

Abstract
Most cutaneous wounds heal with scar formation. Ideally, an inconspicuous normotrophic scar is formed, but an abnormal scar (hypertrophic scar or keloid) can also develop. A major challenge to scientists and physicians is to prevent adverse scar formation after severe trauma (e.g. burn injury) and understand why some individuals will form adverse scars even after relatively minor injury. Currently, many different models exist to study scar formation, ranging from simple monolayer cell culture to 3D tissue-engineered models even to humanized mouse models. Currently, these high-/medium-throughput test models avoid the main questions referring to why an adverse scar forms instead of a normotrophic scar and what causes a hypertrophic scar to form rather than a keloid scar and also, how is the genetic predisposition of the individual and the immune system involved. This information is essential if we are to identify new drug targets and develop optimal strategies in the future to prevent adverse scar formation. This viewpoint review summarizes the progress on in vitro and animal scar models, stresses the limitations in the current models and identifies the future challenges if scar-free healing is to be achieved in the future.
Keywords: hypertrophic, keloid, in vitro, organotypic, organ-on-a-chip, scar
Introduction
Wound healing starts directly at the time when the initial injury occurs. The healed skin always results in a scar, and therefore, for both the patient and the physician, a major outcome parameter in wound healing is the quality of the final scar (Fig.1). In general, after superficial injury, the scar is barely or may not even be visible to the naked eye. In the case of a deeper wound, the scar is often visible but is seen as a smooth, pale and flattened scar known as a normotrophic scar. However, in predisposed individuals and on some predilection sites on the body (e.g. sternum, ear lobe), scar formation can result in increased fibrosis, which in turn can result in adverse scar formation (hypertrophic scar or keloid). A major challenge to scientists and physicians is to prevent increased fibrosis and understand why some individuals will form abnormal scars even after relatively minor injury.
Figure 1.
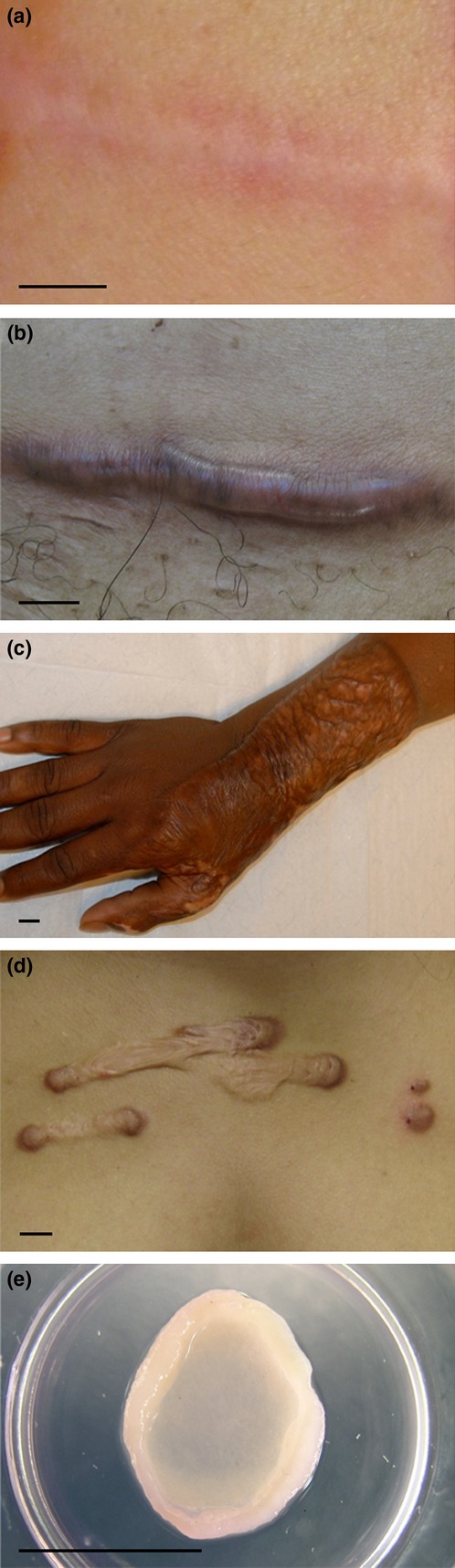
Macroscopic photographs of different scar tissues. (a) Normotrophic scar developed after incision wound (breast). (b) Hypertrophic scar developed after incision wound (abdomen). (c) Hypertrophic scar developed after extreme 3rd degree burn injury (hand). (d) Keloid scar formed from pustule (sternum) (e) In vitro hypertrophic scar model: skin equivalent of reconstructed epidermis on adipose-tissue-derived mesenchymal stem cells populated matrix. Bars = 1 cm.
To develop optimal therapeutic strategies for the different types of scar, it is essential to understand the pathology underlying these different scar types. Clinically, the distinction between a hypertrophic scar and a keloid remains difficult (1). Both hypertrophic scars and keloids can be firm, raised, itchy and painful. Both can have a significant physiological (limited joint mobility in particular with hypertrophic scars) and psychological (especially the face) impact on quality of life of the patient. The main clinical difference between the two adverse scars is that hypertrophic scars generally remain confined to the original wound borders, whereas keloids extend beyond the boundaries of the original lesion (2). Keloids may also develop years after the initial injury, almost never regress, are more common among the darker pigmented skin (up to 6–10% in African populations) and may have a genetic background (3). In contrast, hypertrophic scars occur within 4–8 weeks after injury, may diminish in time and are found in almost all patients when trauma is extensive (up to 91% following large deep burn injury) (3,4). However, a significant group of patients (34–64%) undergoing standard surgical procedures will also develop a hypertrophic scar after closure of the incision wound (5,6). All in all this indicates that, in addition to the standard response to extreme trauma, certain individuals are genetically predisposed to adverse scar formation. If this is indeed the case, then this needs to be taken into account when developing physiologically relevant human scar models. Furthermore, it is important to maintain the clinical distinction between hypertrophic scars and keloids by developing distinct physiologically relevant models for each type of abnormal scar.
Wound healing
Numerous reviews describe cutaneous wound healing as an interactive process involving not only skin residential cells and stem cells, but also infiltrating cells (7,8). Upon tissue damage, inflammation is initiated by the release of cytokines and chemokines from the damaged tissue. Immune cells (granulocytes, monocytes, lymphocytes) are drawn into the wound bed, and neighbouring skin residential cells and regenerative stem cells start to proliferate, migrate and differentiate to close the wound (7–10). Granulation tissue is deposited and extracellular matrix synthesized (7,10,11). Therefore, the early immune response must be involved in the early development of the scar and must play a role in the final quality of the scar. Indeed, adverse scars are thought to arise from an increased and prolonged inflammation. However, the type of immune response involving, for example, mast cells, neutrophils, macrophages, T lymphocytes (especially T helper 2 cells) and Langerhans cells is also thought to be important (3,12–16). Evidence also suggests that intrinsic aberrations in the immune system of those who form keloids exist. Peripheral blood mononuclear cells isolated from keloid-forming patients showed an altered secretion profile of growth factors and cytokines, an increased ability to induce fibroblast proliferation, and were more inclined to differentiate into fibrocytes when compared to patients who form normotrophic scars (17–19). Contradictory results suggest differences found between researchers could be due to the dynamics of wound healing, and therefore, the time of sample collection is very important (16).
Taken together, literature suggests that the i) genetic predisposition of the individual and ii) the extent and type of the initial inflammatory response are key players in scar formation. Both of these are extremely difficult to investigate in current in vitro and animal models. To understand the mechanisms underlying scar formation, scientists have turned from conventional submerged monolayer culture models to tissue-engineered models and even humanized mouse models (human skin is transplanted onto the animal). The progress made to date using these scar models is described below.
Current models and the need for improvements
Scar models are essential to investigate the pathogenesis of adverse scar formation, identify new drug targets and to test new therapeutics. Nowadays, animal models and in vitro cell culture and tissue-engineered models are used with varying degrees of success to represent human scars. Examples are shown in Tables1 and 2 (see extensive Tables S1 and S2 for more information and for references). Patient studies remain essential and shall always be necessary to validate potential novel antiscar therapeutics identified in animal and in vitro scar models. Human individuals are rarely used to explore the pathogenesis of adverse scar formation, probably due to ethical issues, logistical problems and also due to patient variation with regards to extent and duration of trauma. To overcome the problems confronted by patient studies, researchers have tried to extrapolate results from animal studies. Despite the large number of studies describing pigs, mice, rabbits, and other animals as models to investigate hypertrophic scarring or keloid formation, the basic skin physiology, immunology and therefore the wound healing process are markedly different with the result that animals do not develop scars which are comparable with adverse scars in humans (20–23). To humanize the mouse more, a hypertrophic scar model has been described in which a healthy human split-thickness skin graft is transplanted onto the back of a nude mouse (24,25). In a similar manner, to try to gain insight into the pathogenesis of keloid formation, keloid skin (full thickness or dermis only) has been directly transplanted onto nude mice (26–30). The greatly reduced number of T cells in these mice reduces the chance of graft rejection. In this mouse model, the immune component of wound healing and scar formation is severely compromised due to the immune-deficient phenotype of the nude mouse. This is also supported by reports showing that mouse models in general poorly mimic human inflammatory events (e.g. burn wound trauma) (23). The only human immune cells present are derived from the transplanted skin itself as human immune cells from the blood are absent (31). The obvious solution would be a physiologically relevant and fully standardized in vitro human model in which different key cell types, thought to be responsible for excessive scar formation, can be added under controlled conditions.
Table 1.
Overview of hypertrophic scar models and scar-forming parameters that can be assessed. For more extensive information, limitations and references, see Table S1
| Dermal thickness | ECM synthesis | Contraction | No. of vessels | No. of cells | Epith. | Epidermal thickness | Rete ridges | Hair follicles | GF&C | Apoptosis | Fib. proliferation | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In vivo human HT scar formation | + | + | + | + | + | + | + | + | + | + | + | ± |
| In vivo animal models | ||||||||||||
| Grafting split-thickness human skin onto animal | + | + | − | + | + | − | + | + | + | + | + | − |
| Grafting HT scar to animal | + | + | − | − | − | − | − | − | − | ± | − | − |
| Induction of HT scar: full-thickness wounds | + | + | ± | + | + | − | + | + | + | ± | + | ± |
| Induction of HT scar mechanical stress to full-thickness wound | + | + | − | + | + | − | + | + | + | − | − | − |
| In vitro models | ||||||||||||
| Human healthy cells | ||||||||||||
| Monolayer of Fib (+/−scratch) | − | + | − | − | − | − | − | − | − | + | − | − |
| DE: FPL (+/−mechanical stress) | − | + | + | − | − | − | − | − | − | + | + | − |
| SE: reconstructed epidermis of KC on a dermal matrix containing ASC | − | + | + | − | − | + | + | − | − | + | + | − |
| Human HT scar cells | ||||||||||||
| Monolayer of Fib | − | + | − | − | − | − | − | − | − | + | + | − |
| DE: FPL | − | + | + | − | − | − | − | − | − | + | + | + |
| SE: reconstructed epidermis of KC on a self-assembled matrix of Fib | + | + | − | − | − | − | + | − | − | − | − | − |
| Ex vivo | ||||||||||||
| HT scar biopsies (+/−mechanical stress) | + | + | − | − | − | − | + | − | − | − | − | + |
ASC, adipose-tissue-derived mesenchymal cells; DE, dermal equivalent; Epith, Epithelization; Fib, fibroblast; FPL, fibroblast populated lattice; GF & C, growth factors & cytokines; HT scar, hypertrophic scar; KC, keratinocytes; SE, skin equivalent; +, marker can be assessed in model; –, marker is not yet studied or cannot be assessed in model; ±, contradictory results.
Table 2.
Overview of keloid models and scar-forming parameters that can be assessed. For more extensive information, limitations and references, see Table S2
| Dermal thickness | ECM synthesis | Volume/weight | Contraction | No. of vessels | No. of cells | Epidermal thickness | GF&C | Proliferation | Apoptosis | Migration | Invasion | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In vivo Human Keloid formation | + | + | + | + | ± | + | + | + | ± | + | ± | ± |
| In vivo animal models | ||||||||||||
| Grafting Kscar into animal | − | + | + | − | ± | − | − | ± | − | − | − | − |
| Induction of Kscar | − | − | − | − | − | − | − | − | − | − | − | − |
| In vitro human models | ||||||||||||
| Human healthy cells | ||||||||||||
| NF co-cultured with CD14+ cells from keloid patients | − | − | − | − | − | − | − | + | + | − | − | − |
| Human Kscar cells | ||||||||||||
| Monolayer of keratinocytes | − | − | − | − | − | − | − | − | − | − | + | − |
| Monolayer of fibroblasts | − | + | − | − | − | − | − | ± | + | + | + | + |
| DE: FPCL | − | + | − | + | − | − | − | + | − | − | − | − |
| Indirect co-culture of KK with KF | − | + | − | − | − | − | − | + | + | + | − | − |
| NK epidermis on KF-populated matrix | + | + | − | − | − | − | + | − | − | − | − | − |
| Kscar explants | ||||||||||||
| Air-exposed biopsy embedded in collagen gel (6 weeks) | − | + | + | − | − | + | + | + | − | − | − | − |
DE, dermal equivalent; FPCL, fibroblast populated collagen 1 lattice; GF & C, growth factors and cytokines; KF, keloid scar fibroblasts; KK: keloid scar keratinocytes; Kscar, keloid scar; NF, normal skin fibroblasts; NK, normal skin keratinocytes; +, marker can be assessed in model; –, marker is not yet studied or cannot be assessed in model; ±, contradictory results.
In vitro cell culture models have been used for many years to gain insight into different aspects of scar pathogenesis, but almost never to test potential scar treatments. Early models, using conventional monolayer cell cultures, compared normal and scar-derived fibroblasts, or tried to induce a scar phenotype from healthy fibroblasts (32–34). Although being simple, fast and inexpensive, skin comprises more than just the fibroblasts. Indirect co-cultures of keratinocytes (monolayer or differentiated epidermis) and fibroblast monolayers using transwell systems enabled the study of keratinocyte–fibroblast interactions and the evaluation of the effects on either cell type separately (35–39). However, the lack of physiological relevance was obvious due to the absence of any resemblance with the 3D macroscopic fibrotic tissue structure typical of a scar. The expression of biomarkers derived from studies on gene and protein expression is most probably greatly influenced by the 3D structure present in a native scar. It was noticed that the introduction of a more physiologically relevant 3D environment (collagen or fibrin gel) and mechanical load positively influenced the behaviour of fibroblasts towards the scar phenotype (40). By enabling fibroblasts to produce their own matrix, an even more in vivo like situation was created (41). The realization that an extensive crosstalk between keratinocytes within the epidermis and fibroblasts within the dermis occurs to regulate the synthesis of extracellular dermal matrix (42) led to the introduction of organotypic skin equivalents being used to investigate scar pathogenesis. 3D skin equivalent models have been described using keloid fibroblasts in combination with normal skin-derived keratinocytes (35,43). This latter is considered a relevant limitation in the model as keloid keratinocytes have been described to be intrinsically different to normal skin-derived keratinocytes (36–38,44–47). Using a similar method, a fully differentiated epidermis constructed from keratinocytes isolated from hypertrophic scars on a fibroblast (healthy)-populated dermal matrix was able to exhibit a few characteristics of an adverse scar (e.g. dermal thickness, epidermal thickness, collagen I) and illustrated the role of keratinocytes in hypertrophic scar formation (48). Extensive implementation of these models for testing therapeutics is, however, limited by the lack of robust validated biomarkers and their dependence on excised scar tissue. This led to a recent development in our laboratory in which we were able to show that mesenchymal stromal (stem) cells derived from subcutaneous adipose (ASC) can be used to construct a tissue-engineered hypertrophic scar model. The model consists of a reconstructed epidermis derived from normal healthy human keratinocytes on a dermal matrix populated with ASC (49) (Fig.1). The hypertrophic scar model not only exhibits many characteristics of hypertrophic scars (e.g. increased collagen I secretion, contraction and epidermal thickness; decreased epithelization, Il-6 and CXCL8 secretion), but also enabled relevant and quantifiable hypertrophic scar parameters to be identified and validated with antiscar therapeutics (e.g. 5-fluorouracil, triamcinolone). Although this model is definitely a clear advancement, it is only representative of hypertrophic scar formation caused by severe trauma (e.g. burns) where the adipose tissue is exposed. It is not representative of hypertrophic scar formation resulting after skin closure of an excision wound after routine surgery, nor of keloid formation, which can develop years after relatively minor injury.
Multipotent keloid-derived mesenchymal-like stem cells, found in the pathological niche of the scar, have also been implicated in keloid formation (50–54). Therefore, keloid explant models are interesting as they allow these cells to remain in their pathological niche. Ex vivo biopsies have been cultured at air–liquid interface, embedded in collagen gels (55,56). The keloid phenotype persisted in culture as demonstrated by the maintenance of collagen I and III expression, immune cell fraction (T cells, B cells, NK cells, mast cells, neutrophils, Langerhans cells), mesenchymal cells and endothelial cells. The functionality of this model was further confirmed by the reduced epidermal thickness and scar volume after treatment with the dexamethasone. While this model certainly shows promising potential, it is entirely dependent on a regular supply of scars that are both freshly excised and sufficiently large, which prevents widespread implementation.
Limitations
From the above, we can identify a number of clear limitations in the current available models.
Animal models are not suitable for studying human adverse scar formation
Apart from the ethical issues described in the 7th directive (3Rs – reduction, refinement, replacement), the physiology of animal skin and their immune system are so different from humans (23) that pivotal factors responsible for differences between normotrophic, hypertrophic and keloid scar formation are impossible to identify.
Human cell culture models are still limited by their extreme simplicity
A scar is generated by a complex cascade of cellular interactions starting at the initial time of injury. The numerous cell types that are involved such as fibroblasts, endothelial cells, keratinocytes, immune cells (e.g. mast cells, monocytes, macrophages, neutrophils, T cells, dendritic cells) to name but a few are not yet incorporated into relevant human culture models (16,57–59). Furthermore, mechanical loading is not taken into account in current models.
The genetic predisposition factor is not taken into account
With the exception of extreme burn trauma that nearly always results in hypertrophic scarring, an important pivotal factor that is not taken into account is the genetic predisposition of the individual. This predisposition will influence the entire process of scar formation from the inflammatory response to the tissue remodelling and final scar formation.
Scar models have a limited duration of days/weeks, whereas human scars develop over a period of months/years
This means that while scar models will enable us to identify genes and proteins (biomarkers) reflecting the early stages of scar formation, the macroscopic raised but at the same time contracted fibrotic structure of the scar is rarely pronounced to the extent that is characteristic of an adverse scar.
In our efforts to develop high-/medium-throughput test models to study the beginnings of scar formation, we have sacrificed the essence of the subject – why does an adverse scar form instead of a normotrophic scar and what causes a hypertrophic scar to form rather than a keloid scar. This information is essential if we are to develop optimal strategies in the future to prevent adverse scar formation.
Challenges and future perspectives
It is always easy to identify the limitations of a model. However, the solution to the limitations is more difficult and will be an extremely inspiring challenge to scientists. Advancements with constructing TERT-immortalized cell lines should be exploited, making it possible to maintain cell strains representative of patients with different predispositions to normotrophic, hypertrophic scar and keloid as well as solving logistical and ethical limitations concerning freshly excised tissue. Recently, an exciting new multidisciplinary scientific field, ‘organ-on-a-chip’, has been developing for organ and disease models, which may also be suitable for in vitro scar models. Organ-on-a-chip involves engineered tissues, which closely mimic their in vivo counterparts and consist of multiple different cell types adjacent to and interacting with each other under closely controlled conditions and are importantly grown in a microfluidic chip. These controlled conditions will make it possible to mimic the environment of the skin (humidity, temperature, pH, oxygen levels), the elasticity of the skin, and the complex structures and cellular interactions within and between the different cell types of the skin. Importantly, the microfluidics compartment, in addition to possibly prolonging the lifespan of the cultures, will mimic the blood and lymph vasculature enabling incorporation of immune cells into the model. Early examples are ‘lung-on-a-chip’, ‘intestine-on-a-chip’, ‘lymph node-on-a-chip’ and ‘vasculature-on-a-chip’, used to study physiology, pathophysiology and to develop and discover drug targets (60–63). Once a ‘scar-on-a-chip’ model has been established, it should be possible to generate abnormal scar models with different genetic predispositions to a normotrophic, hypertrophic and keloid scar using (e.g. TERT immortalized) skin and immune cells derived from patients. Such an approach will not only enable investigation of the general pathophysiology of scar formation, but will also allow research into effects of genetic influences on the disease process. Ultimately, this will make it possible to develop a medium-throughput drug target discovery and development platform, comprising a library of different genetic backgrounds to be used as ‘in vitro’ clinical trials.
Acknowledgments
This work has been financed in part by the Dutch Government (ZonMw programme Animal-Free Research Techniques project nr: 114021003). We acknowledge Prof Paul van Zuijlen for the photograph of the hypertrophic scar. LJ and GC performed literature study and made Tables1 and 2. LJ and SG wrote the manuscript, which was edited by all authors.
Conflict of interest
The authors have declared no conflicting interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1.Hypertrophic scar models and parameters.
Table S2.Keloid scar models and parameters.
References
- 1.Juckett G, Hartman-Adams H. Am Fam Physician. 2009;80:253–260. [PubMed] [Google Scholar]
- 2.Niessen FB, Spauwen PH, Schalkwijk J, et al. Plast Reconstr Surg. 1999;104:1435–1458. doi: 10.1097/00006534-199910000-00031. [DOI] [PubMed] [Google Scholar]
- 3.Gauglitz GG, Korting HC, Pavicic T, et al. Mol Med. 2011;17:113–125. doi: 10.2119/molmed.2009.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kose O, Waseem A. Dermatol Surg. 2008;34:336–346. doi: 10.1111/j.1524-4725.2007.34067.x. [DOI] [PubMed] [Google Scholar]
- 5.Niessen FB, Spauwen PH, Robinson PH, et al. Plast Reconstr Surg. 1998;102:1962–1972. doi: 10.1097/00006534-199811000-00023. [DOI] [PubMed] [Google Scholar]
- 6.van der Veer WM, Ferreira JA, de Jong EH, et al. Ann Plast Surg. 2010;65:321–325. doi: 10.1097/SAP.0b013e3181c60f88. [DOI] [PubMed] [Google Scholar]
- 7.Broughton G, Janis JE, Attinger CE. Plast Reconstr Surg. 2006;117:12S–34S. doi: 10.1097/01.prs.0000225430.42531.c2. [DOI] [PubMed] [Google Scholar]
- 8.Barrientos S, Stojadinovic O, Golinko MS, et al. Wound Repair Regen. 2008;16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 9.Kroeze KL, Jurgens WJ, Doulabi BZ, et al. J Invest Dermatol. 2009;129:1569–1581. doi: 10.1038/jid.2008.405. [DOI] [PubMed] [Google Scholar]
- 10.Le Y, Zhou Y, Iribarren P, et al. Cell Mol Immunol. 2004;1:95–104. [PubMed] [Google Scholar]
- 11.Gillitzer R, Goebeler M. J Leukoc Biol. 2001;69:513–521. [PubMed] [Google Scholar]
- 12.Armour A, Scott PG, Tredget EE. Wound Repair Regen. 2007;15(Suppl 1):S6–17. doi: 10.1111/j.1524-475X.2007.00219.x. [DOI] [PubMed] [Google Scholar]
- 13.Boyce DE, Ciampolini J, Ruge F, et al. Br J Plast Surg. 2001;54:511–516. doi: 10.1054/bjps.2001.3638. [DOI] [PubMed] [Google Scholar]
- 14.Shaker SA, Ayuob NN, Hajrah NH. Appl Immunohistochem Mol Morphol. 2011;19:153–159. doi: 10.1097/PAI.0b013e3181efa2ef. [DOI] [PubMed] [Google Scholar]
- 15.Shaw TJ, Kishi K, Mori R. Endocr Metab Immune Disord Drug Targets. 2010;10:320–330. doi: 10.2174/1871530311006040320. [DOI] [PubMed] [Google Scholar]
- 16.van der Veer WM, Bloemen MC, Ulrich MM, et al. Burns. 2009;35:15–29. doi: 10.1016/j.burns.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 17.Liao WT, Yu HS, Arbiser JL, et al. Exp Dermatol. 2010;19:e142–e150. doi: 10.1111/j.1600-0625.2009.01021.x. [DOI] [PubMed] [Google Scholar]
- 18.Naylor MC, Lazar DA, Zamora IJ, et al. Wound Repair Regen. 2012;20:277–283. doi: 10.1111/j.1524-475X.2012.00782.x. [DOI] [PubMed] [Google Scholar]
- 19.McCauley RL, Chopra V, Li YY, et al. J Clin Immunol. 1992;12:300–308. doi: 10.1007/BF00918154. [DOI] [PubMed] [Google Scholar]
- 20.Hillmer MP, MacLeod SM. J Cutan Med Surg. 2002;6:354–359. doi: 10.1007/s10227-001-0121-y. [DOI] [PubMed] [Google Scholar]
- 21.Ramos ML, Gragnani A, Ferreira LM. J Burn Care Res. 2008;29:363–368. doi: 10.1097/BCR.0b013e3181667557. [DOI] [PubMed] [Google Scholar]
- 22.Seo BF, Lee JY, Jung SN. Biomed Res Int. 2013;2013:423147. doi: 10.1155/2013/423147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seok J, Warren HS, Cuenca AG, et al. Proc Natl Acad Sci USA. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Momtazi M, Kwan P, Ding J, et al. Wound Repair Regen. 2013;21:77–87. doi: 10.1111/j.1524-475X.2012.00856.x. [DOI] [PubMed] [Google Scholar]
- 25.Yang DY, Li SR, Wu JL, et al. Plast Reconstr Surg. 2007;119:104–109. doi: 10.1097/01.prs.0000244828.80490.62. [DOI] [PubMed] [Google Scholar]
- 26.Estrem SA, Domayer M, Bardach J, et al. Laryngoscope. 1987;97:1214–1218. doi: 10.1288/00005537-198710000-00018. [DOI] [PubMed] [Google Scholar]
- 27.Ishiko T, Naitoh M, Kubota H, et al. J Dermatol. 2013;40:380–383. doi: 10.1111/1346-8138.12116. [DOI] [PubMed] [Google Scholar]
- 28.Kischer CW, Pindur J, Shetlar MR, et al. J Trauma. 1989;29:672–677. doi: 10.1097/00005373-198905000-00023. [DOI] [PubMed] [Google Scholar]
- 29.Shetlar MR, Shetlar CL, Hendricks L, et al. Proc Soc Exp Biol Med. 1985;179:549–552. doi: 10.3181/00379727-179-rc3. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Smith P, Pu LL, et al. J Surg Res. 1999;87:194–200. doi: 10.1006/jsre.1999.5757. [DOI] [PubMed] [Google Scholar]
- 31.Butler PD, Longaker MT, Yang GP. J Am Coll Surg. 2008;206:731–741. doi: 10.1016/j.jamcollsurg.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Kim WS, Lee JS, Bae GY, et al. Exp Dermatol. 2013;22:69–71. doi: 10.1111/exd.12063. [DOI] [PubMed] [Google Scholar]
- 33.Moon H, Yong H, Lee AR. Arch Pharm Res. 2012;35:383–388. doi: 10.1007/s12272-012-0220-x. [DOI] [PubMed] [Google Scholar]
- 34.Phan TT, Sun L, Bay BH, et al. J Trauma. 2003;54:1212–1224. doi: 10.1097/01.TA.0000030630.72836.32. [DOI] [PubMed] [Google Scholar]
- 35.Butler PD, Ly DP, Longaker MT, et al. Am J Surg. 2008;195:144–148. doi: 10.1016/j.amjsurg.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Funayama E, Chodon T, Oyama A, et al. J Invest Dermatol. 2003;121:1326–1331. doi: 10.1111/j.1523-1747.2003.12572.x. [DOI] [PubMed] [Google Scholar]
- 37.Khoo YT, Ong CT, Mukhopadhyay A, et al. J Cell Physiol. 2006;208:336–343. doi: 10.1002/jcp.20668. [DOI] [PubMed] [Google Scholar]
- 38.Lim CP, Phan TT, Lim IJ, et al. J Invest Dermatol. 2009;129:851–861. doi: 10.1038/jid.2008.337. [DOI] [PubMed] [Google Scholar]
- 39.Phan TT, Lim IJ, Aalami O, et al. J Pathol. 2005;207:232–242. doi: 10.1002/path.1826. [DOI] [PubMed] [Google Scholar]
- 40.Derderian CA, Bastidas N, Lerman OZ, et al. Ann Plast Surg. 2005;55:69–75. doi: 10.1097/01.sap.0000168160.86221.e9. [DOI] [PubMed] [Google Scholar]
- 41.Ahlfors JE, Billiar KL. Biomaterials. 2007;28:2183–2191. doi: 10.1016/j.biomaterials.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 42.Ghaffari A, Kilani RT, Ghahary A. J Invest Dermatol. 2009;129:340–347. doi: 10.1038/jid.2008.253. [DOI] [PubMed] [Google Scholar]
- 43.Chiu LL, Sun CH, Yeh AT, et al. Lasers Surg Med. 2005;37:231–244. doi: 10.1002/lsm.20213. [DOI] [PubMed] [Google Scholar]
- 44.Chua AW, Ma D, Gan SU, et al. J Invest Dermatol. 2011;131:644–654. doi: 10.1038/jid.2010.371. [DOI] [PubMed] [Google Scholar]
- 45.Hahn JM, Glaser K, McFarland KL, et al. Wound Repair Regen. 2013;21:530–544. doi: 10.1111/wrr.12060. [DOI] [PubMed] [Google Scholar]
- 46.Lim IJ, Phan TT, Tan EK, et al. J Biol Chem. 2003;278:40851–40858. doi: 10.1074/jbc.M305759200. [DOI] [PubMed] [Google Scholar]
- 47.Ong CT, Khoo YT, Tan EK, et al. J Pathol. 2007;211:95–108. doi: 10.1002/path.2081. [DOI] [PubMed] [Google Scholar]
- 48.Bellemare J, Roberge CJ, Bergeron D, et al. J Pathol. 2005;206:1–8. doi: 10.1002/path.1737. [DOI] [PubMed] [Google Scholar]
- 49.van den Broek LJ, Niessen FB, Scheper RJ, et al. ALTEX. 2012;29:389–402. doi: 10.14573/altex.2012.4.389. [DOI] [PubMed] [Google Scholar]
- 50.Iqbal SA, Syed F, McGrouther DA, et al. Br J Dermatol. 2010;162:1377–1383. doi: 10.1111/j.1365-2133.2010.09738.x. [DOI] [PubMed] [Google Scholar]
- 51.Iqbal SA, Sidgwick GP, Bayat A. Arch Dermatol Res. 2012;304:665–671. doi: 10.1007/s00403-012-1225-5. [DOI] [PubMed] [Google Scholar]
- 52.Moon JH, Kwak SS, Park G, et al. Stem Cells Dev. 2008;17:713–724. doi: 10.1089/scd.2007.0210. [DOI] [PubMed] [Google Scholar]
- 53.Qu M, Song N, Chai G, et al. Med Hypotheses. 2013;81:807–812. doi: 10.1016/j.mehy.2013.08.033. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Q, Yamaza T, Kelly AP, et al. PLoS ONE. 2009;4:e7798. doi: 10.1371/journal.pone.0007798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duong HS, Zhang Q, Kobi A, et al. Cells Tissues Organs. 2005;181:89–102. doi: 10.1159/000091098. [DOI] [PubMed] [Google Scholar]
- 56.Bagabir R, Syed F, Paus R, et al. Exp Dermatol. 2012;21:376–381. doi: 10.1111/j.1600-0625.2012.01476.x. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Jiao H, Stewart TL, et al. Wound Repair Regen. 2007;15:530–539. doi: 10.1111/j.1524-475X.2007.00261.x. [DOI] [PubMed] [Google Scholar]
- 58.Mahdavian DB, van der Veer WM, van Egmond M, et al. Immunobiology. 2011;216:753–762. doi: 10.1016/j.imbio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Foley TT, Ehrlich HP. Plast Reconstr Surg. 2013;131:1036–1044. doi: 10.1097/PRS.0b013e3182865c3f. [DOI] [PubMed] [Google Scholar]
- 60.Baker M. Nature. 2011;471:661–665. doi: 10.1038/471661a. [DOI] [PubMed] [Google Scholar]
- 61.Giese C, Lubitz A, Demmler CD, et al. J Biotechnol. 2010;148:38–45. doi: 10.1016/j.jbiotec.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 62.Huh D, Matthews BD, Mammoto A, et al. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korin N, Kanapathipillai M, Matthews BD, et al. Science. 2012;337:738–742. doi: 10.1126/science.1217815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.Hypertrophic scar models and parameters.
Table S2.Keloid scar models and parameters.