

Abstract
Background
Whole and partial chromosome losses or gains and structural chromosome changes are hallmarks of human tumors. Guanine-rich DNA, which has a potential to form a G-quadruplex (G4) structure, is particularly vulnerable to changes. In Caenorhabditis elegans, faithful transmission of G-rich DNA is ensured by the DOG-1/FANCJ deadbox helicase.
Results
To identify a spectrum of mutations, after long-term propagation, we combined whole genome sequencing (WGS) and oligonucleotide array Comparative Genomic Hybridization (oaCGH) analysis of a C. elegans strain that was propagated, in the absence of DOG-1 and MDF-1/MAD1, for a total of 470 generations, with samples taken for long term storage (by freezing) in generations 170 and 270. We compared the genomes of F170 and F470 strains and identified 94 substitutions, 17 InDels, 3 duplications, and 139 deletions larger than 20 bp. These homozygous variants were predicted to impact 101 protein-coding genes. Phenotypic analysis of this strain revealed remarkable fitness recovery indicating that mutations, which have accumulated in the strain, are not only tolerated but also cooperate to achieve long-term population survival in the absence of DOG-1 and MDF-1. Furthermore, deletions larger than 20 bp were the only variants that frequently occurred in G-rich DNA. We showed that 126 of the possible 954 predicted monoG/C tracts, larger than 14 bp, were deleted in unc-46 mdf-1 such-4; dog-1 F470 (JNC170).
Conclusions
Here, we identified variants that accumulated in C. elegans’ genome after long-term propagation in the absence of DOG-1 and MDF-1. We showed that DNA sequences, with G4-forming potential, are vulnerable to deletion-formation in this genetic background.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1402-y) contains supplementary material, which is available to authorized users.
Keywords: C. elegans, Whole genome sequencing (WGS), oligonucleotide array Comparative Genomic Hybridization (oaCGH), Mutation accumulation (MA), Genomic variation (GV), Spindle assembly checkpoint (SAC), dog-1/FANCJ, G-quadruplex (G4) structure
Background
Genome integrity is crucial for survival of all living organisms. Chromosomal instability (CIN), marked by whole or segmental aneuploidy is hallmark of human tumors, and can drive abnormal proliferation of cancer cells [1]. In Caenorhabditis elegans, mdf-1(gk2) has an essential mdf-1/MAD-1 component of the spindle assembly checkpoint (SAC) missing and this leads to accumulation of genetic errors and ultimately death by the third generation [2]. The checkpoint prevents CIN by inhibiting anaphase-promoting complex/cyclosome (APC/C), and delaying anaphase onset until all the chromosomes have achieved proper attachment to the spindle [3].
While MDF-1 prevents both loss and gain of whole chromosomes during mitosis, DOG-1 prevents segmental aneuploidies by ensuring proper replication of guanine(G)-rich DNA [4-6]. G-rich DNA can adopt a four-stranded helical G-quadruplex (G4) DNA structure [7-9], which can pose a barrier to replication fork progression if left unresolved. The ability to form G4 structures makes the corresponding G-rich DNA sequences particularly vulnerable to chromosomal rearrangements. Studies using C. elegans, as a model organism, were the first to show the striking genomic instability of G-rich DNA sequences when DOG-1, a functional ortholog of the deadbox helicase FANCJ [10], was non-functional [4]. When DOG-1 is functional, G-rich DNA sequences are stable and deletions affecting these regions are not observed [4,5]. Genome-wide bioinformatic analysis of the human genome had identified more than 300,000 DNA sites with G4-forming potential [11,12]. In humans, mutations in FANCJ/dog-1 have been identified in Fanconi anemia (FA) complementation group J patients [13-15], which is a severe, autosomal recessive, disorder with increased spontaneous and DNA crosslink-induced CIN showing a wide range of clinical manifestations [13], and also in early onset breast cancer patients [16,17].
Knowledge of the mutational spectra is crucial for deciphering the cause of heritable genetic disorders as well as the progression of events relevant to cancers. Traditionally, analyses of mutation spectra and rates have been based on a small portion of phenotypically and molecularly detectable loci. In C. elegans, the mutational spectrum of dog-1(gk10) (knockout allele of dog-1) strains, was analyzed using either PCR-based assays [4,10,18] or oligonucleotide array Comparative Genomic Hybridization (oaCGH) [5,6,19]. The rapid advances in “next-generation” DNA sequencing technologies now allows us to perform comprehensive genome-wide analyses of mutational spectra by sequencing whole genomes [20-23].
Here we undertook a whole genome approach in order to analyze mutational events in a C. elegans strain that is defective for both mdf-1/MAD1 and dog-1/FANCJ. This strain was propagated for 470 generations and samples were stored frozen at generations 170 and 270. Phenotypic analysis of the strain revealed striking fitness recovery, indicating that accumulated mutations cooperate to bypass the MDF-1 checkpoint requirement and thus achieve long-term population survival. We performed whole-genome sequencing (WGS) and oaCGH analyses of the strain at three different generations (170, 270 and 470). We identified substitutions, InDels, and copy number variants (CNVs) larger than 20 bp, and compared their accumulation over the generations. We showed that only deletions, which are larger than 20 bp, frequently initiated in G-rich DNA (88% of all of the deletions). Consistent with the fitness recovery observed in this strain, rather than a decline in fitness, the mutation spectrum reported here reflects variants that are either advantageous or neutral in this specific genetic background.
Results and discussion
Whole genome analysis
In order to propagate mdf-1; dog-1 homozygous worms, it is first necessary to isolate a suppressor of mdf-1(gk2) sterility and lethality that occurs in the double mutant [24]. Previously, such-4(h2168) was isolated, which allowed propagation of mdf-1; dog-1 beyond the third generation (Figure 1) [24]. The such-4 suppressor allows for an approximately five-fold increase in fertile hermaphrodite progeny of mdf-1; dog-1 [24]. This increase in fertility occurs in the generations immediately after isolation of the suppressor. We outcrossed one worm to obtain KR4233 [unc-46(e177) mdf-1(gk2) such-4(h2168)] [24,25] (Figure 1). A second line was isolated and maintained in the dog-1(gk10) background for 470 generations, with storage by freezing at generations 170 and 270 (Figure 1). To mark the presence of mdf-1(gk2), unc-46(e177) (a visible marker) was used, which is present in all of our strains (Figure 1). While propagating unc-46 mdf-1 such-4; dog-1 homozygotes, we observed a further increase in reproductive fitness. This increase was significant, 59% of F470unc-46 mdf-1 such-4; dog-1 progeny develop into fertile hermaphrodites, compared to only 10% of the unc-46 mdf-1 such-4 progeny and 2% of unc-46 mdf-1 mutants. Detailed phenotypic analysis of these strains as well as genetic dissection of suppressors has recently been reported [26]. To identify the genomic variations (GVs) that had accumulated in mdf-1 such-4; dog-1 F470 worms after long-term propagation, the genome was sequenced to a depth of 70× genome equivalents and aligned to the C. elegans reference genome WS235 available at WormBase [27]. We also sequenced the unc-46 mdf-1 such-4; dog-1 strains that were frozen at generations 170 and 270 and compared the progress of mutation accumulation (Additional file 1: Figure S1).
Figure 1.
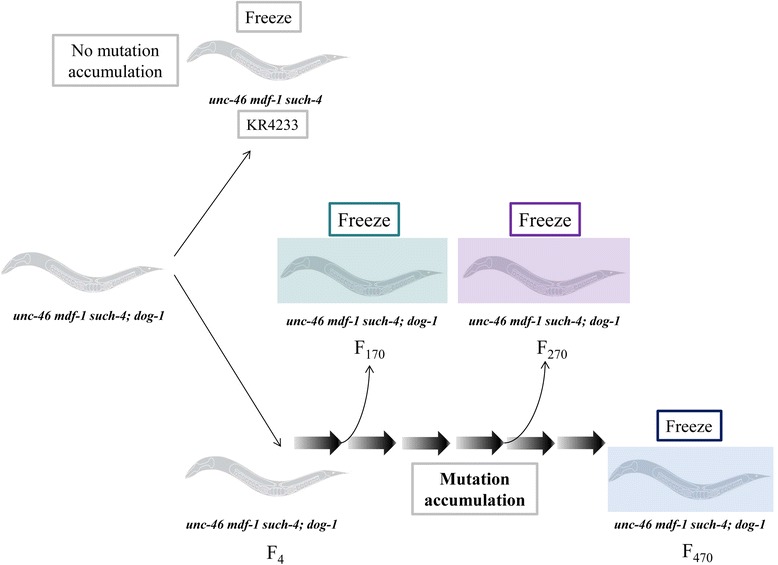
A schematic representation of the long-term propagation of unc-46 mdf-1 such-4; dog-1 homozygotes for 470 generations. We isolated a suppressor (such-4) in F4 from the only plate that we set up that propagated in the absence of MDF-1 and DOG-1 for longer than three generations. We generated a strain (KR4233) by crossing away dog-1(gk10). A second line was propagated for the total of 470 generations. The worms were frozen at generations 170 (green), 270 (purple) and 470 (blue). Note that unc-46 (used as a visible marker) is tightly linked to mdf-1(gk2) in all of our strains.
Single Base Substitutions (SBSs) do not occur within G4-DNA
We used the variant caller VarScan2; version 2.3.2 [28] to identify 525 homozygous SBSs that occurred with a variant frequency (VF) of 90% or higher in the mdf-1 such-4; dog-1F470 genome (Additional file 2: Table S1A). All 42 tested substitutions were confirmed by Sanger re-sequencing (30 were randomly selected; 12 additional substitutions were confirmed by re-sequencing as a result of being adjacent to randomly selected SBSs or as being non-randomly selected as candidates in later analyses), indicating a false positive rate of less than 5%. WGS analysis of the unc-46 mdf-1 such-4; dog-1 strain, which had been frozen at generation 170 (Figure 1), revealed that the majority (431/525) of the SBSs present in generation 470 are also present in generation 170 (Additional file 1: Figure S1A; Table 1). However, we did observe that 32 additional substitutions had accumulated between F170 and F270, and 62 more between F270 and F470 (Table 1). Large number of substitutions observed in F170 indicate the possibility that the original unc-46 mdf-1 such-4; dog-1 strain (Figure 1) had a large number of single nucleotide differences from the reference genome (WS235). One way to test this possibility is to estimate the mutation rates based on available data. If we consider the 94 substitutions that had accumulated in 300 generations, between F170 and F470, we estimate the rate of μbs = 3.1 × 10-9/base/generation. This estimate is similar to the previously reported spontaneous rate of base substitution in C. elegans, 2.7 ± 0.4 × 10−9 /base/generation [21] and other model organisms, 3.5 × 10−9 /base/generation in Drosophila melanogaster [23] and 7.1 ± 0.7 × 10−9 /base/generation in Arabidopsis thaliana [22]. Furthermore, analysis of the 94 substitutions on mutation bias revealed very similar mutation spectrum to the spontaneous mutation spectra in N2 (C. elegans wild-type) [21] (Figure 2), and our analysis of transition bias (transition/transversion – Ts/Tv – base substitution ratios) revealed the Ts/Tv ratio = 0.54, which is within the range observed for spontaneous mutations in multiple mutation accumulation lines (average 0.45; range 0.19 – 0.79) [21]. Therefore, the similarity of the substitution rate over the last 300 generations to the previously reported μbs in N2 strongly implies that the majority of the 431 substitutions identified in the F170 generation are variants originally present in the starting strain; therefore, we focused our analysis on GVs that had occurred between F170 and F470 (Additional file 2: Table S1B).
Table 1.
Summary of the variants identified in the unc-46 mdf-1 such-4; dog-1 strains at generations F 170 , F 270 and F 470
| SBSs | InDels ≤ 20 bp | Duplications | Deletions >20 bp | |
|---|---|---|---|---|
| F170 | 431 | 133 | 3 | 57 |
| F270 | 32 | 8 | 3* | 45 |
| F470 | 62 | 9 | – 1 | 94 |
| Total F170-F470 | 94 | 17 | 3 | 139 |
*Two new duplications and amplification of the cyb-3 locus to three copies.
Figure 2.
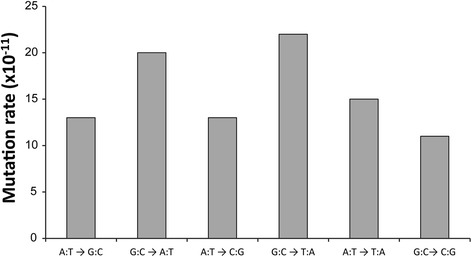
Mutation rate estimates. The variants analyzed are the 94 SBSs that were identified between generations 170 and 470.
Previous studies showed that G4 DNA secondary structure is mutagenic in the absence of DOG-1 [4-6,19]. Using the G4 DNA signature (G3+N1-7G3+N1-7G3+N1-7G3+), we identified 2,372 such sites in C. elegans’ genome (Additional file 2: Table S2). Next, we tested to see if any of the 94 substitutions occurred within the G4 DNA signature sequence and observed that none of them did (Additional file 2: Table S1B), indicating that the identified substitutions most likely arose spontaneously and were not due to lack of functional DOG-1.
Small InDels do not occur within G4-DNA
We applied VarScan2 [28] to identify 150 homozygous InDels that were 20 bp or less in mdf-1 such-4; dog-1F470 (Additional file 2: Table S3A). We randomly selected 25 of the InDels and confirmed all 25 by Sanger re-sequencing, indicating a false positive rate of less than 5% (Additional file 2: Table S3A). Analysis of InDels in the unc-46 mdf-1 such-4; dog-1 strains propagated for 170 and 270 generations (Additional file 1: Figure S1B) revealed that 88% of the F470 InDels (133/150) existed in the F170 generation (Table 1), which again indicates that the majority of InDels were already present in our starting strain. In fact, we only observe eight additional InDels in F270 (Table 1) and another nine InDels accumulated between F270 and F470 (Table 1). Based on the last 300 generations of propagation, we estimate a mutation rate for InDels to be 17/(1 × 108bases × 300generations) = 5.7 × 10-10/base/generation. The ratio of InDels to SBSs observed was 0.18 or one InDel per 5.5 substitutions, which is much lower than the 1.31 ratio reported previously in C. elegans [29]. However, the lower number of InDels to SBSs that we observe in the unc-46 mdf-1 such-4; dog-1 background is comparable to analyses in yeast [30], A. thaliana [22] and human [31]. Namely, WGS analysis in yeast had revealed ratio of InDels to SBSs of ∼ 0.03 [30], which was consistent with previous findings of one InDel per 33 SBSs [20]. Furthermore, analysis on spontaneous occurrence of InDels in A. thaliana revealed the ratio of 0.13 of InDels to SBSs [22]. It may be possible that the small number of InDels, over the last 300 generations, in our strain may be a result of a mutation that was acquired by propagation; however, it may also be that the spontaneous mutation rate of InDels in C. elegans is comparable to that of other organisms.
Next, we tested to see if any of the 17 InDels occurred within the G4 DNA signature sequence and observed that none of them did (Additional file 2: Table S3B). Thus, we believe that these InDels arose spontaneously and are not due to a lack of functional DOG-1.
Duplications do not occur within G4-DNA
Analysis of gene copy-number variant accumulation after long-term propagation in C. elegans using the oaCGH has provided evidence for a high rate of spontaneous gene duplications in this multi-cellular eukaryote [32]. Previously, using the oaCGH we showed that the such-4 suppressor genome contains a large tandem duplication on Chromosome V (LGV) [6]. Here, we used both Pindel [33] and oaCGH [34], and identified four sites with copy number increases in the mdf-1 such-4; dog-1F470 genome (Additional file 2: Table S4A), including a previously identified large tandem duplication [6]. One of the duplications involves a two-copy addition, making the final count of five duplication events and four different duplication sites. Analysis of the CNVs in the F170 and F270 genomes captured a dynamic property of duplications (Additional file 1: Figure S1C). In F170, we observed three duplications (Additional file 2: Table S4A). One is the large tandem duplication located on LGV that amplifies 62 protein-coding genes, which we have described previously [6]. In generation F270, we detected duplications of two new loci, as well as further amplification of the LGV region to three copies (triplication) (Additional file 1: Figure S1C and Additional file 2: Table S4A). In F470, we did not find any new duplications (Additional file 2: Table S4A), but did observe that the duplication on LGI was lost, resulting in a wild-type copy number for this region (Additional file 2: Table S4A). Thus, the LGV duplication exemplifies the property of duplications to further amplify; while LGI duplication shows that a duplicated region can revert back to a normal copy number.
The gene duplication rate for C. elegans was recently estimated to be 3.4 × 10-7/gene/generation [32]. Our data, based on the last 300 generations (from F170 and F470), also revealed a comparably high rate of gene duplication (6.5 × 10-7/gene/generation), specific to the unc-46 mdf-1 such-4; dog-1 background, when a large duplication on LGV is excluded from the analysis. The LGV duplicated region contains 62 protein-coding genes, including cyb-3 [6]. We observed a correlation between increased dosage of CYB-3 and a striking fitness increase in our strains; thus, our experimental protocol selected for LGV amplification (detailed experimental analysis on these findings has been recently published [26]). Importantly, like SBSs and InDels, none of the duplications occurred in the vicinity of the G4 DNA signature sequences (Additional file 2: Table S4B), also indicating that these CNVs arose spontaneously and were not due to a lack of functional DOG-1.
Deletions frequently initiate at G4 DNA sites
The major type of mutation observed, in the absence of DOG-1, is a deletion of 300 bp or smaller, initiating at either the 5′-end of C- or the 3′-end of G-tracts [4,5,10,19]. Using the unique alignments generated by Novoalign and Pindel [33], we identified 183 homozygous deletions larger than 20 bp in the mdf-1 such-4; dog-1F470 genome (Additional file 2: Table S5A), including the known mdf-1(gk2) and dog-1(gk10) deletions. We randomly selected 28 of the deletions, and confirmed all of them using PCR (Additional file 2: Table S5A). To identify deletions, which may have been missed, we also used oaCGH [34]. We confirmed all of the deletions, predicted by Pindel, that were covered by oaCGH probes and identified an additional 13 deletions not detected by Pindel (Additional file 2: Table S5A), making the final count 196 homozygous deletions in the mdf-1 such-4; dog-1F470 genome (Additional file 2: Table S5A).
We observed 57 deletions in F170, indicating that 139 deletions had accumulated in 300 generations (between F170 and F470) propagated in the absence of MDF-1 and DOG-1 (Additional file 1: Figure S1D and Table 1). We found that the majority of the deletions (123 of 139) initiated in G4 DNA (Additional file 2: Table S5A). Previous analysis in C. elegans [32] estimated the spontaneous rate of deletions to be 2.2 × 10-7/gene/generation respectively. The 139 deletions accumulating between the generations F170 and F470 affect 19 protein-coding genes which allowed us to calculate the mutation rate of deletions over the 300 generations to be 19/(20,400protein-coding genes × 300generations) = 3.1 × 10-6/gene/generation in the unc-46 mdf-1 such-4; dog-1 strain, which is approximately 10-fold higher than the estimated rate in N2 [32]. This is similar to the estimated forward mutation frequency of eT1-balanced lethal mutations in dog-1(gk10) background [6]. To determine if the elevated mutation rate of deletions is due to DOG-1 deficiency, we compared mutation rates in non-G4 sites versus mutation rate in the G4 sites. While the mutation rate of deletions affecting the non-G4 sites, 4.9 × 10-7/gene/generation or 5.0 × 10-10/base/generation, is comparable to the previously reported spontaneous rate of deletions [32]; the mutation rate based on G4 sites, 1.7 × 10-4 /G4site/generation, illustrates the striking vulnerability of these DNA regions when DOG-1 is absent. Therefore, deletions larger than 20 bp are the only variants in the unc-46 mdf-1 such-4; dog-1 strain that frequently occurred in the G4-DNA sites and had significantly higher mutation rate than the spontaneous rate reported previously for the strains with normal DOG-1 function.
monoG/C tracts larger than 14 bp are frequently deleted when DOG-1 is not functional
In recent years, the G4 DNA has been implicated in diverse biological processes, such as gene expression [35] and DNA replication initiation [35]. Consistent with the established role of DOG-1, we found that the majority (114) of the homozygous deletions that we identified between F170 and F470 (139) initiated in the previously proposed G4 DNA signature G3+N1-7G3+N1-7G3+N1-7G3+ [5] (Additional file 2: Table S5B) where the G-tract was almost completely removed together with 5′ flanking DNA sequence (Figure 3A and B). In agreement with a previous study [5], we found that the majority of deletions initiate at monoG tracts larger than 14 bp (93 deletions) (Figure 3A), while 11 deletions initiate at monoG-like structures with no more than two nucleotides that interrupt the homopolymer (Figure 3B), and 10 deletions initiate at sequences that interrupt the homopolymers by three or more nucleotides (e.g., GGGtGGGGaagttatGGGaGGG) (Additional file 2: Table S5B). MonoG/C tracts larger than 14 bp have the highest potential of forming the G-quartet structure. In fact, we find here that the unc-46 mdf-1 such-4; dog-1 F470 genome has 13.2% of all the predicted monoG/C tracts larger than 14 bp deleted. An interesting question to be addressed with future research would be to determine how mutation rate changes with decreasing numbers of available targets. Furthermore, it would be also important to see how many of the G4-DNA sites could be deleted in a strain and still maintain viability of the animals.
Figure 3.
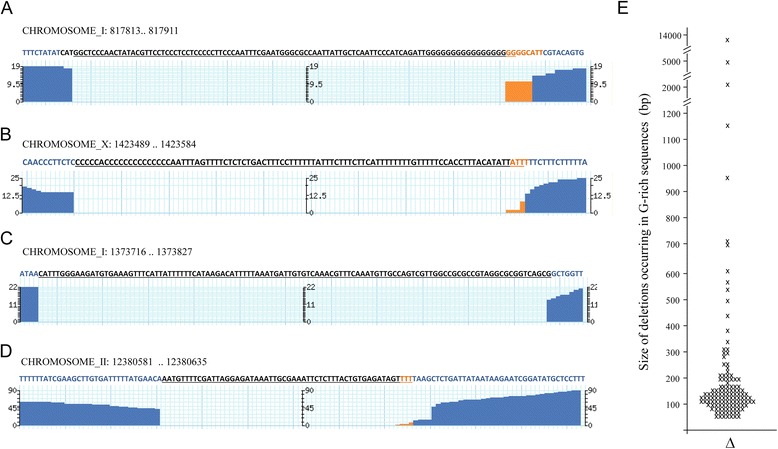
The majority of large deletions initiates at G-rich DNA. Figures (A) through (D) are Genome Browser snapshots of single deletions; x-axis represents genome location, while y-axis represents number of reads that cover the region; blue reads depict coverage of more than 10, while orange depicts 10 or less; the gaps depict no coverage, which is indicative of deletions. The reference sequence is depicted with deleted bases underlined. (A) A 99 bp deletion that initiates at G20 homopolymer. (B) A 96 bp deletion that initiates at G14TG6AGAAG3 sequence. (C) A 112 bp deletion that initiates at a G-rich sequence, G2N3G2N5G2N2G2. (D) A 55 bp deletion that initiates at a non G-rich sequence. (E) Size distribution of homozygous deletions that occur at G-rich sequences (n = 123).
To investigate whether there may be additional sequences, which are vulnerable in the absence of DOG-1, we analyzed the 25 deletions that do not initiate in G4 DNA signature sequences to see if there are common patterns. We found that eight of these deletions initiate at G-rich sequences that correspond to the G2+N1-7G2+N1-7G2+N1-7G2+ signature (four stretches of two or more guanines, alternated with one to seven nucleotides of any type), while one had a G2+N1-7G2+N1-7G2+ signature at the breakpoint (Figure 3C and Table 2). Although, it may be possible that our strain had gained a mutation in an unknown gene important for genome stability, it is also possible that additional DNA sequences may be vulnerable to rearrangements in the absence of DOG-1.
Table 2.
Schematic representation of the nine deletions that initiate at G-rich regions within sequences that deviate from the G4 DNA signature, G 3+ N 1-7 G 3+ N 1-7 G 3+ N 1-7 G 3+
| LG | Locus | Signature |
|---|---|---|
| I | 464950..465071 | G2NG12 |
| I | 815471..815611 | G3NG3NG3N2G2 |
| I | 3747905..3748027 | G2NG2NG2NG2NG2NG2NG2NG2NG2 |
| I | 1373716..1373827 | G2N3G2N5G2N2G2 |
| II | 12687460..12687546 | G2NG14NG2 |
| III | 460436..460942 | G11 |
| IV | 3298588..3298711 | G14 |
| V | 17778167..17778449 | G3NG3NG3N5G2NG2NG3 |
| X | 2802061..2802223* | G2NG3N6G3 |
*Analysis of the sequences revealed that nine match the G2+N1-7G2+N1-7G2+N1-7G2+ signature, while one (marked with *) matches G2+N1-7G2+N1-7G2+ signature at the breakpoint.
Previously, it was found that the deletion sizes in viable lines detected by PCR [4,18] or oaCGH methods [5,6] were predominately smaller than 300 bp. In this study, the 123 deletions that occurred at G-rich sequences ranged between 49 and 10,228 base-pairs with the majority of the deletions (86%) removing less than 300 bp (Figure 3E). These findings are in agreement with the deletion distribution sizes revealed by the study of a 69 G-tract deletion set [5]. However, larger deletions, initiating at G-tracts, have been recovered as lethal mutations [6], consistent with these regions containing essential genes [6]. We also found that 16 deletions, which occurred at non-G-rich sites, removed small regions of less than 300 bp in size (Additional file 2: Table S5B).
101 protein-coding genes are affected by the 253 GVs
To determine an effect of all the identified variants on protein-coding genes, after long-term propagation in the absence of MDF-1 and DOG-1, we used CooVar [36], a tool developed by our group for annotating variants. Many of the G-rich DNA sites are located in close proximity to protein-coding genes in C. elegans [19,37]. Using CooVar [36], we predicted that 19 genes would be affected by 18 of the deletions that had accumulated between generations F170 and F470 (Additional file 2: Table S6). The majority of the deletions (14) represent the first knockout alleles for those genes and thus provide a genetic resource for studying their functions (Additional file 2: Table S6). We found that the majority of the InDels are not located in protein-coding regions, and the one that affects a protein-coding gene is an in-frame deletion (Additional file 2: Table S6). We found that the majority of the detected SBSs are located within non-protein coding regions. However, there are 15 SBSs that fall within protein-coding regions that are predicted to result in missense mutations (Additional file 2: Table S6). In total, 66 protein-coding genes were affected by the duplications (Additional file 2: Table S6). The majority of the genes (62) are affected by the large tandem duplication on LGV (Additional file 2: Table S6). In addition to the 253 GVs that had accumulated between generations F170 and F470, we had also performed analysis of the effect on protein-coding genes of all the variants observed in the mdf-1such-4; dog-1F470 genome (Additional file 2: Table S7).
Mutations are a source of genetic variation that confers to an organism either advantage due to a beneficial change, disadvantage due to a deleterious alteration, or neither due to a neutral change. Considering the ~30-fold fitness recovery in this strain [26], the identified GVs are expected to be either advantageous or neutral in this genetic background. In fact we showed, in a parallel study, that three mutational events discovered in this strain cooperate to increase fitness when MDF-1 absent [26].
Conclusion
In this study we undertook a genomics approach to identify variants that accumulate in the C. elegans genome after long-term propagation in the absence of two genome-guardians, DOG-1/FANCJ and MDF-1/MAD1. Combining WGS analysis with oaCGH analysis allowed us to comprehensively analyze both the small-scale variants (SBSs and InDels of 20 bp or smaller) and large-scale variants (CNVs larger than 20 bp). Freezing the strain, for long term storage, at three different time points, F170, F270 and F470, allowed us to compare and visualize mutation accumulation over the generations. We were able to estimate the mutation rates in this strain for different types of mutations over the 300 generations (F170 to F470). We observed a significantly elevated rate of deletions, larger than 20 bp, that initiated at G/C-rich DNA sequence. Our approach had allowed us to show that, in the absence of DOG-1, DNA sequences with G4-forming potential are vulnerable to deletion-formations. Previous studies were dependent only on PCR or oaCGH analysis; the two approaches are limited in their ability to detect small-scale variants like SBSs and InDels. Furthermore, genome analysis of the strains at F170, F270 and F470 allowed us to capture and visualize an intriguing property of CNVs (Additional file 1: Figure S1). We did not find any reversions of deletions, substitutions, or InDels. As expected, once fixed, these types of mutations are propagated indefinitely (Additional file 1: Figure S1). However, we captured a dynamic property of duplications: amplification of a region on LGV from one-to-two-to-three copies and reversion of the LGI duplication back to a normal copy number (Additional file 1: Figure S1C).
This is the first extensive analysis of a strain that had been propagated in the absence of DOG-1 helicase for hundreds of generations. We identified 954 monoG/C tracts larger than 14 bp in the C. elegans genome WS325; the polyG/C DNA sequence is the sequence with the highest potential of forming G-quartet. We showed that 13% of these 954 sites are deleted in the mdf-1 such-4; dog-1F470 genome. This finding raises an important question on the changes in mutation rate when number of mutagenic targets is decreased. Another important question is regarding to the role of G4 DNA in normal development. Recently, G4 DNA has been implicated in a variety of biological processes including telomere maintenance, gene expression, epigenetic regulation, and DNA replication [38]. One question to consider is how many G4 DNA sites could be removed from a genome yet still maintain viability of the animals.
Methods
C. elegans strains
The following mutant alleles were used in this work: unc-46(e177), mdf-1(gk2), dog-1(gk10), and such-4(h2168). The following strains were used in this work: KR4233 [unc-46(e177) mdf-1(gk2) such-4(h2168)] and KR3627 [unc-46(e177) mdf-1(gk2) V/nT1[let-?(m435)])]; VC13 [dog-1(gk10)]. Additional strains used in this work were generated in this study. Strains were maintained using standard protocol on nematode growth media (NGM) plates seeded with OP50 bacteria [39]. The strains were maintained at 20°C.
Mutation accumulation procedure and phenotypic analysis
The first suppressor, such-4, was isolated as previously described [24]. Briefly, VC13 was backcrossed to N2 ten times to remove any mutations present in VC13. Then the outcrossed dog-1(gk10) males were used to construct unc-46(e177) mdf-1(gk2) +/+ + nT1[let-X]; dog-1(gk10)/dog-1(gk10). Note that unc-46(e177) is linked mdf-1(gk2) and used as a visible marker to track mdf-1(gk2). F1unc-46 mdf-1 homozygotes (n = 40) were picked and plated individually and a single worm, from a plate containing fertile worms in the third generation, was isolated as a suppressor candidate (such-4) [24]. We outcrossed one worm from this strain to establish KR4233 mdf-1(gk2) such-4(h2168) [6,24], while we maintained a second line at 20°C for 470 generations (strain JNC170). Each generation 5 L4 hermaphrodites were transferred to a fresh plate. We also froze the worms at generations 170 (strain JNC168) and 270 (strain JNC169). The phenotypic analysis was performed as previously described [24].
Whole genome sequencing and computational analysis of the unc-46 mdf-1 such-4; dog-1F470 genome
Genomic DNA was prepared from JNC170 following a standard protocol (http://genetics.wustl.edu/tslab/protocols/genomic-stuff/worm-genomic-dna-prep/) originally set up by the Andy Fire Lab. The library was prepared with average insert size of 300 bp and the genome was then sequenced using Illumina Solexa sequencing (at Canada’s Michael Smith Genome Sciences Centre) and 92,282,948 reads of 101 bp in length were obtained. The reads were then aligned to the C. elegans reference genome WS235 (hosted at WormBase) [27] in paired end manner (46,141,474 pairs) using the Novoalign alignment tool. 73,482,133 (79.63%) of the total reads were of base quality 30 or more and were uniquely mapped, generating 70-fold coverage of the genome.
SBSs were detected using the uniquely mapped reads and pileup2snp function of the variant caller VarScan2; version 2.3.2 [28]. We used SAMtools [40] to generate the mpileup file necessary as input for VarScan. We filtered out the SBSs that did not meet the following criteria: depth of coverage > 5 and ≤ 200, variant frequency ≥ 0.9 and base quality ≥ 30. After these filtering steps, we identified 776 substitutions in the mdf-1 such-4; dog-1F470 genome. However, comparison with the sequenced N2 strains from CGC and Horvitz lab (sequencing reads were kind gift from Dr. Bob Waterston) revealed 525 homozygous substitutions that are unique to mdf-1 such-4; dog-1F470 genome. The 251 SBSs were not included in our analysis as they did not accumulate during the course of our experiment. To determine rate of false positives in our set, we randomly selected 30 SBSs (Additional file 2: Table S1A), designed primers flanking the predicted substitution sites, amplified the fragments and re-sequenced using Sanger sequencing method at Genewiz, Inc. All 30 substitutions were confirmed by Sanger re-sequencing, suggesting a false positive rate of less than 5% (Additional file 2: Table S1A). Also, 12 additional substitutions were confirmed by re-sequencing as a result of being adjacent to randomly selected SBSs or as being non-randomly selected as candidates in later analyses.
InDels were detected using the uniquely mapped reads and pileup2indel function of the variant caller VarScan2; version 2.3.2 [28]. We used SAMtools [40] to generate the mpileup file necessary as input for VarScan2. We filtered out the InDels that did not meet the following criteria: depth of coverage > 5 and ≤ 200, variant frequency ≥ 0.9 and base quality ≥ 30. Additionally, we re-evaluated and filtered the output so that each co-ordinate corresponds to one variant. After these filtering steps, we identified 556 InDels in the mdf-1 such-4; dog-1F470 genome. r, comparison with the sequenced N2 strains from CGC and Horvitz lab (sequencing reads were kind gift from Dr. Bob Waterston) revealed 150 homozygous InDels that are unique to mdf-1 such-4; dog-1F470 genome. The 406 InDels were not included in our analysis as they did not accumulate during the course of our experiment. To determine rate of false positives in our set, we have randomly selected 25 InDels (Additional file 2: Table S3A), designed primers flanking the predicted InDel sites, amplified the fragments and re-sequenced using Sanger sequencing method at Genewiz, Inc. All of the 25 InDels were confirmed by Sanger re-sequencing, suggesting a false positive rate of less than 5% (Additional file 2: Table S3A).
These unique-mapping reads were then used, together with Pindel [33], to identify deletions. The final set of fixed/homozygous deletions was selected based on the following criteria: a = the number of unique reads supporting the breakpoints of the deletion; b = the number of reads within the deleted region; select the predicted deletion if is larger than 0.5 and the size of the deletion is larger than 20 bp. From the candidate deletions, 28 were randomly selected and the regions were PCR-amplified using the same genomic DNA that was used for the WGS and primers designed in the flanking regions of the computationally identified deletions. All of the 28 randomly selected predicted deletion sizes were confirmed using DNA electrophoresis gels, suggesting the rate of false positives of less than 5%.
Whole genome sequencing and computational analysis of the unc-46 mdf-1 such-4; dog-1F170 and mdf-1 such-4; dog-1F270 strains
The genomic DNA was prepared from JNC168 and JNC169 following a standard protocol (http://genetics.wustl.edu/tslab/protocols/genomic-stuff/worm-genomic-dna-prep/). The genomic DNA was sheared to generate 500 bp fragment and the library was prepared using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina®. The library was then sequenced using Illumina Solexa sequencing (at Simon Fraser University) and 9,950,748 (F170) and 6,225,488 (F270) reads of 250 bp length were obtained. The reads were then aligned to the C. elegans reference genome WS235 (hosted at WormBase) [27] in paired end manner using the Novoalign alignment tool as described above to achieve 20-fold (F170) and 14-fold (F270) coverage. For both strains, we used Pindel [33] to identify deletions and duplications, and VarScan2; version 2.3.2 [28] to identify small InDels and substitutions, as described above. We also used sequenced N2 strains from CGC and Horvitz lab to remove variants existing in the N2 strain. Then, we compared the variants present in the F170 and F270 genomes with the ones identified in the F470 genome.
oaCGH analysis of the unc-46 mdf-1 such-4; dog-1 strains
To perform oaCGH analysis, we used the same genomic DNA from the mdf-1 such-4; dog-1 lines (F170, F270 and F470) that were used for the WGS and the reference N2 DNA that was prepared following a standard protocol. oaCGH analysis was performed as described by Maydan and colleagues [41] with a newly designed microarray. The 3-plex microarray with design name 120618_Cele_WS230_JK_CGH was manufactured by Roche NimbleGen Inc. with each individual sub-array comprising 720 k 50-mer oligonucleotide probes. The filters used to select the probes primarily followed Maydan and colleagues [41] without focusing on coding regions in order to provide a more uniform coverage of the genome (WormBase release WS230). In regions where unique probes could not be designed selection filters were slightly relaxed in order to allow the inclusion of probes with possible cross-hybridization to at most one other location in the genome.
Availability of supporting data
Whole Genome Sequencing (WGS) data, fastq files, for the three strains: JNC168, JNC169 and JNC170 are available in the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under the BioProject accession number SRP053517 (PRJNA275156) with the fastq files: (170_R1.fastq, 170_R2.fastq, 270_R1.fastq, 270_R2.fastq, 470_R1.fastq and 470_R2.fastq) under the SRR1797354 accession number.
Acknowledgements
We thank the C. elegans Gene Knockout Consortium for generating the deletion mutants, the Caenorhabditis Genetics Center (CGC) for providing the strains, and the Michael Smith Genome Sciences Centre for Illumina sequencing. We also thank Bob Waterston for the sequencing reads of N2 strains; Robert Johnsen, Steven Jones, Harald Hutter and Nancy Hawkins for comments. This work was supported by the Canadian Institutes for Health Research (CIHR) and Fanconi Anemia Fellowship to MTG, and the Discovery Grant from the Natural Science and Engineering Research Council (NSERC) to NC. NC is a Michael Smith Foundation for Health Research (MSFHR) Scholar and a CIHR New Investigator. Work in the laboratory of DGM is supported by a grant from CIHR. DGM is a Senior Fellow of the Canadian Institute for Advanced Research (CIFAR). Work in the laboratory of AMR is supported by grants from CIHR and NSERC.
Abbreviations
- SAC
Spindle assembly checkpoint
- APC/C
Anaphase promoting complex or cyclosome
- CIN
Chromosome instability
- WGS
Whole genome sequencing
- FA
Fanconi anemia
- oaCGH
oligonucleotide array Comparative Genomic Hybridization
- G4
G-quartet
- SBS
Single base substitution
- GV
Genome variant
Additional files
Mutation accumulation in unc-46 mdf-1 such-4; dog-1 strains. Accumulation of mutations was visualized using Circos [42]. Note that due to the limited resolution, data points that occur close in the genome may appear as a single line or a single link. F170 chromosomes and variants originating at F170 are depicted in green; F270 chromosomes and variants originating at F270 are depicted in purple, while F470 chromosomes and variants unique to F470 are depicted in blue. The outer circle is a plot of all the variants present at a specific generation, while inner links are depicting propagation of the variants from one generation time-point to the next. The following variants are shown: (A) SBSs, (B) InDels, (C) Duplications, (D) Deletions.
Homozygous SBSs identified in the F170; F270 and F470 genomes. Table S1B. Homozygous SBSs identified after propagating mdf-1; dog-1; such-4 for 300 generations. Table S2. Location of the G3-5 N1-7G3-5 N1-7G3-5 N1-7G3-5 sequences in the C. elegans genome. Table S3A. Homozygous InDels smaller than or equal to 20 bp identified in the F170; F270 and F470 genomes. Table S3B. Homozygous InDels smaller than or equal to 20 bp after propagating mdf-1; dog-1; such-4 for 300 generations. Table S4A. Duplications identified in the F170; F270 and F470 genomes. Table S4B. Duplications identified after propagating mdf-1; dog-1; such-4 for 300 generations. Table S5A. Homozygous deletions larger than 20 bp identified in the F170; F270 and F470 genomes. Table S5B. Homozygous deletions larger than 20 bp identified after propagating mdf-1; dog-1; such-4 for 300 generations. Table S6. Accumulation of mutations predicted to disrupt protein-coding genes after 300 generations of propagation. Table S7. All mutations predicted to disrupt protein-coding genes in the mdf-1; dog-1; such-4 F470 genome.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MTG made the strains; MTG and TW wrote Perl scripts; MTG and ZQ prepared DNA for WGS and oaCGH. SF and JT generated oaCGH data. MTG analyzed the WGS data. MTG and SF analyzed the oaCGH data. MTG, DGM, AMR and NC led the study. MTG wrote the manuscript. All authors read, edited and approved the final manuscript.
Contributor Information
Maja Tarailo-Graovac, Email: maja@cmmt.ubc.ca.
Tammy Wong, Email: tammywong112@gmail.com.
Zhaozhao Qin, Email: qzhaozhao@gmail.com.
Stephane Flibotte, Email: sflibotte@gmail.com.
Jon Taylor, Email: jon.cr.taylor@gmail.com.
Donald G Moerman, Email: moerman@zoology.ubc.ca.
Ann M Rose, Email: annroseubc@gmail.com.
Nansheng Chen, Email: chenn@sfu.ca.
References
- 1.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–45. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 2.Kitagawa R, Rose AM. Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat Cell Biol. 1999;1:514–21. doi: 10.1038/70309. [DOI] [PubMed] [Google Scholar]
- 3.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 4.Cheung I, Schertzer M, Rose A, Lansdorp PM. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nat Genet. 2002;31:405–9. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 5.Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, Tijsterman M. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr Biol. 2008;18:900–5. doi: 10.1016/j.cub.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Zhao Y, Tarailo-Graovac M, O’Neil NJ, Rose AM. Spectrum of mutational events in the absence of DOG-1/FANCJ in Caenorhabditis elegans. DNA Repair. 2008;7:1846–54. doi: 10.1016/j.dnarep.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 7.Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–6. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 8.Sen D, Gilbert W. A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature. 1990;344:410–4. doi: 10.1038/344410a0. [DOI] [PubMed] [Google Scholar]
- 9.Gellert M, Lipsett MN, Davies DR. Helix formation by guanylic acid. Proc Natl Acad Sci U S A. 1962;48:2013–8. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Youds JL, Barber LJ, Ward JD, Collis SJ, O’Neil NJ, Boulton SJ, et al. DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol Cell Biol. 2008;28:1470–9. doi: 10.1128/MCB.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huppert JL, Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005;33:2908–16. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Todd AK, Johnston M, Neidle S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005;33:2901–7. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nat Genet. 2005;37:934–5. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 14.Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–3. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 15.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–65. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–41. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 17.Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–60. doi: 10.1016/S0092-8674(01)00304-X. [DOI] [PubMed] [Google Scholar]
- 18.Youds JL, O’Neil NJ, Rose AM. Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics. 2006;173:697–708. doi: 10.1534/genetics.106.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pontier DB, Kruisselbrink E, Guryev V, Tijsterman M. Isolation of deletion alleles by G4 DNA-induced mutagenesis. Nat Methods. 2009;6:655–7. doi: 10.1038/nmeth.1362. [DOI] [PubMed] [Google Scholar]
- 20.Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 2008;105:9272–7. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denver DR, Dolan PC, Wilhelm LJ, Sung W, Lucas-Lledó JI, Howe DK, et al. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci U S A. 2009;106:16310–4. doi: 10.1073/pnas.0904895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG, et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–4. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keightley PD, Trivedi U, Thomson M, Oliver F, Kumar S, Blaxter ML. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res. 2009;19:1195–201. doi: 10.1101/gr.091231.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarailo M, Kitagawa R, Rose AM. Suppressors of spindle checkpoint defect (such) mutants identify new mdf-1/MAD1 interactors in Caenorhabditis elegans. Genetics. 2007;175:1665–79. doi: 10.1534/genetics.106.067918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tarailo-Graovac M, Wang J, Tu D, Baillie DL, Rose AM, Chen N. Duplication of cyb-3 (cyclin B3) suppresses sterility in the absence of mdf-1/MAD1 spindle assembly checkpoint component in Caenorhabditis elegans. Cell Cycle Georget Tex. 2010;9:4858–65. doi: 10.4161/cc.9.24.14137. [DOI] [PubMed] [Google Scholar]
- 26.Tarailo-Graovac M, Wong T, Qin Z, Flibotte S, Tylor J, Moerman DG, et al. Cyclin B3 and dynein heavy chain cooperate to increase fitness in the absence of mdf-1/MAD1 in Caenorhabditis elegans. Cell Cycle. 2014;13:1–11. doi: 10.4161/15384101.2014.949491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yook K, Harris TW, Bieri T, Cabunoc A, Chan J, Chen WJ, et al. WormBase 2012: more genomes, more data, new website. Nucleic Acids Res. 2012;40(Database issue):D735–41. doi: 10.1093/nar/gkr954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denver DR, Morris K, Lynch M, Thomas WK. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature. 2004;430:679–82. doi: 10.1038/nature02697. [DOI] [PubMed] [Google Scholar]
- 30.Zhu YO, Siegal ML, Hall DW, Petrov DA. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 2014;111:E2310–8. doi: 10.1073/pnas.1323011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J-Q, Wu Y, Yang H, Bergelson J, Kreitman M, Tian D. Variation in the ratio of nucleotide substitution and indel rates across genomes in mammals and bacteria. Mol Biol Evol. 2009;26:1523–31. doi: 10.1093/molbev/msp063. [DOI] [PubMed] [Google Scholar]
- 32.Lipinski KJ, Farslow JC, Fitzpatrick KA, Lynch M, Katju V, Bergthorsson U. High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr Biol. 2011;21:306–10. doi: 10.1016/j.cub.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–21. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 35.Verma A, Yadav VK, Basundra R, Kumar A, Chowdhury S. Evidence of genome-wide G4 DNA-mediated gene expression in human cancer cells. Nucleic Acids Res. 2009;37:4194–204. doi: 10.1093/nar/gkn1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vergara IA, Frech C, Chen N. CooVar: co-occurring variant analyzer. BMC Res Notes. 2012;5:615. doi: 10.1186/1756-0500-5-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y, O’Neil NJ, Rose AM. Poly-G/poly-C tracts in the genomes of Caenorhabditis. BMC Genomics. 2007;8:403. doi: 10.1186/1471-2164-8-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarsounas M, Tijsterman M. Genomes and G-quadruplexes: for better or for worse. J Mol Biol. 2013;425:4782–9. doi: 10.1016/j.jmb.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 39.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maydan JS, Flibotte S, Edgley ML, Lau J, Selzer RR, Richmond TA, et al. Efficient high-resolution deletion discovery in Caenorhabditis elegans by array comparative genomic hybridization. Genome Res. 2007;17:337–47. doi: 10.1101/gr.5690307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]