with aura and primary amenorrhoea with no relevant family history. Neurological examination was normal except for the left homonymous hemianopia. Blood test showed lactic acidosis. Serology to diverse germs (neurotropic virus, Syphilis, Mycoplasma, Borrelia, Chlamydia, Brucella and Rickettsia) was negative. Brain MRI revealed right temporoparietal-occipital involvement, with significant diffusion restriction (stroke-like) and left parieto-occipital involvement without diffusion restriction. The echocardiogram ruled out cardiomyopathy (left ventricle with good contractility and preserved ejection fraction, right cavities also normal).
A possible mitochondrial encephalopathy MELAS type was suspected. This diagnosis was confirmed by genetic testing (which detected the presence of heteroplasmy in the A3224G mutation).
The patient was referred to our practice due to a decrease in visual acuity to rule out possible ophthalmological lesions. Examination revealed left homonymous hemianopia and best corrected visual acuity of 20/40 in the right eye and of 20/70 in the left eye. Anterior segment biomicroscopy did not reveal significant data. Intraocular pressure was 15 mm Hg in both eyes. The ophthalmoscopy revealed linear pigmentation surrounding the macula with areas of hypopigmentation, consistent with pigment epithelium atrophy, more evident in the right eye (figure 1). Fluorescein angiography demonstrated mottled hyperfluorescent and hypofluorescent central areas indicating multiple window defects in the RPE, with greater involvement of the right eye (figure 2). Electroretinogram showed a reduction of photopic and scotopic b-wave amplitudes with normal implicit times. Therefore, macular pattern dystrophy was diagnosed. Figure 3 shows the results of the autofluorescence and optical coherence tomography images. Figure 4 shows the MRI and a visual field test carried out 2 weeks after the stroke-like episode.
Figure 1.
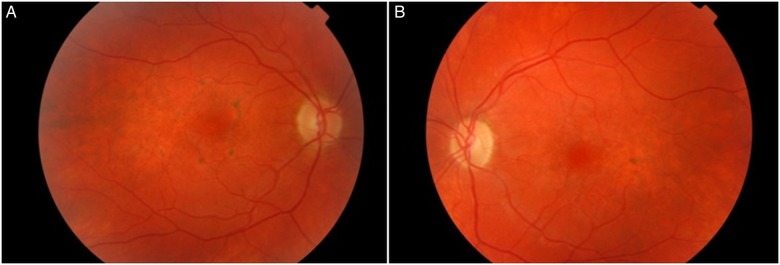
Colour fundus photograph. (A) Right eye. (B) Left eye.
Figure 2.
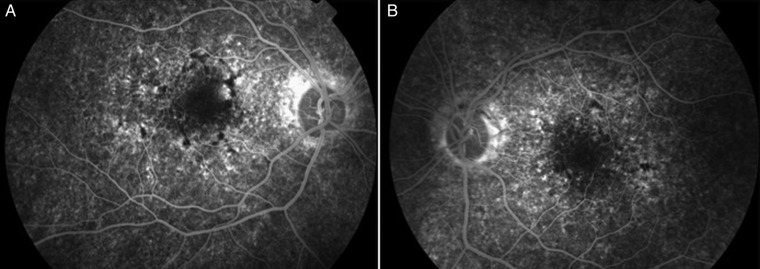
Fluorescein angiography. (A) Right eye. (B) Left eye.
Figure 3.

Autofluorescence imaging/optical coherence tomography imaging.
Figure 4.

MRI/Humphrey visual field test performed 2 weeks after the stroke-like episode showing left quadrantanopsia.
Discussion
The clinical criteria for the diagnosis of MELAS syndrome include episodes that mimic stroke and do not correspond to vascular territories; they consist of focal neurological deficits of sudden onset, with a predilection for the occipital area, causing visual defects; however, motor, sensory or language disorders may appear as well. Other symptoms and signs of this syndrome are muscle weakness, seizures, dementia, episodic vomiting, lactic acidosis, positive family history, ragged red fibres, spongiform degeneration, basal ganglia calcification, short stature, sensorineural hearing loss of cochlear origin and hemicranial headache.3
This disorder is characterised by impaired energy production due to oxidative phosphorylation dysfunction. Mitochondria participate in the synthesis of ATP and in the initiation of apoptosis.2 Affected organs are those with a high-energy demand such as the heart, retina and central nervous system.
Histopathological changes include depigmentation and hyperpigmentation of the RPE, degeneration of the choroid and the outer segment of photoreceptors and a reduction in the number of ganglion cells. The photoreceptor degeneration has been reported to be secondary to pigment epithelium loss. Pigment epithelium is involved in the phagocytosis of the shed tips of photoreceptor outer segments and regulation of subretinal volume via ion-coupled fluid absorption. Ions and water are actively transported by this layer and ATP is necessary to maintain this function. A deficiency in intracellular ATP would compromise the normal function of these cells.4 5
Pigmentary retinopathy occurs late in life and is a non-specific finding of mitochondrial disorder. Ocular fundus changes in MELAS syndrome range from mild ‘salt and pepper’ retinopathy to severe bone spicule-shaped pigment with atrophy of the pigment epithelium.6–10 In our case, a pattern dystrophy was found.
Latkany et al1 and Adjadj et al2 reported cases of geographic maculopathy in MELAS syndrome. In maternal inherited diabetes and deafness (MIDD), bilateral pattern dystrophy was present in 30 of the 35 patients with MIDD (85.7%). However, only one of the three patients with MELAS syndrome had pattern dystrophy.11 In the studies of Sue et al8 and Latvala et al12 pattern macular dystrophy was not found.
Our patient presented with an unusual association, left homonymous hemianopia (caused by the stroke-like episode) and pattern dystrophy of the pigment epithelium. To the best of our knowledge, this is the first time the association has been reported in the same patient, although different types of atrophy of the pigment epithelium have been reported. Pigment epithelium abnormalities are less common than sensorineural hearing impairment.2 11
Learning points.
Retinal manifestations are not uncommon in mitochondrial diseases.
It is necessary to perform a complete ophthalmological examination in every patient with mitochondrial disease.
Patients must also undergo periodic monitoring for the possible development of lesions, which are mostly asymptomatic and detected by ophthalmoscopy.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Latkany P, Ciulla TA, Cacchillo PF et al. Mitochondrial maculopathy: geographic atrophy of the macula in the MELAS associated A to G 3243 mitochondrial DNA point mutation. Am J Ophtalmol 1999;128:112–14. 10.1016/S0002-9394(99)00057-4 [DOI] [PubMed] [Google Scholar]
- 2.Adjadj E, Mansouri K, Borruat FX. Mitochondrial DNA (mtDNA) A 3243G mutation associated with an annular perimacular retinal atrophy. Klin Monbl Augenheilkd 2008;225:462–4. 10.1055/s-2008-1027257 [DOI] [PubMed] [Google Scholar]
- 3.Lombes A, Bonilla E, Dimauro S. Mitochondrial encephalomyopathies. Rev Neurol 1989;145:671–89. [PubMed] [Google Scholar]
- 4.Defoe DM, Ahmad A, Chen W et al. Membrane polarity of the NA(+)-K+ pump in primary cultures of Xenopus retinal pigment epithelium. Exp Eye Res 1994;59:587–96. 10.1006/exer.1994.1144 [DOI] [PubMed] [Google Scholar]
- 5.Hughes BA, Takahira M. ATP-dependent regulation of inwardly rectifyin K+ current in bovine retinal pigment epitelial cells. Am Physiol Soc 1998;275(5 Pt 1):C1372–83. [DOI] [PubMed] [Google Scholar]
- 6.Villafruela IM, Tejada P, García Silva MT, et al. Hallazgos oftalmológicos en pacientes con enfermedades mitocondriales. Published in: http://nexusediciones.com/np_ao_1997_7_2_002.htm.
- 7.Mojon D. Eye diseases in mitocondrial encephalomyopathies. Ther Umsch 2001;58:49–55. 10.1024/0040-5930.58.1.49 [DOI] [PubMed] [Google Scholar]
- 8.Sue CM, Mitchell P, Crimmins DS et al. Pigmentary retinopathy associated with the mitochondrial DNA 3243 point mutation. Neurology 1997;49:1013–17. 10.1212/WNL.49.4.1013 [DOI] [PubMed] [Google Scholar]
- 9.Jones M, Mitchell P, Wang JJ et al. MELAS A3243G mitochondrial DNA mutation and age related maculopathy. Am J Ophtalmol 2004;138:1051–3. 10.1016/j.ajo.2004.06.026 [DOI] [PubMed] [Google Scholar]
- 10.Isashiki Y, et al. Retinal manifestations in mitochondrial diseases associated with mitochondrial DNA mutations. Acta Ophtalmol Scand 1998;76:6–13. 10.1034/j.1600-0420.1998.760103.x [DOI] [PubMed] [Google Scholar]
- 11.Massin P, Virally-Monod M, Vialettes B et al. Prevalence of macular pattern dystrophy in maternally inherited diabetes and deafness. GEDIAM Group. Ophthalmology 1999;106:1821–7. 10.1016/S0161-6420(99)90356-1 [DOI] [PubMed] [Google Scholar]
- 12.Latvala T, Mustonen E, Uusiato R et al. Pigmentary retinopathy in patients with the MELAS mutation 3243A-G in mitochondrial DNA. Graefes Arch Clin Exp Ophthalmol 2002;240:795–801. 10.1007/s00417-002-0555-y [DOI] [PubMed] [Google Scholar]