

Abstract
RhoGDI2 (ARHGDIB) suppresses metastasis in a variety of cancers but the mechanism is unclear; thus, hampering development of human therapeutics. RhoGDI2 is a guanine nucleotide dissociation inhibitor (GDI) for the Rho family of GTPases thought to primarily bind to Rac1; however, Rac1 activation was not decreased by RhoGDI2 expression in bladder cancer cells. To better understand the GTPase binding partners for RhoGDI2, a mass spectrometry-based proteomic approach was employed in bladder cancer cells. As expected endogenous RhoGDI2 co-immunoprecipitates with Rac1 and unexpectedly so does RhoC. Further analysis demonstrated that RhoGDI2 negatively regulates RhoC, as knockdown of RhoGDI2 increased RhoC activation in response to serum stimulation. Conversely, overexpression of RhoGDI2 decreased RhoC activation. RhoC promoted bladder cancer cell growth and invasion, as knockdown increased cell doubling time, decreased invasion through Matrigel, and decreased colony formation in soft agar. Importantly, RhoC knockdown reduced in vivo lung colonization by bladder cancer cells following tail vein injection in immune compromised mice. Finally, unbiased transcriptome analysis revealed a set of genes regulated by RhoGDI2 over-expression and RhoC knockdown in bladder cancer cells.
Keywords: Guanine Nucleotide Dissociation Inhibitors, Rho GDP dissociation inhibitor 2, RhoC, lung colonization, metastasis
INTRODUCTION
Rho family GTPases are widely associated with a variety of cellular responses, including cell morphology and motility (1), and inappropriate activation of Rho GTPases has been correlated with increased migration, invasion and metastasis of cancer cells (2,3). Guanine nucleotide dissociation inhibitors (GDIs) bind to GTPases, sequester them in the cytosol, and negatively regulate GTPases by occluding the binding site of other GTPase regulatory proteins (4). The Rho family of GTPases is regulated by three related GDI proteins, RhoGDI1, 2 and 3. Reduced expression of RhoGDI2 in human bladder cancer and in other cancers is a prognostic indicator of metastasis and is associated with shorter disease-free survival times (5–7), and experimental expression of RhoGDI2 suppresses bladder cancer metastasis in animal models (8).
The signaling pathways through which RhoGDI2 suppresses bladder cancer metastasis are gradually being elucidated. A recent study found that RhoGDI2 suppressed the expression of the proteoglycan versican, and that versican was necessary to promote inflammation in the metastatic niche to enable tumor cell survival (9). Similarly, RhoGDI2 suppresses expression of endothelin (10), a vasoconstricting factor shown to be pro-metastatic in bladder (11). However, the signaling mechanisms for RhoGDI2 negative regulation of versican or endothelin expression are unknown.
Rac1 has been shown to be the primary binding partner of RhoGDI2 in co-expression assays in HEK293 cells (12). Binding of RhoGDI2 to RhoA or Cdc42 is as much as ten times lower than that of RhoGDI1 (13,14). Modulation of RhoGDI2 levels in bladder cancer cells did not cause a decrease in Rac1 activation as would be expected, but rather an increase in Rac1 activation (15), suggesting that RhoGDI2 does not suppress bladder cancer metastasis through Rac1. Given this, and the canonical function of the RhoGDI protein family, we hypothesized that RhoGDI2 may be binding to alternative Rho family GTPases to suppress metastasis. Here we identify RhoC as a prominent binding partner of RhoGDI2 and show that RhoGDI2 modulates RhoC activity in cells. We further show that RhoC promotes lung colonization of bladder cancer cells providing a new mechanism for how RhoGDI2 functions as a metastasis suppressor in bladder cancer and thus a rationale for the development of therapeutics that target RhoC-activated kinases for potential use in the adjuvant setting in patients with advanced primary tumors.
MATERIALS AND METHODS
Cell Culture
Cells were maintained as follows: RT4 cells, McCoy’s 5A supplemented with 10% fetal bovine serum (FBS) (Gibco); UMUC3 and LuL2 cells, MEM supplemented with 10% FBS and 1mM sodium pyruvate; T24 and T24T cells, DMEM/F12 supplemented with 5% FBS. All cells were cultured in a humidified 5% CO2 atmosphere at 37°C. UMUC3 cells expressing GFP or GFP-GDI2 were described previously (15). LuL2 cells were previously described (16).
RhoGDI2 Immunoprecipitation
2.5 x 106 RT4 cells seeded on a 10cm dish were grown to confluency for 72 hrs. Cells were lysed in buffer (50mM HEPES pH 7.1, 5mM MgCl2, 1mM EGTA, 0.5% Triton X-100), and lysates were passed 10 times through an 18 G needle, then clarified at 100K g for 30 min via ultracentrifugation. The clarified lysate was incubated for 2 h at 4°C with rotation with RhoGDI2 antibody (Spring Bioscience) crosslinked to Protein A agarose, then beads were washed and centrifuged 3 times with lysis buffer.
Mass Spectrometry
RhoGDI2 immunoprecipitates were eluted with Lamelli sample buffer and resolved on a 4–20% Criterion TGX gel (Biorad). Gel was silver stained with ProteoSilver™ Plus Silver stain kit (Sigma) according to manufacturer’s protocol, and a gel piece of ~23-15 kDa was excised. Sample was transferred to a siliconized tube and washed and destained in 200 μL 50% methanol overnight. Sample was dehydrated in acetonitrile, rehydrated in 30 μL of 10 mM dithiolthreitol in 0.1 M ammonium bicarbonate and reduced at room temperature for 0.5 h. Sample was alkylated in 30 μL 50 mM iodoacetamide in 0.1 M ammonium bicarbonate at room temperature for 0.5 h, dehydrated in 100 μL acetonitrile, then rehydrated in 100 μL 0.1 M ammonium bicarbonate. Sample was dehydrated in 100 μL acetonitrile, then completely dried by vacuum centrifugation. Sample was rehydrated in 20 ng/μL trypsin in 50 mM ammonium bicarbonate on ice for 10 min, then 20 μL 50 mM ammonium bicarbonate added. Sample was digested overnight at 37°C and peptides extracted from the polyacrylamide in two 30 μL aliquots of 50% acetonitrile/5% formic acid. Extracts were combined and evaporated to 15 μL for MS analysis.
The LC-MS system consisted of a Thermo Electron Orbitrap Velos ETD mass spectrometer system with a Protana nanospray ion source interfaced to a self-packed 8 cm x 75 um id Phenomenex Jupiter 10 um C18 reversed-phase capillary column. 7 μL aliquots of the extract were injected and the peptides eluted from the column by an acetonitrile/0.1 M acetic acid gradient at a flow rate of 0.5 μL/min over 0.5 hours. The nanospray ion source was operated at 2.5 kV. The digest was analyzed using the double play capability of the instrument acquiring full scan mass spectra to determine peptide molecular weights followed by product ion spectra to determine amino acid sequence in sequential scans. Data were analyzed using the Sequest search algorithm against Human IPI.
Western Blot
Western blot images were acquired on an Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE, USA) using anti-Rac1 and anti-RhoG (Millipore); anti-Cdc42, anti-RhoA, and anti-RhoC (Cell Signaling); anti-D4-GDI (RhoGDI2) (Spring Bioscience); anti-RhoGDI1 (Santa Cruz Biotechnology); and anti-tubulin (Sigma).
Rho-GTP Pull-down Assays
Serum starved cells treated as indicated were lysed in ice cold buffer (50mM Tris HCl, pH 7.4, 2mM MgCl2, 1% Triton-X100, 10% glycerol, 100mM NaCl, 1mM DTT) containing 15 ug/reaction GST-Rhotekin-RBD protein (Cytoskeleton). Extracts were clarified at 13,000 rpm for 6 minutes, and incubated with glutathione sepharose beads for 45 min at 4°C with rotation. Pull downs were washed thrice with ice cold lysis buffer and subjected to SDS-PAGE and western blotting.
Generation of shRNA Stable Cell Lines
T24 cell lines stably expressing control or RhoGDI2-targeted shRNAs and T24T cell lines stably expressing control or RhoC-targeted shRNAs were generated using lentiviral transduction particles as previously reported (17). Following lentiviral infection, T24 cells were selected using 1 μg/ml of puromycin and T24T cells were selected with 100 μg/ml hygromycin. The following vectors were used: pLKO.1 control (RHS4080) and shRhoGDI2 (TRCN0000047413) (Thermo Scientific). The following RhoC shRNAs were used: shRhoC-1 sense, GAACUAUAUUGCGGACAUU; shRhoC-1 anti-sense, CUUGAUAUAACGCCUGUAA; shRhoC-2 sense, CCAGCACUUUAUACACUUC; shRhoC-2 antisense, GGUCGUGAAAUAUGUGAAG.
Doubling Time, Invasion and Soft Agar Assays
Doubling time was determined by counting cells every 24 h for a 96 h period. For invasion assays, 4 x 105 cells in serum free media were seeded in the top chamber of a Corning® BioCoat™ Matrigel® Invasion Chamber (Corning) and allowed to migrate towards media containing 5% FBS for 16 h. Cells were removed from the upper well. Invaded cells were fixed and stained with Differential Quik Stain Kit staining solution (Polysciences Inc.), then counted using a gridded cover slip. For soft agar assays, 4 x 104 cells were seeded in growth media with 0.4% agarose above a layer of 0.6% agarose in growth media. After 3 weeks incubation at 37°C and 5% CO2, colonies were stained by incubation with 0.5 mg/mL MTT for 4 h, then scanned and quantified using Image J software.
Experimental Metastasis Assay
Five-week-old female athymic NCr nu/nu mice (Charles River), maintained in accordance with University of Virginia ACUC guidelines, were inoculated via tail vein injection with 2 × 106 T24T cells or 1 × 106 LuL2 cells expressing the indicated shRNAs. 7 or 4 weeks after injection of T24T cells or LuL2 cells respectively, mice were sacrificed and human tumor burden in the lung was assessed by quantitative real-time PCR with a human-specific 12p TaqMan probe and primer set using 3μg of genomic DNA isolated from the left lung lobe (16,18). Animal experiments were performed twice for control and shRhoC-1 cells. Results of experiments were combined and statistical analysis was performed. Incidence was analyzed using Bernard’s exact test. For analysis of non-zero samples only or both 0 and non-zero samples, the Mann Whitney test was applied.
Microarray Analysis
Total RNA from exponentially growing T24T cells expressing shControl or shRhoC-1 was purified using the RNeasy Mini Kit (Qiagen). RNA was hybridized to GeneChip Human Transcriptome Array 2.0 arrays (Affymetrix). Microarray data was analyzed as described (10), and sorted based on a false discovery rate (FDR) P < 0.05.
Expression of selected genes was analyzed in two patient cohorts with publicly available microarray data. Gene expression data was obtained from publication (19) and from Gene Expression Omnibus, Accession #GSE13507 (20). Microarray probes were converted to gene symbols based on Affymetrix or Illumina annotation. If a gene had multiple probes, we selected the probe with the highest mean expression across all samples (21). Differential expression was assessed using the non-parametric Wilcoxon-Rank Sum Test and the fold change (FC) of average gene expression in the tumor samples relative to average expression in the normal samples was reported. The RhoC/RhoGDI2 signature was analyzed by normalizing each gene in the signature to have a mean of 0 and a standard deviation of 1. A signature score was then calculated by subtracting the sum of the (normalized) expression values of all up-regulated genes from the sum of the expression values of all down-regulated genes. Therefore, a sample with a lower RhoC/RhoGDI2 signature score indicates greater similarity to RhoC knockdown or RhoGDI2 expression than a sample with a higher score. The ability of the signature score to distinguish between normal and tumor samples was evaluated using the area under the receiver operating characteristic curve (AUC), with AUC > 0.50 indicating the signature score is higher in tumor than normal samples. The Wilcoxon-Rank Sum Test was used to assess whether the AUC differed from 0.5 (i.e., what would be expected by random chance).
Quantitative Real Time PCR
Total RNA from was purified from cells using the RNeasy Mini Kit, and reverse transcribed with Superscript III kit (Invitrogen) according to manufacturer’s instructions. The resulting first-strand cDNA was subjected to quantitative PCR using the SYBR green detection system (Bio-Rad). The sequences of primers used were: MREG-CAGGTTCGAAGGGAAGTAAGAA and TCACTGATGGTGCTGAGTTTAG; KRT8-GCAGAACAAGATGCTGGAGA and CCGCCTAAGGTTGTTGATGTA. Conditions for amplification were: 95°C for 3 minutes, followed by 40 cycles of 95°C for 10 seconds and 60°C for 30 seconds. KRT8 or MREG expression was normalized to endogenous ACTB and analyzed according to the 2−ΔΔCt method (22).
Statistical Analysis
All data were analyzed using two-tailed Student’s t-test unless otherwise specified.
RESULTS
RhoGDI2 Preferentially Binds to Rac1 and RhoC in Bladder Cancer Cells
RhoGDI2 is an established suppressor of bladder cancer metastasis that is thought to predominantly bind to Rac1, in comparison to other GTPases, such as RhoA or Cdc42, that it binds with much less affinity (12,23). Knockdown or overexpression of RhoGDI2 protein in bladder cells does not decrease Rac1 or RhoA activation (15), suggesting that other GTPases may be responsible for the metastasis suppressor actions of RhoGDI2. To determine which GTPases are binding partners for RhoGDI2 in bladder cells, endogenous RhoGDI2 was immunoprecipitated from RT4 cells, a non-invasive, non-metastatic human bladder cancer cell line (24). Proteins co-immunoprecipitating with RhoGDI2 were separated by SDS-PAGE and silver stained, and a band spanning ~15–23 kDa in size was excised from the gel and analyzed by mass spectrometry. Seven Rho family GTPases were identified, including Rac1, Rac2, Cdc42, RhoA, RhoB, RhoC and RhoG (Supplementary Table 1).
To quantitatively determine the fraction of each GTPase bound to RhoGDI2, western blotting was performed on RhoGDI2 immunoprecipitates along side a standard curve of increasing amounts of total cell extract (Figure 1A, Left). Blots were quantified by fluorescent staining and the amount of each GTPase present in the immunoprecipitate was fit to the corresponding standard curve. We normalized for recovery of RhoGDI2 in the immunoprecipitate to calculate the percent of the total GTPase in the extract that was bound to RhoGDI2 (Figure 1A, Right). In agreement with published reports, more Rac1 co-immunoprecipitated with RhoGDI2 compared to the other GTPases (~38% of total Rac1). Interestingly, RhoC was second among the GTPases in terms of fraction co-immunoprecipitated with RhoGDI2 (27% of total). Only small fractions of RhoA and RhoG associated with RhoGDI2, and Cdc42 was not detected in immunoprecipitates, likely due to the low level of Cdc42 associated with RhoGDI2. Rac2 and RhoB were not detected in total extracts or immunoprecipitates, which may reflect the low abundance of these proteins in RT4 cells or a lack of antibody sensitivity. These results suggested that RhoGDI2 may be an unappreciated negative regulator of RhoC activation in bladder cells. This is especially pertinent because RhoC is proposed to be pro-metastatic in a variety of cell types (25–28), and RhoGDI2 may serve to block those actions.
Figure 1. RhoGDI2 binds Rac1 and RhoC in bladder cells.

A. Endogenous RhoGDI2 was immunoprecipitated from RT4 bladder cancer cell extracts, and immunoprecipitates were probed by western blot for the recovery of the indicated GTPases. Increasing amounts of total protein were used to create a standard curve to quantify the relative amount of each GTPase co-immunoprecipitating with RhoGDI2. Left, representative western blots. Right, results expressed as percent of total GTPase bound to RhoGDI2 presented as the mean±S.E. (n=3). P<0.001, one-way ANOVA.
B. Endogenous RhoGDI1 was immunoprecipitated from RT4 bladder cancer cell extracts and analyzed for GTPase co-immunoprecipitation as above. Left, representative western blots. Right, results expressed as percent of total GTPase bound to RhoGDI1 presented as the mean±S.E. (n=3). P<0.01, one-way ANOVA.
C, D. Endogenous RhoGDI2 from T24 bladder cancer cells (C) or GFP-RhoGDI2 expressed in UMUC3 cells (D) was immunoprecipitated and analyzed by western blot for co-immunoprecipitation of the indicated GTPases. Representative western blots of 3 independent experiments shown.
We explored the GTPase binding partners of RhoGDI1 in bladder cells as a comparison to RhoGDI2. RhoGDI2 primarily affects metastasis while the ubiquitously expressed RhoGDI1 protein affects primary growth of bladder cancer cells (15). Co-immunoprecipitation experiments using RT4 bladder cancer cells showed that RhoGDI1 exhibited a different specificity profile from RhoGDI2, binding large fractions of RhoA and RhoC, and to lesser extents, Rac1, Cdc42 and RhoG (Figure 1B). These results illustrate the differential binding affinities of RhoGDI1 and RhoGDI2 for GTPases in bladder cells and may in part account for the differential effects of RhoGDI1 and RhoGDI2 on bladder cancer growth and metastasis.
To verify that the RhoC/RhoGDI2 interaction was not cell type specific, we also assayed Rho family GTPase binding to RhoGDI2 in two other human bladder cancer cell lines. RhoC and Rac1 co-immunoprecipitated with endogenous RhoGDI2 in T24 non-metastatic human bladder cancer cells (Figure 1C) and with ectopically expressed GFP-RhoGDI2 in UMUC3 metastatic bladder cancer cells that do not express appreciable levels of endogenous RhoGDI2, indicating that RhoGDI2 forms a stable complex with RhoC in multiple bladder cancer cell lines (Figure 1D). Co-immunoprecipitation of RhoA, RhoG, and Cdc42 with RhoGDI2 was not detected in T24 or UMUC3 cell lines, thus leading us to focus our analysis on the functional consequence of the RhoC/RhoGDI2 interaction. Interaction of RhoC with RhoGDI2 appears to be cell type specific as co-immunoprecipitation of RhoC with RhoGDI2 was detected in MDA-MB-231 breast cells, but not Jurkat lymphocyte cells or OVCAR-4 ovarian cells (Supplementary Figure 1).
RhoGDI2 Modulates Activation of RhoC in Bladder Cancer Cells
To determine if RhoGDI2 binding to RhoC has a functional effect on RhoC activation in bladder cancer cells, we performed RhoC pull-down assays utilizing a GST-Rhotekin fusion protein that preferentially binds to the GTP-bound as opposed to the GDP-bound form of Rho proteins. Knock down of RhoGDI2 in T24 bladder cancer cells increased FBS stimulated RhoC activation by 30% when compared to control cells (Figure 2A). Conversely, expression of RhoGDI2 in UMUC3 cells resulted in a 30–40% decrease in the amount of FBS stimulated RhoC activation relative to control cells (Figure 2B). These results show that RhoGDI2 negatively regulates RhoC activation, likely by controlling the amount of free RhoC that is available for activation.
Figure 2. RhoGDI2 suppresses RhoC activation in bladder cells.

A. Extracts from T24 bladder cancer cells with stable expression of control shRNA or shRNA for RhoGDI2 were assayed for levels of RhoC-GTP in response to FBS stimulation (2.5%, 5 min) using Rhotekin pull-down assays. Top, representative western blots. Bottom, results expressed as percent RhoC activation relative to shControl cells treated with FBS presented as the mean± S.E. (n=3). **P<0.01.
B. Extracts from UMUC3 cells with stable expression of GFP or GFP-RhoGDI2 were assayed for levels of RhoC-GTP in response to FBS stimulation (5%, 5 min) using Rhotekin pull-down assays. Top, representative western blots. Bottom, results expressed as percent RhoC activation relative to GFP cells treated with FBS presented as the mean± S.E. (n=3). **P<0.01.
RhoC Promotes Lung Colonization of Bladder Cancer Cells
To link metastasis suppressor activity of RhoGDI2 to suppression of RhoC activation, we first asked whether RhoC is pro-metastatic in bladder cancer cells. Different shRNAs targeting unique sequences were used to knock down RhoC in T24T metastatic bladder cancer cells (Figure 3A). RhoC shRNA #1 (herein identified as shRhoC-1) stably depleted RhoC by approximately 50% in T24T cells, without affecting the expression of RhoA, Rac1 or Cdc42. RhoC shRNA #2 (herein identified as shRhoC-2) almost completely eliminated RhoC in T24T cells, but also triggered a compensatory increase in RhoA protein levels. This compensatory phenomenon has been seen in other cell types (29), and to avoid the potentially confounding effects of increased RhoA expression, we chose to perform the majority of experiments with cells expressing shRhoC-1. These cells would more accurately model the effect of RhoGDI2 in bladder cancer cells as our results show that modulation of RhoGDI2 levels in bladder cancer cells reduced RhoC activation by ~30%. Activation of Rac1 in response to EGF stimulation (Figure 3B) or of RhoA in response to FBS stimulation (Figure 3C) was not affected by RhoC knock down in shRhoC-1 cells, further indicating that knock down of RhoC with shRhoC-1 did not interfere with signaling of these GTPases. Knock down of RhoC slowed down growth and increased the doubling time of cells (15 ± 0.4 h for control versus 16 ± 0.3 h for shRhoC-1 cells, p<0.05) (Figure 3D), significantly decreased invasion of T24T cells through Matrigel (p<0.05) (Figure 3E), and reduced growth in soft agar (p<0.05) (Figure 3F). Similar reductions in invasion and growth in soft agar were seen upon RhoC knock down in Lul2 cells, a second metastatic bladder cancer cell line (Supplementary Figure 2).
Figure 3. Knockdown of RhoC reduces invasion and colony formation of bladder cells.

A. Expression of Rho family GTPases in T24T cells following stable knock down of RhoC by shRNA with different sequences. Representative western blots shown.
B, C. Extracts from T24T bladder cancer cells with stable knockdown of RhoC were assayed for levels of (B) Rac1-GTP in response to EGF stimulation (100 ng/mL, 1 min) or (C) RhoA-GTP in response to FBS stimulation (5% FBS, 5 min) using pull-down assays. Top, representative western blots. Bottom, results expressed as percent Rac1 or RhoA activation relative to shControl cells treated with EGF or FBS presented as the mean± S.E. (n=3). n.s., not significant.
D. Doubling time for T24T cells expressing shCont or shRhoC-1 was determined by counting cells at 24 h intervals for 96 h. Bargraph shows the mean doubling time± S.E. (n=3). *P<0.05.
E. Invasion of T24T cells expressing shCont or shRhoC-1 through matrigel was determined using a transwell assay. Results expressed as percent invasion relative to shControl cells presented as the mean± S.E. *P<0.05. Independent experiments (n=3) were performed in triplicate.
F. Colony formation in soft agar was assessed in T24T cells expressing shCont or shRhoC-1. Top, bargraph indicates the number of colonies formed and is presented as the mean± S.E. *P<0.05. Bottom, representative pictures of colonies. Independent experiments (n=3) were performed in triplicate.
T24T cells expressing either non-targeting control shRNA or shRhoC-1 were injected via tail vein into nude mice and human tumor cell burden in the lung was assessed after 7 weeks. Genomic DNA isolated from mouse lung was analyzed by quantitative real time PCR measurement of the 12p arm of human DNA. This method has been shown to accurately reflect the extent of lung colonization in experimental metastasis assays (16,18). The incidence of lung colonization in mice injected with T24T cells with stable RhoC knock down was significantly less than that for mice injected with T24T cells expressing non-targeting shRNA (4/15 mice for shRhoC-1 versus 10/16 mice for shCont, p=0.05) (Figure 4A). Both zero and non-zero values of human genomic DNA present in the lungs of each animal injected were combined and analyzed. Injection of RhoC knock down cells significantly decreased the amount of human tumor DNA present in all lungs (p=0.028, analysis of 0 and non-zero samples by Mann Whitney test). Figure 4B shows the distribution of non-zero values of human DNA present in mouse lungs injected with T24T cells expressing non-targeting shRNA versus shRhoC-1. The amount of human tumor DNA present in lungs was not statistically significant (p=0.28, analysis of non-zero samples only by Mann Whitney test). A similar trend was seen in animals injected with T24T cells expressing shRhoC-2 (Supplementary Figure 3). We also performed experimental metastasis assays with LuL2 cells expressing non-targeting control shRNA or shRhoC-1 (Supplementary Figure 2A), and we saw a similar decrease in the incidence of lung colonization (3/10 mice for shRhoc-1 versus 9/10 mice for shCont, p=0.003) (Figure 4C). Taken together, we concluded that RhoC knock down reduced lung colonization of bladder cancer cells. This implies some positive role for RhoC in metastasis. Given the effect of RhoC knock down on increasing cell doubling time and decreasing growth in soft agar, we would also predict that RhoC may play a role in primary tumor formation.
Figure 4. Depletion of RhoC reduces incidence of lung colonization of bladder cancer cells.

A. Graph representing incidence of lung colonization 7 weeks after tail vein injection of T24T bladder cancer cells expressing shControl or shRhoC-1. p = 0.05, Barnard’s exact test. Open bar, mice with no lung colonization; shaded bar, mice with lung colonization.
B. Plot comparing lung burden in animals injected with T24T cells expressing shControl or shRhoC-1 as measured by human DNA-specific qPCR amplification of lung genomic DNA. Mean is indicated on a log scale. p=0.28, analysis of non-zero values only, Mann Whitney test.
C. Graph representing incidence of lung colonization 4 weeks after tail vein injection of LuL2 bladder cancer cells expressing shControl or shRhoC-1. p = 0.003, Barnard’s exact test. Open bar, mice with no lung colonization; shaded bar, mice with lung colonization.
Identification of Genes Associated with RhoGDI2 and RhoC in Bladder Cancer Tumors
Our hypothesis was that RhoGDI2 exerts its metastasis suppressor effects on bladder cancer cells at least in part through binding of RhoC and suppression of RhoC activation. In this scenario, knock down of RhoC in bladder cancer cells should phenocopy, to some extent, expression of RhoGDI2 in bladder cancer cells. To identify genes that are commonly regulated by both RhoC and RhoGDI2, we performed gene microarray analysis of RNA extracted from T24T cells expressing shControl or shRhoC-1. The results of this microarray analysis were compared to results generated from RNA extracted from T24T cells expressing a pcDNA3 control plasmid or RhoGDI2 (10). Twelve genes were found to be commonly down-regulated and 50 genes were found to be commonly up-regulated in T24T cells by RhoC knockdown versus RhoGDI2 expression (Figure 5A). Of the 12 down-regulated genes, 3 genes were significantly up-regulated in human bladder tumor specimens, in two cohorts. There were also 14 genes of the 50 up-regulated genes that were significantly down-regulated in human bladder tumor specimens (Supplementary Table 2). Using the 62 genes that were commonly up-regulated and down-regulated by RhoGDI2 expression or RhoC knock down as a signature, we determined that expression of these genes correlate with bladder cancer tumors in two data sets (Figure 5B).
Figure 5. A RhoC/RhoGDI2 signature gene set correlates with human bladder tumors.
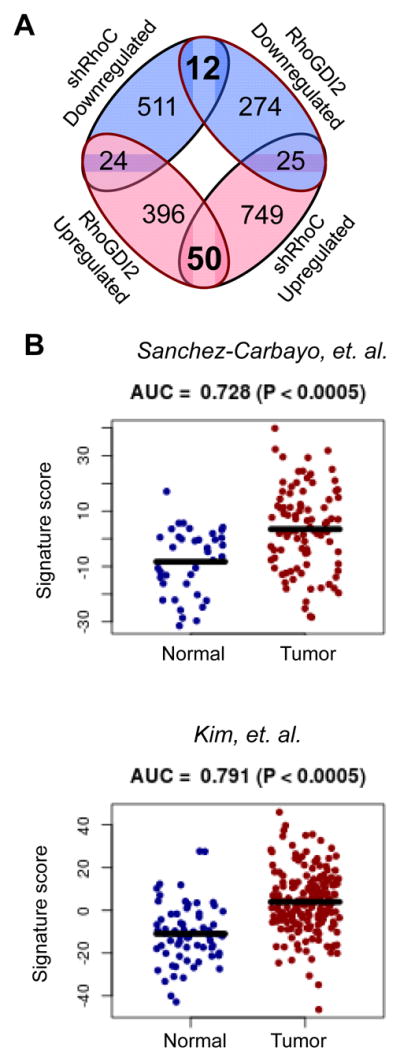
A. Venn diagram illustrating the number of commonly up- and down-regulated genes identified by microarray upon RhoC knock down or RhoGDI2 expression in T24T cells. Blue indicates decreased expression and red indicates increased expression.
B. Ability of the RhoC/RhoGDI2 signature score to distinguish between normal urothelial and bladder tumor samples in the Sanchez-Carbayo et al (n = 129) and Kim et al (n = 255) cohorts. AUC, area under the receiver operating characteristic curve. P-values were calculated by Wilcoxon-Rank Sum Test.
We looked in more detail at the genes commonly down-regulated by both RhoC knock down and RhoGDI2 overexpression, as these genes would be up-regulated when RhoGDI2 is lost in bladder cancer and could potentially be biomarkers for bladder cancer. Of the 12 genes commonly down-regulated, two genes, KRT8, which encodes the protein keratin 8, and MREG, which encodes the protein melanoregulin, were significantly upregulated in tumors with a high fold change (>2) in at least one dataset. Both KRT8 (Figure 6A) and MREG (Figure 6B) were found to have increased expression in bladder cancer tumors when compared to normal bladder samples in two different studies. RNA expression levels of both KRT8 and MREG were down-regulated in T24T cells upon depletion of RhoC or expression of RhoGDI2 (Figure 6C) and in a second cell line, UMUC3, upon expression of RhoGDI2 (Figure 6D), verifying the results of the shRhoC microarray experiment in T24T cells and extending these results to a second cell line.
Figure 6. KRT8 and MREG expression in human normal urothelial tissue and bladder tumor samples.

A, B. Expression of KRT8 (A) and MREG (B) between normal urothelial and bladder tumor samples in Sanchez-Carbayo et al (n = 129) and Kim et al (n = 255) patient cohorts. P-values were calculated by Wilcoxon Rank-Sum Test.
C, D. mRNA expression of KRT8 and MREG in T24T cells with RhoC knock down or RhoGDI2 expression compared to the corresponding control cells (C) or in UMUC3 cells with control or RhoGDI2 expression (D) was determined by quantitative real time PCR. Bar graphs represent the mean± S.E. (n 3) *P<0.05, **P<0.01, ***P<0.001.
DISCUSSION
Here we present evidence that in bladder cancer cells RhoGDI2 binds to RhoC and negatively regulates RhoC activation. RhoC knock down slowed growth of bladder cells, reduced invasion through Matrigel, and reduced anchorage independent growth in soft agar. RhoC has been shown to promote metastasis in a variety of other cell types and models including melanoma, mammary, prostate, and head and neck cancer (25–27,30,31). Here we show that RhoC knockdown decreases the incidence of lung colonization of bladder cancer cells, suggesting that active RhoC promotes metastasis of bladder cancer. The downstream effectors through which RhoC promotes metastasis in bladder cancer are currently unclear. Interestingly, Src and caveolin were shown to positively regulate metastasis in bladder cancer cells through two parallel pathways that converge on RhoA and RhoC to promote Rho-associated serine-threonine kinase (ROCK) activation. Indeed, treatment of animals with the inhibitor Y-27632 concomitantly with tail vein injection of bladder cancer cells abrogated colony formation in lung, supporting the idea that ROCK plays a role in bladder cancer metastasis (32). Further, high levels of RhoC, ROCKI and ROCKII in human bladder cancer specimens were shown to correlate with muscle invasion, lymph node metastasis, and shorter disease free survival times (33). Given that ROCK is a well-known regulator of migration and invasion (34), and that RhoC activates ROCK (35), it is likely that RhoC may activate ROCK to promote lung colonization in our bladder cancer model. Other RhoC effectors besides ROCK such as the kinase PKN3 also have been shown to regulate RhoC mediated metastasis (36). Delineation of the RhoC signaling pathways in bladder cancer and metastasis will be the subject of future studies.
A previous study reported that RhoGDI2 over-expression suppresses lung colonization in the same system used here (8). When RhoGDI2 expression is lost in metastatic bladder cancer, we believe that an increase in RhoC activation may help to drive metastasis. It is interesting to note that mRNA levels of RhoC do not change upon modulation of RhoGDI2 in bladder cancer cells (10), nor do we observe changes in RhoC protein levels upon expression or knock down of RhoGDI2 in bladder cancer cells (Figure 2). This would indicate that RhoGDI2 is not regulating RhoC degradation in contrast to what has been shown for RhoGDI1 with other GTPases (37), nor is RhoGDI2 likely affecting RhoC transcription or translation. Loss of RhoGDI2 in bladder cancer cells would be a way to modulate RhoC activation without changing RhoC expression.
We have identified a set of genes and in particular KRT8 as a gene that is down-regulated upon RhoC depletion or RhoGDI2 expression. Our data show that keratin 8 expression is higher in bladder cancer tumors compared to normal bladder. This is especially interesting as there is some literature linking keratin 8 to cancer (38). For example, keratin 8 expression in circulating tumor cells in patients with renal clear cell carcinoma correlated with the presence of metastasis and poor prognosis used as a prognostic marker in kidney and prostate cancer (39). The presence of keratin 8, 18 and 19 positive prostate cancer cells detected in bone marrow aspirates of patients before surgery also predicted metastatic disease (40). In bladder, Keratin 8 was enriched in proteomic screen of urinary proteins in patients with bladder cancer when compared to normal patients, suggesting that keratin 8 could be a urinary biomarker for bladder cancer (41). Given this, further experiments investigating the link between keratin 8 expression in bladder cancer and development of metastasis are currently warranted, and our experiments thus far point to keratin 8 as a potential prognostic marker for bladder cancer.
Supplementary Material
IMPLICATIONS.
RhoGDI2 suppresses bladder cancer metastatic colonization via negative regulation of RhoC activity, providing a rationale for the development of therapeutics that target RhoC signaling.
Acknowledgments
Financial Support: Supported by National Institutes of Health grant CA143971 to DT, CA910935 to support E.M.G. and the Paul Mellon Urological Cancer Research Institute at the University of Virginia.
Mass spectrometry was performed by Dr. Nicholas E. Sherman of the W.M. Keck Biomedical Mass Spectrometry Laboratory, which is funded by a grant from the University of Virginia Pratt Fund through the School of Medicine. We thank Dr. Stephen D. Turner of the University of Virginia Bioinformatics Core for microarray analysis.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Hall A. Rho family GTPases. Biochem Soc Trans. 2012;40(6):1378–82. doi: 10.1042/BST20120103. [DOI] [PubMed] [Google Scholar]
- 2.Rathinam R, Berrier A, Alahari SK. Role of Rho GTPases and their regulators in cancer progression. Front Biosci (Landmark Ed) 2011;16:2561–71. doi: 10.2741/3872. [DOI] [PubMed] [Google Scholar]
- 3.Ridley AJ. RhoA, RhoB and RhoC have different roles in cancer cell migration. J Microsc. 2013;251(3):242–9. doi: 10.1111/jmi.12025. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12(8):493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Theodorescu D, Sapinoso LM, Conaway MR, Oxford G, Hampton GM, Frierson HF. Reduced expression of metastasis suppressor RhoGD12 is associated with decreased survival for patients with bladder cancer. Clin Cancer Res. 2004;10(11):3800–06. doi: 10.1158/1078-0432.CCR-03-0653. [DOI] [PubMed] [Google Scholar]
- 6.Ma L, Xu G, Sotnikova A, Szczepanowski M, Giefing M, Krause K, et al. Loss of expression of LyGDI (ARHGDIB), a rho GDP-dissociation inhibitor, in Hodgkin lymphoma. Br J Haematol. 2007;139(2):217–23. doi: 10.1111/j.1365-2141.2007.06782.x. [DOI] [PubMed] [Google Scholar]
- 7.Niu H, Li H, Xu C, He P. Expression profile of RhoGDI2 in lung cancers and role of RhoGDI2 in lung cancer metastasis. Oncol Rep. 2010;24(2):465–71. doi: 10.3892/or_00000880. [DOI] [PubMed] [Google Scholar]
- 8.Gildea JJ, Seraj MJ, Oxford G, Harding MA, Hampton GM, Moskaluk CA, et al. RhoGD12 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 2002;62(22):6418–23. [PubMed] [Google Scholar]
- 9.Said N, Smith SC, Sanchez-Carbayo M, Theodorescu D. RhoGDI2 suppresses metastasis via reduction of versican expression and macrophage infiltration. J Clin Invest. 2012;122(4):1503–18. doi: 10.1172/JCI61392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Titus B, Frierson HF, Conaway M, Ching K, Guise T, Chirgwin J, et al. Endothelin axis is a target of the lung metastasis suppressor gene RhoGD12. Cancer Res. 2005;65(16):7320–27. doi: 10.1158/0008-5472.CAN-05-1403. [DOI] [PubMed] [Google Scholar]
- 11.Said N, Smith S, Sanchez-Carbayo M, Theodorescu D. Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. J Clin Invest. 2011;121(1):132–47. doi: 10.1172/JCI42912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y, Moissogiu K, Wang H, Wang X, Frierson HF, Schwartz MA, et al. Src phosphorylation of RhoGDI2 regulates its metastasis suppressor function. Proc Natl Acad Sci U S A. 2009;106(14):5807–12. doi: 10.1073/pnas.0810094106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorvel JP, Chang TC, Boretto J, Azuma T, Chavrier P. Differential properties of D4/LyGDI versus RhoGDI: phosphorylation and rho GTPase selectivity. FEBS Lett. 1998;422(2):269–73. doi: 10.1016/s0014-5793(98)00020-9. [DOI] [PubMed] [Google Scholar]
- 14.Scherle P, Behrens T, Staudt LM. LY-GDI, A GDP-Dissociation Inhibitor of the RhoA GTP-Binding Protein, is Expressed Preferentially in Lymphocytes. Proc Natl Acad Sci U S A. 1993;90(16):7568–72. doi: 10.1073/pnas.90.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moissoglu K, McRoberts KS, Meier JA, Theodorescu D, Schwartz MA. Rho GDP Dissociation Inhibitor 2 Suppresses Metastasis via Unconventional Regulation of RhoGTPases. Cancer Res. 2009;69(7):2838–44. doi: 10.1158/0008-5472.CAN-08-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Overdevest JB, Thomas S, Kristiansen G, Hansel DE, Smith SC, Theodorescu D. CD24 offers a therapeutic target for control of bladder cancer metastasis based on a requirement for lung colonization. Cancer Res. 2011;71(11):3802–11. doi: 10.1158/0008-5472.CAN-11-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124(6):1283–98. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 18.Nitz MD, Harding MA, Theodorescu D. Invasion and metastasis models for studying RhoGDI2 in bladder cancer. Methods Enzymol. 2008;439:219–33. doi: 10.1016/S0076-6879(07)00417-X. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24(5):778–89. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 20.Kim WJ, Kim EJ, Kim SK, Kim YJ, Ha YS, Jeong P, et al. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Mol Cancer. 2010;9:3. doi: 10.1186/1476-4598-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller JA, Cai C, Langfelder P, Geschwind DH, Kurian SM, Salomon DR, et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics. 2011;12:322. doi: 10.1186/1471-2105-12-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Platko JV, Leonard DA, Adra CN, Shaw RJ, Cerione RA, Lim B. A single residue can modify target-binding affinity and activity of the functional domain of the Rho-subfamily GDP dissociation inhibitors. Proc Natl Acad Sci U S A. 1995;92(7):2974–8. doi: 10.1073/pnas.92.7.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Theodorescu D, Cornil I, Fernandez BJ, Kerbel RS. Overexpression of normal and mutated forms of HRAS induces orthotopic bladder invasion in a human transitional cell carcinoma. Proc Natl Acad Sci U S A. 1990;87(22):9047–51. doi: 10.1073/pnas.87.22.9047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406(6795):532–5. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 26.Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, et al. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19(17):1974–9. doi: 10.1101/gad.1310805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iiizumi M, Bandyopadhyay S, Pai SK, Watabe M, Hirota S, Hosobe S, et al. RhoC promotes metastasis via activation of the Pyk2 pathway in prostate cancer. Cancer Res. 2008;68(18):7613–20. doi: 10.1158/0008-5472.CAN-07-6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenthal DT, Zhang J, Bao L, Zhu L, Wu Z, Toy K, et al. RhoC impacts the metastatic potential and abundance of breast cancer stem cells. PLoS One. 2012;7(7):e40979. doi: 10.1371/journal.pone.0040979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho TTG, Stultiens A, Dubail J, Lapiere CM, Nusgens BV, Colige AC, et al. RhoGDI alpha-dependent balance between RhoA and RhoC is a key regulator of cancer cell tumorigenesis. Mol Biol Cell. 2011;22(17):3263–75. doi: 10.1091/mbc.E11-01-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Islam M, Lin G, Brenner JC, Pan Q, Merajver SD, Hou Y, et al. RhoC expression and head and neck cancer metastasis. Mol Cancer Res. 2009;7(11):1771–80. doi: 10.1158/1541-7786.MCR-08-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Unsal-Kacmaz K, Ragunathan S, Rosfjord E, Dann S, Upeslacis E, Grillo M, et al. The interaction of PKN3 with RhoC promotes malignant growth. Mol Oncol. 6(3):284–98. doi: 10.1016/j.molonc.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas S, Overdevest JB, Nitz MD, Williams PD, Owens CR, Sanchez-Carbayo M, et al. Src and Caveolin-1 Reciprocally Regulate Metastasis via a Common Downstream Signaling Pathway in Bladder Cancer. Cancer Res. 2011;71(3):832–41. doi: 10.1158/0008-5472.CAN-10-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamai T, Tsujii T, Arai K, Takagi K, Asami H, Ito Y, et al. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin Cancer Res. 2003;9(7):2632–41. [PubMed] [Google Scholar]
- 34.Rath N, Olson MF. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012;13(10):900–8. doi: 10.1038/embor.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301(1):43–9. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 36.Unsal-Kacmaz K, Ragunathan S, Rosfjord E, Dann S, Upeslacis E, Grillo M, et al. The interaction of PKN3 with RhoC promotes malignant growth. Mol Oncol. 2012;6(3):284–98. doi: 10.1016/j.molonc.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12(5):477–U136. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karantza V. Keratins in health and cancer: more than mere epithelial cell markers. Oncogene. 2011;30(2):127–38. doi: 10.1038/onc.2010.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bluemke K, Bilkenroth U, Meye A, Fuessel S, Lautenschlaeger C, Goebel S, et al. Detection of circulating tumor cells in peripheral blood of patients with renal cell carcinoma correlates with prognosis. Cancer Epidemiol Biomarkers Prev. 2009;18(8):2190–4. doi: 10.1158/1055-9965.EPI-08-1178. [DOI] [PubMed] [Google Scholar]
- 40.Weckermann D, Polzer B, Ragg T, Blana A, Schlimok G, Arnholdt H, et al. Perioperative activation of disseminated tumor cells in bone marrow of patients with prostate cancer. J Clin Oncol. 2009;27(10):1549–56. doi: 10.1200/JCO.2008.17.0563. [DOI] [PubMed] [Google Scholar]
- 41.Lei T, Zhao X, Jin S, Meng Q, Zhou H, Zhang M. Discovery of potential bladder cancer biomarkers by comparative urine proteomics and analysis. Clin Genitourin Cancer. 2013;11(1):56–62. doi: 10.1016/j.clgc.2012.06.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.