

Abstract
Current recommendations for resuscitation of the critically injured patient are limited by a lack of point-of-care (POC) assessment of coagulation status. Accordingly, the potential exists for indiscriminant blood component administration. Furthermore, although thromboembolic events have been described shortly after injury, the time sequence of post-injury coagulation changes is unknown. Our current understanding of hemostasis has shifted from a classic view, in which coagulation was considered a chain of catalytic enzyme reactions, to the cell-based model (CBM), representing the interplay between the cellular and plasma components of clot formation. Thromboelastography (TEG), a time-sensitive dynamic assay of the viscoelastic properties of blood, closely parallels the CBM, permitting timely, goal-directed restoration of hemostasis via POC monitoring of coagulation status. TEG-based therapy allows for goal-directed blood product administration in trauma, with potential avoidance of the complications resulting from overzealous component administration, as well as the ability to monitor post-injury coagulation status and thromboprophylaxis. This overview addresses coagulation status and thromboprophylaxis management in the trauma patient and the emerging role of POC TEG.
Keywords: Coagulopathy, hypercoagulability, trauma, cell-based model, thromboelastography, massive transfusion, transfusion ratio
Restoration and subsequent maintenance of normal coagulation function is essential for survival of the severely injured bleeding patient. During the acute phase of trauma, efforts are focused on resuscitation and hemorrhage control, with attempts at restoration of normal blood clotting. Furthermore, after initial stabilization, trauma patients paradoxically face the dangers of a hypercoagulable state, demanding accurate risk stratification and chemoprophylaxis for prevention of highly morbid thromboembolic events (TEs).
The coagulation system involves a fine balance between the procoagulant (potentially prothrombotic) and fibrinolytic systems. Such a homeostatic state prevents catastrophic hemorrhage at the site of injury, with simultaneous control of indiscriminant thrombosis elsewhere. Massive tissue injury and shock overwhelm the body’s endogenous regulatory mechanisms, placing the critically injured patient at risk for both excessive bleeding and unwanted thrombosis. Early recognition of such dysregulation is a prerequisite toward the evolution of effective management strategies.
Thrombelastography (TEG), a point-of-care (POC) assay of the viscoelastic properties of blood, provides a comprehensive real-time analysis of hemostasis, from initial thrombin burst to fibrinolysis, permitting improved transfusion strategies resulting in the potential for goal-directed therapy of coagulation abnormalities following injury. This review describes the coagulation abnormalities associated with trauma, underscores the limitations of the traditional approach to post-injury coagulopathy, and highlights the emerging role of POC TEG in addressing these limitations.
COAGULATION DISTURBANCES IN THE TRAUMA PATIENT
Hypocoagulability
Hemorrhage accounts for up to 40% of all trauma-related deaths1,2 and is compounded by the presence of coagulopathy.3,4 Furthermore, many trauma patients presenting with massive hemorrhage succumb to refractory coagulopathy despite surgical control of bleeding.3,5,6 In fact, continuing hemorrhage due to coagulopathy remains the most common indication for damage control procedures,2 but the acute coagulopathy of trauma remains poorly characterized.7,8 Traditionally, post-injury coagulopathy was thought to originate as a result of the depletion and dilution of coagulation factors resulting from hemorrhage and subsequent fluid resuscitation, termed the “bloody vicious cycle” by our institution in 1982.5 In the face of ongoing hemorrhage, hypothermia and acidosis compound this coagulopathy, leading to a worsening coagulopathy culminating in death.9,10
The coagulopathy of trauma is a complex process, with tissue injury and hypoperfusion initiating a cascade of events resulting in a hypocoagulable state.8,11,12 Although clotting factor depletion, dilutional effects from crystalloid resuscitation, hypothermia, and acidosis have significant roles, current evidence suggests that tissue injury, hypoperfusion, accelerated fibrinolysis, and inflammatory mechanisms may be more pivotal inciting events.
Immunoactivation is an important component of this coagulopathic process, particularly as it relates to endothelial cell activation, neutrophil priming, and subsequent release of inflammatory mediators from the innate immune system via activation of the complement cascade.13 Brohi et al8 suggest that the acute coagulopathy of trauma is mediated by thrombomodulin via activated protein C, leading to impaired thrombin generation and increased fibrinolysis. Such a process may be protective teleologically by inducing an “auto-anticoagulation” state that protects the perfusion of tissue beds from thrombosis in the milieu of an activated coagulation system. These authors suggest that fibrinolysis results from activated protein C’s inactivation of plasminogen activator inhibitor (PAI)-1. However, an established mechanistic link of fibrinolysis to the acute coagulopathy of trauma has not been shown, and its occurrence in trauma patients has been variably reported, from 15% to 20%,14 but is likely underestimated due to a lack of accurate and timely laboratory tests for this entity. Others believe that acute coagulopathy after injury is a variant of disseminated intravascular coagulation, with accelerated fibrinolysis mediated by PAI-1 consumption.15
Fig. 1 depicts our current view of the “bloody vicious cycle,” which emphasizes that acute endogenous coagulopathy exists immediately after injury, is unrelated to clotting factor deficiency, and is thus resistant to factor replacement. Recent evidence from our group12,16 and others8,17 supports this theory, suggesting that more than a third of multiply injured patients are coagulopathic within 30 minutes from injury. This subset of patients has an increased incidence of multiple organ failure (MOF) and death. Of note, although the acute coagulopathy of trauma predominates in the immediate post-injury phase, subsequent uncontrolled hemorrhage and disrupted tissue consumption of clotting factors eventuates in factor depletion, leading to a state of systemic coagulopathy. In contrast to the acute endogenous coagulopathy of trauma, the resulting systemic coagulopathy requires prompt repletion of factors to restore hemostatic integrity.
Figure 1.

The updated bloody vicious cycle. This schematic depicts an updated view of the multiple factors contributing to ongoing post-injury coagulopathy. Recent observations suggest that acute endogenous coagulopathy occurs early after traumatic injury. The subsequent development of progressive systemic coagulopathy is time dependent, resulting from persistent hypotension, acidosis, and cellular shock. FFP, fresh-frozen plasma; RBC, red blood cells. Reproduced with permission from Kashuk et al.12
Understanding how these factors contribute to the development and perpetuation of coagulopathy is essential to designing optimal treatment strategies. The complex interactions of thrombin, fibrinogen, platelets, protein clotting factors, calcium ions, inflammatory mediators, and the endothelium18 create a challenge for designing a comprehensive approach to the management of acute coagulopathy of trauma.
Hypercoagulability
The oscillation in coagulation status after injury has been recognized since Walter Cannon’s studies in 1917.19 During World War I, Cannon et al studied the physiology of traumatic shock, reporting brief hypercoagulability immediately after injury, followed by hypocoagulability, ultimately returning to a hypercoagulable state. Current clinical evidence confirms that a transition from a hypocoagulable to a hypercoagulable state occurs in patients who survive the acute phase of trauma.20 Our work21 suggests that the delayed hypercoagulable state predisposes trauma patients to subsequent TE events. Currently, little is known about the timing of this transition. The etiology of the hypercoagulable state is likely multifactorial, involving endothelial injury, circulatory stasis, platelet activation, decreased levels of endogenous anticoagulants, and impaired fibrinolysis. Deep vein thrombosis has been reported to occur in 10 to 80% of patients following trauma.22 Pulmonary embolism has been observed in 2 to 22% of injured patients, and fatal pulmonary embolisms are the third most common cause of death in those who survive the first 24 hours.23
Although endothelial injury and platelet activation are known contributors to arterial thromboemboli,18 the sequence of events leading to venous thrombosis is not as clear. According to the long-standing triad of Virchow, three major factors contribute to the development of venous TE: stasis of blood flow, hypercoagulability, and damaged endothelium. In addition, several lines of evidence suggest that activation of platelets contributes to the development and propagation of venous thrombi.24,25 In an analysis of 50 venous thrombi obtained from femoral valve pockets, Sevitt et al identified areas that were characterized by fibrin and erythrocytes, mostly located in proximity of the endothelium, as well as areas dominated by a platelet-fibrin network, located more distally in the growing thrombus.26 Of note, scanning electron microscopy studies show aggregated platelets attached to venous endothelium.27 Moreover, inhibition of P-selectin on the surface of an activated platelet results in impaired thrombus formation both in an experimental model of venous thrombosis and in vivo.28 Accordingly, platelets likely contribute significantly to the formation of venous thrombi. Finally, proinflammatory activation of endothelium has been implicated in the genesis of venous thrombi.13 Risk stratification models for post-injury hypercoagulability should therefore incorporate both enzymatic and platelet components of hemostasis.
LIMITATIONS OF CURRENT CONCEPTS AND TESTING
Classic versus Cell-Based Model of Coagulation
Our current understanding of the scientific basis of the acute coagulopathy of trauma is limited by the fact that the classic laboratory tests of coagulation function, such as prothrombin time/international normalized ratio (PT/INR) and activated partial thromboplastin time (aPTT), are largely designed for assessment of anticoagulation therapy, and their applicability to the acutely injured patient has never been proven. Consequently, no consensus exists regarding the definition of either coagulopathy or subsequent hypercoagulable states, and currently there is a regrettable lack of scientific evidence to support preventive diagnostic and therapeutic strategies.
The classic model of hemostasis suggests a central role of protein coagulation factors directing the hemostatic process, with the cells surface serving primarily to provide an anionic phospholipid region for procoagulant complex assembly.29 Accordingly, the “cascade” model of coagulation (extrinsic and intrinsic pathways) is structured as a series of proteolytic reactions. Although this model supports traditional laboratory tests of coagulation by plasma-based algorithms, it does not correlate with current concepts of hemostasis occurring in vivo.18
The two laboratory tests performed traditionally to assess coagulation are PT/INR and aPTT. These plasma-based assays are performed by recalcifying a sample of citrated plasma in the presence of animal-derived or recombinant thromboplastin (PT) or a negatively charged substrate (aPTT), which results in factor activation, thereby initiating coagulation via the extrinsic (PT/INR) or intrinsic (aPTT) clotting pathway.30 The end point for these tests is the time (in seconds) until the earliest formation of a fibrin clot is detected by visual, optical, or electromechanical means.
The applicability of these laboratory tests in the trauma setting has not been validated. Furthermore, the time required to conduct these assays in most trauma centers is ~45 minutes, limiting usefulness in the setting of hemorrhagic shock.31 Indeed, the entire blood volume of the bleeding trauma patient may have been exchanged several times during this time interval, rendering the results of these laboratory tests obsolete by the time they are available. Furthermore, both the PT/INR and aPTT are performed on platelet-poor plasma, and these tests are sensitive only to the earliest initiation of clot formation. However, >95% of thrombin generation occurs after the initial polymerization of fibrinogen.32 Finally, these tests are performed in an artificial environment (pH = 7.40, temperature = 38°C), irrespective of the patient’s core body temperature and pH. Impairment of coagulation by acidosis and hypothermia has been thoroughly documented both in vitro and in vivo.33,34 Thus coagulation testing performed at a normal pH and temperature will not reflect in vivo derangements responsible for coagulopathy.9
Diagnosis of fibrinolysis is also problematic. The euglobulin lysis time (ELT) has been used for estimating the functional fibrinolytic capacity of plasma.14 Unfortunately, ELT remains a complex and time-consuming procedure that can take >180 minutes; hence it is not a practical test in the acutely bleeding patient. Other techniques used to diagnose hyperfibrinolysis, such as plasmin-antiplasmin complex, PAI-1, thrombin-activatable fibrinolytic inhibitor, and D-dimers, suffer from similar limitations. Gando et al failed to validate D-dimer, plasmin-antiplasmin complexes, fibrinopeptide-B β 15–42, or PAI-1 as indicators for increased fibrinolysis after trauma.15
The limitations of classic measurements of coagulation among trauma patients have been documented in several clinical studies. Over 30 years ago, Counts et al,35 from the Puget Sound Blood Center, performed extensive prospective analyses of coagulation function in patients receiving massive transfusions (MTs), where 69% of packed red blood cells (PRBCs) were administered within 24 hours of hospitalization. In this cohort, the predictive value of the PT, aPTT, and bleeding time was poor for assessment of generalized bleeding. Further, based on animal studies of hypovolemic shock and resuscitation, Lucas and Ledgerwood36 observed that PT and aPTT changes were infrequent unless transfusions in excess of an equivalent of 15 units of whole blood occurred.
The classic view of the intrinsic and extrinsic coagulation cascades has been replaced recently by a cell based model (CBM) of coagulation.29 This model recognizes the important interactions of the cellular and plasma components to clot formation.9 The CBM suggests that procoagulant properties result from expression of a variety of cell-based features, including protein receptors, which activate components of the coagulation system at specific cell surfaces. This model also facilitates our understanding of potential mechanistic links of cross-talk among inflammation and coagulation components because interaction between platelet receptors, endothelial cells, and cytokines have important roles in coagulation.37 This model also considers the red blood cells (RBC) and their interactions with the hemostatic process.38 For example, the provision of adenosine diphosphate from RBCs is known to contribute to platelet activation.39 Furthermore, the rheologic margination of platelets toward the vessel’s perimeter (areas with greatest shear stress) and subsequent adhesion after endothelial injury appears dependent on the quantity and size of RBCs.40
In sum, the CBM represents a major paradigm shift from a theory that views coagulation as being controlled by concentrations and kinetics of coagulation proteins to one that considers the process to be controlled by diverse cellular interactions.
Presumptive Blood Component Therapy for Post-Injury Coagulopathy
Consumption and dilution of clotting factors via crystalloid resuscitation and other factor-poor blood products perpetuates the acute coagulopathy of trauma. Coagulation factors present in plasma contained in PRBCs have minimal activity due to prolonged storage and associated short coagulation factor half-lives.41 Isolated administration of PRBCs in the absence of plasma will therefore potentiate the acute coagulopathy of trauma. Accordingly, most MT protocols advocate early replacement of factors and platelets. However, the definition of MT, and the timing and ratio of specific factor replacement, remains widely debated, due at least in part to differences in protocols as well as inherent flaws in retrospective data analysis.
A valid definition of MT is lacking. MT has been defined as administration of ≥10 units of PRBCs to an individual or the transfusion of more than one blood volume (70 mL/kg) in 24 hours. Other dynamic definitions of MT are currently used, particularly to design appropriate MT protocols. Such definitions include transfusion >10 units of PRBCs in a 24-, 12- or 6-hour period. Our group recently reviewed transfusion practices in severely injured patients at risk for post-injury coagulopathy.42 We noted that >85% of transfusions were accomplished within 6 hours post-injury, suggesting this is the critical period to assess the impact of preemptive factor replacement, rather than the 24-hour time period others have emphasized.43
Current clinical MT protocols promoting “damage control resuscitation” (i.e., preemptive transfusion of plasma, platelets, and fibrinogen) assume that patients presenting with life-threatening hemorrhage at risk for post-injury traumatic coagulopathy should receive component therapy in amounts approximating those found in whole blood during the first 24 hours.43–46 The U.S. military experience in Iraq suggesting improved survival based on a 1:1:1 fresh-frozen plasma (FFP)-to-RBC-to platelet ratio44 has led to recommendations of fixed ratios of these blood products during the first 24 hours post-injury in civilian trauma centers.43,45,46
In contrast, our results12 as well as those of others47 suggest that the optimal survival ratio appears to be in the range of a 1:2 to 1:3 FFP-to-RBC ratio. Of note, a prospective study by Scalea et al48 showed no benefit to a standardized 1:1 RBC-to-FFP transfusion protocol for critically injured trauma patients, and their study did not specifically address the early (<6 hour) time frame. We believe the reported benefits from a 1:1 strategy likely represent a surrogate marker of survival. Specifically, those patients who survive injury are simply able to receive more plasma transfusions, as opposed to those who die from acute hemorrhagic shock early after injury. Two recent reports provide further evidence of this survival bias. Snyder et al49 as well as Magnotti et al50 analyzed sequential timing of transfusions administered for post-injury traumatic resuscitation and found evidence that early (<6 hour) assessment of bleeding is critical for analysis of post-injury traumatic coagulopathy. Furthermore, they noted that the absence of time-varying assessment of blood component administration in trauma likely results in a survivor bias in studies assessing cumulative transfusion ratios. These reports further question the accuracy of prior studies using a cumulative ratio calculated at 24 hours. Pending further data, most trauma surgeons recommend a preemptive product transfusion of the critically injured patient in hemorrhagic shock using a RBC-to-FFP ratio of 1:2.
The role of early platelet transfusion in the setting of hemorrhagic shock also remains debated. As with FFP, recent military reports51 have suggested routine administration of apheresis platelets to the injured patient. However, a similar survival bias has been suggested to explain the apparent benefit of early platelet administration. Furthermore, studies from more than 2 decades ago31,52 evaluating clotting factor and platelet counts in massively transfused patients concluded that a platelet count of 100,000/mm3 is the threshold for diffuse bleeding, and that thrombocytopenia was not a clinically significant problem until transfusions exceeded 15 to 20 units of blood. Specifically, patients with a platelet count >50,000/mm3 had only a 4% chance of developing diffuse bleeding. The authors of these reports concluded that routine platelet administration was not warranted in this group of patients.31,52 Hiippala and colleagues53 assessed the changes in platelet and fibrinogen concentrations after major surgical bleeding and noted that a platelet count of 50,000/mm3 was only reached late in the course of blood loss, and the changes in individual patients were unpredictable.
Although the classic threshold for platelet transfusion has been 50,000/mm3, a higher target level of 100,000/mm3 has been suggested for multiply injured patients and patients with massive hemorrhage.53 However, the relationship of platelet count to hemostasis and the contribution of platelets to formation of a stable clot in the injured patient remain largely unknown. Furthermore, platelet function, irrespective of number, is also of crucial importance. The complex relationship of thrombin generation to platelet activation requires dynamic evaluation of clot function,54,55 as opposed to isolated measurements of platelet count, or older methods of clot assessment, such as the bleeding time, which is of no use in the trauma setting. Thus the significance of a decreased platelet count should be interpreted in the patient’s specific clinical context and with knowledge of platelet function. At this time, no evidence supports an absolute trigger for platelet transfusions in trauma.
Concerns over high ratios of blood component therapy stem in large part from a growing body of evidence documenting the adverse effects of transfusion. The association of massive transfusion of PRBCs with nosocomial pneumonia, acute lung injury, and acute respiratory distress syndrome (ARDS) has been documented.56,57 Regarding transfusion of plasma-rich components after injury, Watson et al58 evaluated independent risks associated with administration of FFP, platelets, and cryoprecipitate in a multicenter cohort study. Although early transfusion of these products was associated with decreased mortality within the first 48 hours, it was noted that every unit of FFP given was independently associated with a 2.1% and 2.5% increased risk of MOF and ARDS, respectively. Our group and others have found a similar association between transfusion of FFP and organ failure.59,60 Similar to FFP and PRBC administration, platelet transfusion is also associated with immunological complications, with a reported incidence of >200 per 100,000 transfused patients.61 Additionally, blood products are an expensive resource and of limited supply.
A major limiting factor of current MT protocols is the lack of a real-time assessment of coagulation function. Temporal and logistical limitations of conventional measurements of coagulation have precluded true goal-directed therapy of hemorrhagic shock, with current treatment algorithms advocating preemptive replacement of products. Accordingly, the role of goal-directed therapy specifically addressing coagulation abnormalities remains to be established. Furthermore, current treatment algorithms fail to consider the complex temporal and pathophysiological dynamics of coagulation. Preemptive coagulation factor replacement is essential for the initial resuscitation of the acutely bleeding patient. We remain concerned, however, regarding the continued administration of FFP and platelets via protocols that lack POC assessment of ongoing blood component requirements.
Diagnosis and Treatment of Hypercoagulability of Traumatic Critical Illness
Thromboprophylaxis remains a challenge because objective measurements of hypercoagulability are lacking. Designed originally to assess hypocoagulable states, classic coagulation tests can neither identify hypercoagulability nor predict TEs.62,63 Current treatment guidelines are thus generally based on clinical risk stratification (Table 1). Most protocols advocate a combination of subcutaneous heparinoids and sequential compression devices.64 Unfortunately, generic dosing of both unfractionated heparin (UH) and low molecular weight heparin (LMWH) does not account for variable pharmacokinetics between patients, which in turn depend on several clinical parameters, including weight, mobility, and degree of inflammation. Furthermore, response to thromboprophylaxis is not routinely monitored. Currently, the test used for monitoring LMWH effect is the anti-factor Xa activity level (anti-Xa) in plasma. However, anti-Xa measurement is expensive and labor intensive, and it has not been implemented routinely given recent controversy regarding the lack of correlation with TE events.
Table 1.
Independent Risk Factors for Venous Thromboembolism after Trauma (Multivariate Logistic Regression)
| Risk Factor | OR (95% CI) | p Value |
|---|---|---|
| Age ≥40 years | OR = 2.01 (1.74–2.32) | <0.0001 |
| Lower extremity fracture (AIS ≥3) | OR = 1.92 (1.64–2.26) | <0.0001 |
| Head injury (AIS ≥3) | OR = 1.24 (1.05–1.46) | 0.0125 |
| Ventilator days >3 | OR = 8.08 (6.86–9.52) | <0.0001 |
| Vascular injury | OR = 3.56 (2.25–5.72) | <0.0001 |
| Major operative procedure | OR = 1.53 (1.30–1.80) | <0.0001 |
OR, odds ratio; CI, confidence interval; AIS, abbreviated injury scale. Reproduced with permission from Knudson et al.101
Another current limitation of chemoprophylaxis is the failure to assess platelet hypercoagulability. Current thromboprophylactic regimens using UH or LMWH decrease thrombin levels through antithrombin potentiation and by direct factor Xa inhibition.65 Although thrombin is the main platelet activator, given the multiplicity of factors known to activate platelets and the direct interactions between the platelet and the endothelium, solely targeting thrombin production via current regimens may not adequately prevent platelet activation.18 Accordingly, a hypercoagulable state may persist despite “therapeutic” anticoagulation. The addition of pharmacological inhibition of platelet activation and aggregation could further reduce the incidence and propagation of thrombus growth and lead to reduction of subsequent TEs, although the use of platelet antagonists in the acutely injured patient requires precise monitoring of hemostasis due to the serious and potentially devastating consequences of recurrent hemorrhage.
Given the limitations just described, it is not surprising that several studies have reported that the frequency of TEs has not changed despite adoption of current thromboprophylactic guidelines.66–68 In a multi-center study involving 12 medical and surgical intensive care units (ICUs), Patel et al concluded that most venous thromboembolisms (VTEs) occurred due to prophylaxis failure rather than failure to provide prophylaxis.66 In their study, 65% of VTEs occurred in patients receiving standard pharmacological thromboprophylaxis.
POINT-OF-CARE THROMBOELASTOGRAPHY
Historical Perspective
Hellmut Hartert conceived TEG in Germany, at the Heidelberg University School of Medicine in 1948.69 Introduced initially for the evaluation of clotting factor deficiencies, TEG was applied increasingly throughout Europe during the 1950s, validating its use as a reliable assessment of anticoagulant effect, the presence of thrombocytopenia, and fibrinolysis.70 The availability of TEG in the United States was limited until the 1980s, at which time it was found to be useful during liver transplantation due to the frequency of fibrinolysis during the anhepatic phase.71 From this initial experience, TEG evolved for use in monitoring patients undergoing cardiopulmonary bypass. In this group, it was primarily useful for monitoring intraoperative heparinization and was also found beneficial for predicting postoperative bleeding.72 TEG gained additional acceptance as a tool for assessment of coagulation in the general surgical patient, permitting differentiation of coagulopathy from surgical bleeding.73,74
Technique and Interpretation
The TEG analyzer is composed of two mechanical parts separated by a blood specimen: a plastic cup or cuvette, into which a 0.36-mL blood specimen is pipetted, and a plastic pin attached to a torsion wire and suspended within the specimen (Fig. 2). Once the sample within the cuvette is placed on the TEG analyzer, the temperature is adjusted to that of the patient. The cup then oscillates slowly through an angle of 4°45′. Initially, movement of the cuvette does not affect the pin, but as clot develops, resistance from the developing fibrin strands couples the pin to the motion of the cuvette. In turn, the torsion wire generates a signal that is amplified and records the characteristic tracing seen in Fig. 2. In earlier iterations, this tracing was recorded on heat-sensitive paper moving at a rate of 2 mm/minute. More recent computer technology has allowed for automatic calculation of TEG variables. At our institution, a dynamic TEG tracing is transmitted directly to the operating room or ICU via computer within minutes, enabling prompt interpretation.
Figure 2.
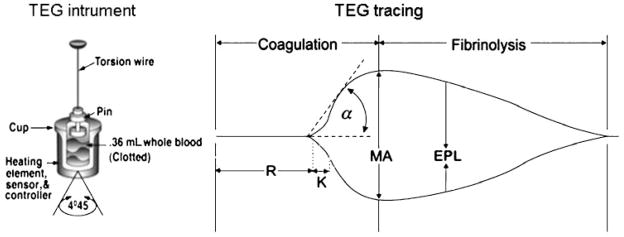
Thrombelastography instrument and tracing. The instrument diagram depicts the cuvette where a whole blood sample is placed and the pin attached to a torsion wire. Once the assay is initiated, a tracing is produced and an initial linear segment (zone of precoagulation) extends from the beginning of the test to the formation of the first fibrin strand, causing the tracing to split (split point). The progressive divergence of the tracing reflects the formation of the clot. The R time is reached when the onset of clotting provides enough resistance to produce a 2-mm amplitude reading. The K time is a measurement of the time interval from the R time to the point where fibrin cross-linking provides enough clot resistance to produce a 20-mm amplitude reading. The α angle is the angle formed by the slope of a tangent line traced from the R to the K time measured in degrees. The maximum amplitude, MA, indicates the point at which clot strength reaches its maximum measure in millimeters. Estimated percentage lysis, EPL, is a measure of fibrinolysis and reflects the slow yet progressive decrease in clot strength once MA is reached. Reproduced with permission from Haemonetics Corporation.
Based on time-resistance correlations, the following seven TEG parameters have been defined: (1) split point (SP, minutes), (2) reaction time (R, minutes), (3) coagulation time (K, minutes), (4) α angle (α, degrees), (5) maximum amplitude (MA, mm), (6) clot strength (G, dynes/cm2), and (7) estimated percentage lysis (EPL, %). The various components of the TEG tracing are depicted in Fig. 2. The SP time is a measure of the time to initial clot formation, interpreted from the earliest resistance detected by the TEG analyzer causing the tracing to split; this is the point at which all other platelet-poor plasma clotting assays (e.g., PT and aPTT) fail to progress. The R value is defined as the time elapsed from the initiation of the test until the point where the onset of clotting provides enough resistance to produce a 2-mm amplitude reading on the TEG tracing. Of note, in the rapid-TEG (R-TEG) assay (discussed later, due to the acceleration of clotting initiation, the R time is represented by a TEG-derived activated clotting time (TEG-ACT). The R time and TEG-ACT are most representative of the initiation phase of enzymatic clotting factors. Whereas a prolonged R time or TEG-ACT is diagnostic of hypocoagulability, decreased values suggest hypercoagulability. The K time is a measurement of the time interval from the R time to the point where fibrin cross-linking provides enough clot resistance to produce a 20-mm amplitude reading. The α angle is the angle formed by the slope of a tangent line traced from the R to the K time measured in degrees. Both the K time and the α-angle denote the rate at which the clot strengthens and are most representative of thrombin’s cleaving of available fibrinogen into fibrin. The MA indicates the point at which clot strength reaches its maximum measure in millimeters on the TEG tracing, and it reflects the end result of maximal platelet-fibrin interaction via glycoprotein (GP) IIb-IIIa receptors.75 Finally, G is a calculated measure of total clot strength derived from amplitude (A, mm); G = (5000 × A)/(100 × A). Due to its exponential relationship with A, G is a more realistic representation of overall clot strength.76 Table 2 summarizes the various TEG components and their significance.
Table 2.
Thromboelastrography Parameters
| Parameter | Significance |
|---|---|
| SP time | Earliest activity of enzymatic factors, causing tracing to split |
| R time | Initiation phase of enzymatic factor activity |
| K time | Potentiation phase of enzymatic factors yielding clot strengthening |
| Alpha-angle | Rate of clot strengthening through polymerization of available fibrinogen |
| MA | Platelet functional contribution to clot strength through GP IIb-IIIa receptor interaction with fibrin |
| G | Overall total clot strength resulting from all coagulation interactions, calculated from amplitude (A), G = (5000 × A)/(100 × A). |
| EPL | Fibrinolytic activity, derived from percentage decrease in clot strength after MA is reached. |
SP, split point, minutes; R, reaction time, minutes; K, coagulation time, minutes; α angle, degrees; MA, maximum amplitude, millimeters; G, clot strength, dynes/cm2; EPL, estimated percent agelysis, %.
Blood coagulation in TEG is initiated by addition of an activating solution consisting of kaolin, phospholipids, and buffered stabilizers, which requires an activation phase of several minutes before coagulation starts. To expedite time to generate results (e.g., in the setting of hemorrhagic shock), clotting initiation can be further prompted by the addition of tissue factor. This permits the earliest tracings via R-TEG to be viewed within 10 minutes. Furthermore, when heparinized patients are evaluated, the effects of heparin on the TEG tracing may be delineated via a simultaneous analysis of the heparinized sample on a conventional cuvette and another containing heparinase. This strategy is particularly useful for assessment of the efficacy of chemoprophylaxis. Similarly, TEG platelet mapping assesses the patient’s percentage platelet inhibition against their own maximum platelet function, as measured by changes in the MA secondary to anti-platelet therapy (e.g., acetylsalicylic acid, dipyridamole, clopidogrel).77
Specific coagulation derangements produce characteristic TEG patterns. Fig. 3 depicts these deranged profiles along with a normal TEG tracing. Anticoagulation causes enzymatic inhibition of coagulation factors and produces a prolonged R time due to delayed initiation of clot formation, along with normal fibrinogen (normal K time and α angle) and platelet function (normal MA) (Fig. 3B). Platelet dysfunction is noted by a tracing with a normal R time and a primarily decreased MA, as seen in Fig. 3C. We have defined clinically significant fibrinolysis when EPL values exceed 15%7 resulting in a characteristic tapering of the TEG tracing immediately after the MA is reached (Fig. 3D). Depicted in Fig. 3E is a characteristic tracing of hypercoagulability, identifiable by decreased R and K times along with an elevated α angle and MA.
Figure 3.

Normal and abnormal thrombelastographic patterns. (A) Normal tracing. (B) Prolonged clotting time seen with coagulation factor deficiency or inhibition by anticoagulation. (C) Decreased maximum amplitude (MA), seen during platelet dysfunction or pharmacological inhibition. (D) Increased percentage lysis in a fibrinolytic tracing. (E) Decreased clotting and clotting formation time, elevated α angle, and increased MA represent a hypercoagulable state. Reproduced with permission from Haemonetics Corporation.
Coagulopathic tracings seen in the severely injured patient involve a combination of the examples just described. A prolonged TEG-ACT and R time early after injury typically characterize an evolving coagulopathy. Further resuscitation with crystalloid and PRBCs may lead to decreased platelet function (reduced MA), as seen in Fig. 3C.
The basic TEG tracing has been further studied to generate novel TEG parameters that may be useful markers of coagulation abnormalities.78–81 Using the CBM,29 Nielsen described a clot lifespan model (CLSM) of coagulation.78 Employing a TEG-based approach with a standardized clotting and fibrinolytic stimulus, he assessed parametric, elastic modulus-based parameters of clot formation. The model provides mechanistic insight into hemostasis through clotting and fibrinolytic stimuli that would not otherwise be explained by coagulation factor activity alone. The CLSM was able to differentiate enzymatic inhibition from cellular-controlled noncatalytic inhibition of protein–protein interactions responsible for changes in both clot growth and disintegration during hemodilution. Observing this CLSM of coagulation, Sorensen et al noted that thrombelastographic coagulation kinetics correlated with thrombin generation in vivo.79 This correlation resulted from calculating the first derivative of changes in clot resistance expressed as a change in amplitude per unit of time (mm × 100/second), representing the maximum velocity of clot formation. Based on these derivatives, clot-velocity curves (V curve), depicted in Fig. 4, may be plotted using standard TEG software. Rivard et al demonstrated that the V-curve parameter “time to maximum rate of thrombus generation” (TMRTG), calculated from the time of clot initiation to the point where maximum velocity of thrombus growth is reached, correlated with thrombin-antithrombin levels,80 validating TMRTG as a potential marker of thrombin generation.
Figure 4.

Thrombus velocity curve. Sample of a thrombus velocity curve (V curve, in gray), calculated from the first derivative of changes in clot resistance, depicted here over a standard thromboelastographic tracing, showing relationship of delta (R to SP) with thrombus generation parameters. MRTG, maximum rate of thrombus generation; R, reaction time; SP, split point; TMRTG, time to maximum rate of thrombus generation; TTG, total thrombus generation; Δ, delta.
We recently studied V-curve parameters in a cohort of surgical intensive care unit patients at risk for TE complications and demonstrated a linear correlation (r = 0.94) between a novel parameter delta and TMRTG.81 Delta (depicted in Fig. 4) is calculated from the same time interval as TMRTG by subtracting SP from the R time. A decreased delta indicates increased thrombin generation, as seen in a hypercoagulable state, whereas an increased delta indicates decreased thrombin generation, as seen in hypocoagulability or enzymatic inhibition of coagulation factors. In our pilot study, 30% of patients receiving prophylactic LMWH had an appropriately increased delta but remained hypercoagulable according to increased total clot strength (G) and MA. Delta may thus aid in differentiation of enzymatic from platelet hypercoagulability.
The various TEG values, derived from a single measurement of whole blood coagulation, are not independent parameters but rather a continuum of blood coagulation with interactions between all components. For instance, thrombin liberates fibrinopeptides from fibrinogen, allowing association with other fibrinogen molecules for soluble fibrin and subsequently thrombin-activated factor XIII converts soluble into cross-linked fibrin. This impacts the rate of clot strength as represented by the α angle and K time. Furthermore, thrombin exerts known effects on platelet function due to interactions with factor VIII and von Willebrand factor; accordingly, such changes mainly impact MA and G.
Recent data suggest the superiority of TEG as compared with both aPTT and PT/INR for assessment of the acute coagulopathy of trauma. Kheirabadi et al82 showed in a rabbit model that TEG is a more sensitive indicator of dilutional hypothermic coagulopathy than PT. A recent clinical study of trauma patients surviving the first 24 hours reported that TEG detected hypercoagulability (by an increased MA and G) whereas the PT and aPTT did not.83 In a retrospective review of penetrating injuries, Plotkin et al84 recently demonstrated hypocoagulation based on delayed propagation of the clot (increased K time and reduced α angle) and decreased clot strength (reduced MA), where MA correlated with blood product use (r = 0.57, p <0.01). These findings emphasize the limitations of classic coagulation tests and their lack of effectiveness, particularly in post-injury coagulopathy.
POC testing has also proved useful for the detection of fibrinolysis. In a cohort of trauma patients, Schöchl et al85 determined fibrinolysis as detected by ROTEM as a better predictor of mortality than injury severity score. The primary difference between ROTEM and TEG relates to hardware; whereas TEG operates by oscillating a cup filled with a sample that engages a pin/wire transduction system as clot formation occurs, the ROTEM involves an immobile cup in which the pin/wire transduction system slowly oscillates.86 Parameters and diagnostic nomenclature among these two assays are similar. In our recent experience with trauma patients requiring MT, fibrinolysis was identified by TEG in 18% of patients and occurred <1 hour post-injury.7 Among patients requiring MT, 65% had fibrinolysis, with the risk of death correlating significantly with the presence of fibrinolysis (p = 0.027).
Goal-Directed Resuscitation of Post-Injury Coagulopathy
Goal-directed transfusion therapy guided by TEG tailors blood product administration to the pathophysiological state of the individual patient. At our institution, this approach has become an integral part of resuscitation.87 Using this technology, a variety of coagulation abnormalities have been noted that in the past would have been overlooked. Our current protocol of component transfusion therapy emphasizes a goal-directed approach, permitting stepwise correction of coagulation dysfunction by repeated assessment via TEG. Fig. 5 shows a clinical example of this stepwise goal-directed approach to post-injury coagulopathy.
Figure 5.

Coagulopathic tracing after injury and subsequent correction via goal-directed therapy. The black thromboelastographic tracing was obtained upon admission and shows delayed clot initiation, decreased rate of clot formation (decreased angle), and poor platelet contribution to clot strength (decreased maximum amplitude). Administration of fresh-frozen plasma, cryoprecipitate, and platelets gradually achieved a normal profile, as seen by the subsequent tracings in gray.
With results available within 10 minutes, an initial hemostatic assessment with R-TEG identifies patients at risk for post-injury coagulopathy upon arrival. Blood component therapy is then tailored to address each deranged phase of clotting in a specific manner, and subsequent reassessment allows the evaluation of response until a set threshold is reached. This strategy also permits improved communication with the blood bank; based on initial assessment and response to component therapy, more accurate estimations of component requirements can be made.
Fig. 6 depicts our current goal-directed approach to coagulopathy. Reflecting the initiation phase of enzymatic factor activity, a prolonged TEG-ACT value is the earliest indicator of coagulopathy; when the value is above threshold, FFP is administered. K time and α angle follow and are most dependent on the availability of fibrinogen to be cleaved into fibrin while in the presence of thrombin. If indicated by K and α angle, cryoprecipitate is administered, providing a concentrated form of fibrinogen (150 to 250 mg/10 mL).88 MA is then noted, considering the relationship between fibrin generated during the initial phases of hemostasis and platelets via GP IIb-IIIa receptor interaction.75 Platelets are administered based on an MA <54 mm, which reflects the platelets’ functional contribution to clot formation. Of note, in this protocol, platelet transfusion is not based on platelet count but on functional contribution to clotting, reflected by the MA.
Figure 6.

Algorithm of goal-directed approach to post-traumatic coagulopathy. α angle, rate of clot formation; ACA, aminocaproic acid; ACT, activated clotting time; EPL, estimated percentage lysis; FFP, fresh-frozen plasma; G, clot strength; MA, maximum amplitude; r-TEG, rapid thromboelastography; TEG, thromboelastography.
Antifibrinolytic drugs such as aprotinin or aminocaproic acid have proven effective in hemorrhage during cardiac surgery and hepatic transplantation.89,90 However, both cost and morbidity associated with indiscriminant use mandate an accurate diagnosis of fibrinolysis. Specifically, these drugs have been associated with an increased risk of renal failure and thrombotic events such as stroke and myocardial infarction.91 Of note, TEG is the only current test able to establish a diagnosis of fibrinolysis rapidly and reliably in the acutely bleeding patient.92,93 After the tracing has reached MA, an EPL index is obtained based on the decreasing rate of clot strength. Epsilon-aminocaproic acid is indicated in the presence of significant fibrinolysis (EPL >15%). Fig. 7 shows a clinical example of correction of a fibrinolytic TEG tracing in a hemorrhaging patient following administration of aminocaproic acid. Furthermore, we have observed a post-fibrinolysis consumptive coagulopathy, which represents diffuse clotting factor deficiency secondary to massive consumption after fibrinolysis. The resulting severe thrombin deficiency may be an indication for recombinant factor VIIa, and we have noted rapid improvement with normalization of TEG patterns after such treatment.
Figure 7.

Fibrinolytic tracing. Profound fibrinolysis (tracing in black) seen in hemorrhaging patient after injury. Subsequent correction of coagulation profile is achieved (tracing in gray) after administration of aminocaproic acid.
In summary, implementation of a goal-directed approach to post-injury coagulopathy offers the following potential benefits: (1) reduction of transfusion volumes via specific goal-directed treatment of identifiable coagulation abnormalities, (2) earlier correction of coagulation abnormalities with more efficient restoration of physiological hemostasis, (3) improved survival in the acute hemorrhagic phase due to improved hemostasis from correction of coagulopathy, and (4) improved outcomes in the later phase due to attenuation of immuno-inflammatory complications, including ARDS and MOF.
Current retrospective data and pilot studies support a reduction in MT rates, decreased need for multiple and repeated classic coagulation tests, and decreased morbidity and mortality after implementation of TEG in trauma care.21,81,85,94 We recently compared pre-TEG to post-TEG outcomes of patients at risk of post-injury coagulopathy admitted to our trauma center.95 The TEG G value was significantly associated with survival (p = 0.03), whereas PT/INR and PTT did not discriminate between survivors and nonsurvivors. Further validation studies are ongoing at our institution as well as other trauma centers.
Post-Injury Hypercoagulability: Risk Stratification
Because POC TEG characterizes dynamic hypercoagulability and simultaneously reflects the antithrombotic effect of chemoprophylaxis, it may serve as a template for designing tailored thromboprophylactic regimens. We anticipate that TEG will prove useful for optimizing prophylactic LMWH dosing and monitoring, and for identifying those patients requiring antiplatelet therapy in addition to, or in lieu of, LMWH therapy for the prevention of TE events. Additionally, recent experience gained in cardiovascular medicine with TEG platelet mapping for dosing and monitoring of platelet inhibitors96–98 suggests its usefulness for guiding prophylactic antiplatelet therapy in this subgroup.
Our recent work has validated that TEG hypercoagulable G values were predictive of TE events in a cohort of critically ill surgical patients receiving standard chemoprophylaxis.21 Despite chemoprophylaxis, 60% of patients remained hypercoagulable as measured by G. In a multiple regression analysis, for every 1-point increase in G there was a 25% increased risk of TE complications, when controlling for the presence of chemoprophylaxis (odds ratio: 1.25, 95% confidence interval, 1.12–1.39). None of the patients with a normal coagulation TEG profile developed a TE event (p <0.001).
Recently, an in vivo study evaluating the effect of LMWH on TEG parameters reported that TEG parameters correlated significantly with anti-Xa levels. Among these parameters, the TEG R time had the best correlation with anti-Xa levels with the most favorable combination of sensitivity and specificity for the therapeutic range of anti-Xa levels (r = 0.82; p <0.0001; sensitivity 68%, specificity 100%).62
As mentioned previously, one prominent limitation of current thromboprophylactic regimens is the absence of platelet inhibition; no differentiation between enzymatic and platelet contribution to hypercoagulability can be made based on classic coagulation tests. Delta’s linear correlation with TMRTG may allow for identification of thrombin’s important contribution to clot formation.81 Using delta as a measure of enzymatic activity may allow for differentiation of the enzymatic contribution to hypercoagulability from that of platelet reactivity.
LIMITATIONS AND FUTURE DIRECTIONS
Introduction of TEG into clinical practice has multiple challenges. Although the routine operational cost of a TEG assay is comparable with that of a single classic coagulation test (PT/INR, aPTT), the cost of the TEG analyzer is significantly higher than that of the equipment required for classic coagulation testing. However, reductions in financial and administrative costs have been documented at institutions using TEG in cardiac surgery patients because decreased blood transfusion and surgical reexploration rates have decreased after implementation of TEG.99,100 Despite this, no documented financial benefit has yet been established prospectively comparing TEG with routine clinical practice when caring for trauma patients. The potential benefits of TEG are contingent on accurate interpretation of results. This comes with a significant learning curve for the clinician to translate these potential benefits into clinical practice. Our initial experience with goal-directed therapy via TEG and our current preliminary studies have led us to initiate a randomized prospective clinical trial to evaluate more accurately the usefulness of TEG as a screening test for hypercoagulability and monitoring of thromboprophylaxis. If confirmed, an improved risk stratification of surgical patients based on the presence, degree, and differentiation of the hypercoagulable state could facilitate changes to our current thromboprophylaxis regimens. We have also initiated a randomized prospective clinical trial evaluating the efficacy of our current TEG protocol in managing post-injury coagulopathy in the context of MT. These studies will help elucidate the usefulness of this promising technology.
CONCLUSION
Successful management of the severely injured patient requires knowledge of the dynamic process of coagulation. Impaired platelet function and coagulation factor activity are not predicted by dilution or hemorrhagic shock alone. Rather, the coagulopathy of trauma is a complex multifactorial event. Furthermore, complications resulting from hypercoagulable states developing after post injury remain significant continuing challenges after successful resuscitation has been accomplished. Current thromboprophylactic regimens incompletely address the pathophysiology of venous thrombosis, which likely reflects multiple complex interactions involving coagulation proteins, platelets, endothelial cells, and inflammatory mediators. Recent experience with TEG suggests that this technology could serve as a template for clinical applications of the CBM of coagulation in the injured patient. Subsequent treatment protocols are now being tailored based on specific evaluation of clot formation as an ex vivo representative assay of the coagulation process.
References
- 1.Sauaia A, Moore FA, Moore EE, et al. Epidemiology of trauma deaths: a reassessment. J Trauma. 1995;38:185–193. doi: 10.1097/00005373-199502000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Kauvar DS, Lefering R, Wade CE. Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. J Trauma. 2006;60(6, Suppl):S3–S11. doi: 10.1097/01.ta.0000199961.02677.19. [DOI] [PubMed] [Google Scholar]
- 3.Cosgriff N, Moore EE, Sauaia A, Kenny-Moynihan M, Burch JM, Galloway B. Predicting life-threatening coagulopathy in the massively transfused trauma patient: hypothermia and acidoses revisited. J Trauma. 1997;42(5):857–861. doi: 10.1097/00005373-199705000-00016. discussion 861–862. [DOI] [PubMed] [Google Scholar]
- 4.MacLeod JB, Lynn M, McKenney MG, Cohn SM, Murtha M. Early coagulopathy predicts mortality in trauma. J Trauma. 2003;55(1):39–44. doi: 10.1097/01.TA.0000075338.21177.EF. [DOI] [PubMed] [Google Scholar]
- 5.Kashuk JL, Moore EE, Millikan JS, Moore JB. Major abdominal vascular trauma—a unified approach. J Trauma. 1982;22(8):672–679. doi: 10.1097/00005373-198208000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Tieu BH, Holcomb JB, Schreiber MA. Coagulopathy: its pathophysiology and treatment in the injured patient. World J Surg. 2007;31(5):1055–1064. doi: 10.1007/s00268-006-0653-9. [DOI] [PubMed] [Google Scholar]
- 7.Kashuk JL, Moore EE, Sawyer M, et al. Primary fibrinolysis is integral in the pathogenesis of acute coagulopathy of trauma. Paper presented at: American Surgical Association 130th Annual Meeting; 2010; Chicago, IL. [Google Scholar]
- 8.Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg. 2007;245(5):812–818. doi: 10.1097/01.sla.0000256862.79374.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossaint R, Cerny V, Coats TJ, et al. Key issues in advanced bleeding care in trauma. Shock. 2006;26(4):322–331. doi: 10.1097/01.shk.0000225403.15722.e9. [DOI] [PubMed] [Google Scholar]
- 10.Spivey M, Parr MJ. Therapeutic approaches in trauma-induced coagulopathy. Minerva Anestesiol. 2005;71(6):281–289. [PubMed] [Google Scholar]
- 11.MacLeod JB. Trauma and coagulopathy: a new paradigm to consider. Arch Surg. 2008;143(8):797–801. doi: 10.1001/archsurg.143.8.797. [DOI] [PubMed] [Google Scholar]
- 12.Kashuk JL, Moore EE, Johnson JL, et al. Postinjury life threatening coagulopathy: is 1:1 fresh frozen plasma:packed red blood cells the answer? J Trauma. 2008;65(2):261–270. doi: 10.1097/TA.0b013e31817de3e1. discussion 270–271. [DOI] [PubMed] [Google Scholar]
- 13.Amara U, Rittirsch D, Flierl M, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. 2008;632:71–79. doi: 10.1007/978-0-387-78952-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapsch DN, Metzler M, Harrington M, Mitchell FL, Silver D. Fibrinolytic response to trauma. Surgery. 1984;95(4):473–478. [PubMed] [Google Scholar]
- 15.Gando S, Tedo I, Kubota M. Posttrauma coagulation and fibrinolysis. Crit Care Med. 1992;20(5):594–600. doi: 10.1097/00003246-199205000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Moore EE, Moore FA, Fabian TC, et al. PolyHeme Study Group. Human polymerized hemoglobin for the treatment of hemorrhagic shock when blood is unavailable: the USA multicenter trial. J Am Coll Surg. 2009;208(1):1–13. doi: 10.1016/j.jamcollsurg.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 17.Maegele M, Lefering R, Yucel N, et al. AG Polytrauma of the German Trauma Society (DGU) Early coagulopathy in multiple injury: an analysis from the German Trauma Registry on 8724 patients. Injury. 2007;38(3):298–304. doi: 10.1016/j.injury.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359(9):938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 19.Cannon WB, Fraser J, Cowell E. The preventive treatment of wound shock. JAMA. 1918;70:618–621. [Google Scholar]
- 20.Schreiber MA, Differding JS, Thorborg P, Mayberry JC, Mullins RJ. Hypercoagulability is most prevalent early after injury and in female patients. J Trauma. 2005;58(3):475–480. doi: 10.1097/01.ta.0000153938.77777.26. discussion 480–481. [DOI] [PubMed] [Google Scholar]
- 21.Kashuk JL, Moore EE, Sabel A, et al. Rapid thrombelastography (r-TEG) identifies hypercoagulability and predicts thromboembolic events in surgical patients. Surgery. 2009;146(4):764–772. doi: 10.1016/j.surg.2009.06.054. discussion 772–774. [DOI] [PubMed] [Google Scholar]
- 22.Attia J, Ray JG, Cook DJ, Douketis J, Ginsberg JS, Geerts WH. Deep vein thrombosis and its prevention in critically ill adults. Arch Intern Med. 2001;161(10):1268–1279. doi: 10.1001/archinte.161.10.1268. [DOI] [PubMed] [Google Scholar]
- 23.O’Malley KF, Ross SE. Pulmonary embolism in major trauma patients. J Trauma. 1990;30(6):748–750. doi: 10.1097/00005373-199006000-00018. [DOI] [PubMed] [Google Scholar]
- 24.Ramacciotti E, Myers DD, Jr, Wrobleski SK, et al. P-selectin/PSGL-1 inhibitors versus enoxaparin in the resolution of venous thrombosis: a meta-analysis. Thromb Res. 2010;125(4):e138–e142. doi: 10.1016/j.thromres.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.López JA, Kearon C, Lee AY. Deep venous thrombosis. Hematology Am Soc Hematol Educ Program. 2004:439–456. doi: 10.1182/asheducation-2004.1.439. [DOI] [PubMed] [Google Scholar]
- 26.Sevitt S. The structure and growth of valve-pocket thrombi in femoral veins. J Clin Pathol. 1974;27(7):517–528. doi: 10.1136/jcp.27.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim Y, Nakase H, Nagata K, Sakaki T, Maeda M, Yamamoto K. Observation of arterial and venous thrombus formation by scanning and transmission electron microscopy. Acta Neurochir (Wien) 2004;146(1):45–51. doi: 10.1007/s00701-003-0156-5. discussion 51. [DOI] [PubMed] [Google Scholar]
- 28.Myers DD, Jr, Rectenwald JE, Bedard PW, et al. Decreased venous thrombosis with an oral inhibitor of P selectin. J Vasc Surg. 2005;42(2):329–336. doi: 10.1016/j.jvs.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman M, Monroe DM., III A cell-based model of hemostasis. Thromb Haemost. 2001;85(6):958–965. [PubMed] [Google Scholar]
- 30.McPherson MA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 21. Philadelphia, PA: W.B. Saunders; 2006. [Google Scholar]
- 31.Lucas CE, Ledgerwood AM. Clinical significance of altered coagulation tests after massive transfusion for trauma. Am Surg. 1981;47(3):125–130. [PubMed] [Google Scholar]
- 32.Brummel KE, Paradis SG, Butenas S, Mann KG. Thrombin functions during tissue factor-induced blood coagulation. Blood. 2002;100(1):148–152. doi: 10.1182/blood.v100.1.148. [DOI] [PubMed] [Google Scholar]
- 33.Martini WZ. Coagulopathy by hypothermia and acidosis: mechanisms of thrombin generation and fibrinogen availability. J Trauma. 2009;67(1):202–208. doi: 10.1097/TA.0b013e3181a602a7. discussion 208–209. [DOI] [PubMed] [Google Scholar]
- 34.Ferrara A, MacArthur JD, Wright HK, Modlin IM, McMillen MA. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg. 1990;160(5):515–518. doi: 10.1016/s0002-9610(05)81018-9. [DOI] [PubMed] [Google Scholar]
- 35.Counts RB, Haisch C, Simon TL, Maxwell NG, Heimbach DM, Carrico CJ. Hemostasis in massively transfused trauma patients. Ann Surg. 1979;190(1):91–99. doi: 10.1097/00000658-197907000-00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lucas CE, Ledgerwood AM. Clinical significance of altered coagulation tests after massive transfusion for trauma. Am Surg. 1981;47(3):125–130. [PubMed] [Google Scholar]
- 37.Opal SM, Esmon CT. Bench-to-bedside review: functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Crit Care. 2003;7(1):23–38. doi: 10.1186/cc1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lier H, Krep H, Schroeder S, Stuber F. Preconditions of hemostasis in trauma: a review. The influence of acidosis, hypocalcemia, anemia, and hypothermia on functional hemostasis in trauma. J Trauma. 2008;65(4):951–960. doi: 10.1097/TA.0b013e318187e15b. [DOI] [PubMed] [Google Scholar]
- 39.Quaknine-Orlando B, Samama CM, Riou B, et al. Role of the hematocrit in a rabbit model of arterial thrombosis and bleeding. Anesthesiology. 1999;90(5):1454–1461. doi: 10.1097/00000542-199905000-00031. [DOI] [PubMed] [Google Scholar]
- 40.Turitto VT, Weiss HJ. Red blood cells: their dual role in thrombus formation. Science. 1980;207(4430):541–543. doi: 10.1126/science.7352265. [DOI] [PubMed] [Google Scholar]
- 41.Aucar JA, Isaak E, Anthony D. The effect of red blood cell age on coagulation. Am J Surg. 2009;198(6):900–904. doi: 10.1016/j.amjsurg.2009.05.034. [DOI] [PubMed] [Google Scholar]
- 42.Kashuk JL, Moore EE, Johnson JL, et al. Postinjury life threatening coagulopathy: is 1:1 fresh frozen plasma:packed red blood cells the answer? J Trauma. 2008;65(2):261–270. doi: 10.1097/TA.0b013e31817de3e1. discussion 270–271. [DOI] [PubMed] [Google Scholar]
- 43.Holcomb JB, Wade CE, Michalek JE, et al. Increased plasma and platelet to red blood cell ratios improves outcome in 466 massively transfused civilian trauma patients. Ann Surg. 2008;248(3):447–458. doi: 10.1097/SLA.0b013e318185a9ad. [DOI] [PubMed] [Google Scholar]
- 44.Borgman MA, Spinella PC, Perkins JG, et al. The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J Trauma. 2007;63(4):805–813. doi: 10.1097/TA.0b013e3181271ba3. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez EA, Moore FA, Holcomb JB, et al. Fresh frozen plasma should be given earlier to patients requiring massive transfusion. J Trauma. 2007;62(1):112–119. doi: 10.1097/01.ta.0000250497.08101.8b. [DOI] [PubMed] [Google Scholar]
- 46.Gunter OL, Jr, Au BK, Isbell JM, Mowery NT, Young PP, Cotton BA. Optimizing outcomes in damage control resuscitation: identifying blood product ratios associated with improved survival. J Trauma. 2008;65(3):527–534. doi: 10.1097/TA.0b013e3181826ddf. [DOI] [PubMed] [Google Scholar]
- 47.Davenport R, De’Ath H, Manson J, et al. Coagulation response to different plasma:red cell ratios in trauma transfusion. Paper presented at: Annual Meeting of the Eastern Association for the Surgery of Trauma; 2010; Naples, FL. [Google Scholar]
- 48.Scalea TM, Bochicchio KM, Lumpkins K, et al. Early aggressive use of fresh frozen plasma does not improve outcome in critically injured trauma patients. Ann Surg. 2008;248(4):578–584. doi: 10.1097/SLA.0b013e31818990ed. [DOI] [PubMed] [Google Scholar]
- 49.Snyder CW, Weinberg JA, McGwin G, Jr, et al. The relationship of blood product ratio to mortality: survival benefit or survival bias? J Trauma. 2009;66(2):358–362. doi: 10.1097/TA.0b013e318196c3ac. discussion 362–364. [DOI] [PubMed] [Google Scholar]
- 50.Magnotti LJ, Zarzaur BL, Croce MA, et al. Improved survival after hemostatic resuscitation: does the emperor have no clothes? J Trauma. 2010 doi: 10.1097/TA.0b013e3182051691. In press. [DOI] [PubMed] [Google Scholar]
- 51.Perkins JG, Cap AP, Andrew CP, et al. An evaluation of the impact of apheresis platelets used in the setting of massively transfused trauma patients. J Trauma. 2009;66 (4, Suppl):S77–S84. doi: 10.1097/TA.0b013e31819d8936. discussion S84–S85. [DOI] [PubMed] [Google Scholar]
- 52.Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol. 1987;67:365–368. doi: 10.1111/j.1365-2141.1987.tb02359.x. [DOI] [PubMed] [Google Scholar]
- 53.Hiippala ST, Myllylä GJ, Vahtera EM. Hemostatic factors and replacement of major blood loss with plasma-poor red cell concentrates. Anesth Analg. 1995;81(2):360–365. doi: 10.1097/00000539-199508000-00026. [DOI] [PubMed] [Google Scholar]
- 54.Zuckerman L, Cohen E, Vagher JP, Woodward E, Caprini JA. Comparison of thrombelastography with common coagulation tests. Thromb Haemost. 1981;46(4):752–756. [PubMed] [Google Scholar]
- 55.Ellis TC, Nielsen VG, Marques MB, et al. Thrombelastographic measures of clot propagation: a comparison of alpha with the maximum rate of thrombus generation. Blood Coagul Fibrinolysis. 2007;18:45–48. doi: 10.1097/MBC.0b013e3280111a8e. [DOI] [PubMed] [Google Scholar]
- 56.Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008;36(9):2667–2674. doi: 10.1097/CCM.0b013e3181844677. [DOI] [PubMed] [Google Scholar]
- 57.Moore FA, Moore EE, Sauaia A. Blood transfusion. An independent risk factor for postinjury multiple organ failure. Arch Surg. 1997;132(6):620–624. discussion 624–625. [PubMed] [Google Scholar]
- 58.Watson GA, Sperry JL, Rosengart MR, et al. Inflammation and Host Response to Injury Investigators. Fresh frozen plasma is independently associated with a higher risk of multiple organ failure and acute respiratory distress syndrome. J Trauma. 2009;67(2):221–227. doi: 10.1097/TA.0b013e3181ad5957. discussion 228–230. [DOI] [PubMed] [Google Scholar]
- 59.Johnson JL, Moore EE, Kashuk JL. Early transfusion of FFP is independently associated with postinjury multiple organ failure. Arch Surg. 2010 doi: 10.1001/archsurg.2010.216. In press. [DOI] [PubMed] [Google Scholar]
- 60.Khan H, Belsher J, Yilmaz M, et al. Fresh-frozen plasma and platelet transfusions are associated with development of acute lung injury in critically ill medical patients. Chest. 2007;131(5):1308–1314. doi: 10.1378/chest.06-3048. [DOI] [PubMed] [Google Scholar]
- 61.Flesland O. A comparison of complication rates based on published haemovigilance data. Intensive Care Med. 2007;33(Suppl 1):S17–S21. doi: 10.1007/s00134-007-2875-1. [DOI] [PubMed] [Google Scholar]
- 62.Artang R, Frandsen NJ, Nielsen JD. Application of basic and composite thrombelastography parameters in monitoring of the antithrombotic effect of the low molecular weight heparin dalteparin: an in vivo study. Thromb J. 2009;10(7):14. doi: 10.1186/1477-9560-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van PY, Cho SD, Underwood SJ, Morris MS, Watters JM, Schreiber MA. Thrombelastography versus AntiFactor Xa levels in the assessment of prophylactic-dose enoxaparin in critically ill patients. J Trauma. 2009;66(6):1509–1515. doi: 10.1097/TA.0b013e3181a51e33. discussion 1515–1517. [DOI] [PubMed] [Google Scholar]
- 64.Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2008;133(6 Suppl):381S–453S. doi: 10.1378/chest.08-0656. [DOI] [PubMed] [Google Scholar]
- 65.Weitz JI. Low-molecular-weight heparins. N Engl J Med. 1997;337(10):688–698. doi: 10.1056/NEJM199709043371007. [DOI] [PubMed] [Google Scholar]
- 66.Patel R, Cook DJ, Meade MO, et al. Burden of Illness in venous ThromboEmbolism in Critical care (BITEC) Study Investigators; Canadian Critical Care Trials Group. Burden of illness in venous thromboembolism in critical care: a multicenter observational study. J Crit Care. 2005;20(4):341–347. doi: 10.1016/j.jcrc.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 67.Ibrahim EH, Iregui M, Prentice D, Sherman G, Kollef MH, Shannon W. Deep vein thrombosis during prolonged mechanical ventilation despite prophylaxis. Crit Care Med. 2002;30(4):771–774. doi: 10.1097/00003246-200204000-00008. [DOI] [PubMed] [Google Scholar]
- 68.Geerts WH, Jay RM, Code KI, et al. A comparison of low-dose heparin with low-molecular-weight heparin as prophylaxis against venous thromboembolism after major trauma. N Engl J Med. 1996;335(10):701–707. doi: 10.1056/NEJM199609053351003. [DOI] [PubMed] [Google Scholar]
- 69.Hartert H. Blutgerinnungsstudien mit der Thrombelastographie, einem neuen Untersuchungsverfahren. Klin Wochenschr. 1948;16(37–38):577–583. doi: 10.1007/BF01697545. [DOI] [PubMed] [Google Scholar]
- 70.De Nicola P. Thromboelastography. Oxford, United Kingdom: Blackwell Scientific; 1957. [Google Scholar]
- 71.Kang YG, Martin DJ, Marquez J, et al. Intraoperative changes in blood coagulation and thrombelastographic monitoring in liver transplantation. Anesth Analg. 1985;64(9):888–896. [PMC free article] [PubMed] [Google Scholar]
- 72.Tuman KJ, Spiess BD, McCarthy RJ, Ivankovich AD. Comparison of viscoelastic measures of coagulation after cardiopulmonary bypass. Anesth Analg. 1989;69(1):69–75. [PubMed] [Google Scholar]
- 73.Gibbs NM, Crawford GP, Michalopoulos N. Thrombelastographic patterns following abdominal aortic surgery. Anaesth Intensive Care. 1994;22(5):534–538. doi: 10.1177/0310057X9402200506. [DOI] [PubMed] [Google Scholar]
- 74.Chen A, Teruya J. Global hemostasis testing thromboelastography: old technology, new applications. Clin Lab Med. 2009;29(2):391–407. doi: 10.1016/j.cll.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 75.Khurana S, Mattson JC, Westley S, O’Neill WW, Timmis GC, Safian RD. Monitoring platelet glycoprotein IIb/IIIa-fibrin interaction with tissue factor-activated thromboelastography. J Lab Clin Med. 1997;130(4):401–411. doi: 10.1016/s0022-2143(97)90040-8. [DOI] [PubMed] [Google Scholar]
- 76.Nielsen VG, Geary BT, Baird MS. Evaluation of the contribution of platelets to clot strength by thromboelastography in rabbits: the role of tissue factor and cytochalasin D. Anesth Analg. 2000;91(1):35–39. doi: 10.1097/00000539-200007000-00007. [DOI] [PubMed] [Google Scholar]
- 77.Michelson AD. Methods for the measurement of platelet function. Am J Cardiol. 2009;103(3, Suppl):20A–26A. doi: 10.1016/j.amjcard.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 78.Nielsen VG. Beyond cell based models of coagulation: analyses of coagulation with clot “lifespan” resistance-time relationships. Thromb Res. 2008;122(2):145–152. doi: 10.1016/j.thromres.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 79.Sørensen B, Johansen P, Christiansen K, Woelke M, Ingerslev J. Whole blood coagulation thrombelastographic profiles employing minimal tissue factor activation. J Thromb Haemost. 2003;1(3):551–558. doi: 10.1046/j.1538-7836.2003.00075.x. [DOI] [PubMed] [Google Scholar]
- 80.Rivard GE, Brummel-Ziedins KE, Mann KG, Fan L, Hofer A, Cohen E. Evaluation of the profile of thrombin generation during the process of whole blood clotting as assessed by thrombelastography. J Thromb Haemost. 2005;3(9):2039–2043. doi: 10.1111/j.1538-7836.2005.01513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gonzalez E, Kahuk JL, Moore EE, Silliman C. Differentiation of enzymatic from platelet hypercoagulability using the novel thrombelastography parameter delta. J Surg Res. 2010 Apr 21; doi: 10.1016/j.jss.2010.03.058. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kheirabadi BS, Crissey JM, Deguzman R, Holcomb JB. In vivo bleeding time and in vitro thrombelastography measurements are better indicators of dilutional hypothermic coagulopathy than prothrombin time. J Trauma. 2007;62(6):1352–1359. doi: 10.1097/TA.0b013e318047b805. discussion 1359–1361. [DOI] [PubMed] [Google Scholar]
- 83.Park MS, Martini WZ, Dubick MA, et al. Thromboelastography as a better indicator of hypercoagulable state after injury than prothrombin time or activated partial thromboplastin time. J Trauma. 2009;67(2):266–275. doi: 10.1097/TA.0b013e3181ae6f1c. discussion 275–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Plotkin AJ, Wade CE, Jenkins DH, et al. A reduction in clot formation rate and strength assessed by thromboelastography is indicative of transfusion requirements in patients with penetrating injuries. J Trauma. 2008;64(2 Suppl):S64–S68. doi: 10.1097/TA.0b013e318160772d. [DOI] [PubMed] [Google Scholar]
- 85.Schöchl H, Frietsch T, Pavelka M, Jámbor C. Hyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J Trauma. 2009;67(1):125–131. doi: 10.1097/TA.0b013e31818b2483. [DOI] [PubMed] [Google Scholar]
- 86.Nielsen VG. A comparison of the Thrombelastograph and the ROTEM. Blood Coagul Fibrinolysis. 2007;18(3):247–252. doi: 10.1097/MBC.0b013e328092ee05. [DOI] [PubMed] [Google Scholar]
- 87.Stahel PF, Moore EE, Schreier SL, Flierl MA, Kashuk JL. Transfusion strategies in postinjury coagulopathy. Curr Opin Anaesthesiol. 2009;22(2):289–298. doi: 10.1097/ACO.0b013e32832678ed. [DOI] [PubMed] [Google Scholar]
- 88.Bolliger D, Szlam F, Molinaro RJ, Rahe-Meyer N, Levy JH, Tanaka KA. Finding the optimal concentration range for fibrinogen replacement after severe haemodilution: an in vitro model. Br J Anaesth. 2009;102(6):793–799. doi: 10.1093/bja/aep098. [DOI] [PubMed] [Google Scholar]
- 89.Vorweg M, Hartmann B, Knüttgen D, Jahn MC, Doehn M. Management of fulminant fibrinolysis during abdominal aortic surgery. J Cardiothorac Vasc Anesth. 2001;15(6):764–767. doi: 10.1053/jcan.2001.28337. [DOI] [PubMed] [Google Scholar]
- 90.Kang Y. Thromboelastography in liver transplantation. Semin Thromb Hemost. 1995;21(Suppl 4):34–44. [PubMed] [Google Scholar]
- 91.Ray WA, Stein CM. The aprotinin story—is BART the final chapter? N Engl J Med. 2008;358(22):2398–2400. doi: 10.1056/NEJMe0803514. [DOI] [PubMed] [Google Scholar]
- 92.Ide M, Bolliger D, Taketomi T, Tanaka KA. Lessons from the aprotinin saga: current perspective on antifibrinolytic therapy in cardiac surgery. J Anesth. 2010;24(1):96–106. doi: 10.1007/s00540-009-0866-9. [DOI] [PubMed] [Google Scholar]
- 93.Stamou SC, Reames MK, Skipper E, et al. Aprotinin in cardiac surgery patients: is the risk worth the benefit?. Presented at: The Society of Critical Care Medicine; January 2008; Nashville, TN. [Google Scholar]
- 94.Zumwinkle L, Kashuk J, Moore EE. Rapid thrombelastography differentiates primary from secondary fibrinolysis. Crit Care Med. 2008;36:187–190. [Google Scholar]
- 95.Kashuk JL, Moore EE, Wohlauer M, et al. Point of care rapid thrombelastography improves management of life threatening post-injury coagulopathy. Paper presented at: American Association for the Surgery of Trauma 68th Annual Meeting; October 2009; Pittsburgh, PA. [Google Scholar]
- 96.Kaur J, Jones N, Mallett S. Thrombelastography Platelet Mapping is a useful preoperative tool in surgical patients taking antiplatelet medication. Br J Anaesth. 2009;103(2):304. doi: 10.1093/bja/aep184. author reply 304–305. [DOI] [PubMed] [Google Scholar]
- 97.Agarwal S, Coakley M, Coakely M, Reddy K, Riddell A, Mallett S. Quantifying the effect of antiplatelet therapy: a comparison of the platelet function analyzer (PFA-100) and modified thromboelastography (mTEG) with light transmission platelet aggregometry. Anesthesiology. 2006;105(4):676–683. doi: 10.1097/00000542-200610000-00011. [DOI] [PubMed] [Google Scholar]
- 98.Tantry US, Bliden KP, Gurbel PA. Overestimation of platelet aspirin resistance detection by thrombelastograph platelet mapping and validation by conventional aggregometry using arachidonic acid stimulation. J Am Coll Cardiol. 2005;46(9):1705–1709. doi: 10.1016/j.jacc.2005.05.090. [DOI] [PubMed] [Google Scholar]
- 99.Spalding GJ, Hartrumpf M, Sierig T, Oesberg N, Kirschke CG, Albes JM. Bedside thrombelastography. Cost reduction in cardiac surgery [in German] Anaesthesist. 2007;56(8):765–771. doi: 10.1007/s00101-007-1200-2. [DOI] [PubMed] [Google Scholar]
- 100.Spiess BD, Gillies BS, Chandler W, Verrier E. Changes in transfusion therapy and reexploration rate after institution of a blood management program in cardiac surgical patients. J Cardiothorac Vasc Anesth. 1995;9(2):168–173. doi: 10.1016/S1053-0770(05)80189-2. [DOI] [PubMed] [Google Scholar]
- 101.Knudson MM, Ikossi DG, Khaw L, Morabito D, Speetzen LS. Thromboembolism after trauma: an analysis of 1602 episodes from the American College of Surgeons National Trauma Data Bank. Ann Surg. 2004;240(3):490–496. doi: 10.1097/01.sla.0000137138.40116.6c. discussion 496–498. [DOI] [PMC free article] [PubMed] [Google Scholar]