

Abstract
Prostatic small cell neuroendocrine carcinoma (SCNC) is a rare but aggressive form of prostate cancer (PCa) that is negative for androgen receptor (AR) and not responsive to hormonal therapy. The molecular etiology of this PCa variant is not well understood; however, mutation of the p53 (TP53) tumor suppressor in prostate neuroendocrine cells inactivates the IL8-CXCR2-p53 pathway that normally inhibits cellular proliferation, leading to the development of SCNC. SCNC also overexpresses Aurora kinase A (AURKA) which is considered to be a viable therapeutic target. Therefore, the relationship of these two molecular events was studied and we show that p53 mutation leads to increased expression of miR-25 and down-regulation of the E3 ubiquitin ligase FBXW7, resulting in elevated levels of Aurora kinase A. This study demonstrates an intracellular pathway by which p53 mutation leads to Aurora kinase A expression, which is critically important for the rapid proliferation and aggressive behavior of prostatic SCNC.
Keywords: p53, Aurora Kinase A, miR-25, Fbxw7, Small cell neuroendocrine carcinoma
Introduction
Prostate cancer is the leading cause of cancer-related death for men in western countries. Understanding the molecular mechanisms of prostate carcinogenesis and progression is the foundation and a challenge for the development of effective therapy. Patients with low grade and early stage of prostate cancers can be cured by surgery or radiation. For those with advanced and metastatic prostate cancers that are not amenable for local therapies, hormonal therapy targeting androgen receptor (AR) pathway has been the treatment of choice for many decades. Unfortunately, this therapy is not curative, and the cancer invariably progresses to castration-resistant state with few therapeutic options.
The majority of human prostate cancers are classified as adenocarcinoma with the bulk tumor cells showing luminal differentiation including the expression of AR and prostate specific antigen (PSA). Interestingly, all adenocarcinomas of the prostate contain some neuroendocrine cells (1, 2). Unlike the bulk tumor cells, the scattered NE tumor cells are usually quiescent. In contrast, an important histologic variant prostate cancer called small cell neuroendocrine carcinoma (SCNC) is composed of NE tumor cells that are highly proliferative and aggressive. Although SCNC is occasionally diagnosed in patients without any previous history of prostate cancer, it more commonly occurs as a recurrent tumor in patients with a history of adenocarcinoma who have received hormonal therapy. It has been suggested that the novel drugs abiraterone and enzalutamide (formerly known as MDV3100) that further inhibit AR signaling will induce even more cases of SCNC.
We recently demonstrated that IL8/CXCR2/p53 signaling pathway keeps the NE cells in adenocarcinoma in a quiescent state, and mutant p53 inactivates this pathway, leading to hyper-proliferation of NE cells and the development of SCNC (3). Meanwhile, previous study also found that Aurora kinase A was overexpressed in the majority of cases of SCNC, indicating a potential role of Aurora Kinase A in the development of SCNC (4).
In this study, we provide evidence showing that p53 mutation leads to elevated expression of Aurora kinase A through regulation of miR-25 and Fbxw7, thus revealing a potential molecular mechanism of p53 mutation in promoting the rapid proliferation and aggressive behavior of NE tumor cells in prostatic SCNC.
Material and Methods
Cell lines
Human prostate LNCaP Clone FGC, PC-3 and NCI-H660 cells were from American Type Culture Collection (ATCC, Manassas, VA) and were authenticated utilizing short tandem repeat (STR) profiling. LNCaP Clone FGC cells were cultured in ATCC-formulated RPMI-1640 Medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin and 100 μg/ml streptomycin in a humidified atmosphere of 5% CO2 maintained at 37°C. PC-3 cells were cultured in ATCC-formulated F-12K Medium with 10% fetal bovine serum. NCI-H660 cells were cultured in RPMI-1640 medium with 0.005 mg/ml Insulin, 0.01 mg/ml Transferrin, 30nM Sodium selenite, 10 nM Hydrocortisone, 10 nM beta-estradiol, 4mM L-glutamine, and 5% fetal bovine serum (HITES medium). NE1.8 cells were provided by Dr. Ming-Fong Lin (5), and were cultured in phenol red-free RPMI 1640 supplemented with 10% charcoal-stripped FBS.
Nucleic Acids
Small interference RNA for p53 was purchased from IDT (Coralville, Iowa) as predesigned siRNA: sense strand 5′rCrCrArCrCrArUrCrCrArCrUrArCrArArCrUrArCrArUrGT3′, antisense strand 5′rCrArCrArUrGrUrArGrUrUrGrUrArGrUrGrGrArUrGrGrUrGrGrUrA3′. Small interference RNA for Fbxw7: sense strand 5′-rGrGrArGrUrGrGrArCrCrArGrArGrArArArUrUrGrCrUrUG C-3′, antisense strand 5′-rGrCrArArGrCrArArUrUrUrCrUrCrUrGrGrUrCrCrArCrUrCrCrArG-3′. cDNA of firefly luciferase was cloned into pCI-Neo vector followed by 3′UTR of Fbxw7, which was joined by two separate PCR fragments (left fragment: 5′CTAGTCTAGAAGAGCAGAAAAGATGAATTT3′ and 5′TTAGAGGCACAGATGGCTCA3′, right fragment: 5′TTGTCCCAACCCTGTACTGTA3′ and 5′CATGAAAAAACACATTTTATTGCACTTAAGTTATAAG3′) after restriction digestion with EcoRI followed by ligation. Mutant 3′UTR of Fbxw7 was constructed by QuickChange method to change the sequence of consensus miR-25 seed sequence of 5′UGCAAU3′ at two locations into a mutant sequence of 5′GGAUCC3′. Plasmid DNA encoding wild-type p53 (pCDNA3.1-p53wt) was described previously (6). Plasmid DNA encoding R175H mutation of p53 (DNp53) was generated by mutagenesis PCR. These constructs were verified through restriction digestion and sequencing analysis.
Lentivirus
p53 (R175H) was subcloned into the EcoRI site of FUCRW lentiviral vectors (7). This construct was verified through restriction digestion and sequencing analysis. The lentivirus was prepared and titered as described (8). LNCaP cells were spin infected at 1,800 rpm for 45 minutes at room temperature. All procedures were performed under University of California, Los Angeles, safety regulations for lentivirus usage.
Antibodies
Anti-Aurora A kinase antibody was from Cell Signaling (Beverly, MA), anti-Fbxw7 antibody was from Bethyl Laboratories (Montgomery, TX), anti-p53 antibody and anti-c-Myc was from Santa Cruz Biotechnology (Santa Cruz, CA), anti-MYCN antibody was from Abgent (San Diego, CA), anti-GAPDH antibody was from GeneTex, Inc (Irvine, CA).
Immunoblot Assay
Cells were washed with PBS and lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS) containing SigmaFAST™ Protease Inhibitor Cocktail (Sigma-Aldrich, St. Louis, MO) for 15 min at 4°C. Cell lysates were centrifuged and supernatants were collected. Equivalent amounts of proteins as measured by Bradford assay were resolved on SDS-PAGE gels and transferred to PVDF membranes. The resulting blots were blocked in 5% nonfat dry milk in PBS for 30 minutes followed by incubation with primary antibody in 5% BSA overnight. Appropriate HRP-conjugated secondary antibodies and Supersignal West Femto chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA) were used to visualize antigen-antibody complexes.
siRNA transfection
Transfections were performed with siRNA control, p53 or fbxw7 siRNA (IDT, Coralville, Iowa) using the Xfect™ siRNA Transfection Reagent (Clontech, Mountain View, CA), according to the manufacturer’s protocol.
Quantitative RT-PCR
Total RNA or miRNA was extracted from cells using the RNeasy Mini Kit (Qiagen, Valencia, CA) per the manufacturer’s instructions. Conversion to cDNA was achieved through the PrimeScript™ RT Master Mix (Takara, Mountain View, CA). Quantitative RT-PCR was carried out using SYBR Premix Ex Taq II (Takara, Mountain View, CA), 0.4 μmol/L oligonucleotide primers, and 0.1 μg cDNA. All primer sets for quantitative RT-PCR were illustrated in Supplementary Table S1. miRNA quantification was performed using miRCURY LNA™ Universal RT microRNA PCR Starter Kit (Exiqon, Woburn, MA). Relative fold change in mRNA levels were calculated after normalization to β-actin using the comparative Ct method (9).
Immunohistochemistry
For immunohistochemical analysis of p53 and Aurora A kinase, tissue sections were deparaffinized with xylene and rehydrated through graded ethanol. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol for 10 min. Heat-induced antigen retrieval (HIER) was carried out for all sections in 0.01 M citrate buffer, pH 6.0 using a vegetable steamer at 95°C for 25min. Mouse monoclonal anti-p53 antibody, clone 1801 (EMD, OP09-100UG) was diluted with BSA to a concentration of 1:50 and applied to the sections. Incubation was for 45 minutes at room temperature followed by anti-mouse secondary antibody (MACH 2 Mouse HRP-Polymer, Biocare Medical, MHRP520L) incubation for 30 minutes at room temperature. Rabbit monoclonal Aurora kinase A antibody (Abcam, 1800-1) was diluted with BSA to a concentration of 1:50 and applied to the sections. Incubation was for 1 hour at room temperature followed by anti-rabbit secondary antibody (Dakocytomation Envision System Labelled Polymer HRP anti rabbit, Cat.# 4003) incubation for 30 minutes at room temperature. Diaminobenzidine was then applied for 10 minutes at room temperature to visualize p53 and Aurora Kinase A. Sections were counterstained with hematoxylin, dehydrated through graded alcohols, and coverslipped. Immunohistochemical semi-quantitation were performed using Quick score (Q) method (10). Results are scored by multiplying the percentage of positive cells (P) by the intensity (I) (0 = no staining, 1 = weak staining, 2 = moderate staining, 3 = strong staining). Formula: Q = P x I; Maximum = 300.
Immunofluorescence Double Staining
Slides were deparaffinized with xylene and rehydrated through graded ethanol. Heat-induced antigen retrieval (HIER) was carried out in 0.01 M citrate buffer, pH 6.00, using a vegetable steamer at 95°C for 25 min. Sections were permeabilized for 10 minutes with 0.25% Triton X-100 and rinsed with PBS. Blocking was done with 2% BSA for 30 minutes at room temperature. Primary antibody mixtures (Aurora Kinase A 1:100 BSA + p53 1:25 BSA) were applied for 1 hour at room temperature. Slides were rinsed with PBS and the secondary antibody mixture (goat anti-Mouse-Alexa Fluor® 488 + goat anti-rabbit-Alexa Fluor® 568, both 1:500 BSA) was applied for 1 hour at room temperature. Slides were rinsed with PBS and coverslipped using VECTASHIELD® HardSet Mounting Medium with DAPI (Vector, H-1500).
Statistical analysis
Statistical analysis were performed using the Student’s t-test with the Excel 2013 software. Error bars indicate standard deviation calculated from three independent experiments.
Results
P53 mutation leads to increased expression of miRNA-25 in prostate cancer cells
We previously demonstrated that the quiescent NE cells in prostatic adenocarcinoma contain wild-type p53, while the rapidly proliferating NE tumor cells of SCNC often contain mutated p53 (3). We proposed that p53 mutation may play a critical role in the development of aggressive behavior of prostatic SCNC, but the detailed mechanisms were unclear. P53 can regulate miRNA expression in cancer cells (11). In glioblastoma cells, for example, p53 has been reported to repress the expression of miR-25 and -32 (12). Thus, it is quite interesting whether there is also a relationship between p53 expression/function and the expression of miRNAs, such as miR-25 and/or -32, and the interaction then contribute to the biological behavior of prostatic SCNC. We therefore tested this hypothesis in prostate cancer PC-3 and LNCaP cells. Our previous study has shown that LNCaP cells are typical prostate adenocarcinoma cells, and PC-3 cells are characteristic of SCNC (13). In addition, LNCaP cells express wild-type p53 protein and PC-3 cells contain truncated p53 mutation which leads to absence of p53 protein expression.
We expressed either wild-type p53 protein in PC-3 cells, or mutant p53 that is defective in DNA binding (R175H) in LNCaP cells. We found that expression of wild-type p53 protein repressed miR-25 expression in PC-3 cells, and expression of R175H mutant p53 protein increased miR-25 level in LNCaP cells, both with statistical significances (Figure 1A). However, we noticed that expression of wild-type p53 protein in PC-3 cells did not cause obvious changes in the expression of miR-32 (data not shown). In prostate neuroendocrine (NE) cancer cell line NCI-H660 that contains wild-type p53, we also observed that knockdown of p53 by siRNA or expression of the dominant negative p53 mutant both resulted in enhanced miR-25 expression (Figure 1B).
Figure 1.

p53 regulates miR-25, Fbxw7 and Aurora kinase A. A and B miR-25 expression changes in response to p53 expression and mutation status. Plasmids encoding wild-type p53 (left panel) or mutant p53 (right panel) were transfected into PC-3 or LNCaP cells for 48hrs. RNAs were then isolated for quantification of miR-25 using qRT-PCR. Two independent triplicate experiments were performed, and results are presented as mean ± SD. The ratio of the miR-25 over miR-191 as an internal control was plotted (A). siRNA for p53, or lentiviral vector for dominant negative p53 were introduced into NCI H660 cells for 48hrs, and miR-25 was quantitated as in A (B). Asterisks indicate significant differences (p<0.05). C. Immunoblot analysis results showing the changes of proteins in response to p53 regulation for 48hrs in NCI H660 cells.
We further examined whether the changes of miR-25 level in these cells were associated with potential regulation of cell cycles induced by changes of p53 status. We found that expression of Dominant negative p53 (DNp53) did not cause obvious changes of cell cycle distribution in LNCaP cells, although miR-25 level changes observed; In PC-3 cells, however, expression of wild-type p53 (WTp53) did induce G2/M transition 24 hours later after transfection of mammalian expressive WTp53 construct. Interestingly, we observed statistically significant reduction of miR-25 levels in cells at this time point (Supplementary Figure S1).
Taken together, these results suggest that p53 can regulate miR-25 expression in prostate cancer cells, whereas mutant p53 or loss of p53 functions cause elevated expression of miR-25 expression.
P53 mutation leads to decreased expression of Fbxw7 and overexpression of Aurora kinase A
miR-25 has many potential targets including Fbxw7 and Wwp2 (14). Indeed, we observed that overexpression of miR-25 could lead to reduced levels of both Fbxw7 and Wwp2 in PC-3 cells, as well as in NE1.8 cells, a variant of LNCaP cells that resemble neuroendocrine cells (5) (Supplementary Figure S2). Fbxw7 encodes an E3 ubiquitin ligase whose substrates include several positive cell cycle regulators such as MYCN (15), MYC (16, 17), Cyclin E (18, 19) and Aurora kinase A (20, 21). Of them, Aurora Kinase A is overexpressed in prostate small cell carcinoma and may play important roles in the development of aggressive prostate tumor (4). We thus further tested whether p53 mutation or loss of function could cause changes of Aurora Kinase A expression via regulation of miR-25 and Fbxw7. Indeed, as shown in Figure 1C, we found that knocking down p53 with siRNA in NCI-H660 cells resulted in increase of Aurora kinase A expression. And expression of DNp53 in NCI-H660 led to a change of Aurora kinase A expression similar to p53 knockdown. In addition, we observed reduced expression of Fbxw7 in NCI-H660 cells with these manipulations of p53 expression.
Next, we determined if change of Fbxw7 expression would affect the protein level of Aurora kinase A. In NCI-H660 and PC-3 cells, we found transfections of Fbxw7 siRNA decreased the expression of Fbxw7 protein, and increased levels of Aurora kinase A. Similar results were also observed in NE1.8 cells (Figure 2). In these cells, silencing of Fbxw7 also resulted in elevated protein levels of its targets C-MYC and MYCN. Thus, these results suggested that Fbxw7 may also function as the ubiquitin E3 ligase targeting Aurora kinase A.
Figure 2.
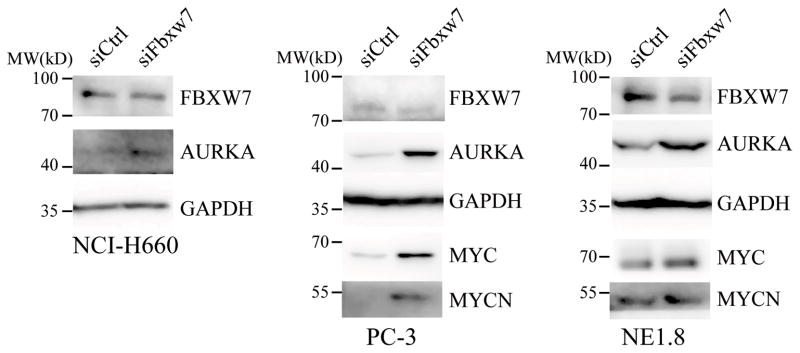
Fbxw7 regulates Aurora kinase A expression. Control or Fbxw7 siRNA at 100 nM was transfected into NE1.8, PC-3 or NCI-H660 cells. Aurora kinase A and Fbxw7 were examined via immunoblot analysis. MYCN and MYC are included as known validated targets of Fbxw7.
miR-25 mediates mutant p53-induced expression of Aurora A kinase
We next determined the potential role of miR-25 in the regulations of Aurora kinase A upon loss of p53 function in these prostate cancer cells. For this, we co-transfected p53 siRNA with miR-25 inhibitor into LNCaP cells. Figure 3A and B showed that, miR-25 inhibitor prevented p53 knockdown-induced downregulation of Fbxw7 and upregulation of Aurora kinase A in these LNCaP cells, indicating that loss of function of p53 may regulate Aurora kinase A expression through a linear pathway involving miR-25 and Fbxw7. To further clarify the roles of miR-25 in this process, we transfected miR-25 into NE1.8 cells. As expected, our results showed that overexpression of miR-25 reduced Fbxw7 level and increased Aurora kinase A protein expression (Figure 3C).
Figure 3.

miR-25-regulated Aurora kinase A is dependent on loss of p53 function. A. Westernblot analysis showing that miR-25 inhibitor eliminate the effect of p53 knockdown on regulation of FBXW7 and AURKA. B. Quantification of the densitometry for western blot bands using ImageJ 1.47t. C. The effects of miR-25 on expressions of AUKUA and FBXW7 proteins. Control or miR-25 mimic at 100 nM was transfected into NE1.8 cells, a variant of LNCaP cells that resemble neuroendocrine cells. Aurora kinase A and Fbxw7 were examined via immunoblot analysis. D. A graphic representation for expression plasmids containing firefly luciferase with wild-type 3′ UTR of human Fbxw7 and the mutant 3′UTR without the target sequence for miR-25 (top). These plasmids were transfected into NE1.8 cells together with control or miR-25 mimic, luciferase activities were measured 48hrs later and plotted (bottom). Three independent experiments were performed, and results are presented as mean ± SD.
To determine whether miR-25 can regulate Fbxw7 expression through its likely target in its 3′UTR region, we generated a reporter construct where coding sequence for firefly luciferase was following linked by a 3′UTR sequence of Fbxw7, or by a mutant 3′UTR sequence of Fbxw7 where the target sequences of miR-25 were destroyed. As shown in Figure 3D, exogenous expression of miR-25 caused ~50% reduction of the luciferase activity with the wild-type 3′UTR of Fbxw7, while no obvious changes of luciferase activity was observed with the mutant 3′UTR in response to the miRNA expression. Thus, our results indicated that miR-25 could directly regulate Fbxw7 expression through the miRNA binding site in its 3′UTR.
We also tested the potential roles of miR-25 in the biological behaviors of SCNC PC-3 cells. Our results showed that, Inhibition of miR-25 in PC-3 cells attenuates cell proliferation and reduced invasion capability (Supplementary Figure S3).
Taken together, our results suggested a likely signaling pathway through which p53 mutation induces upregulation of miR-25 and downregulation of Fbxw7, eventually leading to the overexpression of Aurora kinase A which may promote rapid proliferation of the NE tumor cells of SCNC.
Co-Expression of nuclear p53 and Aurora kinase A in human SCNC tissue
We further verified the co-expression of p53 and Aurora Kinase A in 12 cases of human prostate small cell carcinoma and an equal number of cases of prostate adenocarcinoma. Our immunohistochemistry study showed that, eight of the 12 small cell carcinoma cases had positive nuclear p53 staining, which usually results from mutation of p53 with increased p53 protein stability; Nine of the 12 cases also showed overexpression of Aurora kinase A; Of them, 6 cases were positive for both nuclear p53 and Aurora kinase A overexpression (Figure 4A). We also noticed that all 12 cases of adenocarcinoma were negative for both p53 nuclear staining and Aurora kinase A overexpression. Furthermore, we found that the co-expression of nuclear p53 and Aurora kinase A in these SCNC tissues (Figure 4B). These results supported our hypothesis that p53 mutation might lead to the overexpression of Aurora Kinase A in prostatic SCNC.
Figure 4.

Expressions of p53, Aurora kinase A and miR-25 in human prostatic small cell carcinoma. A. Representative images showing the IHC staining with anti-p53 and anti-Aurora kinase A antibodies in human prostatic small cell carcinoma B. Representative images showing the co-expression of nuclear p53 and Aurora kinase A in these human prostatic small cell carcinoma using immunofluorescence double staining. C. miR-25 was quantitated in small cell carcinoma or adenocarcinoma into Box-and-Whisker Plots, the ratio of the miR-25 over miR-191 as an internal control was plotted. D. Differential expression of Aurora kinase A in human prostatic adenocarcinoma or small cell carcinoma. Representative image showing the results for Immunohistochemistry staining with antibody for Aurora kinase A in these different types of prostate cancers. E. Semi-quantitation of AURKA IHC staining were performed using Quick score method.
We have shown previously that p53 mutation is common in prostatic SCNC and rare in untreated adenocarcinoma (3). Similarly, Rubin’s group (22) showed that Aurora kinase A is overexpressed in prostatic SCNC but not in adenocarcinoma. Therefore, we performed analysis for miR-25 expression between the two types of tumors. In this study, total RNA was isolated from the paraffin section of the cancerous area from 12 cases of SCNC and an equal number of adenocarcinoma cases. The levels of miR-25 were measured and normalized to miR-191 as the internal control, and the ratios of these two miRNAs were then plotted. The Box-and-Whisker Plots showed that almost half of the cases of small cell carcinomas have higher levels of miR-25 expression when compare to prostate adenocarcinoma (Figure 4C). Examples of expression of AURKA as assessed using immunohistochemistry in prostatic adenocarcinoma and small cell neuroendocrine carcinoma tissues (Figure D and E), demonstrating high expression of AURKA in prostatic SCNC. Data from the human prostate cancer tissues also support the notion that p53 mutation in small cell carcinoma may leads to a higher miR-25 expression that contributes to the higher expression of Aurora kinase A.
Discussion
Prostatic small cell carcinoma is an under-diagnosed entity because patients with widely metastatic disease after hormonal therapy usually do not undergo biopsy or resection for histologic diagnosis. Its incidence is expected to rise after the recent approval of super-blockers of AR signaling pathway such as abiraterone and enzalutamide. In addition to a lack of effective therapy, the molecular mechanisms driving the development of SCNC remain unclear.
We recently showed that p53 mutation may be a critical molecular responsible for the aggressive behavior of SCNC (23), and the study from Rubin’s group reported gene amplification and overexpression of MYCN and Aurora kinase A in these tumors (4). Aurora kinase A is an evolutionarily conserved serine/threonine kinase critical for mitotic regulation (23). It can phosphorylate multiple mitosis-associated proteins (e.g., Tacc and Ndel1) thus modulating their activities (24, 25) and orchestrating centrosome maturation, spindle assembly, and mitotic entry. Aurora kinase A can also regulate protein translation through CPEB phosphorylation (26, 27). Thus, Aurora kinase A plays important roles in cell proliferation, and has been considered a potential therapeutic target for prostatic SCNC. In addition, it has been also reported that wild-type p53 suppresses the expression of miR-25 (12), and mutant p53 or loss of p53 function resulted in increased miR-25 expression. The relationship between mutant p53 and overexpression of Aurora Kinase A in SCNC is thus quite interesting. In present study, we show that expression of mutant p53 protein lead to enhanced expression of Aurora kinase A in prostate cancer cells, and this was most likely mediated by increased miR-25 expression and decreased expression of Fbxw7 subsequent to p53 mutation.
miR-25 is a well-studied oncogenic miRNA. It is 22 nucleotides long, localized in the minichromosome maintenance protein-7 (MCM7) gene, and transcribed as part of the miR-106b~25 polycistron. It is overexpressed in several human cancers, including pediatric brain tumors (28), gastric adenocarcinoma (29), epidermal growth factor receptor-positive lung adenocarcinoma (30) and prostate carcinoma (31), and has been reported to target different regulators of the apoptotic pathway, such as BIM (32), PTEN (31) and TRAIL (33). miR-25 also affects Ca2+ homeostasis by regulating mitochondria calcium efflux through targeting the mitochondria calcium uniporter (34), causing a strong decrease in mitochondrial Ca2+ uptake and, likely, conferring resistance to Ca2+-dependent apoptotic stimuli. We found that p53 mutation-induced miR-25 overexpression down-regulates the expression of ubiquitin E3 ligase Fbxw7. Fbxw7 is a potent ubiquitin E3 ligase that can degrade Aurora Kinase A (35), lower levels of Fbxw7 thus leads to increased protein level of Aurora kinase A. In PC-3 and NE1.8 cells, we noticed that transfection with miR-25 reduced mRNA expressions of Aurora kinase A, but significantly increased protein levels of Aurora kinase A which can be affected by exposure to cycloheximide, a protein synthesis inhibitor (Supplementary Figure S4), suggesting that the elevated protein level of aurora kinase A in these cells was caused by the Fbxw7-induced blockage of aurora kinase A degradation. Inactivation of Fbxw7 has also been noticed to be critical for the proliferation of leukemic stem cells, and contributes to the development of leukemia (36, 37). Thus, our results suggest a potential signaling pathway that how mutant p53 regulate level of Aurora Kinase A in prostate cancer cells that may be correlated to rapid proliferation and aggressive behavior of prostatic SCNC.
Although our data suggests the presence of a linear pathway of p53 -> miR-25 -> Fbxw7 -> Aurora kinase A in SCNC, there are likely other important players in the pathogenesis of prostatic SCNC. In a significant number of SCNC cases, Aurora kinase A overexpression is associated with gene amplification, which involves different genetic events. Of note, Rubin’s group has shown that MYCN is also amplified and overexpressed in prostatic SCNC (4). Since expression of Fbxw7 can also cause degradation of MYC, it would be interesting to study if p53 mutation also leads to MYC overexpression. In addition, Collins’ group reported that downregulation of the REST transcription complex may lead to the development of SCNC. These diverse findings suggest that the pathogenesis of SCNC is a complex process that may involve different players and multiple signaling pathways.
Supplementary Material
Figure 5.

A diagram depicting multiple pathways that leads to overexpression of Aurora kinase A in human prostatic small cell carcinoma.
Implications.
The pathogenesis of prostatic SCNC involves a p53 and Aurora Kinase A signaling mechanism, both potentially targetable pathways.
Acknowledgments
We thank Dr. Ming-Fong Lin for sharing NE1.8 cells. Research supported by a Stand Up to Cancer - Prostate Cancer Foundation - Prostate Dream Team Translational Cancer Research Grant. This research grant is made possible by the generous support of the Movember Foundation. Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Grant Support
J.H. is supported by UCLA SPORE in Prostate Cancer (PI: Reiter), Department of Defense Prostate Cancer Research Program W81XWH-11-1-0227 (PI: Huang), W81XWH-12-1-0206 (PI: L. Wu), NIH 1R01CA158627-01 (PI: Marks), 1R01CA172603-01A1 (PI: Jiaoti Huang), Prostate Cancer Foundation Honorable A. David Mazzone Special Challenge Award (PI: Robert Reiter), and Stand–up-to-Cancer Dream Team Award (PI: Small and Witte).
Footnotes
Potential conflicts of interest: The authors disclose no potential conflicts of interest.
Authors’ Contributions
Conception and design: Y. Sun, J. Huang
Development of methodology: Y. Sun, Z. Li, X. Chen, J. Squires
Acquisition of data: Z. Li, Y. Sun, X. Chen, J. Squires
Analysis and interpretation of data: Z. Li, Y. Sun, X. Chen, J. Squires, B. Nowroozizadeh
Writing, review, and/or revision of the manuscript: Z. Li, Y. Sun, X. Chen, J. Huang, C. Liang
Administrative, technical, or material support: Y. Sun, Z. Li, X. Chen
Study supervision: Y. Sun, X. Chen, J. Huang
References
- 1.Abrahamsson PA, Falkmer S, Falt K, Grimelius L. The course of neuroendocrine differentiation in prostatic carcinomas. An immunohistochemical study testing chromogranin A as an “endocrine marker”. Pathology, research and practice. 1989;185:373–80. doi: 10.1016/S0344-0338(89)80016-0. [DOI] [PubMed] [Google Scholar]
- 2.Abrahamsson PA, Wadstrom LB, Alumets J, Falkmer S, Grimelius L. Peptide-hormone- and serotonin-immunoreactive tumour cells in carcinoma of the prostate. Pathology, research and practice. 1987;182:298–307. doi: 10.1016/S0344-0338(87)80065-1. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Sun Y, Wu C, Magyar CE, Li X, Cheng L, et al. Pathogenesis of prostatic small cell carcinoma involves the inactivation of the P53 pathway. Endocrine-related cancer. 2012;19:321–31. doi: 10.1530/ERC-11-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer discovery. 2011;1:487–95. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang XQ, Kondrikov D, Yuan TC, Lin FF, Hansen J, Lin MF. Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cancer cells. Oncogene. 2003;22:6704–16. doi: 10.1038/sj.onc.1206764. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Wong JY, Wong P, Radany EH. Low-dose valproic acid enhances radiosensitivity of prostate cancer through acetylated p53-dependent modulation of mitochondrial membrane potential and apoptosis. Molecular cancer research: MCR. 2011;9:448–61. doi: 10.1158/1541-7786.MCR-10-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7789–94. doi: 10.1073/pnas.0602567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nature protocols. 2006;1:241–5. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 9.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 10.Detre S, Saclani Jotti G, Dowsett M. A “quickscore” method for immunohistochemical semiquantitation: validation for oestrogen receptor in breast carcinomas. Journal of clinical pathology. 1995;48:876–8. doi: 10.1136/jcp.48.9.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–33. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 12.Suh SS, Yoo JY, Nuovo GJ, Jeon YJ, Kim S, Lee TJ, et al. MicroRNAs/TP53 feedback circuitry in glioblastoma multiforme. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5316–21. doi: 10.1073/pnas.1202465109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tai S, Sun Y, Squires JM, Zhang H, Oh WK, Liang CZ, et al. PC3 is a cell line characteristic of prostatic small cell carcinoma. The Prostate. 2011;71:1668–79. doi: 10.1002/pros.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu D, Davis MP, Abreu-Goodger C, Wang W, Campos LS, Siede J, et al. MiR-25 regulates Wwp2 and Fbxw7 and promotes reprogramming of mouse fibroblast cells to iPSCs. PloS one. 2012;7:e40938. doi: 10.1371/journal.pone.0040938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. The EMBO journal. 2004;23:2116–25. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–7. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 19.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413:316–22. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, et al. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS letters. 2012;586:1409–18. doi: 10.1016/j.febslet.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–9. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- 22.Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia. 2013;15:1–10. doi: 10.1593/neo.121550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. Journal of cell science. 2007;120:2987–96. doi: 10.1242/jcs.013136. [DOI] [PubMed] [Google Scholar]
- 24.Barros TP, Kinoshita K, Hyman AA, Raff JW. Aurora A activates D-TACC-Msps complexes exclusively at centrosomes to stabilize centrosomal microtubules. The Journal of cell biology. 2005;170:1039–46. doi: 10.1083/jcb.200504097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori D, Yano Y, Toyo-oka K, Yoshida N, Yamada M, Muramatsu M, et al. NDEL1 phosphorylation by Aurora-A kinase is essential for centrosomal maturation, separation, and TACC3 recruitment. Molecular and cellular biology. 2007;27:352–67. doi: 10.1128/MCB.00878-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD. Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature. 2000;404:302–7. doi: 10.1038/35005126. [DOI] [PubMed] [Google Scholar]
- 27.Mendez R, Murthy KG, Ryan K, Manley JL, Richter JD. Phosphorylation of CPEB by Eg2 mediates the recruitment of CPSF into an active cytoplasmic polyadenylation complex. Molecular cell. 2000;6:1253–9. doi: 10.1016/s1097-2765(00)00121-0. [DOI] [PubMed] [Google Scholar]
- 28.Birks DK, Barton VN, Donson AM, Handler MH, Vibhakar R, Foreman NK. Survey of MicroRNA expression in pediatric brain tumors. Pediatric blood & cancer. 2011;56:211–6. doi: 10.1002/pbc.22723. [DOI] [PubMed] [Google Scholar]
- 29.Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer cell. 2008;13:272–86. doi: 10.1016/j.ccr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Dacic S, Kelly L, Shuai Y, Nikiforova MN. miRNA expression profiling of lung adenocarcinomas: correlation with mutational status. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2010;23:1577–82. doi: 10.1038/modpathol.2010.152. [DOI] [PubMed] [Google Scholar]
- 31.Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Science signaling. 2010;3:ra29. doi: 10.1126/scisignal.2000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, Zuo Z, Lu X, Wang L, Wang H, Zhu Z. MiR-25 regulates apoptosis by targeting Bim in human ovarian cancer. Oncology reports. 2012;27:594–8. doi: 10.3892/or.2011.1530. [DOI] [PubMed] [Google Scholar]
- 33.Razumilava N, Bronk SF, Smoot RL, Fingas CD, Werneburg NW, Roberts LR, et al. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology. 2012;55:465–75. doi: 10.1002/hep.24698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Current biology: CB. 2013;23:58–63. doi: 10.1016/j.cub.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujii Y, Yada M, Nishiyama M, Kamura T, Takahashi H, Tsunematsu R, et al. Fbxw7 contributes to tumor suppression by targeting multiple proteins for ubiquitin-dependent degradation. Cancer science. 2006;97:729–36. doi: 10.1111/j.1349-7006.2006.00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153:1552–66. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer cell. 2013;23:347–61. doi: 10.1016/j.ccr.2013.01.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.