

Abstract
Objective
Previously, we reported that a novel variant, p.Ser707Tyr, in phospholipase Cγ2 (PLCγ2) is the cause of a dominantly inherited autoinflammatory disease, APLAID. The hypermorphic mutation enhances the PLCγ2 activity and causes an increase in intracellular Ca2+ release from ER stores. As increased intracellular Ca2+ signaling has been associated with NLRP3 inflammasome activation, we studied the role of the NLRP3 inflammasome in the pathogenesis of this disease.
Methods
Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy controls and two affected patients. Inflammasome activation was analyzed by Western blotting. Intracellular Ca2+ levels were measured with the FLIPR Calcium 4 assay kit.
Results
Patients’ cells had elevated basal levels of intracellular Ca2+ and the intracellular Ca2+ flux triggered by extracellular CaCl2 was substantially enhanced. Patients’ PBMCs secreted IL-1β in response to LPS priming alone, and this effect was attenuated by use of a PLC inhibitor, intracellular Ca2+ blockers, or an adenylate cyclase activator.
Conclusion
Our findings suggest that the inflammation in patients with APLAID is partially driven by the activation of the NLRP3 inflammasome. These data link two seemingly distinct molecular pathways and provide new insights into the pathogenesis of APLAID and autoinflammation.
Keywords: autoinflammatory disease, inflammasome, phospholipase Cγ2, IL-1β, pathogenesis
INTRODUCTION
Monogenic autoinflammatory diseases are caused by aberrant activation of innate immune cells, primarily neutrophils and macrophages. At the present time, mutations in more than 15 genes mediating several distinct pathways have been associated with autoinflammatory syndromes.1 Discovering the genetic causes of monogenic autoinflammatory diseases permitted their recognition as disorders many of which are mediated by the release of proinflammatory cytokines such as interleukin (IL)-1β.
Recently, we identified a gain-of-function mutation in PLCG2 as the cause of a dominantly inherited disorder, APLAID (autoinflammation and PLCγ2-associated antibody deficiency and immune dysregulation), in a small family with only two affected members.2 Both father (Patient 1) and daughter (Patient 2) suffered from early onset recurrent blistering skin lesions, pulmonary disease, arthralgia, inflammatory eye and bowel disease, and mild immunodeficiency. Lymphocyte phenotyping revealed a near complete absence of class-switched memory B-cells, potentially explaining the increased propensity to develop bacterial infections. Both patients were refractory to treatments with NSAIDs and their symptoms were partially responsive to steroids, although they experienced many of the adverse side effects of high dose steroids.
PLCG2 encodes phospholipase Cγ2 (PLCγ2), an enzyme of the phospholipase C family, which cleaves the membrane phospholipid phosphatidyl inositol-4,5-biphosphate into the second messenger molecules inositol-1,4,5-triphosphate (InsP3) and diacylglycerol (DAG). InsP3 increases intracellular calcium levels by inducing the release of endoplasmic reticulum (ER) calcium stores. The APLAID-associated p.Ser707Tyr mutation disrupts the highly conserved C-terminal copy of a tandem pair of SH2 autoinhibitory domains (cSH2), causing an increase in production of intracellular InsP3 and increased intracellular Ca2+ release.2 Together, in vitro and ex vivo experiments demonstrated evidence that the PLCγ2 signaling pathway is more active in mutant cells.
IL-1β is a proinflammatory cytokine that plays an important role in host defense and inflammatory disease, fever, and septic shock.3 The maturation and secretion of IL-1β are mediated by caspase-1, which is activated by inflammasomes (cytoplasmic multiprotein platforms) in response to cellular infection or stress.4 The NLRP3 (also known as NALP3 or cryopyrin) inflammasome has been shown to be activated by a wide range of pathogen-associated or danger-associated molecular patterns, such as ATP, endogenous urate, cholesterol crystals, silica, and asbestos particles. Missense mutations in the NLRP3 protein are associated with a spectrum of dominantly inherited autoinflammatory diseases, which are called cryopyrin-associated periodic syndromes (CAPS).5,6 Recently, we showed in murine cells that the calcium sensing receptor activates the NLRP3 inflammasome through PLC, which catalyzes InsP3 production and thereby induces release of Ca2+ from ER stores.7 The increased cytoplasmic Ca2+ promotes the assembly of inflammasome components, and intracellular Ca2+ is required for spontaneous inflammasome activity in cells from CAPS patients.
The leukocytes from patients with APLAID showed enhanced PLCγ2 activity. However, the question remains regarding the molecular basis of systemic inflammation in these patients. Since InsP3-mediated Ca2+ release from the ER triggers NLRP3 inflammasome activation in murine cells, here we studied NLRP3 inflammasome activity in leukocytes from APLAID patients.
MATERIALS AND METHODS
Cell preparation
Blood specimens from healthy controls and APLAID patients were drawn after obtaining informed consent under a protocol approved by the NIAMS/NIDDK Institutional Review Board. Human PBMCs were isolated by LSM-Lymphocyte Separation Medium (50494, MP Biomedicals, Santa Ana, CA).
Inflammasome activation or inhibition
Inflammasome activation experiments were performed in two stages, LPS priming for 3h and activation (within 1h). PBMCs (2×106 cells/well) were plated in 12-well plates and then primed with 1 μg ml−1 LPS in RPMI 1640 (Invitrogen) containing 10% FBS. For NLRP3 inflammasome activation, the medium was replaced with RPMI 1640 supplemented with ATP (2 mM), CaCl2 (1 mM, resulting in a total Ca2+ concentration of 1.42 mM), or m-3M3FBS (1–50 μM). For the inhibition of inflammasome activation, LPS-primed PBMCs were treated with U73122 (1–10 μM), U73343 (10 μM), BAPTA-AM (10 or 50 μM), 2-APB (10–50 μM), or NKH477 (500 μM) in the presence or absence of extracellular NLRP3 inflammasome activators (ATP or CaCl2). After 30 to 50 min of treatment, supernatants and cell lysates were collected for immunoblot analysis.
Immunoblots
Immunoblots were prepared with Novex® Tris-Glycine Gel Systems (Invitrogen) and probed with anti-human IL-1β Ab (AF-201-NA, R&D Systems, Minneapolis, MN) or anti-actin Ab (sc-1615, Santa Cruz Biotechnology).
Intracellular calcium measurements
Human PBMCs were seeded at 5×105 cells per 100 μl loading medium (RPMI 1640, 10% FBS) into 96-Well Black Clear-Bottom Plates (Costar). The cells were incubated for 1h at 37 °C with 1 μg/ml of LPS after which an equal volume of assay loading buffer (FLIPR Calcium 4 assay kit, Molecular Devices) in Hanks’ balanced salt solution supplemented with 20 mM of HEPES and 2 mM of probenecid was added. The cells were incubated for 1h at 37 °C before adding 1 mM of CaCl2 and then the calcium flux peak was measured using a FlexStation 3 (Molecular Devices). The data were analyzed with SOFT Max Pro 5.2 (Molecular Devices). Data are shown as fluorescent counts and the y-axis labeled as iLm1×1000.
RESULTS
Correlation between the PLC activity and the presence of active IL-1β has been reported in LPS-primed murine bone marrow derived macrophages (BMDMs).7 Here, we examined the role of PLC in NLRP3 inflammasome activation in human PBMCs. First, we found that activation and secretion of IL-1β from LPS-primed and ATP- or CaCl2-stimulated human PBMCs could be blocked in the presence of a known PLC inhibitor, U73122, in a dose-dependent manner (figure 1A and B). But at the highest dose (10 μM), U73122 also reduces the levels of pro-IL-1β and actin, which is probably due to a toxic effect. In contrast, U73343, an inactive analog of U73122, had no effect on the IL-1β secretion. Conversely, 2,4,6-trimethyl-N-[3-(trifluoromethyl)-phenyl]benzenesulphonamide (m-3M3FBS), a direct activator of PLC alone, induced a dose-dependent secretion of IL-1β from LPS-primed human PBMCs in the absence of any additional inflammasome activators (figure 1C). The activation of PLC leads to the production of inositol trisphosphate (InsP3) that subsequently causes an increase in the concentration of cytosolic Ca2+. The increase of cytoplasmic Ca2+ has been shown to mediate the activation of the NLRP3 inflammasome in mouse BMDMs.7 Indeed, ATP or Ca2+ driven IL-1β secretion from LPS-primed human PBMCs is inhibited substantially by BAPTA-AM, an intracellular Ca2+ chelator (figure 1D). These data indicate that PLC-InsP3-mediated Ca2+ release can trigger the activation of the NLRP3 inflammasome in human PBMCs in much the same way as was previously shown in the mouse.7
Figure 1. PLC-InsP3-mediated calcium release from the ER triggers NLRP3 inflammasome activation.
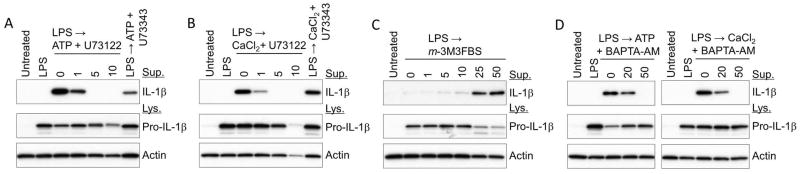
(A, B) LPS (1 mg ml−1 for 3 h)-primed PBMCs from a healthy control were treated with the indicated doses of U73122 or U73343 in the presence of 2 mM ATP or 1 mM CaCl2 for 40 min. (C) LPS-primed PBMCs were treated with the indicated doses of m-3M3FBS for 40 min. (D) LPS-primed PBMCs were treated with the indicated doses of BAPTA-AM in the presence of ATP (2 mM) or CaCl2 (1 mM) for 40 min. Cell culture supernatants and cell lysates were analyzed by immunoblotting for IL-1β. All immunoblot data shown are representative of more than three independent experiments.
The p.Ser707Tyr APLAID mutation disrupts the autoinhibition of PLCγ2, and enhanced PLCγ2 activity was shown in patients’ leukocytes.2 We observed that the basal levels of intracellular Ca2+ in patients’ PBMCs are higher than the Ca2+ levels in controls’ PBMCs (figure 2A). Upon stimulation with extracellular CaCl2, an activator of the NLRP3 inflammasome, LPS-primed patients’ cells release significantly higher amounts of Ca2+ into the cytosol than the cells of healthy controls (figure 2B). Next, we examined IL-1β secretion in LPS-primed PBMCs with or without NLRP3 inflammasome activators. In the absence of inflammasome activators, LPS-primed PBMCs from patients with APLAID secreted IL-1β whereas control LPS-primed PBMCs secreted IL-1β only following the stimulation with CaCl2 (figure 2C). Consistent with the data from figure 2B, higher baseline levels of cytosolic Ca2+ in patients’ PBMCs likely contributed to an enhanced IL-1β secretion from mutant cells.
Figure 2. Increased intracellular Ca2+ flux and IL-1β production of PBMCs from APLAID patients.

(A) Intracellular Ca2+ levels were measured from untreated or LPS-primed PBMCs of APLAID patients and 3 healthy controls. Data are shown as mean ± SEM (n=4). Statistical significance was analyzed by t test; *, P< 0.05, **; P < 0.01. (B) Intracellular Ca2+ fluxes triggered by CaCl2 were measured from PBMCs of APLAID patients and 3 healthy controls. PBMCs were primed with LPS (1 μg/ml) prior to exposure to CaCl2. Changes in intracellular Ca2+ levels were monitored for 600 seconds at 10 second intervals. The data are shown as fluorescent counts and the y-axis labeled as iLm1×1000. Each experimental value is the mean± SEM of four determinations. (C) PBMCs from APLAID patients and healthy control were non-primed (Untreat) or primed with LPS (1 μg ml−1) for 3h and then treated with/without 2 mM ATP or 1 mM CaCl2 for 40 min. Cell lysates and cell culture supernatants were analyzed for IL-1β secretion. All Intracellular Ca2+ measurements and immunoblot data shown are representative of two independent experiments.
To further investigate the mechanism of inflammation in APLAID, we examined the role of PLC-InsP3-mediated Ca2+ release from the ER on IL-1β secretion from patients’ PBMCs. The IL-1β secretion from LPS-primed patients’ PBMCs was blocked in the presence of PLC inhibitor, U73122, and not in the presence of its inactive analog U73433 (figure 3A). In addition, constitutive IL-1β secretion from patients’ PBMCs was substantially reduced in the presence of inhibitors of InsP3-mediated intracellular Ca2+ signaling pathways, 2-APB (inhibitor for InsP3 receptor) and BAPTA-AM (figure 3B and C). Taken together, these data demonstrate PLC-dependent NLRP3 inflammasome activation in APLAID.
Figure 3. The role of intracellular Ca2+ and IL-1β in the pathogenesis of APLAID.

(A–D) LPS-primed PBMCs from APLAID patients were treated with U73122 (10 μg/ml) or U73343 (10 μg/ml) (A), the indicated dose of 2-APB (B), BAPTA-AM (C), or NKH477 (500 μM) (D). Cell lysates and cell culture supernatants were analyzed for IL-1β secretion. All immunoblot data shown are representative of two independent experiments.
Finally, we examined suppressive role of cyclic AMP (cAMP) on the IL-1β secretion from patients’ PBMCs because cAMP has been shown to suppress NLRP3 inflammasome activation (online figure S1).7 Indeed, we found that the constitutive IL-1β secretion from patients PBMCs was substantially reduced by the treatment with NKH477, the water-soluble analog of forskolin, which is a potent activator of adenylyl cyclase (figure 3D). These results suggest cAMP as a potential target for therapy of APLAID and other NLRP3 mediated diseases.
DISCUSSION
This manuscript extends the connection between PLC and activation of the NLRP3 inflammasome, which plays an important role not only in the pathogenesis of relatively rare disorders such as CAPS, but also more common diseases such as gout8, type 2 diabetes mellitus9,10, artherosclerosis11, and Alzheimer’s disease.12 Earlier work from our laboratory demonstrated that the NLRP3 inflammasome is activated by the calcium-sensing receptor, a G-protein coupled receptor that stimulates PLC and inhibits adenylate cyclase.7 Recently we described a novel autoinflammatory disorder, APLAID, that is caused by a missense p.Ser707Tyr substitution in PLCγ2, leading to increased PLC activity. From these previous studies, one would predict increased NLRP3 inflammasome activity in APLAID patients. The present manuscript confirms this prediction.
PLCγ2 is expressed in lymphocytes as well as innate immune cells, and is known to trigger a number of signaling pathways, including protein kinase C. Hence, one would not expect APLAID patients to have a dramatic response to IL-1 inhibitors such as anakinra, and that in fact has been our clinical experience with these patients. Nevertheless, this manuscript contributes to a growing body of data suggesting a possible role for agents that modulate PLC and/or adenylate cyclase activity in the treatment of excessive NLRP3-dependent IL-1β production. Given the frequency of these illnesses, and the cost of biologics, such strategies may eventually have significant impact.
Supplementary Material
LPS-primed PBMCs from healthy control were treated with NKH477 (500 μM) in the presence or absence of 1 mM CaCl2. Cell lysates and cell culture supernatants were analyzed for IL-1β secretion. The immunoblot data shown are representative of three independent experiments.
Acknowledgments
Supported by the Intramural Research Programs of the National Human Genome Research Institute and the National Institute of Allergy and Infectious Diseases.
References
- 1.Gattorno M, Martini A. Beyond the NLRP3 inflammasome: autoinflammatory diseases reach adolescence. Arthritis Rheum. 2013;65:1137–47. doi: 10.1002/art.37882. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Q, Lee GS, Brady J, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. 2012;91:713–20. doi: 10.1016/j.ajhg.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 4.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman HM, Mueller JL, Broide DH, et al. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Gene. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46:3340–8. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee GS, Subramanian N, Kim AI, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–7. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinon F, Petrilli V, Mayor A, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 9.Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou R, Tardivel A, Thorens B, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 11.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LPS-primed PBMCs from healthy control were treated with NKH477 (500 μM) in the presence or absence of 1 mM CaCl2. Cell lysates and cell culture supernatants were analyzed for IL-1β secretion. The immunoblot data shown are representative of three independent experiments.