

Abstract
Equilibrative transporters are potential drug targets, however most functional assays involve radioactive substrate uptake that is unsuitable for high-throughput screens (HTS). We developed a robust yeast-based growth assay that is potentially applicable to many equilibrative transporters. As proof of principle, we applied our approach to Equilibrative Nucleoside Transporter 1 of the malarial parasite Plasmodium falciparum (PfENT1). PfENT1 inhibitors might serve as novel antimalarial drugs since PfENT1-mediated purine import is essential for parasite proliferation. To identify PfENT1 inhibitors, we screened 64,560 compounds and identified 171 by their ability to rescue the growth of PfENT1-expressing fui1Δ yeast in the presence of a cytotoxic PfENT1 substrate, 5-fluorouridine (5-FUrd). In secondary assays, nine of the highest activity compounds inhibited PfENT1-dependent growth of a purine auxotrophic yeast strain with adenosine as the sole purine source (IC50 0.2–2 µM). These nine compounds completely blocked [3H]adenosine uptake into PfENT1-expressing yeast and erythrocyte-free trophozoite-stage parasites (IC50 5–50 nM), and inhibited chloroquine-sensitive and -resistant parasite proliferation (IC50 5–50 µM). Wild-type (WT) parasite IC50 values were up to four-fold lower compared to PfENT1-knockout (pfent1Δ) parasites. pfent1Δ parasite killing showed a delayed-death phenotype not observed with WT. We infer that in parasites, the compounds inhibit both PfENT1 and a secondary target with similar efficacy. The secondary target identity is unknown, but its existence may reduce the likelihood of parasites developing resistance to PfENT1 inhibitors. Our data support the hypothesis that blocking purine transport through PfENT1 may be a novel and compelling approach for antimalarial drug development.
Keywords: malaria, purine transport, drug development
Several widely used drugs target membrane transport proteins including thiazide and loop diuretics, proton pump inhibitors, selective serotonin reuptake inhibitors and sodium-glucose cotransporter inhibitors. Many other membrane transport proteins represent potential drug targets but most functional assays utilize uptake of radiolabeled substrates.1 Particularly for equilibrative transporters, these assays are not suitable for HTS. This has limited the ability to develop drugs to target these transporters.
Malaria is a major public health illness. P. falciparum causes the most severe form of malaria. The emergence of resistance to antimalarial drugs has been a major problem.2 Reports of increasing artemisinin resistance in Southeast Asia suggest that artemisinin resistance will become widespread over time.3–6 Thus, it is critical to develop novel antimalarial drugs.7–9
Plasmodium parasites are purine auxotrophs, so purine import is essential in all life cycle stages. Purine salvage pathway enzymes modify imported purines to form nucleotides required for DNA and RNA synthesis and other cellular metabolic processes.10, 11 Purine import, predominantly hypoxanthine and adenosine, is mediated by equilibrative nucleoside transporters (ENTs).10, 12 The P. falciparum genome encodes four ENT homologues (PfENT1–4).13–15 The function of PfENT2 and PfENT3 are unknown, although PfENT2 is localized in the parasite’s endoplasmic reticulum.16 PfENT4 is a low-affinity purine transporter, but not a major importer for purine salvage because it does not transport hypoxanthine.17
PfENT1 is the major transporter supplying the purine salvage pathway. It is present in the parasite’s plasma membrane.18 It is a low-affinity, high-capacity transporter for a variety of purine and pyrimidine nucleosides and nucleobases.19–23 pfent1Δ parasites are not viable at purine concentrations found in human blood (< 10 µM).24–26 pfent1Δ parasite viability can be rescued using media supplemented with purines at levels 10–100 times higher than their physiological concentration.25 This emphasizes the importance of PfENT1 for purine acquisition and parasite proliferation.
Inhibitors of human ENT1 (hENT1) with nanomolar affinities exist (e.g., dipyridamole and nitrobenzylthioinosine). These molecules do not block PfENT1, which is only about 17% identical to hENT1.20, 21, 23 Thus, ENTs are druggable targets. Based on the genetic, physiological, and biochemical data, we and others hypothesized that PfENT1 may be a viable target for antimalarial drug development, and that high affinity inhibitors with specificity for PfENT1 may be identified.10, 15, 23, 25
Because functional assays for facilitative transporters typically involve the measurement of radioactive substrate fluxes, they are poorly suited for high-throughput screening efforts. We developed a simple, yet robust, yeast-based, high-throughput screen (HTS) to identify PfENT1 inhibitors. The assay relies on the sensitivity of Saccharomyces cerevisiae to the cytotoxic nucleoside analog 5-FUrd.27, 28 Yeast strains deleted for the high-affinity uridine transporter FUI1 (fui1Δ) are viable and resistant to 5-FUrd. PfENT1 expression in fui1Δ strains restores 5-FUrd sensitivity and provides a powerful positive selection for PfENT1 inhibitors: In the presence of 5-FUrd, PfENT1-expressing fui1Δ yeast will only grow in the presence of a PfENT1 inhibitor. We screened 64,560 compounds for inhibitors of PfENT1 function. We identified a diverse group of small-molecule chemotypes that block PfENT1 activity. Importantly, these compounds inhibit P. falciparum proliferation in vitro. The data provides chemical validation that PfENT1 is a viable target for the development of novel antimalarial drugs.
RESULTS AND DISCUSSION
PfENT1-expressing fui1Δ yeast are susceptible to 5-FUrd
Our HTS required functional expression of PfENT1 in S. cerevisiae. The native pfent1 DNA sequence (PlasmoDB ID: PF3D7_1347200) expresses functionally in Xenopus laevis oocytes, with or without a hemagglutinin (HA) epitope-tag (PfENT1-HA),21, 29 however, it did not express in S. cerevisiae. To express PfENT1 in S. cerevisiae, we generated a yeast codon-optimized PfENT1 gene (PfENT1-HA-CO) (Supporting Information (SI) Figure S1). The PfENT1-HA-CO sequence was integrated into the S. cerevisiae genome replacing FUI1 to generate the fui1Δ::PfENT1-HA-CO strain (SI Figure S2). This strain was transformed with a plasmid expressing a mitochondrion-targeted GFP.30 This allowed detection of yeast proliferation by both optical density (OD) and GFP fluorescence intensity. Using both OD and GFP fluorescence to identify hits allowed us to eliminate compounds where interference with one or the other assay readout (e.g., precipitating compounds that increased OD values or compounds with fluorescence properties similar to GFP) might cause false positives.
Using the codon-optimized construct, PfENT1 protein was detectable in transformed yeast cells by Western analysis (Figure 1A). We confirmed functional PfENT1 expression by substrate uptake and 5-FUrd susceptibility. S. cerevisiae lacks an adenosine transporter but adenosine is a PfENT1substrate.19, 20, 23 We measured the [3H]adenosine uptake time course. The fui1Δ::PfENT1-HA-CO strain accumulated radioactivity, while the background fui1Δ::kanMX4 strain did not (Figure 1B). Similarly, the fui1Δ::PfENT1-HA-CO strain was susceptible to 5-FUrd toxicity, while the fui1Δ::kanMX4 strain was resistant (Figure 1C). Yeast cell growth assessed by OD600 or by GFP fluorescence showed 5-FUrd IC50 values of 7 ± 2 µM and 9 ± 4 µM, respectively (Figure 1C). Background growth at 125 µM 5-FUrd, the concentration used in the HTS, was effectively zero.
Figure 1.

Development and characterization of a HTS assay for PfENT1 inhibitors. (A) Western blot probed with anti-HA antibodies shows expression of the codon-optimized PfENT1-HA protein in an fui1Δ S. cerevisiae strain (lane 1, empty pCM189m vector; lane 2, PfENT1-HA [native sequence]/pCM189m vector; lane 3, PfENT1-HA-CO [codon optimized]/pCM189m vector). (B) [3H]adenosine uptake time course into fui1Δ::PfENT1-HA-CO yeast (circles) compared to fui1Δ yeast (triangles). (C) 5-FUrd dose-dependent growth inhibition of PfENT1-HA-CO expressing fui1Δ yeast (circles) compared to a control fui1Δ::kanMX4 strain (triangles). Growth in microtiter plates determined by OD600 (open symbols, dashed lines) and by GFP fluorescence (filled symbols, solid lines). (D) Effect of inosine on growth in the presence (circles) or absence (squares) of 125 µM 5-FUrd in microtiter plates as determined by OD600 (open symbols, dashed lines) and by GFP fluorescence (filled symbols, solid lines). Single experiments with four technical replicates are shown with each point representing mean ± SD.
As a proof of concept that blockade of 5-FUrd uptake could restore growth, we competitively inhibited 5-FUrd uptake by adding inosine, a PfENT1 substrate, to the growth medium.19, 20 High inosine concentrations completely rescued fui1Δ::PfENT1-HA-CO strain growth in 125 µM 5-FUrd (Inosine EC50 = 5.6 ± 2.1 mM [fluorescence]; 3.3 ± 0.2 mM [OD600]) (Figure 1D). In the HTS, we used inosine at concentrations >10 mM as a positive control for the maximal growth achievable upon rescue of 5-FUrd-induced yeast cell death. The inosine competition experiments demonstrated that growth in 5-FUrd can serve as a positive readout for PfENT1 inhibitors.
High-Throughput Screen
We performed the HTS in 384 well plates. Assay robustness was characterized by Z’-scores > 0.831 (SI Figure S3), with the optimal signal obtained 19 h after inoculation of yeast in test media (SI Figure S3E). The compounds were dissolved in DMSO. The 1% DMSO concentration used in the HTS did not affect either yeast growth or PfENT1 function.
We screened 64,560 compounds (each at 10 µM) from several chemical libraries for their ability to rescue 5-FUrd-induced cell death and allow growth of the fui1Δ::PfENT1-HA-CO strain (SI Table S1). Using the criterion that yeast cell growth must be greater than four standard deviations above the no-growth control level, 171 compounds (0.26% hit rate) were identified as positive (SI Table S2).
We chose nine compounds for more extensive characterization in a series of secondary assays. These compounds are identified by their “Rank Number” in SI Table S2. The nine compounds included the top seven hits (SI Table S2), the first hit identified while conducting the screen (19) and the highest ranked quinazolinone (13) (representative of a class that included compounds 18, 34, 90, 141, 144). The nine compounds represent five distinct chemotypes and include four molecules (2, 6, 7, and 19) with a common chemical scaffold. For the nine compounds, chemically related molecules could be identified among the other 162 hits (SI Table S2). Thus, these nine compounds were not isolated hits but represented the best molecules for a given chemotype. The libraries included FDA and EU approved drugs and natural products; 16 were positive but the yeast growth levels were <7% of maximal growth (SI Table S2; compounds #74, 77, 81, 82, 84, 93, 95, 98, 102, 109, 120, 121, 127, 129, 135, 151). As expected, the hENT1 inhibitor dipyridamole was not positive. The screened libraries included the Medicines for Malaria Venture Toolbox;32 none of these molecules were positive in the HTS.
Validation of Compounds as PfENT1 Inhibitors
We purchased the nine compounds from Chembridge Corp. (San Diego, CA) and verified their purity and identity by LC/MS (SI Chemical Synthesis and Characterization). All displayed concentration-dependent rescue of yeast cell growth in the 5-FUrd assay with EC50 values in the low micromolar range (Figure 2A and Table 1). None of these compounds were toxic to yeast at concentrations in the hundreds of micromolar range.
Figure 2.

Validation of nine selected compounds identified in the HTS as PfENT1 inhibitors. (A) Concentration-dependent rescue by the compounds of PfENT1-HA-CO-expressing fui1Δ yeast from 125 µM 5-FUrd induced death (Table 1). (B) Concentration-dependent growth inhibition of PfENT1-CO expressing purine auxotrophic yeast in 1 mM adenosine media. (C) Concentration-dependent inhibition of [3H]adenosine uptake into PfENT1-CO expressing yeast over 15 min. Mean values of technical replicates from single experiments are shown.
Table 1.
Structures and IC50s in the yeast and parasite growth and [3H]adenosine uptake assays for the nine compounds selected from the HTS.
| HTS Rank # |
Structure | Compound Name ChemBridge # |
EC50 5-FUrd growth rescue of fui1Δ::Pf ENT1- HA-CO (µM) |
IC50 ade2Δ + PfENT1- CO adenosine growth inhibition (µM) |
IC50 [3H]- adenosine uptake into ade2Δ + PfENT1- CO yeast (nM) |
IC50 [3H]- adenosine uptake into 3D7 parasites (nM) |
IC50 3D7 parasite viability – low purine media (µM) |
IC50 3D7 parasite viability – high purine media (µM) |
IC50 Dd2 parasite viability – high purine media (µM) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 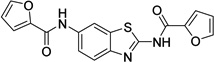 |
N,N'-1,3- benzothiazole-2,6- diyldi(2-furamide) 9001893 |
0.8 ± 0.0 |
0.2 ± 0.1 |
3.0 ± 1.3 |
5.4 ± 2.9 |
31.1 ± 4.7 |
37.7 ± 2.0 |
25.8 ± 1.3 |
| 2 | 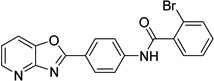 |
2-bromo-N-(4- [1,3]oxazolo[4,5- b]pyridin-2- ylphenyl)benzamide 6718896 |
6.0 ± 1.7 |
2.1 ± 0.9 |
10.1 ± 8.4 |
28.0 ± 12.3 |
31.2 ± 7.7 |
53.4 ± 11.1 |
52.4 ± 5.7 |
| 3 | 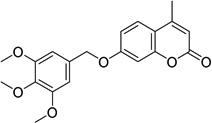 |
4-methyl-7-[(3,4,5- trimethoxybenzyl)oxy] -2H-chromen-2-one 6946484 |
2.3 ± 0.9 |
0.6 ± 0.1 |
2.4 ± 1.5 |
6.4 ± 3.0 |
19.2 ± 4.3 |
41.0 ± 4.3 |
36.2 ± 6.0 |
| 4 | 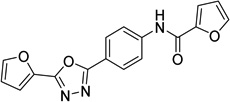 |
N-{4-[5-(2-furyl)-1,3,4- oxadiazol-2- yl]phenyl}-2-furamide 6081106 |
5.7 ± 0.8 |
1.7 ± 0.4 |
13.7 ± 7.5 |
45.8 ± 24.1 |
15.0 ± 1.5 |
14.7 ± 1.9 |
15.3 ± 2.7 |
| 5 | 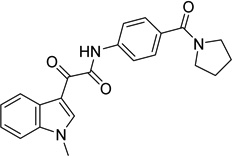 |
2-(1-methyl-1H-indol- 3-yl)-2-oxo-N-[4- (pyrrolidin-1- ylcarbonyl)phenyl]acet amide 9039333 |
6.5 ± 1.5 |
2.0 ± 0.01 |
22.7 ± 7.2 |
38.6 ± 20.1 |
35.5 ± 0.7 |
33.1 ± 0.5 |
28.9 ± 2.6 |
| 6 | 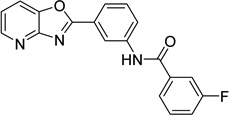 |
3-fluoro-N-(3- [1,3]oxazolo[4,5- b]pyridin-2- ylphenyl)benzamide 9011026 |
5.7 ± 1.3 |
1.1 ± 0.1 |
9.6 ± 7.4 |
19.8 ± 12.9 |
34.8 ± 2.4 |
33.7 ± 5.0 |
38.8 ± 4.9 |
| 7 | 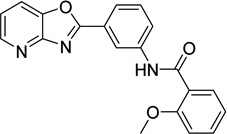 |
2-methoxy-N-(3- [1,3]oxazolo[4,5- b]pyridin-2- ylphenyl)benzamide 6736283 |
6.4 ± 0.9 |
1.6 ± 0.01 |
10.9 ± 6.8 |
24.8 ± 17.4 |
43.6 ± 4.4 |
44.8 ± 3.8 |
31.0 ± 0.6 |
| 13 |  |
2-[2-(2- methylphenyl)vinyl]- 4(3H)-quinazolinone 6517398 |
4.7 ± 2.9 |
0.6 ± 0.1 |
3.9 ± 1.6 |
21.6 ± 14.2 |
6.9 ± 0.4 |
6.7 ± 0.5 |
2.6 ± 0.2 |
| 19 | 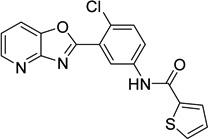 |
N-(4-chloro-3- [1,3]oxazolo[4,5- b]pyridin-2-ylphenyl)- 2- thiophenecarboxamid e 9011680 |
5.4 ± 0.8 |
1.7 ± 0.2 |
38.4 ± 16.5 |
17.9 ± 12.7 |
43.2 ± 2.9 |
42.9 ± 2.8 |
37.4 ± 1.6 |
Mean ± SD are shown, N ≥ 3 biological replicates for all data.
HTS Rank # from SI Table S2 based on efficacy in the primary HTS.
In the primary assay, yeast growth is likely due to inhibition of 5-FUrd uptake through PfENT1. To rule out alternative explanations, we created a secondary orthogonal assay in which inhibition of cell growth was the indicator of PfENT1 inhibition. Yeast strains deleted for ADE2 are purine auxotrophs since they lack a critical enzyme for de novo purine biosynthesis.33 Adenine transport via the endogenous FCY2-encoded purine-cytosine permease permits growth of ade2Δ yeast in minimal medium supplemented with adenine.34 In contrast, ade2Δ strains cannot proliferate with adenosine as the sole purine source because they lack an endogenous adenosine transporter. Since PfENT1 can transport adenosine, PfENT1 expression in ade2Δ yeast rescues growth in adenosine-containing media. We observed maximal growth of PfENT1-CO-expressing yeast (PfENT1-Codon-Optimized, no HA tag) in media supplemented with ≥1 mM adenosine (SI Figure S4A). This strain accumulated [3H]adenosine, confirming functional expression (SI Figure S4B). In media where adenosine was the sole purine source, PfENT1 inhibitors blocked adenosine import exposing the purine auxotrophy of PfENT1-CO-expressing ade2Δ yeast. Thus, in contrast to the primary assay where PfENT1 inhibitors allowed growth, in the secondary assay PfENT1 inhibitors prevented growth. PfENT1 expression was the only common link between the two assays. All nine compounds inhibited growth of PfENT1-CO-expressing ade2Δ yeast with IC50 values ranging from 200 nM to 2 µM (Figure 2B and Table 1).
All nine compounds had significantly different, but well correlated EC50 and IC50 values for the two yeast assays (SI Figure S5). The 5-FUrd concentration was chosen to minimize yeast proliferation and thus false positives in the HTS. The adenosine concentration was chosen to maximize assay sensitivity to PfENT1 inhibition. Thus, the 5-FUrd assay was more stringent (SI Figure S5). The differences in the EC50 and IC50 values and the opposite endpoint, cell growth vs death, argue strongly that in the yeast cell assays the efficacy target for the compounds is PfENT1 and not a secondary yeast target.
Furthermore, all nine compounds inhibited [3H]adenosine uptake into PfENT1-CO-expressing ade2Δ yeast with IC50 values in the 2 to 40 nM range (Figure 2C, Table 1). Thus, the compounds identified in our yeast-based growth assays inhibit PfENT1-mediated transport. We tested whether compound 3 was specific for PfENT1 vs PfENT4. Compound 3 did not inhibit [14C]2-deoxyadenosine uptake into PfENT4-expressing Xenopus oocytes at concentrations up to 50 µM (SI Figure S6). Thus, compound 3 is specific for PfENT1 over PfENT4 (~17% sequence identical).
Effects on parasite purine uptake and growth
Mild saponin treatment releases Plasmodium parasites from their red blood cell (RBC) hosts while preserving parasite plasma membrane integrity. All nine compounds inhibited [3H]adenosine uptake into RBC-free trophozoite-stage parasites with IC50 values in the low nanomolar range, similar to the observed inhibition of [3H]adenosine uptake into PfENT1-expressing yeast (Figure 3C and Table 1).
Figure 3.

Interaction of nine selected compounds with P. falciparum. (A) Concentrationdependent inhibition of [3H]adenosine uptake into RBC-free trophozoite-stage P. falciparum over 15 min. Mean values from technical replicates of single experiments are shown. (B and C) Concentration-dependent inhibition of P. falciparum 3D7 strain proliferation by the compounds in the presence of extracellular hypoxanthine at (B) 367 µM or (C) 10 µM. (D) DNA quantification at the indicated time points for the cultures used in panel (E). (E) Parasite morphology from control and 10× IC50 AKR-122 (3) treated cultures at the indicated times. CQ, chloroquine.
In 72-h growth assays, all nine compounds inhibited the proliferation of chloroquine-sensitive 3D7 and chloroquine-resistant Dd2 parasites, in growth media containing 10 µM hypoxanthine. IC50 values were in the 5 to 55 µM range (Figure 3A, 3B and Table 1) and were similar for 3D7 and Dd2 parasites (Table 1 and SI Figure S7). Microscopic examination of parasite cultures treated with ~10× the IC50 concentration of compound 3 showed that during the first 24 h of culture the compound-treated parasites failed to progress beyond the ring stage (Figure 3E). After a 24-h treatment, only hyperchromic spots remained in infected RBCs (Figure 3E) consistent with the absence of DNA replication in the treated culture (Figure 3D). The control culture progressed to the schizont stage by 48 h and again by 96 h.
P. falciparum culture media formulations often include supplemental hypoxanthine, at 50 mg/L (367 µM) to facilitate parasite culture, much higher than the <10 µM purine concentration found in human blood.26, 35, 36 This is ~30 times the hypoxanthine concentration necessary to achieve maximal proliferation (SI Figure S9A). The compounds inhibited parasite growth with similar efficacy regardless of whether the media contained low (10 µM) or high (367 µM) concentrations of hypoxanthine (Figure 3A, 3B and Table 1). Because pfent1Δ parasites are viable in media containing high hypoxanthine, we expected that in high hypoxanthine the parasites might proliferate in the presence of PfENT1 inhibitors.25 The fact that proliferation was inhibited even in 367 µM media may reflect the difference between a genetic and chemical knockout. Perhaps an alternative purine transport pathway is overexpressed in pfent1Δ parasites. Experiments are in progress to test this hypothesis.
The [3H]adenosine uptake assays represented a direct, short-term test of a compound’s ability to inhibit PfENT1. In contrast, the yeast and parasite growth assays are indirect tests of efficacy; they depend on the number of expressed transporters and their substrate transport capacity relative to the purine needs of the growing cells. It is notable that the IC50 values in the [3H]adenosine uptake assays were in the ten nanomolar range but the IC50 values in the growth assays are significantly higher, in the hundreds of nanomolar to micromolar range. We infer that to inhibit growth a high percentage of the purine transport capacity must be blocked in either yeast or parasites.
Efficacy target validation in parasites
The difference between IC50 values for [3H]adenosine uptake into parasites and for the parasite cytotoxicity raised two questions. First, do the compounds reach PfENT1 in the parasite inside the RBC? Second, are the compounds killing parasites by inhibiting PfENT1 or by interaction with a secondary target?
Xanthine vs hypoxanthine as purine source
To address the first question, we compared the compound IC50 values when the media purine source was xanthine vs hypoxanthine. RBCs cannot metabolize xanthine; however, they can convert hypoxanthine into other purine compounds and into nucleotides such as IMP. In order for parasites to utilize xanthine as a purine source, they must import it into the parasite cytoplasm. We generated pfent1Δ parasites in the Dd2 background (SI Figure S8). In agreement with El-Bissati et al. (2008), WT parasites grew with xanthine as the sole purine source, but pfent1Δ parasites did not (SI Figure S9A). This implies that PfENT1 is the sole entry pathway for xanthine into parasites and that the RBC cannot convert xanthine to another purine form that the pfent1Δ parasites can utilize.
While WT parasites can import [3H]hypoxanthine, pfent1Δ parasites do not transport [3H]hypoxanthine (SI Figure S9B).24 However, pfent1Δ parasites can grow on hypoxanthine supplemented media, albeit at concentrations significantly higher than required by WT parasites (SI Figure S9A).25 The inability of pfent1Δ parasites to transport hypoxanthine implies that they must import an RBC-generated hypoxanthine-derived purine via an alternate transport pathway, i.e., not via PfENT1. The nature of the hypoxanthine-derived purine and the transporter are unknown. This implies that hypoxanthine can support WT parasite purine requirements either by direct import via PfENT1 or by RBC metabolism to an alternative purine that can be imported via an alternative transport pathway.
We determined IC50 values for five of the compounds (1, 2, 3, 5, 13; one from each chemotype) in WT Dd2 parasites with xanthine as the purine source. For compounds 1, 2, 3, and 5, the IC50 values with xanthine as the purine source were significantly lower than the IC50 values with hypoxanthine (Figure 4A). The higher compound IC50 values with hypoxanthine suggest that import of RBC-generated, hypoxanthine-derived purines via the alternate purine transport pathway can partially compensate for compound inhibition of hypoxanthine uptake via PfENT1. This contributes to the higher compound IC50 values observed with hypoxanthine compared to xanthine. The differences in the compound IC50 values for inhibition of parasite growth with xanthine vs hypoxanthine as the sole purine source supports the hypothesis that the compounds were able to interact with PfENT1 in parasites in situ. Furthermore, the different IC50 values supports the hypothesis that the compounds kill parasites by inhibiting PfENT1.
Figure 4.

Efficacy target validation for parasitocidal effects of PfENT1 inhibitors. (A) IC50 values for five PfENT1 inhibitor compounds, one from each chemotype, with either 10 µM hypoxanthine (gray bars) or 90 µM xanthine (black bars) as the sole purine source in the media. Mean ± SD shown. (B) Ratio of the IC50 value in pfent1Δ parasites to the IC50 value in WT Dd2 parasites for the five compounds. Dashed line indicates a ratio of 1, no difference in the IC50 values. (C, D) IC50 values for the effects of the five compounds on (C) pfent1Δ and (D) WT Dd2 parasites measured 48 (black bars), 72 (light gray bars) and 96 (dark gray bars) h after application of compound. Note the progressive decrease in the IC50 values with time for pfent1Δ but not for Dd2 parasites. Mean ± SD (n=3).
PfENT1 knockout parasites
To address the second question regarding potential secondary targets, we used pfent1Δ parasites (SI Figure S9). We tested the efficacy of compounds 1, 2, 3, 5, 13 against the pfent1Δ parasites. For compounds, 1, 2, and 3, the IC50 values were 3–4 fold higher with the pfent1Δ parasites compared to WT Dd2 and 1.9-fold higher for 13 (Figure 4B). For compound 5, the IC50 values were similar. In contrast to WT parasites, for the pfent1Δ parasites, the measured IC50 values for all five compounds decreased with increasing duration of incubation in compound from 48 to 96 h (Figure 4C). This delayed death phenotype was not observed with the WT parasites (Figure 4D). This suggests two things: 1) The compounds kill pfent1Δ parasites by interaction with a different target than in WT parasites, and 2) All five compounds may be interacting with the same secondary target.
The pfent1Δ parasites provide a more definitive proof of the compounds’ efficacy target. For four of the five compounds tested, the IC50 values were significantly lower for WT parasites compared to pfent1Δ parasites (Figure 4B). Therefore, for these four compounds the primary efficacy target for killing parasites is PfENT1. Interestingly, at higher concentrations the compounds all killed pfent1Δ parasites. This implies that they also have a secondary, lower efficacy target (except for compound 13 where the IC50 values for PfENT1 and the secondary target is similar). At present the identity of the secondary target remains unknown. For all five compounds, the secondary target revealed in the pfent1Δ parasites displayed a delayed death phenotype, with compound effectiveness increasing with increased duration of exposure from 48 to 96 h. Similar delayed death phenotypes have been observed with parasite killing by bacterial antibiotics, which appear to act via destruction of the apicoplast, as well as agents that act independently of the apicoplast37–41 (Ekland and Fidock, unpublished data).
The absence of the delayed death phenotype in killing of WT parasites argues that death of WT parasites is most likely due to inhibition of PfENT1 and is not primarily mediated by the secondary target. The similar efficacy in killing WT and pfent1Δ parasites suggests that the secondary target binding site may be structurally similar to the binding site in PfENT1. The existence of two targeted proteins may make this an excellent target to avoid the development of drug resistance because both targets would have to mutate in order for the parasites to develop resistance. This provides strong support for PfENT1 as a target for antimalarial drug development.
Chemical confirmation and structure-activity
For compound 3, a coumarin that was one of the most potent inhibitors, we confirmed its structure through independent synthesis, NMR spectroscopy and mass spectrometry (SI Chemical Synthesis and Characterization). The activity of our resynthesized compound 3 (referred to as AKR-122 to distinguish it from the purchased compound, 3, in the Tables) was similar across all screening assays (SI Table S3). The activity of AKR-122 at high purity supports the library structure assignment and diminishes concern that activity in the original screening assays represented the activity of unidentified degradation products and/or impurities.
The library contained a cluster of coumarins of lesser activity than compound 3 (SI Figure S10 and Table S2). From the structures of these compounds we deduce that their activity is markedly diminished by substitutions at R1, R2, R3, R4, and R6 (SI Figure S10). We synthesized six analogs of 3 for exploratory structure-activity studies (SI Chemical Synthesis and Characterization). These experiments demonstrated that an alkoxy-/methoxybenzyloxy group at R5 was essential for activity (SI Figure S10 and Table S3). We observed that the 3,4,5-trimethoxybenzl substitution pattern is most potent. Removal of any methoxyl, or substitution with halogen atoms or alkanes, decreased activity in both yeast and parasite growth assays. Similarly, substitutions at the coumarin ring 4-position with a bulky or longer-chain hydrocarbon, also decreased activity (SI Table S3).
All nine compounds that we chose for more extensive characterization, representing five distinct chemotypes, inhibited P. falciparum proliferation in vitro (Figure 3 and Table 1). Four of the compounds studied, 2, 6, 7, and 19, represent a single chemotype (SI Table S2). In addition, we have evaluated a preliminary SAR of compound 3 (SI Table S3). Thus, we obtained similar results with compounds of diverse chemical structures. This further supports the hypothesis that their parasitocidal mechanism of action involves inhibition of PfENT1.
Some compounds have been noted to cause pan-assay interference (PAINS) in HTS.42 Compounds 3 and 13 have PAINS substructures, but the fact that 3 has different IC50 values depending on whether xanthine or hypoxanthine is the purine source (Figure 4A) or between WT and pfent1Δ parasites in the case of 13 (Figure 4B) suggests that in our assays they are truly interacting with PfENT1. We also note that 47 medicines approved for clinical use by the Australian Medical Board have PAINS substructures, so having a PAINS substructure does not necessarily preclude their potential as therapeutic leads.42
Summary
We have developed a HTS that identified inhibitors of PfENT1. The 171 positive compounds (0.26% hit rate) represent several structurally unique chemical scaffolds. These molecules provide diverse starting points for medicinal chemistry efforts to identify more potent derivatives. Despite the varied structures, all nine compounds from the HTS were active in all of the secondary assays including yeast and parasite proliferation and [3H]adenosine uptake assays. Thus, the HTS effectively identified PfENT1 inhibitors.
Our assay overcomes difficulties of standard transporter-based assays for use in HTS. By employing a positive readout, cell growth, to indicate PfENT1 inhibition, we avoided selecting as false positives, compounds inherently toxic to yeast. The assay’s robustness arises in part from the rapid doubling time of yeast: In the presence of a potent PfENT1 inhibitor, the ~80,000 yeast cells inoculated into each 384-well plate well yielded >107 yeast cells after an overnight incubation. The basic requirements for the assay are: 1) the transporter (drug target) must import a substance that is cytotoxic to yeast, 2) it must be feasible to generate yeast strains that do not import the cytotoxic substance via endogenous transporters, and 3) one must be able to functionally express the transporter in yeast. Given the simplicity and robustness of this approach, similar HTS assays could identify inhibitors of other transporters, especially purine transporters from other purine auxotrophic parasites.43
We hypothesize that PfENT1 inhibitors kill P. falciparum parasites by purine starvation. During the 48-h intraerythrocytic life cycle, RNA and DNA synthesis acts as a sink for cytoplasmic purines that will likely deplete the amount of cellular ATP if purine import is blocked. We hypothesize that ATP depletion leads to parasite death. In the presence of a PfENT1 inhibitor, the parasites do not progress beyond the ring stage, perhaps because they are unable to make the RNA needed for ribosome synthesis and thus protein synthesis is impeded (Figure 3E).
The fact that the compounds all kill pfent1Δ parasites with a similar but lower efficacy implies that they also interact with a secondary target. The existence of two proteins, PfENT1 and the secondary target, with similar IC50 values for the identified PfENT1 inhibitors, may make this an even better drug development target. Resistance is less likely to develop if mutations must arise in both proteins.
In summary, our HTS identified compounds that inhibit an essential malaria parasite transport protein, PfENT1, and kill P. falciparum parasites in culture. These newly described compounds may represent therapeutic leads for the development of a new generation of antimalarial drugs. This assay may also be useful to identify chemical leads for the development of drugs to treat other parasitic diseases caused by purine auxotrophic organisms.
METHODS
Generation of yeast strains, HTS conditions, other yeast- and parasite-based assays, and generation of pfent1Δ parasites
Described in SI Methods.
Supplementary Material
ACKNOWLEDGMENTS
We thank A. Bresnick, A. Finkelstein and V. Schramm for helpful discussions and D. Pierce for expert technical assistance.
Funding
IJF and RD were supported in part by the NIGMS Medical Scientist Training Program grant T32-GM007288. This work was supported by Albert Einstein College of Medicine funds (MHA), NIGMS grant GM085177 (IMW), NIAID grant AI085584 (DAF) and NYSTEM grant SDH C026715 SC3 (DWL)
Footnotes
ASSOCIATED CONTENT
Supporting Information Supporting information contains Figures S1 to S10, Tables S1 to S3, descriptions of the chemical synthesis and compound characterization, and detailed Methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
IJF and MHA conceived the HTS and designed the experiments. RDM and IMW provided yeast strains, plasmids and advice on construction of PfENT1- and GFP-expressing yeast strains. IJF and FK performed the HTS. IJF and RD performed the yeast- and parasite-based assays. AR, SD and DWL designed the chemical derivatives. AR synthesized and characterized the synthesized compounds. SHA, OC-F, and DAF generated the pfent1Δ parasites. IJF, RD and MHA analyzed the data. All authors participated in writing and editing the manuscript.
The authors declare potentially competing financial interests: A patent application is pending on the yeast-based assays as a method to identify nucleoside transporter inhibitors. A second pending patent covers the use of the 171 compounds as PfENT1 inhibitors and as potential antimalarial drugs. The patents have not been licensed and no negotiations are in progress.
REFERENCES
- 1.Xie H. Activity assay of membrane transport proteins. Acta Biochim Biophys Sin (Shanghai) 2008;40:269–277. doi: 10.1111/j.1745-7270.2008.00400.x. [DOI] [PubMed] [Google Scholar]
- 2.Wellems TE. Plasmodium chloroquine resistance and the search for a replacement antimalarial drug. Science. 2002;298:124–126. doi: 10.1126/science.1078167. [DOI] [PubMed] [Google Scholar]
- 3.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phyo AP, Nkhoma S, Stepniewska K, Ashley EA, Nair S, McGready R, ler Moo C, Al-Saai S, Dondorp AM, Lwin KM, Singhasivanon P, Day NP, White NJ, Anderson TJ, Nosten F. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379:1960–1966. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miotto O, Almagro-Garcia J, Manske M, Macinnis B, Campino S, Rockett KA, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Duong S, Nguon C, Chuor CM, Saunders D, Se Y, Lon C, Fukuda MM, Amenga-Etego L, Hodgson AV, Asoala V, Imwong M, Takala-Harrison S, Nosten F, Su XZ, Ringwald P, Ariey F, Dolecek C, Hien TT, Boni MF, Thai CQ, Amambua-Ngwa A, Conway DJ, Djimde AA, Doumbo OK, Zongo I, Ouedraogo JB, Alcock D, Drury E, Auburn S, Koch O, Sanders M, Hubbart C, Maslen G, Ruano-Rubio V, Jyothi D, Miles A, O'Brien J, Gamble C, Oyola SO, Rayner JC, Newbold CI, Berriman M, Spencer CC, McVean G, Day NP, White NJ, Bethell D, Dondorp AM, Plowe CV, Fairhurst RM, Kwiatkowski DP. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat Genet. 2013;45:648–655. doi: 10.1038/ng.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale JC, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burrows JN, Chibale K, Wells TN. The state of the art in anti-malarial drug discovery and development. Curr Top Med Chem. 2011;11:1226–1254. doi: 10.2174/156802611795429194. [DOI] [PubMed] [Google Scholar]
- 8.Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med. 2013;19:156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burrows JN, van Huijsduijnen RH, Mohrle JJ, Oeuvray C, Wells TN. Designing the next generation of medicines for malaria control and eradication. Malar J. 2013;12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Downie MJ, Kirk K, Mamoun CB. Purine salvage pathways in the intraerythrocytic malaria parasite Plasmodium falciparum. Eukaryot Cell. 2008;7:1231–1237. doi: 10.1128/EC.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassera MB, Zhang Y, Hazleton KZ, Schramm VL. Purine and pyrimidine pathways as targets in Plasmodium falciparum. Curr Top Med Chem. 2011;11:2103–2115. doi: 10.2174/156802611796575948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young JD, Yao SY, Sun L, Cass CE, Baldwin SA. Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica. 2008;38:995–1021. doi: 10.1080/00498250801927427. [DOI] [PubMed] [Google Scholar]
- 13.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin RE, Henry RI, Abbey JL, Clements JD, Kirk K. The 'permeome' of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum. Genome Biol. 2005;6:R26. doi: 10.1186/gb-2005-6-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baldwin SA, McConkey GA, Cass CE, Young JD. Nucleoside transport as a potential target for chemotherapy in malaria. Curr Pharm Des. 2007;13:569–580. doi: 10.2174/138161207780162845. [DOI] [PubMed] [Google Scholar]
- 16.Downie MJ, El Bissati K, Bobenchik AM, Nic Lochlainn L, Amerik A, Zufferey R, Kirk K, Ben Mamoun C. PfNT2, a permease of the equilibrative nucleoside transporter family in the endoplasmic reticulum of Plasmodium falciparum. J Biol Chem. 2010;285:20827–20833. doi: 10.1074/jbc.M110.118489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frame IJ, Merino EF, Schramm VL, Cassera MB, Akabas MH. Malaria parasite type 4 equilibrative nucleoside transporters (ENT4) are purine transporters with distinct substrate specificity. Biochem J. 2012;446:179–190. doi: 10.1042/BJ20112220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rager N, Mamoun CB, Carter NS, Goldberg DE, Ullman B. Localization of the Plasmodium falciparum PfNT1 nucleoside transporter to the parasite plasma membrane. J Biol Chem. 2001;276:41095–41099. doi: 10.1074/jbc.M107037200. [DOI] [PubMed] [Google Scholar]
- 19.Carter NS, Drew ME, Sanchez M, Vasudevan G, Landfear SM, Ullman B. Cloning of a novel inosine-guanosine transporter gene from Leishmania donovani by functional rescue of a transport-deficient mutant. J Biol Chem. 2000;275:20935–20941. doi: 10.1074/jbc.M002418200. [DOI] [PubMed] [Google Scholar]
- 20.Parker MD, Hyde RJ, Yao SY, McRobert L, Cass CE, Young JD, McConkey GA, Baldwin SA. Identification of a nucleoside/nucleobase transporter from Plasmodium falciparum, a novel target for anti-malarial chemotherapy. Biochem J. 2000;349:67–75. doi: 10.1042/0264-6021:3490067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Downie MJ, Saliba KJ, Howitt SM, Broer S, Kirk K. Transport of nucleosides across the Plasmodium falciparum parasite plasma membrane has characteristics of PfENT1. Mol Microbiol. 2006;60:738–748. doi: 10.1111/j.1365-2958.2006.05125.x. [DOI] [PubMed] [Google Scholar]
- 22.Downie MJ, Saliba KJ, Broer S, Howitt SM, Kirk K. Purine nucleobase transport in the intraerythrocytic malaria parasite. Int J Parasitol. 2008;38:203–209. doi: 10.1016/j.ijpara.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Riegelhaupt PM, Cassera MB, Frohlich RF, Hazleton KZ, Hefter JJ, Schramm VL, Akabas MH. Transport of purines and purine salvage pathway inhibitors by the Plasmodium falciparum equilibrative nucleoside transporter PfENT1. Mol Biochem Parasitol. 2010;169:40–49. doi: 10.1016/j.molbiopara.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El Bissati K, Downie MJ, Kim SK, Horowitz M, Carter N, Ullman B, Ben Mamoun C. Genetic evidence for the essential role of PfNT1 in the transport and utilization of xanthine, guanine, guanosine and adenine by Plasmodium falciparum. Mol Biochem Parasitol. 2008;161:130–139. doi: 10.1016/j.molbiopara.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Bissati K, Zufferey R, Witola WH, Carter NS, Ullman B, Ben Mamoun C. The plasma membrane permease PfNT1 is essential for purine salvage in the human malaria parasite Plasmodium falciparum. Proc Natl Acad Sci U S A. 2006;103:9286–9291. doi: 10.1073/pnas.0602590103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 27.Wagner R, de Montigny J, de Wergifosse P, Souciet JL, Potier S. The ORF YBL042 of Saccharomyces cerevisiae encodes a uridine permease. FEMS Microbiol Lett. 1998;159:69–75. doi: 10.1111/j.1574-6968.1998.tb12843.x. [DOI] [PubMed] [Google Scholar]
- 28.Vickers MF, Yao SY, Baldwin SA, Young JD, Cass CE. Nucleoside transporter proteins of Saccharomyces cerevisiae. Demonstration of a transporter (FUI1) with high uridine selectivity in plasma membranes and a transporter (FUN26) with broad nucleoside selectivity in intracellular membranes. J Biol Chem. 2000;275:25931–25938. doi: 10.1074/jbc.M000239200. [DOI] [PubMed] [Google Scholar]
- 29.Riegelhaupt PM, Frame IJ, Akabas MH. Transmembrane segment 11 appears to line the purine permeation pathway of the Plasmodium falciparum equilibrative nucleoside transporter 1 (PfENT1) J Biol Chem. 2010;285:17001–17010. doi: 10.1074/jbc.M110.115758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westermann B, Neupert W. Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast. 2000;16:1421–1427. doi: 10.1002/1097-0061(200011)16:15<1421::AID-YEA624>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 31.Iversen PW, Beck B, Chen Y-F, Dere W, Devanarayan V, Eastwood BJ, Farmen MW, Iturria SJ, Montrose C, Moore RA, Weidner JR, Sittampalam GS. HTS Assay Validation. In: Sittampalam GS, Gal-Edd N, Arkin M, Auld D, Austin C, Bejcek B, Glicksman M, Inglese J, Lemmon V, Li Z, McGee J, McManus O, Minor L, Napper A, Riss T, Trask OJ, Weidner J, editors. Assay Guidance Manual. Bethesda MD: 2004. [Google Scholar]
- 32.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TN, Willis P. The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One. 2013;8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 34.Polak A, Grenson M. Evidence for a common transport system for cytosine, adenine and hypoxanthine in Saccharomyces cerevisiae and Candida albicans. Eur J Biochem. 1973;32:276–282. doi: 10.1111/j.1432-1033.1973.tb02608.x. [DOI] [PubMed] [Google Scholar]
- 35.Reber-Liske R. A labour-saving method for the in vitro culture of Plasmodium falciparum. Acta Trop. 1983;40:39–43. [PubMed] [Google Scholar]
- 36.Divo AA, Jensen JB. Studies on serum requirements for the cultivation of Plasmodium falciparum. 2. Medium enrichment. Bull World Health Organ. 1982;60:571–575. [PMC free article] [PubMed] [Google Scholar]
- 37.Dahl EL, Shock JL, Shenai BR, Gut J, DeRisi JL, Rosenthal PJ. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob Agents Chemother. 2006;50:3124–3131. doi: 10.1128/AAC.00394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramya TN, Mishra S, Karmodiya K, Surolia N, Surolia A. Inhibitors of nonhousekeeping functions of the apicoplast defy delayed death in Plasmodium falciparum. Antimicrob Agents Chemother. 2007;51:307–316. doi: 10.1128/AAC.00808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pradel G, Schlitzer M. Antibiotics in malaria therapy and their effect on the parasite apicoplast. Curr Mol Med. 2010;10:335–349. doi: 10.2174/156652410791065273. [DOI] [PubMed] [Google Scholar]
- 40.Ekland EH, Schneider J, Fidock DA. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J. 2011;25:3583–3593. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011;9:e1001138. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 43.Landfear SM. Nutrient transport and pathogenesis in selected parasitic protozoa. Eukaryot Cell. 2011;10:483–493. doi: 10.1128/EC.00287-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.