

Abstract
It is well documented that metabolic syndrome (i.e. a group of risk factors, such as abdominal obesity, elevated blood pressure, elevated fasting plasma glucose, high serum triglycerides and low cholesterol level in high-density lipoprotein), which raises the risk for heart disease and diabetes, is associated with increased reactive oxygen and nitrogen species (ROS/RNS) generation. ROS/RNS can modulate cardiac NO signalling and trigger various adaptive changes in NOS and antioxidant enzyme expressions/activities. While initially these changes may represent protective mechanisms in metabolic syndrome, later with more prolonged oxidative, nitrosative and nitrative stress, these are often exhausted, eventually favouring myocardial RNS generation and decreased NO bioavailability. The increased oxidative and nitrative stress also impairs the NO-soluble guanylate cyclase (sGC) signalling pathway, limiting the ability of NO to exert its fundamental signalling roles in the heart. Enhanced ROS/RNS generation in the presence of risk factors also facilitates activation of redox-dependent transcriptional factors such as NF-κB, promoting myocardial expression of various pro-inflammatory mediators, and eventually the development of cardiac dysfunction and remodelling. While the dysregulation of NO signalling may interfere with the therapeutic efficacy of conventional drugs used in the management of metabolic syndrome, the modulation of NO signalling may also be responsible for the therapeutic benefits of already proven or recently developed treatment approaches, such as ACE inhibitors, certain β-blockers, and sGC activators. Better understanding of the above-mentioned pathological processes may ultimately lead to more successful therapeutic approaches to overcome metabolic syndrome and its pathological consequences in cardiac NO signalling.
Linked Articles
This article is part of a themed section on Pharmacology of the Gasotransmitters. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-6
Tables of Links
| TARGETS | |
|---|---|
| Enzymesa | GPCRsb |
| AMPK | Angiotensin AT1 receptor |
| Caspase-3 | β1-adrenoceptor |
| DPP-4, dipeptidyl peptidase-4 | β3-adrenoceptor |
| eNOS | Ion channelsc |
| iNOS | ATP-sensitive K+ channels, Kir6.x |
| nNOS | RyR, ryanodine receptors |
| PDE1 | Transportersd |
| PDE2 | GLUT 4, glucose transporter (SLC2A4) |
| PDE5 | Nuclear hormone receptorse |
| PKG | PPARα |
| ROCK, Rho kinase | PPARβ/δ |
| SERCA | PPARγ |
| sGC, soluble guanylate cyclase | |
| Xanthine oxidase/dehydrogenase |
| LIGANDS | |
|---|---|
| 15D-PGJ2, 15-deoxy-Δ12,14-prostaglandin J2 | Leptin |
| ADMA, asymmetric dimethylarginine | L-NAME |
| Allopurinol | Lovastatin |
| Angiotensin II | Melatonin |
| Ataciguat | Metformin |
| Atenolol | Nebivolol |
| Atorvastatin | Niacin, nicotinic acid |
| Atrial natriuretic peptide | NO |
| BH4, tetrahydrobiopterin | Pioglitazone |
| Captopril | Pravastatin |
| cGMP | Rosiglitazone |
| Cinaciguat | Rosuvastatin |
| Enalapril | Sildenafil |
| Fasudil | Simvastatin |
| Fluvastatin | Tadalafil |
| GIP | Vardenafil |
| GLP-1 | Vitamin C |
| GW7647 | Zofenopril |
| Insulin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,d,eAlexander et al., 2013a,b,c,d,e).
Introduction
Although NO was discovered decades ago, scientific interest in this gasotransmitter is continuously increasing. Enzymic and non-enzymic formation of NO and cGMP-dependent and independent NO signalling has been reviewed in detail in the current Themed Issue (Csonka et al., 2015) and elsewhere (Ferdinandy and Schulz, 2003; Stasch et al., 2011; Tang et al., 2013; Rassaf et al., 2014). Intercellular and intracellular NO signalling is very complex, reflecting its many pathways and interactions with other free radicals to form additional signalling molecules. Reactive oxygen species (ROS), especially the superoxide anion radical, can react with NO non-enzymically with an extremely high-rate constant limited only by diffusion. These reactions produce peroxynitrite (ONOO−) and other highly reactive oxygen and nitrogen species (ROS/RNS), which in concert with NO act as signalling molecules and also account for oxidative, nitrative and nitrosative stress (Ferdinandy, 2006; Pacher et al., 2007; Pechanova and Simko, 2009). Most techniques available for the measurement of NO and its reactive metabolites have numerous technical limitations (reviewed in this Themed Issue by Csonka et al., 2015) which further complicates the interpretation of results on the role of NO signalling in physiology and pathology.
NO plays an important role in the regulation of cardiovascular functions in health and disease by, for example, promoting vasodilation, inhibiting vascular smooth muscle cell growth, platelet aggregation, and leukocyte adhesion, apart from by regulating myocardial function and providing cardioprotection (see Pacher et al., 2007; Ferdinandy and Schulz, 2003; and reviewed in this Themed Issue by Andreadou et al., 2015). The metabolic syndrome, comprising hypertension, hyperlipidaemia and insulin resistance/diabetes, is the major cardiovascular risk factor and thus accounts for leading causes of morbidity and mortality in industrialized societies. Publications on the role of NO-related pathways in these pathologies are continuously growing. In this review, we attempt to summarize the knowledge related to the role of NO signalling in the heart in the presence of the major cardiovascular risk factors that are associated with the metabolic syndrome. Our review focuses on the effect of the metabolic syndrome on NO signalling in the non-ischaemic heart.
The role of NO in myocardial ischaemia/reperfusion injury and cardioprotection by ischaemic conditioning in the healthy heart and in different co-morbidities is reviewed in detail elsewhere (Ferdinandy and Schulz, 2003; Andreadou et al., 2015). In brief, NO itself protects the heart against ischaemia/reperfusion injury. However, accumulation of excess NO during prolonged ischaemia contributes to reperfusion injury via an increased oxidative/nitrative stress. The role of endogenous NO in cardioprotection induced by ischaemic preconditioning is still controversial (Csonka et al., 1999; Nakano et al., 2000; Post et al., 2000). Nevertheless, it seems that mild oxidative/nitrative stress induced by exogenous or endogenous NO is necessary to trigger both pre- and post-conditioning (Nakano et al., 2000; Csonka et al., 2001; Heusch, 2001; Kupai et al., 2009).
NO signalling in the heart
In the heart tissue, coronary and endocardial endothelial cells and cardiac myocytes are major sources of NO. However, NO may also derive from intracardiac ganglia and some nerve fibres located close to cardiac blood vessels. Endothelial NOS (eNOS) is expressed typically in the coronary and cardiac endothelium, whereas neuronal NOS (nNOS) is mainly located in the cardiac myocytes (see Pacher et al., 2007; Tirziu and Simons, 2008). In coronary vascular endothelial cells, the eNOS-caveolin-1 interaction in the caveolae is important for normal eNOS activity (Feron and Balligand, 2006). The physiological triggers for NO release from endothelial cells are the flow-induced shear stress and mechanical deformations of the endothelium during the cardiac cycle (Michel, 2010). In cardiac myocytes, eNOS is co-localized with caveolin-3 in the T tubules of plasmalemmal caveolae, nNOS is localized in the sarcoplasmic reticulum (Shah and MacCarthy, 2000), and the putative mitochondrial NOS (mtNOS) in cardiac mitochondria (Dedkova and Blatter, 2009). The normal intracellular function of eNOS and nNOS in cardiomyocytes depends on discrete coupling mechanisms in the local cytosolic environments. These mechanisms can be affected by altered metabolism due to the metabolic syndrome (see Huang, 2009; Pechanova and Simko, 2010).
In fact, NO generated by inducible NOS (iNOS) may have its origin in the myocytes or neutrophils that migrate in the proximity of myocytes during inflammation and also in activated fibroblasts. iNOS, when expressed in cardiac myocytes, can regulate the response to β-adrenoceptor stimulation. However, as the neutrophils migrate to sites close to the myocytes, iNOS becomes essential for the ability of neutrophils to damage myocytes (Poon et al., 2003). Indeed, an increase in iNOS expression in the heart with substrate limitation leads to uncoupled iNOS producing superoxide anions and contributing to contractile dysfunction (Heusch et al., 2010). These findings demonstrate that cellular source and local cytosolic environment strongly modulate the effects of different NOS isoforms, as reviewed elsewhere (Tirziu and Simons, 2008; Huang, 2009; Pechanova and Simko, 2010).
NO may affect myocytes in a number of different ways. NO signalling via cGMP-dependent or independent pathways modulates the function of downstream proteins via specific post-translational modifications, such as phosphorylation by cGMP-dependent PK (PKG) or S-nitrosylation. Interestingly, an increase in intracellular cGMP induced by natriuretic peptides or cGMP analogues was recently shown to modulate both sarcolemmal and mitochondrial ATP-sensitive K+ channel opening in ventricular cardiomyocytes suggesting further diverse actions of NO (Burley et al., 2014).
NO also affects mitochondrial function and dynamics, thus regulating cardiac energy metabolism. Under pathological conditions, it may also contribute to the development of myocardial dysfunction and heart failure (Davidson and Duchen, 2006; Azevedo et al., 2013; Dai et al., 2013; Miller et al., 2013). Localization of NO production within mitochondria seems to provide a distinct reciprocal regulation between mtNOS and intramitochondrial Ca2+, pH, L-arginine and oxygen. NO produced by the putative mtNOS may represent a mechanism of fine regulation of the respiratory complexes, enzymes of the citric acid cycle and energy metabolism as well (see Zaobornyj and Ghafourifar, 2012; Csonka et al., 2015; Andreadou et al., 2015). However, the existence of mitochondrial mtNOS is still a controversial issue (Pacher et al., 2007), and NO, which rapidly diffuses into mitochondria from other cellular compartments or cells, is sufficient to efficiently regulate energy metabolism. Despite these facts, very few studies investigated the role of NO signalling in mitochondrial function in hearts with the metabolic syndrome.
The main physiological role of NO derived from eNOS and nNOS includes reduction of contractile frequency of cardiomyocytes, attenuation of cardiac contractility, acceleration of relaxation and increasing distensibility of cardiomyocytes, and improvement of the efficiency of myocardial oxygen consumption. In conditions of enhanced cardiac reserve and cardiac hypertrophy, NO derived from eNOS modulates receptor-mediated signalling which ultimately leads to a moderate inhibition of cardiac contractility (Shah and MacCarthy, 2000; Yue and Yu, 2011). NO derived from the complex of nNOS-ryanodine receptor (RyR) stabilizes RyR calcium release and increases the efficiency of Ca2+ cycling in sarcoplasmic reticulum by the inhibitory effects (Yue and Yu, 2011). In swine, intracoronary infusion of an NO synthesis inhibitor, N-ω-nitro-L-arginine, markedly decreased left ventricle (LV) function, while peak LV pressure and mean coronary arterial pressure were increased (Post et al., 2001). Similarly, in healthy humans, inhibition of endogenous NO release also reduced, whereas replenishment with exogenous NO increased left ventricular function, further emphasizing that NO contributes to normal left ventricular function (Rassaf et al., 2006). Thus, dysfunction of NOS induced by altered expression, location, coupling and activity may contribute to the contractile dysfunction, adverse remodelling and myocardial hypertrophy – changes associated with various cardiac disease conditions, such as heart failure and infarction (Tang et al., 2013).
Interestingly, eNOS expression was not affected by cardiovascular risk factors like hypertension, obesity and insulin resistance (Fulton et al., 2004; Bouvet et al., 2007), and paradoxically was found to be increased in various pathological states associated with oxidative stress (Li et al., 2002; Ding et al., 2007; Zhen et al., 2008). This effect may be partly mediated by limiting the availability of NO, thereby exerting a negative feedback on NOS expression through activation of NF-κB (Zhen et al., 2008; Pechanova and Simko, 2009; 2010,; Vrankova et al., 2009) (Figure 1).
Figure 1.
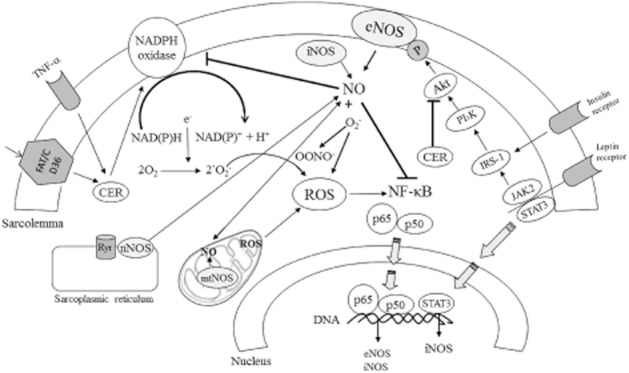
NO signalling and metabolic syndrome-related pathways. ROS generated by NADPH oxidases and other sources (e.g. mitochondria, XO, uncoupled NOS, among others) leads to increased NF-κB activity followed by eNOS and iNOS up-regulation. eNOS produces NO which prevents activation of both NADPH oxidase and NF-κB. The leptin/STAT3 pathway may also up-regulate the gene for iNOS whereas the leptin/JAK2/IRS-1 pathway increases eNOS activity via Akt stimulation, as does insulin. Increased circulating free fatty acids lead to ceramide elevation with increasing effects on NADPH oxidase activity and diminishing effects on Akt activation. NO produced by neuronal NOS (nNOS) and putative mtNOS may affect heart function in metabolic syndrome by different specific routes.
In conclusion, signalling functions of NO produced by specific NOS isoforms seem to be compartmentalized in distinct cellular microdomains and thus modulate cardiac function differently. Moreover, they may be further affected by risk factors of the metabolic syndrome.
NO signalling in the hypertensive heart
Left ventricular remodelling and heart failure represent major pathological consequences of chronic arterial hypertension. During the development of hypertension, differential signals and metabolic abnormalities lead to the structural remodelling of the cardiovascular system, as characterized by myocardial hypertrophy and/or fibrosis, and coronary artery wall hyperplasia which finally result in heart injury known as cardiomyopathy (Kristek and Gerová, 1996; Babal et al., 1997; Pechanova et al., 1997; Tribulova et al., 2000; Cebova and Kristek, 2011). Pathological remodelling of the hypertensive heart is due to an imbalance of stimulatory and inhibitory signals of tissue proliferation. Angiotensin II (Ang II), aldosterone or endothelin, with their vasoconstrictor and pro-proliferative effects, stand on one side of the balance and NO, prostacyclin, bradykinin or atrial natriuretic peptide, exerting vasodilating and antiproliferative activities, provide the counteracting factors (Swynghedauw, 1999; Cuspidi et al., 2006; Pechanova and Simko, 2010). NO antagonizes the effects of Ang II on vascular tone, cell growth and renal sodium excretion, while it down-regulates the synthesis of ACE and angiotensin AT1 receptor. On the other hand, Ang II decreases NO bioavailability by promoting oxidative stress (Zhou et al., 2004). Mice infused with Ang II displayed an increase in blood pressure, cardiac hypertrophy and fibrosis associated with enhanced collagen I content, TGF-β1 activity and endoplasmic reticulum stress markers, which were, however, blunted after endoplasmic reticulum stress inhibition (Kassan et al., 2012). Recently, however, Jin et al. (2012) demonstrated that myocardial nNOS is up-regulated by Ang II which functions as an early adaptive mechanism to attenuate NADPH oxidase activity and facilitate myocardial relaxation by promoting the cGMP/PKG pathway. It was also documented that activation of this pathway by novel soluble guanylate cyclase (sGC) stimulators, including riociguat (BAY 63-2521), attenuates systemic hypertension and systolic dysfunction, as well as fibrotic tissue remodelling in the myocardium in a rodent model of pressure and volume overload (Geschka et al., 2011). This is in line with earlier data showing impaired NO-sGC signalling pathways in hypertension and heart failure, and beneficial effects of sGC stimulators/activators in preclinical models of hypertension in attenuating myocardial hypertrophy and remodelling (Evgenov et al., 2006; Stasch et al., 2011). Validating this concept, recent clinical trials with riociguat in pulmonary hypertension and chronic thromboembolic pulmonary hypertension showed encouraging results, which lead to the FDA approval of the drug for these indications (Ghofrani et al., 2013a,b).
It is generally believed that increased production of ROS plays an important role in the pathology of hypertension, but so far the limited number of clinical studies using non-specific antioxidants yielded mixed results. Complicating the picture, it should also be noted that temporarily increased ROS generation in hypertension is not necessarily harmful, as it may stimulate the activity of the antioxidant defence system and improve the NO signalling pathway, resulting in the establishment of a new equilibrium between increased oxidative load and the stimulated NO pathways, thus maintaining sufficient NO availability (Dröge, 2002). However, in hypertension associated with obesity or diabetes, ROS may favour activation of pro-inflammatory NF-κB-dependent pathways (Figure 1). In these conditions, activation of NF-κB increases levels of cytokines such as IL-6 and TNF-α that may affect the phosphorylation of tyrosine kinases and decrease NOS activity with a final decrease in NO generation (see Belin de Chantemele and Stepp, 2012).
In conclusion, increased ROS formation during hypertension may activate NF-κB and promote pro-inflammatory and pro-oxidant changes (increased expression of TNF-α, COX2, iNOS, NADPH oxidase, etc.) or compensatory adaptive mechanisms (increased expression of eNOS and antioxidant enzymes). Prolonged ROS/RNS formation may also lead to uncoupling of eNOS/iNOS and impaired NO-sGC signalling in hypertensive cardiovascular system.
NO signalling in the obese/hyperlipidaemic heart
In obesity, cardiac output increases to serve the larger body mass of the obese individual (Kardassis et al., 2012). The increase in cardiac output is due to a larger blood volume resulting in elevated venous return and an increased activation of the sympathetic nervous system, both prevalent in the obese population. An increase in cardiac output elevates cardiac oxygen consumption. Consequently, the need for perfusion is increased (Alvarez et al., 2002; Frohlich and Susic, 2008). In mice fed a high-fat diet, obesity suppressed left ventricular ejection fraction, increased left ventricular remodelling, and led to diminished circulating endothelial progenitor cells level and impaired recovery of damaged endothelium (Tsai et al., 2012).
The importance of two adipocyte-derived hormones – leptin and angiotensinogen – in the pathological consequence of obesity has been highlighted (Coatmellec-Taglioni and Ribière, 2003). Leptin regulates energy balance and metabolism by a variety of peripheral and central mechanisms through specific cell surface receptors (Koh et al., 2008). Leptin infusion was shown to reduce blood pressure and heart rate, which may be reversed by an increased NO synthesis (Frühbeck, 1999). In vitro studies demonstrated that leptin elicited endothelium-dependent NO-mediated vasorelaxation in rats (Lembo et al., 2000). In the mouse heart, disruption of leptin signalling may contribute to obesity-related cardiac disease, as leptin-deficient (ob/ob) mice display cardiac hypertrophy, increased cardiac apoptosis and reduced survival. These changes were linked to decreased cardiac expression of nNOS and NO production, with a concomitant increase in xanthine oxidase (XO) activity and oxidative stress, resulting in nitroso-redox imbalance (Saraiva et al., 2007). Furthermore, cardiac β3-adrenoreceptor expression and function were shown to be dependent on leptin as they were severely diminished in the same model (ob/ob mice). It was proposed that diminished β3-adrenoreceptor signalling may be the critical element to explain the direct effects of leptin on the myocardium and suggest an important role of leptin in obesity-related cardiac hypertrophy and heart failure (Larson et al., 2012). Leptin may up-regulate iNOS to generate large amounts of NO that induce nitrosative and nitrative stress and impair endothelial and myocyte functions (Koh et al., 2008). In ventricular myocytes isolated from male Sprague-Dawley rats, leptin-induced NO generation inhibited myocyte contraction which was prevented by the NOS inhibitor L-NAME (Nickola et al., 2000). In addition, hyperleptinaemia may result in the overdrive of hypothalamus-pituitary-adrenal axis (HPA axis) and the sympathetic nervous system, as well as in impaired insulin secretion and insulin resistance. HPA axis overdrive would account for metabolic abnormalities such as central adiposity, hyperglycaemia, dyslipidemia, hypertension and other cardiovascular diseases which are well-known clinical aspects of the metabolic syndrome (Peters et al., 2002).
Cardiac lipotoxicity caused by the accumulation of lipids has been well described in rodent models of obesity, hyperlipidaemia and diabetes (Zhou et al., 2000; Chiu et al., 2001; 2005; Young et al., 2002). Feeding mice a palmitate-rich diet led to the accumulation of medium- and long-chain ceramides and sphingomyelins, which were incorporated into cellular membrane, thus changing the micro-domain structure of the plasma membrane of cardiomyocytes. The palmitate-rich diet also resulted in a decreased expression of caveolins, structural components of plasmalemmal rafts, the caveole (Knowles et al., 2011; 2013). In addition, ceramides may activate NADPH oxidase leading to an increased oxidative stress (Zhang et al., 2003) (Figure 1). In cardiomyocytes, eNOS localizes to caveolae, which contains β-adrenoceptors and L-type calcium channels as well (Garcia-Cardena et al., 1996; Feron and Balligand, 2006). The co-localization of caveolin-3 and eNOS may facilitate both eNOS activation by cell surface receptors as well as NO release at the cell surface for intercellular signalling (Feron and Balligand, 2006). Immunohistochemistry findings in human cardiac tissue samples from obese humans showed a drastic reduction of caveolin-3 expression in cardiomyocytes (Knowles et al., 2013), further signifying the role of caveolin proteins in obesity.
In conclusion, it seems that the dual effect of leptin in the obese heart depends on eNOS or iNOS activation by different mechanisms. Elevation of ceramide levels in obesity may inhibit eNOS activity by decreasing caveolin proteins and promoting oxidative stress.
NO signalling in the hypercholesterolaemic heart
It is well documented that hypercholesterolaemia profoundly affects cardiac NO metabolism. It has been previously reported that in cholesterol-fed rats, cardiac NO level decreases (Ferdinandy et al., 1997; Giricz et al., 2003; Onody et al., 2003) and that hypercholesterolaemia blunts activity of downstream signalling elements of NO as indicated by a lower PKG activity (Giricz et al., 2009). Reports on the effect of hypercholesterolaemia on the phosphorylation of myocardial eNOS, which reflects its activity, however, are controversial. In cholesterol-fed rats, Zhang showed a decreased p-eNOS level in parallel with an elevated apoptosis (Zhang et al., 2012); meanwhile, in hearts of hypercholesterolaemic LDLr(−/−) mice, eNOS phosphorylation was unchanged (Ou et al., 2011). Similarly, eNOS protein concentrations were found to be unchanged in cholesterol-fed rabbits (Rajamannan et al., 2005) and rats (Giricz et al., 2003). These discrepancies might be attributed to the vast differences between the animal models. It has been also uncovered that the decrease in NO content in hypercholesterolaemic animals is supposedly not due to a decreased activity of NOS isoenzymes, but instead a result of an increased clearance of NO, as assessed by elevated markers of oxidative stress, such as dityrosine, nitrotyrosine (Giricz et al., 2003) and superoxide anion formation due to at least, in part, XO activity (Onody et al., 2003), and elevated expression and activity of NADPH oxidase (Onody et al., 2003; Varga et al., 2013). These reports were confirmed by Stokes et al. (2009) who found cardiac S-nitrosothiol (SNO) levels elevated and cardiac nitrite levels decreased in hypercholesterolaemic mice. In genetic models of hypercholesterolaemia, similar findings were reported. In human apoB100 transgenic mice, cholesterol-enriched diet increased cardiac superoxide anion generation and NADPH oxidase expression in parallel with an elevated cardiac nitrotyrosine level (Csont et al., 2007). LDLr(−/−) mice also have a higher net production of ROS and susceptibility to develop membrane permeability transition, and increased ROS production in mitochondria can be observed (Oliveira et al., 2005). These findings strongly emphasize that cardiac NO production is diminished, while its elimination is accelerated in diet-induced and genetic models of hypercholesterolaemia as well. Meanwhile, there is an apparent dearth of reports on the successful pharmacological restoration of hindered NO-related mechanisms: fasudil, a selective Rho-associated PK (ROCK) inhibitor elevated activity of antioxidant enzymes and the expression of eNOS as well as cardiac NO, and elsewhere atorvastatin increased eNOS protein concentrations and serum nitrite concentrations in cholesterol-fed rabbits (Rajamannan et al., 2005). This scarcity of direct evidence is quite interesting, especially in view of the high number of antioxidant and anti-hyperlipidaemic treatments that have been under development recently. Therefore, it is likely that novel pharmacological targets will have to be explored aiming to restore cardiac NO homeostasis in hypercholesterolaemia.
One can speculate that disturbed NO metabolism might affect cardiac function. Indeed, it has been demonstrated in guinea pigs fed with a cholesterol-enriched diet that increased plasma XO activities were associated with a profound myocardial and coronary endothelial dysfunction (Schwemmer et al., 2000). Similarly, cholesterol feeding resulted in the deterioration of cardiac function in rats (Onody et al., 2003). This notion is further supported by other studies where positive chronotropic effect of atropine was selectively lost in genetically hypercholesterolaemic apoE−/− mice, which was restored after a rosuvastatin treatment (Pelat et al., 2003). This latter paper also reported that cardiac expression of caveolin-1 was elevated in apoE−/− mice, further evidencing a disturbed NO metabolism in hypercholesterolaemia. Similarly, LDLr(−/−) mice demonstrated a decrease in left atrial contractility and eNOS expression relative to wild-type mice. Interestingly, LDLr(−/−) mice fed with an atherogenic diet for 15 days showed increased left ventricular mass and enhanced expression of NOS isoforms, which was reversed by the administration of S-nitroso-N-acetylcysteine (Garcia et al., 2008). These results highlight that, although it is well studied, the contribution of disturbed NO signalling to the deteriorated cardiac function in hypercholesterolaemia is not completely understood.
Isolated hypercholesterolaemia in humans is rarely seen; however, it is a major contributor to numerous pathological conditions, such as atherosclerosis and diabetes. NO metabolism in the human heart has been studied in even rarer cases. In hypercholesterolaemic patients, tetrahydrobiopterin (BH4) attenuated acetylcholine (ACh)-induced decrease in coronary diameter and restored ACh-induced increase in coronary blood flow, which was not shown in normocholesterolaemic patients (Fukuda et al., 2002). Asymmetric dimethylarginine (ADMA) is an endogenous NOS inhibitor and an established cardiovascular risk factor in adults (Wu, 2009; Wu et al., 2009). Serum concentration of ADMA is elevated in hypercholesterolaemic adults, which contributes to NO-dependent endothelial dysfunction (Böger et al., 1998; for review, see Horowitz and Heresztyn, 2007), but not in children with hypercholesterolaemia type II, possibly due to an increase in dimethylarginine dimethylaminohydrolase activity (Chobanyan-Jürgens et al., 2012). However, whether ADMA influences NO bioavailability in the heart, it has yet to be assessed.
In addition to decreased NO bioavailability, the NO-sGC signalling is also pathologically impaired in atherosclerosis, which can be successfully restored by novel sGC stimulators/activators in preclinical rodent models of atherosclerosis and restenosis, where these drugs attenuate inflammation and other pathological changes (Evgenov et al., 2006; Stasch et al., 2011).
In conclusion, in animal models and humans, hypercholesterolaemia hinders cardiac NO metabolism and, in these conditions, increased oxidative stress plays a major role. Furthermore, diminished NO availability and, most likely, impaired NO-sGC signalling in the heart tissue manifests in deteriorated cardiac function and would contribute to the development of other cardiovascular pathologies.
NO signalling in the diabetic heart
In diabetic patients, independent of vascular complications, a specific form of cardiomyopathy develops known as diabetic cardiomyopathy. Many factors may contribute to the evolution of this pathology, including metabolic disturbances (glucotoxicity, lipotoxicity), inflammatory processes, mitochondrial uncoupling, enhanced oxidative stress and deteriorated NO signalling (Pacher et al., 2005). Several publications highlight the role of altered NO metabolism in diabetic cardiomyopathy, but surprisingly there is limited information on the direct measurement of cardiac NO levels obtained by strictly NO-specific methods (see Csonka et al., 2015). As assessed by electron paramagnetic resonance spectrometry, a gold standard NO-specific method, NO level was increased in the hearts of streptozotocin-induced diabetic rats (Amour et al., 2007). In line with this finding, an increase in cardiac NO metabolites (nitrite, nitrate) has been reported in the Goto-Kakizaki rat model of type 2 diabetes (Desrois et al., 2010). Although these reports indicate that cardiac NO metabolism is influenced by diabetes, to date no data have been published on cardiac levels or bioavailability of NO from diet-induced animal models, let alone diabetic patients.
Diverse mechanisms have been proposed in diabetes-induced dysfunction of NO signalling. The pivotal role of altered eNOS function as the rate-limiting step in NO bioavailability is emphasized in the pathomechanism (Münzel et al., 2005; Zhang et al., 2011). The mechanisms responsible for eNOS dysfunction, however, remain elusive. Availability of cofactors for the eNOS complex, especially of BH4, determines the ratio of NO or superoxide anion produced by the enzyme (Gielis et al., 2011). Furthermore, a decrease in the dimer to monomer eNOS ratio within the myocardium of diabetic animals has been reported (Zou et al., 2002; Jo et al., 2011). Monomerization and subsequent uncoupling of NOS results in increased oxidative stress and decreased NO bioavailability that has been implicated in the pathophysiology of many cardiovascular diseases.
Of the three major NOS isoforms, two (iNOS and eNOS) are known to be increased in the diabetic heart (Stockklauser-Färber et al., 2000; Farhangkhoee et al., 2003; Jesmin et al., 2006; Rajesh et al., 2012). The increase in NOS expression in the diabetic heart is associated with an increase in lipid peroxidation and nitrotyrosine formation, which might be related to the uncoupled and monomer state of the enzyme. Indeed, inhibition of NOS activity in diabetes (by L-NAME or L-NMMA) improves myocardial function, suggesting that the increased production of superoxide anion and peroxynitrite rather than NO is a major contributor of suppressed contractile function (Smith et al., 1997; Esberg and Ren, 2003). Moreover, it seems that peroxynitrite-induced nitrative stress contributes to inactivation of succinyl-CoA:3-oxoacid CoA transferase causing deterioration of energy metabolism of the diabetic heart (Turko et al., 2001). In addition, restoration of iNOS coupling by BH4 administration improves ischaemic tolerance, reduces iNOS-derived superoxide anion generation, and increases NO bioavailability in the diabetic heart. The authors also imply that iNOS-derived NO-mediated cardioprotection occurs through protein S-nitrosylation but not cGMP-dependent signalling in the diabetic heart (Okazaki et al., 2011). The central role of oxidative stress in impaired NO bioavailability and signalling in diabetic hearts is further substantiated by Rajesh et al. (2009), demonstrating that the XO inhibitor allopurinol not only attenuated the myocardial oxidative stress, but also attenuated the pathologically increased nitrosative/nitrative stress, cell death, remodelling and cardiac dysfunction in diabetic mice hearts (see Ansley and Wang, 2013).
Much less is known about the NO-related downstream pathways (cGMP-PKG and NO-dependent post-translational modifications) in the diabetic heart. Recently, in patients with heart failure with preserved ejection fraction (obese and diabetic subjects), myocardial cGMP content as well as PKG activity is decreased, which might be related to the increase in oxidative/nitrosative stress (van Heerebeek et al., 2012). However, it seems that natriuretic peptide-induced cGMP-PKG signalling is not affected by diabetes, as shown by Rosenkranz et al. (2003). They reported that B-type natriuretic peptide is a suitable anti-hypertrophic strategy in the diabetic myocardium, where NO-dependent (bradykinin – ACE inhibitor) mechanisms fail to positively affect the development of hypertrophy (Rosenkranz et al., 2003). cGMP-independent effects of NO are mainly mediated by S-nitrosylation, the covalent modification of a protein cysteine thiol by an NO group to generate SNO. Puthanveetil et al. reported recently that in the diabetic myocardium, iNOS-dependent S-nitrosylation of GAPDH and caspase-3 contributes to increased poly[ADP-ribose] polymerase-1 (PARP-1) activity, and thereby initiates cell death activation in hyperglycaemia (Puthanveetil et al., 2012). This is also in line with data confirming the central role of PARP in diabetic cardiac complications (Pacher et al., 2002; Pacher and Szabó, 2005).
In conclusion, diabetes markedly decreases NO availability in the heart that is related to increased superoxide (from various sources including uncoupled NOS) and peroxynitrite formation. As a consequence of increased oxidative/nitrosative stress, downstream signalling of NO (cGMP-PKG and protein S-nitrosylation) is also profoundly affected.
Cardiac NO signalling as a pharmacological target
NO donors
NO donors are pharmacologically active substances that spontaneously release NO, or are metabolized to NO or its redox congeners and provide a wide scope for pharmacotherapy in cardiovascular medicine (Ignarro et al., 2002). Several NO donors have been used in clinical settings for decades, such as nitroglycerin and sodium nitroprusside. Nitrate tolerance, however, has become a limiting factor for their clinical use (Kojda et al., 1995; 1998; Csont and Ferdinandy, 2005). The underlying mechanisms responsible for nitrate tolerance may include neurohormonal counter-regulatory factors, intravascular volume or intrinsic abnormalities such as desensitization of the target enzyme guanylate cyclase or a decrease in biotransformation of NO donors (Munzel et al., 1995; Dikalov et al., 1997; 1998; 1999). Molsidomine and pentaerythrityl tetranitrate (PETN) represent more effective tolerance-devoid NO donors with a pharmacodynamically beneficial effect. Molsidomine is one of the sydnonimines and it is metabolized to the active linsidomine. PETN is the nitrate ester of pentaerythritol, structurally very similar to nitroglycerin. It was found to be the most active drug in cGMP production (Hinz et al., 1998; Mollnau et al., 2005). Despite these facts, neither molsidomine nor PETN was able to improve pathological changes of the cardiovascular system in adult spontaneously hypertensive rats (Kristek et al., 2003).
The compound LA-419 is an analogue of isosorbide mononitrate containing a protected thiol group in its molecular structure. Preclinical studies have shown that this compound has anti-atherogenic and antioxidant properties that make it applicable for the treatment of chronic cardiovascular disorders (Megson and Leslie, 2009). Ruiz-Hurtado and Delgado (2010) demonstrated that LA-419 prevents left ventricular remodelling in rats with aortic stenosis at doses not affecting arterial blood pressure. In their experiment, LA-419 even restored cardiac eNOS expression and enhanced the interaction between eNOS and its positive regulator, heat shock protein 90, and re-established the normal cardiac levels of cGMP. The thiol group of LA-419 improved also NO stability by converting NO into nitrosothiols and protecting the formed NO from reaction with ROS (Ruiz-Hurtado et al., 2007; Ruiz-Hurtado and Delgado, 2010).
In conclusion, there are very few data about the effects of NO donors on heart and/or cardiomyocyte functions in the metabolic syndrome. Nevertheless, the beneficial effect of compound LA-419 seems to be a promising therapeutic approach against cardiac remodelling due to the metabolic syndrome and the associated risk factors as well. However, the impaired NO-sGC signalling in the metabolic syndrome by oxidative stress is likely to represent a major obstacle for the success of this approach.
ROS scavengers
ROS are involved in several physiological cellular signalling mechanisms. However, pathological increase in oxidative stress contributes to different pathologies including themetabolic syndrome and cardiovascular disorders. Accordingly, one of the most powerful antioxidants, 4-hydroxy-2,2,6,6,-tetramethylpiperidine-1-oxyl (tempol), prevents cardiovascular damage in different experimental hypertension and diabetes models (Ebenezer et al., 2009; Hasdan et al., 2002; Nagase et al., 2007), decreases hypertrophic responses to atrial natriuretic peptide in neonatal rat cardiac myocytes (Laskowski et al., 2006), and reduces apoptosis in cardiac cells exposed to hyperglycaemia or in diabetic rats (Fiordaliso et al., 2007). Tempol decreased apoptosis in response to increased aldosterone signalling via a non-genomic pathway in cardiomyocytes (Hayashi et al., 2008) and inhibited the Ca2+ transient within cardiac myocytes stimulated by pressure-flow stress (Belmonte and Morad, 2008). Tempol improved insulin sensitivity and dyslipidemia, reduced weight gain and diastolic dysfunction and heart failure in diet-induced preclinical models of the metabolic syndrome (see Wilcox, 2010). Moreover, infusion of tempol into hyperglycaemic dogs normalized their coronary endothelial dysfunction and coronary wall shear stress in type 1 and 2 diabetes models (Gross et al., 2003). Chronic treatment with another antioxidant, N-acetylcysteine (NAC), partially attenuated the increase in blood pressure in young, but not in adult spontaneously hypertensive rats (SHR). The antioxidant action of NAC on lipid peroxidation, inhibition of NF-κB expression and eNOS activation was greater in young than in adult SHR, indicating preventive rather than therapeutic effect of NAC (Pechánová et al., 2006). Melatonin, an indolamine with antioxidant properties, has been shown do decrease blood pressure even in the established form of the spontaneous hypertension. An in vitro study revealed that melatonin lowered the tone of phenylephrine-precontracted femoral artery via both NO-dependent and NO-independent components since vasorelaxation was preserved even after the blockade of sGC by oxadiazolo[4,3-a]quinoxalin-1-one (Pechánová et al., 2007). Melatonin treatment also prevented the development or induced a reversal of left ventricular fibrosis in the model of L-NAME-induced hypertension or in spontaneously hypertensive rats (see Simko and Pechanova, 2010). It has been documented that melatonin reduces blood pressure in patients with hypertension or non-dipping blood pressure (Reiter et al., 2009). Interestingly, melatonin, leptin and insulin have been found to activate the same intracellular signalling pathways, particularly PI3K and STAT-3 (Carvalheira et al., 2001). As a consequence, melatonin may attenuate or reverse insulin resistance in obesity by mimicking the actions of insulin and leptin signalling via crosstalk between these pathways (see Nduhirabandi et al., 2012).
Several studies described positive effects of different polyphenolic compounds on the heart by restoring the balance between ROS and NO production, in hypertension as well as in other components of the metabolic syndrome (Pechánová et al., 2004; Galleano et al., 2010). Sutra et al. (2008) showed the preventive effects of different polyphenolic molecules, like catechin, resveratrol, delphinidin and gallic acid, on cardiac fibrosis associated with the metabolic syndrome. Similarly, protection of ROS/NO balance was suggested to be involved in the beneficial effect of resveratrol. The results of Penumathsa et al. (2008) suggested that the effect of resveratrol is non-insulin-dependent but triggers some of the intracellular insulin signalling components such as eNOS and Akt through the AMPK pathway in the myocardium. Furthermore, resveratrol was shown to regulate the caveolin-1 and caveolin-3 status that might play an essential role in GLUT-4 translocation and glucose uptake in streptozotocin-induced type 1 diabetic myocardium (Penumathsa et al., 2008). Similarly, olive leaf extract containing polyphenols, such as oleuropein and hydroxytyrosol, was shown to reverse chronic inflammation and oxidative stress in rat model of diet-induced obesity and diabetes (Poudyal et al., 2010). Resveratrol also protected against diabetic cardiac dysfunction by inhibiting oxidative/nitrative stress and improving NO availability (Zhang et al., 2010).
Despite the fact that antioxidants represent great promise in the treatment of hypertension and other components of the metabolic syndrome, data from clinical studies and trials with non-specific antioxidants are not conclusive. For example, in the HOPE (Heart Outcomes Prevention Evaluation) study, involving patients with atherosclerotic complications or diabetes mellitus, vitamin E in the dose of 400 IU daily, was not able to reduce blood pressure and morbidity and mortality from cardiovascular reasons (Yusuf et al., 2000; Ward and Croft, 2006). In contrast, a more recent study confirmed that subjects with type 2 diabetes after a 3 month long supplementation of vitamins C and E or their combination demonstrated significantly lower level of hypertension, decreased levels of blood glucose, and increased superoxide dismutase (SOD) and GSH enzyme activity that could probably reduce insulin resistance by attenuating oxidative stress (Rafighi et al., 2013). Vitamin C was also shown to increase BH4 levels by preventing its oxidation, which reduced eNOS uncoupling (Landmesser et al., 2003). Thus, preservation of BH4 may also explain the effects of long-term ascorbate treatment on blood pressure in patients with hypertension (Duffy et al., 1999).
Several studies suggest that imbalance between ROS production and mitochondrial antioxidants also contributes to the pathogenesis of hypertension and associated vascular pathologies. Ito et al. (1995) found that hypertension and cardiac hypertrophy were associated with decreased expression of SOD1 and SOD2 in spontaneously hypertensive rats compared with Wistar-Kyoto rats. Indeed, overexpression of mitochondrial SOD2 and thioredoxin 2 reduced the production of both mitochondrial and cytoplasmic ROS (Widder et al., 2009). SOD2 overexpression also attenuated H2O2-induced apoptosis, decreased lipid peroxidation, reduced age-related decline in mitochondrial ATP levels and decreased blood pressure (see Dikalov and Ungvari, 2013).
Recently, extracellular antioxidant enzymes like EC-SOD or covalent bienzymes like SOD-CHS-CAT conjugate (superoxide dismutase-chondroitin sulphate-catalase) started to be of particular interest, as they demonstrated protective actions against development of hypertension, heart failure and diabetes mellitus in vivo (see Maksimenko and Vavaev, 2012).
Taken together, based on numerous promising preclinical studies with mitochondrial antioxidants, XO and/or NADPH oxidase inhibitors in models of hypertension, diabetes and atherosclerosis, it appears that, instead of using non-specific antioxidants, selectively targeting the sources of ROS with more specific drugs may represent a better approach to overcome metabolic syndrome and its complications.
PDE inhibitors
The cGMP-dependent NO signalling is largely influenced by the family of PDEs that control cGMP levels and therefore affect the downstream effects of NO including PKG stimulation. Several PDEs, including PDE1, PDE2 and PDE5, play a role in the regulation of cGMP in both vascular smooth muscle cells and cardiac myocytes. PDEs are compartmentalized providing selective interactions of a certain source of cGMP and PDE hydrolysis. PDE1 and/or PDE5 are up-regulated in chronic disease conditions such as atherosclerosis, cardiac pressure-load stress, and heart failure, as well as in response to long-term exposure to nitrates. In pathophysiological states with reduced NO availability, such as, for example, diabetes and hyperlipidaemia (see above), using selective PDE inhibitors may be particularly helpful (see Kass et al., 2007). Because PDE-5 is widely distributed in the body, selective PDE-5 inhibitors have been extensively developed. The first PDE-5 inhibitor sildenafil on the market is used for the indication of erectile dysfunction. However, recent studies revealed several beneficial pleiotropic cardiovascular effects of PDE-5 inhibitors in patients with erectile dysfunction and multiple co-morbidities, including coronary artery disease, heart failure, hypertension and diabetes mellitus (see Chrysant and Chrysant, 2012). For example, tadalafil attenuates oxidative stress and inflammation and induces cardioprotection in type 2 diabetic mice models (Varma et al., 2012; Koka et al., 2013). Moreover, vardenafil attenuated diabetes-induced cardiac dysfunction in type 1 diabetic rats (Radovits et al., 2009).
In conclusion, PDE inhibition is a promising tool to restore the downstream signalling pathway of NO in the metabolic syndrome.
sGC stimulators and activators
Activation of sGC has traditionally been achieved with nitrovasodilator drugs extensively used in ischaemic heart disease. However, these drugs are associated with the rapid development of tolerance and potentially deleterious cGMP-independent actions (see Csont and Ferdinandy, 2005). Furthermore, the NO-sGC signalling pathway is impaired in hypertension, heart failure and atherosclerosis by ROS/RNS, limiting the ability of NO to activate its own signalling machinery (Evgenov et al., 2006; Stasch et al., 2011). Therefore, NO- and haem-independent sGC activators have been developed, such as, for example, cinaciguat and ataciguat. These compounds selectively activate the oxidized/haem-free enzyme via binding to the haem pocket of the enzyme, thereby causing strong vasodilatation. Accordingly, activators of sGC may be beneficial in the treatment of a variety of pathologies including systemic and pulmonary hypertension, heart failure, atherosclerosis and peripheral arterial disease (Evgenov et al., 2006; Stasch et al., 2011). Indeed, NO-insensitive sGC activators attenuated left ventricular hypertrophy, preserved cardiac function, and increased survival in spontaneously hypertensive stroke-prone rats with high-salt high-fat diet (Costell et al., 2012), in salt-sensitive Dahl rats (Geschka et al., 2011), as well as in chronic L-NAME-treated rats (Zanfolin et al., 2006). sGC activators have demonstrated beneficial effects not only in hypertension and heart failure models but also in models of atherosclerosis and restenosis (see Evgenov et al., 2006; Stasch et al., 2011). Following successful recent clinical trials, riociguat received FDA approval for the treatment of pulmonary hypertension and chronic thromboembolic pulmonary hypertension in humans, and clinical trials with other similar drugs are ongoing in heart failure.
In conclusion, the pharmacological activation of sGC may be the most promising tool to restore the downstream signalling pathway of NO in the metabolic syndrome, which should be validated in future clinical trials.
Interaction of pharmacological treatment of metabolic syndrome with cardiac NO signalling
Interaction of antihypertensives with cardiac NO signalling
Three approaches have been developed to correct the imbalance between increased oxidative stress and simultaneously decreased NO synthesis in the cardiovascular system: (1) reducing ROS bioavailability by administration of antioxidant compounds; (2) increasing NO levels via administration of NO donors such as nitroglycerin or mono/dinitrates; and (3) reducing ROS production and stimulating NO production, for example, by treatment with statins, ACE inhibitors, angiotensin AT1 receptor antagonists, or β-adrenoceptor antagonists (β-blockers) with NO-dependent properties such as nebivolol (see Münzel et al., 2010).
Among antihypertensives, the third-generation β-blockers with stimulating effect on NOS and/or β3-adrenoceptors have the best described effect on cardiac NO signalling. Nebivolol achieved a marked improvement on cardiac mass, coronary flow, mRNA expression levels of sarcoplasmic reticulum Ca2+ ATPase (SERCA2a), and atrial natriuretic peptide and phospholamban (PLN)/SERCA2a and phospho-PLN/PLN ratio in rats treated with isoprenaline (Ozakca et al., 2013). In Zucker diabetic fatty rats, nebivolol and atenolol showed a comparable reduction in blood pressure; however, nebivolol appeared to achieve a better lipid profile, left ventricular function and less left ventricular hypertrophy, compared with atenolol. Moreover, a reduction in platelet aggregation and an increased endothelium-dependent and endothelium-independent relaxation were observed in the nebivolol group versus the atenolol group. Together with an attenuation of oxidative stress parameters, nebivolol also better preserved antioxidant defence markers (Toblli et al., 2010). Concerning NO signalling, nebivolol has been shown to stimulate endogenous production of NO by inducing phosphorylation of eNOS (Maffei et al., 2006) which determines its favourable effects on cardiac function in patients with heart failure when compared with classical β-blockers. The action of nebivolol on iNOS was also confirmed by real-time PCR experiments, showing cardiac overexpression of iNOS, but not nNOS or eNOS, in male C57BL/6N mice (Maffei et al., 2007).
Among other promising antihypertensives with NO increasing and ROS reducing effect are the ACE inhibitors with a thiol group such as captopril and the newer zofenopril. In our earlier studies, both captopril and enalapril increased NOS activity in the heart of spontaneously hypertensive animals but did not increase the expression of eNOS. Both ACE inhibitors increased the level of cGMP. However, cGMP levels were significantly higher in the captopril group. Captopril, besides inhibition of ACE, prevented hypertension by increasing NOS activity and by simultaneous decrease of oxidative stress which resulted in increase of cGMP concentration (Pechánová, 2007). Most of the clinical studies revealed that captopril, besides decreasing blood pressure, has also vasodilator effects and attenuates left ventricular hypertrophy (Konstam et al., 2000). The SMILE (Survival of Myocardial Infarction Long-term Evaluation) program indicates that zofenopril may favourably affect the prognosis of patients with a recent myocardial infarction (Lombardi et al., 2012) and even of patients with the metabolic syndrome (Borghi et al., 2008). Accordingly, a 12 week zofenopril treatment significantly decreased lipid peroxidation, reduced cardiac hypertrophy and improved NO pathway in patients with essential hypertension (Napoli et al., 2004).
In conclusion, antihypertensive drugs, such as β- blockers with NO-dependent effects and ACE inhibitors with a thiol group, may successfully restore NO signalling in the heart in the metabolic syndrome.
Interaction of antidiabetic drugs with cardiac NO signalling
For the treatment of diabetes, several classes of drugs are available with markedly different mechanisms of action. Besides various synthetic insulin analogues, several other non-insulin-related drugs were developed and marketed in the last years. The mechanism of action involves the stimulation of endogenous insulin secretion, the sensitization of peripheral tissues to insulin or the increase in incretin levels. Although these mechanisms are directly not related to NO signalling, all of these drugs have some degree of interaction with NO-related pathways.
Insulin itself is a strong regulator of cardiac NO level by affecting eNOS phosphorylation. Administration of insulin in vivo to healthy rats activates Akt through a PI3K-dependent mechanism. Phosphorylation of the eNOS and the concurrent increase in NO production is a result of Akt activation (Gao et al., 2002). However, this NO-related effect of insulin is attenuated in the diabetic myocardium (Zakula et al., 2011).
Sulfonylurea drugs are potent stimulators of endogenous insulin secretion by acting on ATP-sensitive K+ channels. Although these drugs do not interact directly with myocardial NO production, experimental and clinical data suggest considerable interaction with NO signalling. Cardioprotection mediated by NO is mainly related to the opening of mitochondrial ATP-sensitive K+ channels (Han et al., 2002; Ljubkovic et al., 2007). The non-selective nature of K+ channel inhibition results in the attenuation of NO-mediated cardioprotection by sulfonylureas, limiting their clinical applicability in diabetic patients with ischaemic heart diseases (Garratt et al., 1999).
Insulin-sensitizing drugs include biguanides (metformin is the most often used) and the thiazolidinedione class of antidiabetic drugs (rosiglitazone and pioglitazone). These drugs mainly act at peripheral tissues by sensitizing them to the action of insulin. Metformin facilitates the activation of AMP-activated PK (AMPK) in the heart that has been shown to be cardioprotective during heart failure. Metformin-induced positive effects were associated with increased AMPK and eNOS phosphorylation, and reductions in insulin, TGF-β1, basic fibroblast growth factor, and TNF-α levels in the circulation and/or in the myocardium (Gundewar et al., 2009; Wang et al., 2011). Withdrawal of rosiglitazone from the market due to adverse cardiovascular effects (increased mortality, accentuation if ischaemic heart diseases) highlighted the controversial cardiovascular effects of thiazolidinediones. In experimental studies, both rosiglitazone (Gonon et al., 2007) and pioglitazone (Ye et al., 2008a) reduced infarct size possibly via increased eNOS phosphorylation. However, the mechanisms that resulted in adverse effects in humans are still not known.
Dipeptidyl peptidase-4 (DPP-4) inhibitors are a relatively new class of antidiabetics. By the inhibition of DPP-4, they increase the level of incretins (GIP and GLP-1), inhibiting glucagon release, which in turn increases insulin secretion, decreases gastric emptying and decreases blood glucose level (Figure 2). DPP-4 inhibitors were proven to be atheroprotective (Matheeussen et al., 2013) and to affect positively diastolic function in the insulin-resistant Zucker diabetic fatty rat model by increasing phosphorylation of eNOS (Ser1177) and the expression of total eNOS (Aroor et al., 2013).
Figure 2.
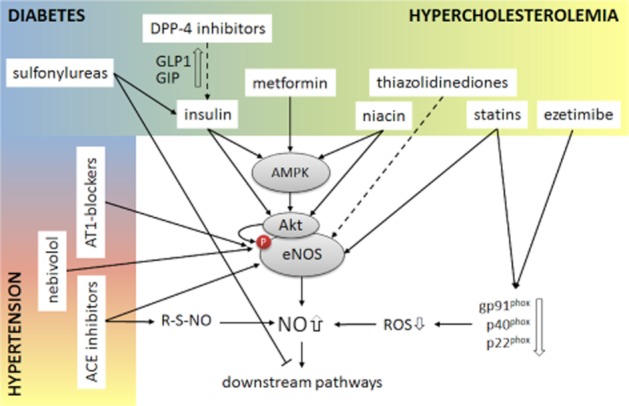
Effect of drugs used in the metabolic syndrome on cardiac NO signalling. Antihypertensives activate primarily eNOS or potentiate the release of NO from SNOs. Antidiabetics activate predominantly the kinases AMPK and Akt upstream of eNOS to induce its phosphorylation. Sulfonylureas may, however, interfere with NO-related downstream effectors (i.e. mitochondrial KATP channels). Anti-hyperlipidaemic drugs have pleiotropic effects on NO signalling, serving as antioxidants and inducers of eNOS phosphorylation.
In conclusion, antidiabetics (except for sulfonylureas) may positively affect tissue NO availability and NO signalling, thereby providing a promising tool to treat cardiac complications of the metabolic syndrome.
Interactions of anti-hyperlipidaemic treatments and the cardiac NO signalling
Statins are the most frequently prescribed anti-hyperlipidaemic medications. Apart from their HMG-CoA reductase inhibitory function, statins reduce cardiovascular risks associated with hypercholesterolemia via a wide range of well-documented pleiotropic effects. For instance, in the heart, atorvastatin increases phosphorylation of a host of mediators associated with NO signalling, such as ERK, PDK-1, Akt and eNOS itself, plausibly via an adenosine receptor-dependent mechanism (Merla et al., 2007; Ye et al., 2008b). Direct modulation of NO signalling by statins downstream of NOS was also suggested by another study, where rosuvastatin administration reverted the elevation in mean arterial blood pressure and cardiac remodelling caused by a treatment with a NOS inhibitor, L-NAME (Baraka et al., 2009). Statins modulate cardiac NO metabolism under hyperlipidaemic conditions as well. In OLETF rats, both atorvastatin and pravastatin up-regulated cardiac eNOS expression compared with their genetic controls (Yu et al., 2004; Chen et al., 2007). Interestingly, not all statins are equally effective in the modulation of cardiac NO metabolism. For example, pravastatin induced eNOS more effectively than atorvastatin (Chen et al., 2007), and we have previously shown that the first-generation statin, lovastatin, does not affect NO production or NOS activity in cholesterol-fed animals (Giricz et al., 2003). Similarly, in spontaneously hypertensive rats, pravastatin treatment failed to modulate the expression of nNOS, eNOS, sGC or the NADPH oxidase subunits p40Phox and Gp91 in myocardial tissue (Herring et al., 2011), which highlights that NO modulation is not a general characteristic of the whole class of statins and the effects are strongly model dependent. Newer statins have been also shown to alter cardiac NO bioavailability in other pathologies unrelated to hyperlipidaemia. For instance, in a hypertension model of rats overexpressing renin, rosuvastatin decreased the accentuated myocardial gp91(phox), p40(phox), p22(phox) expression and reduced the myocardial lipid peroxidation, nitrotyrosine formation and malondialdehyde content, suggesting that it increased NO bioavailability by reducing ROS formation (Habibi et al., 2007). Elsewhere, simvastatin reduced iNOS expression in cytokine-treated H9C2 cardiac myoblasts, which appeared to be related to the cholesterol biosynthesis-modulating effect of statins, since mevalonate, and geranylgeranyl pyrophosphate could reverse these effects (Madonna et al., 2005). However, other statins exerted seemingly opposing effects on the cardiac NO production modulated by pro-inflammatory signals: lipophilic statins fluvastatin and lovastatin increased IL-1β-induced nitrite production by cardiac myocytes, whereas hydrophilic pravastatin did not. Fluvastatin also increased iNOS expression (Ikeda et al., 2001). These data demonstrate clearly that, before initiating a statin treatment, compounds must be evaluated individually in the view of the other coexisting pathologies. Statins might have positive effects on age-related disturbance of cardiac NO metabolism as well. In 20-month-old rats, atorvastatin administration for 4 months reversed the age-related increase in cardiac malondialdehyde and decrease of SOD, catalase and NOS activity (Han et al., 2012). Direct effects of statins on cardiac NO signalling have been studied in humans in a few publications. Perioperative simvastatin therapy of patients undergoing non-coronary cardiac surgery increased nitrite and nitrate levels, expression and phosphorylation of eNOS at Ser1177, phosphorylation of Akt, HSP90, and its association with eNOS in right atrial appendage (Almansob et al., 2012). Furthermore, atorvastatin induced a mevalonate-reversible inhibition of NOX2-NADPH oxidase activity in right atrial samples from patients who developed post-operative atrial fibrillation (AF); however, it did not affect ROS, or NOS uncoupling in patients with permanent AF (Reilly et al., 2011). Although the general notion is that statins improve cardiac NO metabolism, these data also suggest that differences in the biochemical background of diverse pathologies might profoundly influence the beneficial pleiotropic effects of several of the statins.
The PPAR family of nuclear receptors has been a target for numerous antidiabetic and anti-hyperlipidaemic agents, many of which are shown to modulate NO metabolism. GW7647, a potent PPARα inducer, enhanced cardiac eNOS activation in isolated papillary muscles of rat hearts (Xiao et al., 2010). WY-14643, another PPARα agonist, has also been shown to increase the expression of eNOS and iNOS, as well as nitrite/nitrate levels in the ischaemic myocardium of Goto-Kakizaki and Wistar rats (Bulhak et al., 2009). This publication also demonstrated that PPARα activation leads to the induction of the downstream mediators of NO, as shown by an elevated cardiac phosphorylation of Akt at Ser473 and Thr308. However, elsewhere, fenofibrate, also a PPARα inducer, did not alter cardiac NO or its metabolites in LPS-treated Wistar rats (Jozefowicz et al., 2007). Similarly, PPARβ/δ agonist GW0742 reduced the ischaemia/reperfusion-induced increase in the expression of iNOS and normalized the phosphorylation of Akt and glycogen synthase kinase-3β in a rat model of regional myocardial I/R in vivo (Kapoor et al., 2010), demonstrating the involvement of these PPAR isoforms in NO metabolism. More information is available on the effects of PPARγ induction on cardiac NO balance. The endogenous PPARγ ligand, 15-deoxy-Δ12,14-PGJ2 (15D-PGJ2), attenuated the cardiac ischaemia/reperfusion-induced increase in iNOS mRNA expression in rats (Wayman et al., 2002). The inhibition of iNOS expression by 15D-PGJ2, but not by rosiglitazone, a synthetic PPARγ agonist, was confirmed in neonatal cardiomyocytes pretreated with LPS (Hovsepian et al., 2010) or IL-1β (Mendez and LaPointe, 2003). Negative correlation between the activity of PPARγ and NOS enzymes was confirmed in another study, where pioglitazone down-regulated iNOS expression in a murine cardiac allotransplantation model (Hasegawa et al., 2011). However, pioglitazone seems to have opposing effects on eNOS. In diabetic OLETF rats, cardiac expression of eNOS and phosphorylation of Akt was reduced compared with non-diabetic controls, which was reversed by the induction of PPARγ by pioglitazone (Makino et al., 2009). The notion that disturbed eNOS signalling is restored by PPARγ activation seems to be strengthened by other experiments. Phosphorylated eNOS was increased in mice receiving rosiglitazone before ischaemia/reperfusion (Gonon et al., 2007), and in diabetic db/db mice, the reduced dilations of coronary arterioles in response to ACh and the NO donor NONOate were augmented by rosiglitazone (Bagi et al., 2004).
There is only a limited amount of data on the effect of other less frequently prescribed anti-hyperlipidaemic agents on cardiac NO signalling. Although dietary supplementation of niacin is often recommended for obese patients, its effect on the NO-cGMP-PKG system has been revealed indirectly in a single publication. Niacin-bound chromium induced myocardial phosphorylation of Akt, AMPK and eNOS in streptozotocin-induced diabetic rats after ischaemia-reperfusion injury, suggesting that beneficial effects of niacin and chromium are mediated not only through the modulation of metabolic pathways, but via the activation of the NO pathway as well (Penumathsa et al., 2009). Inhibition of cholesterol absorption by ezetimibe, an inhibitor of the intestinal Niemann-Pick C1-like 1 protein, has been shown to decrease cardiac NADPH oxidase-mediated oxidative stress in hyperlipidaemic db/db mice (Fukuda et al., 2010), therefore, plausibly increase NO bioavailability, known to be depressed in hyperlipidaemia (Ferdinandy et al., 1997; Giricz et al., 2003; Onody et al., 2003).
In conclusion, experimental evidence shows that administration of various cholesterol-reducing medications leads to a restoration of the balance in NO metabolism disturbed by hyperlipidaemia.
Conclusions and perspectives
Published data show that NO availability and its signalling in the heart is impaired in the presence of risk factors associated with the metabolic syndrome. The decreased tissue availability of NO is a consequence of increased oxidative and nitrosative/nitrative stress rather than a decreased cardiac NO synthesis. The impaired NO signalling in the heart due to the metabolic syndrome leads to different pathophysiological processes including myocardial hypertrophy, fibrosis and eventually heart failure. Therefore, in addition to treating the individual risk factors related to the metabolic syndrome, restoration of NO signalling in the heart by pharmacological tools may be a promising therapeutic avenue to alleviate cardiac pathologies related to the metabolic syndrome.
Acknowledgments
This study was elaborated within the projects APVV-0742-10, VEGA 2/0183/12 and 2/0144/14, the New Horizons Grant of the European Foundation for the Study of Diabetes, Hungarian Scientific Research Fund (OTKA K109737) and COST Action BM1005, and the Intramural Program of NIH/NIAA. All authors contributed equally on literature search and metabolic syndrome models preparation.
Glossary
- ADMA
asymmetric dimethylarginine
- AF
atrial fibrillation
- DPP-4
dipeptidyl peptidase-4
- PARP-1
poly[ADP-ribose] polymerase-1
- PETN
pentaerythrityl tetranitrate
- PKG
cGMP-dependent PK
- ROCK
Rho-associated PK
- ROS
reactive oxygen species
- SNO
S-nitrosothiol
- Tempol
4-hydroxy-2,2,6,6,-tetramethylpiperidine-1-oxyl
- 15D-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
Conflicts of interest
The authors declare no conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013a;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013c;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013d;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear Hormone Receptors. Br J Pharmacol. 2013e;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almansob MA, Xu B, Zhou L, Hu XX, Chen W, Chang FJ, et al. Simvastatin reduces myocardial injury undergoing noncoronary artery cardiac surgery: a randomized controlled trial. Arterioscler Thromb Vasc Biol. 2012;32:2304–2313. doi: 10.1161/ATVBAHA.112.252098. [DOI] [PubMed] [Google Scholar]
- Alvarez GE, Beske SD, Ballard TP, Davy KP. Sympathetic neural activation in visceral obesity. Circulation. 2002;106:2533–2536. doi: 10.1161/01.cir.0000041244.79165.25. [DOI] [PubMed] [Google Scholar]
- Amour J, Loyer X, Le Guen M, Mabrouk N, David JS, Camors E, et al. Altered contractile response due to increased beta3-adrenoceptor stimulation in diabetic cardiomyopathy: the role of nitric oxide synthase 1-derived nitric oxide. Anesthesiology. 2007;107:452–460. doi: 10.1097/01.anes.0000278909.40408.24. [DOI] [PubMed] [Google Scholar]
- Andreadou I, Iliodromitis EK, Rassaf T, Schulz R, Papapetropoulos A, Ferdinandy P. The role of gaseous transmitters NO, H2S, CO in myocardial ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Br J Pharmacol. 2015;172:1587–1606. doi: 10.1111/bph.12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansley DM, Wang B. Oxidative stress and myocardial injury in the diabetic heart. J Pathol. 2013;229:232–241. doi: 10.1002/path.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroor AR, Sowers JR, Bender SB, Nistala R, Garro M, Mugerfeld I, et al. Dipeptidylpeptidase inhibition is associated with improvement in blood pressure and diastolic function in insulin-resistant male Zucker obese rats. Endocrinology. 2013;154:2501–2513. doi: 10.1210/en.2013-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo PS, Minicucci MF, Santos PP, Paiva SA, Zornoff LA. Energy metabolism in cardiac remodeling and heart failure. Cardiol Rev. 2013;21:135–140. doi: 10.1097/CRD.0b013e318274956d. [DOI] [PubMed] [Google Scholar]
- Babal P, Pechanova O, Bernatova I, Stvrtina S. Chronic inhibition of NO synthesis produces myocardial fibrosis and arterial media hyperplasia. Histol Histopathol. 1997;12:623–629. [PubMed] [Google Scholar]
- Bagi Z, Koller A, Kaley G. PPARgamma activation, by reducing oxidative stress, increases NO bioavailability in coronary arterioles of mice with Type 2 diabetes. Am J Physiol Heart Circ Physiol. 2004;286:H742–H748. doi: 10.1152/ajpheart.00718.2003. [DOI] [PubMed] [Google Scholar]
- Baraka A, Mikhail M, Guemei A, El Ghotny S. Effect of targeting mitogen-activated protein kinase on cardiac remodeling in rats. J Cardiovasc Pharmacol Ther. 2009;14:339–346. doi: 10.1177/1074248409349620. [DOI] [PubMed] [Google Scholar]
- Belin de Chantemele EJ, Stepp DW. Influence of obesity and metabolic dysfunction on the endothelial control in the coronary circulation. J Mol Cell Cardiol. 2012;52:840–847. doi: 10.1016/j.yjmcc.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte S, Morad M. Pressure-flow'-triggered intracellular Ca2+ transients in rat cardiac myocytes: possible mechanisms and role of mitochondria. J Physiol. 2008;586:1379–1397. doi: 10.1113/jphysiol.2007.149294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi C, Cicero AF, Ambrosioni E. Effects of early treatment with zofenopril in patients with myocardial infarction and metabolic syndrome: the SMILE Study. Vasc Health Risk Manag. 2008;4:665–671. doi: 10.2147/vhrm.s2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet C, Belin de Chantemele E, Guihot AL, Vessières E, Bocquet A, Dumont O, et al. Flow-induced remodeling in resistance arteries from obese Zucker rats is associated with endothelial dysfunction. Hypertension. 2007;50:248–254. doi: 10.1161/HYPERTENSIONAHA.107.088716. [DOI] [PubMed] [Google Scholar]
- Böger RH, Bode-Böger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998;98:1842–1847. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- Bulhak AA, Jung C, Ostenson CG, Lundberg JO, Sjöquist PO, Pernow J. PPAR-alpha activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: involvement of the PI3-Kinase/Akt and NO pathway. Am J Physiol Heart Circ Physiol. 2009;296:H719–H727. doi: 10.1152/ajpheart.00394.2008. [DOI] [PubMed] [Google Scholar]
- Burley DS, Cox CD, Zhang J, Wann KT, Baxter GF. Natriuretic peptides modulate ATP-sensitive K(+) channels in rat ventricular cardiomyocytes. Basic Res Cardiol. 2014;109:402. doi: 10.1007/s00395-014-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalheira JB, Siloto RM, Ignacchitti I, Brenelli SL, Carvalho CR, Leite A, et al. Insulin modulates leptin-induced STAT3 activation in rat hypothalamus. FEBS Lett. 2001;500:119–124. doi: 10.1016/s0014-5793(01)02591-1. [DOI] [PubMed] [Google Scholar]
- Cebova M, Kristek F. Age-dependent ultrastructural changes of coronary artery in spontaneously hypertensive rats. Gen Physiol Biophys. 2011;30:364–372. doi: 10.4149/gpb_2011_04_364. [DOI] [PubMed] [Google Scholar]
- Chen Y, Ohmori K, Mizukawa M, Yoshida J, Zeng Y, Zhang L, et al. Differential impact of atorvastatin vs pravastatin on progressive insulin resistance and left ventricular diastolic dysfunction in a rat model of type II diabetes. Circ J. 2007;71:144–152. doi: 10.1253/circj.71.144. [DOI] [PubMed] [Google Scholar]
- Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, et al. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- Chobanyan-Jürgens K, Fuchs AJ, Tsikas D, Kanzelmeyer N, Das AM, Illsinger S, et al. Increased asymmetric dimethylarginine (ADMA) dimethylaminohydrolase (DDAH) activity in childhood hypercholesterolemia type II. Amino Acids. 2012;43:805–811. doi: 10.1007/s00726-011-1136-3. [DOI] [PubMed] [Google Scholar]
- Chrysant SG, Chrysant GS. The pleiotropic effects of phosphodiesterase 5 inhibitors on function and safety in patients with cardiovascular disease and hypertension. J Clin Hypertens. 2012;14:644–649. doi: 10.1111/j.1751-7176.2012.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coatmellec-Taglioni G, Ribière C. Factors that influence the risk of hypertension in obese individuals. Curr Opin Nephrol Hypertens. 2003;12:305–308. doi: 10.1097/00041552-200305000-00013. [DOI] [PubMed] [Google Scholar]
- Costell MH, Ancellin N, Bernard RE, Zhao S, Upson JJ, Morgan LA, et al. Comparison of soluble guanylate cyclase stimulators and activators in models of cardiovascular disease associated with oxidative stress. Front Pharmacol. 2012;3:128. doi: 10.3389/fphar.2012.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csonka C, Szilvássy Z, Fülöp F, Páli T, Blasig IE, Tosaki A, et al. Classic preconditioning decreases the harmful accumulation of nitric oxide during ischemia and reperfusion in rat hearts. Circulation. 1999;100:2260–2266. doi: 10.1161/01.cir.100.22.2260. [DOI] [PubMed] [Google Scholar]
- Csonka C, Csont T, Onody A, Ferdinandy P. Preconditioning decreases ischemia/reperfusion-induced peroxynitrite formation. Biochem Biophys Res Commun. 2001;285:1217–1219. doi: 10.1006/bbrc.2001.5308. [DOI] [PubMed] [Google Scholar]
- Csonka C, Pali T, Bencsik P, Gorbe A, Ferdinandy P, Csont T. Measurement of NO in biological samples. Br J Pharmacol. 2015;172:1620–1632. doi: 10.1111/bph.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csont T, Ferdinandy P. Cardioprotective effects of glyceryl trinitrate: beyond vascular nitrate tolerance. Pharmacol Ther. 2005;105:57–68. doi: 10.1016/j.pharmthera.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Csont T, Bereczki E, Bencsik P, Fodor G, Görbe A, Zvara A, et al. Hypercholesterolemia increases myocardial oxidative and nitrosative stress thereby leading to cardiac dysfunction in apoB-100 transgenic mice. Cardiovasc Res. 2007;76:100–109. doi: 10.1016/j.cardiores.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Cuspidi C, Ciulla M, Zanchetti A. Hypertensive myocardial fibrosis. Nephrol Dial Transplant. 2006;21:20–23. doi: 10.1093/ndt/gfi237. [DOI] [PubMed] [Google Scholar]
- Dai Z, Wu Z, Yang Y, Wang J, Satterfield MC, Meininger CJ, et al. Nitric oxide and energy metabolism in mammals. Biofactors. 2013;39:383–391. doi: 10.1002/biof.1099. [DOI] [PubMed] [Google Scholar]
- Davidson SM, Duchen MR. Effects of NO on mitochondrial function incardiomyocytes: pathophysiological relevance. Cardiovasc Res. 2006;71:10–21. doi: 10.1016/j.cardiores.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Dedkova EN, Blatter LA. Characteristics and function of cardiac mitochondrial nitric oxide synthase. J Physiol. 2009;587(Pt 4):851–872. doi: 10.1113/jphysiol.2008.165423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrois M, Clarke K, Lan C, Dalmasso C, Cole M, Portha B, et al. Upregulation of eNOS and unchanged energy metabolism in increased susceptibility of the aging type 2 diabetic GK rat heart to ischemic injury. Am J Physiol Heart Circ Physiol. 2010;299:H1679–H1686. doi: 10.1152/ajpheart.00998.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S, Skatchkov M, Bassenge E. Quantification of peroxynitrite, superoxide, and peroxyl radicals by a new spin trap hydroxylamine 1-hydroxy-2,2,6,6-tetramethyl-4-oxopiperidine. Biochem Biophys Res Commun. 1997;230:54–57. doi: 10.1006/bbrc.1996.5880. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Fink B, Skatchkov M, Sommer O, Bassenge E. Formation of reactive oxygen species by pentaerithrityl tetranitrate and glyceryl trinitrate in vitro and development of nitrate tolerance. J Pharmacol Exp Ther. 1998;286:938–944. [PubMed] [Google Scholar]
- Dikalov S, Fink B, Skatchkov M, Bassenge E. Comparison of glyceryl trinitrate-induced with pentaerythrityl tetranitrate-induced in vivo formation of superoxide radicals: effect of vitamin C. Free Radic Biol Med. 1999;27:170–176. doi: 10.1016/s0891-5849(99)00066-0. [DOI] [PubMed] [Google Scholar]
- Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol. 2013;305:H1417–H1427. doi: 10.1152/ajpheart.00089.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Aljofan M, Triggle CR. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J Cell Physiol. 2007;212:682–689. doi: 10.1002/jcp.21063. [DOI] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Duffy SJ, Gokce N, Holbrook M, Huang A, Frei B, Keaney JF, Jr, et al. Treatment of hypertension with ascorbic acid. Lancet. 1999;354:2048–2049. doi: 10.1016/s0140-6736(99)04410-4. [DOI] [PubMed] [Google Scholar]
- Ebenezer PJ, Mariappan N, Elks CM, Haque M, Francis J. Diet-induced renal changes in Zucker rats are ameliorated by the superoxide dismutase mimetic TEMPOL. Obesity. 2009;17:1994–2002. doi: 10.1038/oby.2009.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esberg LB, Ren J. Role of nitric oxide, tetrahydrobiopterin and peroxynitrite in glucose toxicity-associated contractile dysfunction in ventricular myocytes. Diabetologia. 2003;46:1419–1427. doi: 10.1007/s00125-003-1183-8. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Pacher P, Schmidt PM, Haskó G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhangkhoee H, Khan ZA, Mukherjee S, Cukiernik M, Barbin YP, Karmazyn M, et al. Heme oxygenase in diabetes-induced oxidative stress in the heart. J Mol Cell Cardiol. 2003;35:1439–1448. doi: 10.1016/j.yjmcc.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P. Peroxynitrite: just an oxidative/nitrosative stressor or a physiological regulator as well? Br J Pharmacol. 2006;148:1–3. doi: 10.1038/sj.bjp.0706693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2003;138:532–543. doi: 10.1038/sj.bjp.0705080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Szilvássy Z, Horváth LI, Csont T, Csonka C, Nagy E, et al. Loss of pacing-induced preconditioning in rat hearts: role of nitric oxide and cholesterol-enriched diet. J Mol Cell Cardiol. 1997;29:3321–3333. doi: 10.1006/jmcc.1997.0557. [DOI] [PubMed] [Google Scholar]
- Feron O, Balligand JL. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc Res. 2006;69:788–797. doi: 10.1016/j.cardiores.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Fiordaliso F, De Angelis N, Bai A, Cuccovillo I, Salio M, Maria SD, et al. Effect of beta-adrenergic and renin-angiotensin system blockade on myocyte apoptosis and oxidative stress in diabetic hypertensive rats. Life Sci. 2007;81:951–959. doi: 10.1016/j.lfs.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Frohlich ED, Susic D. Mechanisms underlying obesity associated with systemic and renal hemodynamics in essential hypertension. Curr Hypertens Rep. 2008;10:151–155. doi: 10.1007/s11906-008-0028-8. [DOI] [PubMed] [Google Scholar]
- Frühbeck G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes. 1999;48:903–908. doi: 10.2337/diabetes.48.4.903. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, et al. Ezetimibe ameliorates cardiovascular complications and hepatic steatosis in obese and type 2 diabetic db/db mice. J Pharmacol Exp Ther. 2010;335:70–75. doi: 10.1124/jpet.110.170373. [DOI] [PubMed] [Google Scholar]
- Fukuda Y, Teragawa H, Matsuda K, Yamagata T, Matsuura H, Chayama K. Tetrahydrobiopterin restores endothelial function of coronary arteries in patients with hypercholesterolaemia. Heart. 2002;87:264–269. doi: 10.1136/heart.87.3.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Harris MB, Kemp BE, Venema RC, Marrero MB, Stepp DW. Insulin resistance does not diminish eNOS expression, phosphorylation, or binding to HSP-90. Am J Physiol Heart Circ Physiol. 2004;287:H2384–H2393. doi: 10.1152/ajpheart.00280.2004. [DOI] [PubMed] [Google Scholar]
- Galleano M, Pechanova O, Fraga CG. Hypertension, nitric oxide, oxidants, and dietary plant polyphenols. Curr Pharm Biotechnol. 2010;11:837–848. doi: 10.2174/138920110793262114. [DOI] [PubMed] [Google Scholar]
- Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, et al. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation. 2002;105:1497–1502. doi: 10.1161/01.cir.0000012529.00367.0f. [DOI] [PubMed] [Google Scholar]
- Garcia JA, dos Santos L, Moura AL, Ricardo KF, Wanschel AC, Shishido SM, et al. S-nitroso-N-acetylcysteine (SNAC) prevents myocardial alterations in hypercholesterolemic LDL receptor knockout mice by antiinflammatory action. J Cardiovasc Pharmacol. 2008;51:78–85. doi: 10.1097/FJC.0b013e31815c39d4. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci U S A. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garratt KN, Brady PA, Hassinger NL, Grill DE, Terzic A, Holmes DR., Jr Sulfonylurea drugs increase early mortality in patients with diabetes mellitus after direct angioplasty for acute myocardial infarction. J Am Coll Cardiol. 1999;33:119–124. doi: 10.1016/s0735-1097(98)00557-9. [DOI] [PubMed] [Google Scholar]
- Geschka S, Kretschmer A, Sharkovska Y, Evgenov OV, Lawrenz B, Hucke A, et al. Soluble guanylate cyclase stimulation prevents fibrotic tissue remodeling and improves survival in salt-sensitive Dahl rats. PLoS ONE. 2011;6:e21853. doi: 10.1371/journal.pone.0021853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC, et al. PATENT-1 Study group. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013a;369:330–340. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- Ghofrani HA, D'Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, et al. CHEST-1 Study Group. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013b;369:319–329. doi: 10.1056/NEJMoa1209657. [DOI] [PubMed] [Google Scholar]
- Gielis JF, Lin JY, Wingler K, Van Schil PE, Schmidt HH, Moens AL. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic Biol Med. 2011;50:765–776. doi: 10.1016/j.freeradbiomed.2010.12.018. [DOI] [PubMed] [Google Scholar]
- Giricz Z, Csonka C, Onody A, Csont T, Ferdinandy P. Role of cholesterol-enriched diet and the mevalonate pathway in cardiac nitric oxide synthesis. Basic Res Cardiol. 2003;98:304–310. doi: 10.1007/s00395-003-0412-0. [DOI] [PubMed] [Google Scholar]
- Giricz Z, Görbe A, Pipis J, Burley DS, Ferdinandy P, Baxter GF. Hyperlipidaemia induced by a high-cholesterol diet leads to the deterioration of guanosine-3′,5′-cyclic monophosphate/protein kinase G-dependent cardioprotection in rats. Br J Pharmacol. 2009;158:1495–1502. doi: 10.1111/j.1476-5381.2009.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonon AT, Bulhak A, Labruto F, Sjöquist PO, Pernow J. Cardioprotection mediated by rosiglitazone, a peroxisome proliferator-activated receptor gamma ligand, in relation to nitric oxide. Basic Res Cardiol. 2007;102:80–89. doi: 10.1007/s00395-006-0613-4. [DOI] [PubMed] [Google Scholar]
- Gross ER, LaDisa JF, Jr, Weihrauch D, Olson LE, Kress TT, Hettrick DA, et al. Reactive oxygen species modulate coronary wall shear stress and endothelial function during hyperglycemia. Am J Physiol Heart Circ Physiol. 2003;284:H1552–H1559. doi: 10.1152/ajpheart.01013.2002. [DOI] [PubMed] [Google Scholar]
- Gundewar S, Calvert JW, Jha S, Toedt-Pingel I, Ji SY, Nunez D, et al. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009;104:403–411. doi: 10.1161/CIRCRESAHA.108.190918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi J, Whaley-Connell A, Qazi MA, Hayden MR, Cooper SA, Tramontano A, et al. Rosuvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, decreases cardiac oxidative stress and remodeling in Ren2 transgenic rats. Endocrinology. 2007;148:2181–2188. doi: 10.1210/en.2006-1355. [DOI] [PubMed] [Google Scholar]
- Han J, Kim N, Joo H, Kim E, Earm YE. ATP-sensitive K(+) channel activation by nitric oxide and protein kinase G in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H1545–H1554. doi: 10.1152/ajpheart.01052.2001. [DOI] [PubMed] [Google Scholar]
- Han L, Li M, Liu Y, Han C, Ye P. Atorvastatin may delay cardiac aging by upregulating peroxisome proliferator-activated receptors in rats. Pharmacology. 2012;89:74–82. doi: 10.1159/000335783. [DOI] [PubMed] [Google Scholar]
- Hasdan G, Benchetrit S, Rashid G, Green J, Bernheim J, Rathaus M. Endothelial dysfunction and hypertension in 5/6 nephrectomized rats are mediated by vascular superoxide. Kidney Int. 2002;61:586–590. doi: 10.1046/j.1523-1755.2002.00166.x. [DOI] [PubMed] [Google Scholar]
- Hasegawa T, Okada K, Okita Y, Pinsky DJ. Antioxidant properties of pioglitazone limit nicotinamide adenine dinucleotide phosphate hydrogen oxidase and augment superoxide dismutase activity in cardiac allotransplantation. J Heart Lung Transplant. 2011;30:1186–1196. doi: 10.1016/j.healun.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Kobara M, Abe M, Tanaka N, Gouda E, Toba H, et al. Aldosterone nongenomically produces NADPH oxidase-dependent reactive oxygen species and induces myocyte apoptosis. Hypertens Res. 2008;31:363–375. doi: 10.1291/hypres.31.363. [DOI] [PubMed] [Google Scholar]
- van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830–839. doi: 10.1161/CIRCULATIONAHA.111.076075. [DOI] [PubMed] [Google Scholar]
- Herring N, Lee CW, Sunderland N, Wright K, Paterson DJ. Pravastatin normalises peripheral cardiac sympathetic hyperactivity in the spontaneously hypertensive rat. J Mol Cell Cardiol. 2011;50:99–106. doi: 10.1016/j.yjmcc.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G. Nitroglycerin and delayed preconditioning in humans: yet another new mechanism for an old drug? Circulation. 2001;103:2876–2878. doi: 10.1161/01.cir.103.24.2876. [DOI] [PubMed] [Google Scholar]
- Heusch P, Aker S, Boengler K, Deindl E, van de Sand A, Klein K, et al. Increased inducible nitric oxide synthase and arginase II expression in heart failure: no net nitrite/nitrate production and protein S-nitrosylation. Am J Physiol Heart Circ Physiol. 2010;299:H446–H453. doi: 10.1152/ajpheart.01034.2009. [DOI] [PubMed] [Google Scholar]
- Hinz B, Kuntze U, Schröder H. Pentaerithrityl tetranitrate and its phase I metabolites are potent activators of cellular cyclic GMP accumulation. Biochem Biophys Res Commun. 1998;253:658–661. doi: 10.1006/bbrc.1998.9845. [DOI] [PubMed] [Google Scholar]
- Horowitz JD, Heresztyn T. An overview of plasma concentrations of asymmetric dimethylarginine (ADMA) in health and disease and in clinical studies: methodological considerations. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;851:42–50. doi: 10.1016/j.jchromb.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Hovsepian E, Penas F, Goren NB. 15-deoxy-Delta12,14 prostaglandin GJ2 but not rosiglitazone regulates metalloproteinase 9, NOS-2, and cyclooxygenase 2 expression and functions by peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms in cardiac cells. Shock. 2010;34:60–67. doi: 10.1097/SHK.0b013e3181cdc398. [DOI] [PubMed] [Google Scholar]
- Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab. 2009;20:295–302. doi: 10.1016/j.tem.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Napoli C, Loscalzo J. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide: an overview. Circ Res. 2002;90:21–28. doi: 10.1161/hh0102.102330. [DOI] [PubMed] [Google Scholar]
- Ikeda U, Shimpo M, Ikeda M, Minota S, Shimada K. Lipophilic statins augment inducible nitric oxide synthase expression in cytokine-stimulated cardiac myocytes. J Cardiovasc Pharmacol. 2001;38:69–77. doi: 10.1097/00005344-200107000-00008. [DOI] [PubMed] [Google Scholar]
- Ito H, Torii M, Suzuki T. Decreased superoxide dismutase activity and increased superoxide anion production in cardiac hypertrophy of spontaneously hypertensive rats. Clin Exp Hypertens. 1995;17:803–816. doi: 10.3109/10641969509033636. [DOI] [PubMed] [Google Scholar]
- Jesmin S, Zaedi S, Maeda S, Yamaguchi I, Goto K, Miyauchi T. Effects of a selective endothelin a receptor antagonist on the expressions of iNOS and eNOS in the heart of early streptozotocin-induced diabetic rats. Exp Biol Med. 2006;231:925–931. [PubMed] [Google Scholar]
- Jin CZ, Jang JH, Wang Y, Kim JG, Bae YM, Shi J, et al. Neuronal nitric oxide synthase is up-regulated by angiotensin II and attenuates NADPH oxidase activity and facilitates relaxation in murine left ventricular myocytes. J Mol Cell Cardiol. 2012;52:1274–1278. doi: 10.1016/j.yjmcc.2012.03.013. [DOI] [PubMed] [Google Scholar]
- Jo H, Otani H, Jo F, Shimazu T, Okazaki T, Yoshioka K, et al. Inhibition of nitric oxide synthase uncoupling by sepiapterin improves left ventricular function in streptozotocin-induced diabetic mice. Clin Exp Pharmacol Physiol. 2011;38:485–493. doi: 10.1111/j.1440-1681.2011.05535.x. [DOI] [PubMed] [Google Scholar]
- Jozefowicz E, Brisson H, Rozenberg S, Mebazaa A, Gelé P, Callebert J, et al. Activation of peroxisome proliferator-activated receptor-alpha by fenofibrate prevents myocardial dysfunction during endotoxemia in rats. Crit Care Med. 2007;35:856–863. doi: 10.1097/01.CCM.0000256843.75446.A0. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Collino M, Castiglia S, Fantozzi R, Thiemermann C. Activation of peroxisome proliferator-activated receptor-beta/delta attenuates myocardial ischemia/reperfusion injury in the rat. Shock. 2010;34:117–124. doi: 10.1097/SHK.0b013e3181cd86d6. [DOI] [PubMed] [Google Scholar]
- Kardassis D, Bech-Hanssen O, Schönander M, Sjöström L, Petzold M, Karason K. Impact of body composition, fat distribution and sustained weight loss on cardiac function in obesity. Int J Cardiol. 2012;159:128–133. doi: 10.1016/j.ijcard.2011.02.036. [DOI] [PubMed] [Google Scholar]
- Kass DA, Takimoto E, Nagayama T, Champion HC. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res. 2007;75:303–314. doi: 10.1016/j.cardiores.2007.02.031. [DOI] [PubMed] [Google Scholar]
- Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32:1652–1661. doi: 10.1161/ATVBAHA.112.249318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles CJ, Dionne M, Cebova M, Pinz IM. Palmitate-induced translocation of caveolin-3 and endothelial nitric oxide synthase in cardiomyocytes. OnLine J Biol Sci. 2011;11:27–36. doi: 10.3844/ojbsci.2011.27.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles CJ, Cebova M, Pinz IM. Palmitate diet-induced loss of cardiac caveolin-3: a novel mechanism for lipid-induced contractile dysfunction. PLoS ONE. 2013;8:e61369. doi: 10.1371/journal.pone.0061369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KK, Park SM, Quon MJ. Leptin and cardiovascular disease: response to therapeutic interventions. Circulation. 2008;117:3238–3249. doi: 10.1161/CIRCULATIONAHA.107.741645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojda G, Stein D, Kottenberg E, Schnaith EM, Noack E. In vivo effects of pentaerythrityl-tetranitrate and isosorbide-5 mononitrate on the development of atherosclerosis and endothelial dysfunction in cholesterol-fed rabbits. J Cardiovasc Pharmacol. 1995;25:763–773. doi: 10.1097/00005344-199505000-00012. [DOI] [PubMed] [Google Scholar]
- Kojda G, Hacker A, Noack E. Effects of nonintermittent treatment of rabbits with pentaerythritol tetranitrate on vascular reactivity and superoxide production. Eur J Pharmacol. 1998;355:23–31. doi: 10.1016/s0014-2999(98)00460-9. [DOI] [PubMed] [Google Scholar]
- Koka S, Das A, Salloum FN, Kukreja RC. Phosphodiesterase-5 inhibitor tadalafil attenuates oxidative stress and protects against myocardial ischemia/reperfusion injury in type 2 diabetic mice. Free Radic Biol Med. 2013;60:80–88. doi: 10.1016/j.freeradbiomed.2013.01.031. [DOI] [PubMed] [Google Scholar]
- Konstam MA, Patten RD, Thomas I, Ramahi T, La Bresh K, Goldman S, et al. Effects of losartan and captopril on left ventricular volumes in elderly patients with heart failure: results of the ELITE ventricular function substudy. Am Heart J. 2000;139:1081–1087. doi: 10.1067/mhj.2000.105302. [DOI] [PubMed] [Google Scholar]
- Kristek F, Gerová M. Long-term NO synthase inhibition affects heart weight and geometry of coronary and carotid arteries. Physiol Res. 1996;45:361–367. [PubMed] [Google Scholar]
- Kristek F, Fáberová V, Varga I. Long-term effect of molsidomine and pentaerythrityl tetranitrate on cardiovascular system of spontaneously hypertensive rats. Physiol Res. 2003;52:709–717. [PubMed] [Google Scholar]
- Kupai K, Csonka C, Fekete V, Odendaal L, van Rooyen J, de Marais W, et al. Cholesterol diet-induced hyperlipidemia impairs the cardioprotective effect of postconditioning: role of peroxynitrite. Am J Physiol Heart Circ Physiol. 2009;297:H1729–H1735. doi: 10.1152/ajpheart.00484.2009. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson JE, Rainer PP, Watts VL, Yang R, Miller KL, Phan A, et al. Dependence of β3-adrenergic signaling on the adipokine leptin in cardiac myocytes. Int J Obes. 2012;36:876–879. doi: 10.1038/ijo.2011.137. [DOI] [PubMed] [Google Scholar]
- Laskowski A, Woodman OL, Cao AH, Drummond GR, Marshall T, Kaye DM, et al. Antioxidant actions contribute to the antihypertrophic effects of atrial natriuretic peptide in neonatal rat cardiomyocytes. Cardiovasc Res. 2006;72:112–123. doi: 10.1016/j.cardiores.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Lembo G, Vecchione C, Fratta L, Marino G, Trimarco V, d'Amati G, et al. Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes. 2000;49:293–297. doi: 10.2337/diabetes.49.2.293. [DOI] [PubMed] [Google Scholar]
- Li H, Wallerath T, Munzel T, Forstemann U. Regulation of endothelial-type NO synthase expression in pathophysiology and in response to drugs. Nitric Oxide. 2002;7:149–164. doi: 10.1016/s1089-8603(02)00111-8. [DOI] [PubMed] [Google Scholar]
- Ljubkovic M, Shi Y, Cheng Q, Bosnjak Z, Jiang MT. Cardiac mitochondrial ATP-sensitive potassium channel is activated by nitric oxide in vitro. FEBS Lett. 2007;581:4255–4259. doi: 10.1016/j.febslet.2007.07.071. [DOI] [PubMed] [Google Scholar]
- Lombardi C, Castrini AI, Metra M, Carubelli V, Lazzarini V, Inama L, et al. Efficacy of ACE inhibitors in patients with recent myocardial infarction. Studies with zofenopril. G Ital Cardiol. 2012;13(10 Suppl. 2):55S–58S. doi: 10.1714/1167.12922. [DOI] [PubMed] [Google Scholar]
- Madonna R, Di Napoli P, Massaro M, Grilli A, Felaco M, De Caterina A, et al. Simvastatin attenuates expression of cytokine-inducible nitric-oxide synthase in embryonic cardiac myoblasts. J Biol Chem. 2005;280:13503–13511. doi: 10.1074/jbc.M411859200. [DOI] [PubMed] [Google Scholar]
- Maffei A, Aretini A, Vecchione C, Poulet R, Marino G, Gentile MT, et al. Characterization of nitric oxide release by nebivolol and its metabolites. Am J Hypertens. 2006;19:579–586. doi: 10.1016/j.amjhyper.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Maffei A, Di Pardo A, Carangi R, Carullo P, Poulet R, Gentile MT, et al. Nebivolol induces nitric oxide release in the heart through inducible nitric oxide synthase activation. Hypertension. 2007;50:652–656. doi: 10.1161/HYPERTENSIONAHA.107.094458. [DOI] [PubMed] [Google Scholar]
- Makino N, Maeda T, Oyama J, Higuchi Y, Mimori K. Improving insulin sensitivity via activation of PPAR-gamma increases telomerase activity in the heart of OLETF rats. Am J Physiol Heart Circ Physiol. 2009;297:H2188–H2195. doi: 10.1152/ajpheart.00421.2009. [DOI] [PubMed] [Google Scholar]
- Maksimenko AV, Vavaev AV. Antioxidant enzymes as potential targets in cardioprotection and treatment of cardiovascular diseases. Enzyme antioxidants: the next stage of pharmacological counterwork to the oxidative stress. Heart Int. 2012;7:e3. doi: 10.4081/hi.2012.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheeussen V, Waumans Y, Martinet W, Van Goethem S, Van der Veken P, Scharpé S, et al. Dipeptidyl peptidases in atherosclerosis: expression and role in macrophage differentiation, activation and apoptosis. Basic Res Cardiol. 2013;108:350. doi: 10.1007/s00395-013-0350-4. [DOI] [PubMed] [Google Scholar]
- Megson IL, Leslie SJ. La-419, a nitric-oxide donor for the treatment of cardiovascular disorders. Curr Opin Investig Drugs. 2009;10:276–285. [PubMed] [Google Scholar]
- Mendez M, LaPointe MC. PPARgamma inhibition of cyclooxygenase-2, PGE2 synthase, and inducible nitric oxide synthase in cardiac myocytes. Hypertension. 2003;42:844–850. doi: 10.1161/01.HYP.0000085332.69777.D1. [DOI] [PubMed] [Google Scholar]
- Merla R, Ye Y, Lin Y, Manickavasagam S, Huang MH, Perez-Polo RJ, et al. The central role of adenosine in statin-induced ERK1/2, Akt, and eNOS phosphorylation. Am J Physiol Heart Circ Physiol. 2007;293:H1918–H1928. doi: 10.1152/ajpheart.00416.2007. [DOI] [PubMed] [Google Scholar]
- Michel T. The complexities of coupling nitric oxide synthase pathways in the heart. Circulation. 2010;121:484–486. doi: 10.1161/CIR.0b013e3181d1e24e. [DOI] [PubMed] [Google Scholar]
- Miller MW, Knaub LA, Olivera-Fragoso LF, Keller AC, Balasubramaniam V, Watson PA, et al. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am J Physiol Heart Circ Physiol. 2013;304:H1624–H1633. doi: 10.1152/ajpheart.00987.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollnau H, Oelze M, August M, Wendt M, Daiber A, Schulz E, et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler Thromb Vasc Biol. 2005;25:2554–2559. doi: 10.1161/01.ATV.0000190673.41925.9B. [DOI] [PubMed] [Google Scholar]
- Munzel T, Sayegh H, Freeman BA, Hink U, Matheis E, Hartmann M. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J Clin Invest. 1995;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Ullrich V, Mülsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol. 2005;25:1551–1557. doi: 10.1161/01.ATV.0000168896.64927.bb. [DOI] [PubMed] [Google Scholar]
- Münzel T, Gori T, Bruno RM, Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J. 2010;31:2741–2749. doi: 10.1093/eurheartj/ehq396. [DOI] [PubMed] [Google Scholar]
- Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T. Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension. 2007;50:877–883. doi: 10.1161/HYPERTENSIONAHA.107.091058. [DOI] [PubMed] [Google Scholar]
- Nakano A, Liu GS, Heusch G, Downey JM, Cohen MV. Exogenous nitric oxide can trigger a preconditioned state through a free radical mechanism, but endogenous nitric oxide is not a trigger of classical ischemic preconditioning. J Mol Cell Cardiol. 2000;32:1159–1167. doi: 10.1006/jmcc.2000.1152. [DOI] [PubMed] [Google Scholar]
- Napoli C, Sica V, de Nigris F, Pignalosa O, Condorelli M, Ignarro LJ, et al. Sulfhydryl angiotensin-converting enzyme inhibition induces sustained reduction of systemic oxidative stress and improves the nitric oxide pathway in patients with essential hypertension. Am Heart J. 2004;148:e5. doi: 10.1016/j.ahj.2004.03.025. [DOI] [PubMed] [Google Scholar]
- Nduhirabandi F, du Toit EF, Lochner A. Melatonin and the metabolic syndrome: a tool for effective therapy in obesity-associated abnormalities? Acta Physiol. 2012;205:209–223. doi: 10.1111/j.1748-1716.2012.02410.x. [DOI] [PubMed] [Google Scholar]
- Nickola MW, Wold LE, Colligan PB, Wang GJ, Samson WK, Ren J. Leptin attenuates cardiac contraction in rat ventricular myocytes. Role of NO. Hypertension. 2000;36:501–505. doi: 10.1161/01.hyp.36.4.501. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Otani H, Shimazu T, Yoshioka K, Fujita M, Katano T, et al. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. J Mol Cell Cardiol. 2011;50:534–544. doi: 10.1016/j.yjmcc.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Oliveira HC, Cosso RG, Alberici LC, Maciel EN, Salerno AG, Dorighello GG, et al. Oxidative stress in atherosclerosis-prone mouse is due to low antioxidant capacity of mitochondria. FASEB J. 2005;19:278–280. doi: 10.1096/fj.04-2095fje. [DOI] [PubMed] [Google Scholar]
- Onody A, Csonka C, Giricz Z, Ferdinandy P. Hyperlipidemia induced by a cholesterol-rich diet leads to enhanced peroxynitrite formation in rat hearts. Cardiovasc Res. 2003;58:663–670. doi: 10.1016/s0008-6363(03)00330-4. [DOI] [PubMed] [Google Scholar]
- Ou ZJ, Chang FJ, Luo D, Liao XL, Wang ZP, Zhang X, et al. Endothelium-derived microparticles inhibit angiogenesis in the heart and enhance the inhibitory effects of hypercholesterolemia on angiogenesis. Am J Physiol Endocrinol Metab. 2011;300:E661–E668. doi: 10.1152/ajpendo.00611.2010. [DOI] [PubMed] [Google Scholar]
- Ozakca I, Arioglu-Inan E, Esfahani H, Altan VM, Balligand JL, Kayki-Mutlu G, et al. Nebivolol prevents desensitization of β-adrenoceptor signaling and induction of cardiac hypertrophy in response to isoprenaline beyond β1-adrenoceptor blockage. Am J Physiol Heart Circ Physiol. 2013;304:H1267–H1276. doi: 10.1152/ajpheart.00352.2012. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabó C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal. 2005;7:1568–1580. doi: 10.1089/ars.2005.7.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabó E, Szabó C. The role of poly (ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- Pacher P, Obrosova IG, Mabley JG, Szabó C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem. 2005;12:267–275. doi: 10.2174/0929867053363207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechánová O. Contribution of captopril thiol group to the prevention of spontaneous hypertension. Physiol Res. 2007;56(Suppl. 2):S41–S48. doi: 10.33549/physiolres.931396. [DOI] [PubMed] [Google Scholar]
- Pechánová O, Bernátová I, Babál P, Martínez MC, Kyselá S, Stvrtina S, et al. Red wine polyphenols prevent cardiovascular alterations in L-NAME-induced hypertension. J Hypertens. 2004;22:1551–1559. doi: 10.1097/01.hjh.0000133734.32125.c7. [DOI] [PubMed] [Google Scholar]
- Pechánová O, Zicha J, Kojsová S, Dobesová Z, Jendeková L, Kunes J. Effect of chronic N-acetylcysteine treatment on the development of spontaneous hypertension. Clin Sci. 2006;110:235–242. doi: 10.1042/CS20050227. [DOI] [PubMed] [Google Scholar]
- Pechánová O, Zicha J, Paulis L, Zenebe W, Dobesová Z, Kojsová S, et al. The effect of N-acetylcysteine and melatonin in adult spontaneously hypertensive rats with established hypertension. Eur J Pharmacol. 2007;561:129–136. doi: 10.1016/j.ejphar.2007.01.035. [DOI] [PubMed] [Google Scholar]
- Pechanova O, Simko F. Chronic antioxidant therapy fails to ameliorate hypertension: potential mechanisms behind. J Hypertens. 2009;27:S32–S36. doi: 10.1097/01.hjh.0000358835.25934.5e. [DOI] [PubMed] [Google Scholar]
- Pechanova O, Simko F. The role of nuclear factor kappa B and nitric oxide interaction in heart remodelling. J Hypertens. 2010;28(Suppl. 1):S39–S44. doi: 10.1097/01.hjh.0000388493.81578.b1. [DOI] [PubMed] [Google Scholar]
- Pechanova O, Bernatova I, Pelouch V, Simko F. Protein remodelling of the heart in NO-deficient hypertension: the effect of captopril. J Mol Cell Cardiol. 1997;29:3365–3374. doi: 10.1006/jmcc.1997.0566. [DOI] [PubMed] [Google Scholar]
- Pelat M, Dessy C, Massion P, Desager JP, Feron O, Balligand JL. Rosuvastatin decreases caveolin-1 and improves nitric oxide-dependent heart rate and blood pressure variability in apolipoprotein E-/- mice in vivo. Circulation. 2003;107:2480–2486. doi: 10.1161/01.CIR.0000065601.83526.3E. [DOI] [PubMed] [Google Scholar]
- Penumathsa SV, Thirunavukkarasu M, Zhan L, Maulik G, Menon VP, Bagchi D, et al. Resveratrol enhances GLUT-4 translocation to the caveolar lipid raft fractions through AMPK/Akt/eNOS signalling pathway in diabetic myocardium. J Cell Mol Med. 2008;12:2350–2361. doi: 10.1111/j.1582-4934.2008.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penumathsa SV, Thirunavukkarasu M, Samuel SM, Zhan L, Maulik G, Bagchi M, et al. Niacin bound chromium treatment induces myocardial Glut-4 translocation and caveolar interaction via Akt, AMPK and eNOS phosphorylation in streptozotocin induced diabetic rats after ischemia-reperfusion injury. Biochim Biophys Acta. 2009;1792:39–48. doi: 10.1016/j.bbadis.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Peters A, Schweiger U, Frühwald-Schultes B, Born J, Fehm HL. The neuroendocrine control of glucose allocation. Exp Clin Endocrinol Diabetes. 2002;110:199–211. doi: 10.1055/s-2002-33068. [DOI] [PubMed] [Google Scholar]
- Poon BY, Raharjo E, Patel KD, Tavener S, Kubes P. Complexity of inducible nitric oxide synthase: cellular source determines benefit versus toxicity. Circulation. 2003;108:1107–1112. doi: 10.1161/01.CIR.0000086321.04702.AC. [DOI] [PubMed] [Google Scholar]
- Post H, Schulz R, Behrends M, Gres P, Umschlag C, Heusch G. No involvement of endogenous nitric oxide in classical ischemic preconditioning in swine. J Mol Cell Cardiol. 2000;32:725–733. doi: 10.1006/jmcc.2000.1117. [DOI] [PubMed] [Google Scholar]
- Post H, Schulz R, Gres P, Heusch G. No involvement of nitric oxide in the limitation of beta-adrenergic inotropic responsiveness during ischemia. Am J Physiol Heart Circ Physiol. 2001;281:H2392–H2397. doi: 10.1152/ajpheart.2001.281.6.H2392. [DOI] [PubMed] [Google Scholar]
- Poudyal H, Campbell F, Brown L. Olive leaf extract attenuates cardiac, hepatic, and metabolic changes in high carbohydrate-, high fat-fed rats. J Nutr. 2010;40:946–953. doi: 10.3945/jn.109.117812. [DOI] [PubMed] [Google Scholar]
- Puthanveetil P, Zhang D, Wang Y, Wang F, Wan A, Abrahani A, et al. Diabetes triggers a PARP1 mediated death pathway in the heart through participation of FoxO1. J Mol Cell Cardiol. 2012;53:677–686. doi: 10.1016/j.yjmcc.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Radovits T, Bömicke T, Kökény G, Arif R, Loganathan S, Kécsán K, et al. The phosphodiesterase-5 inhibitor vardenafil improves cardiovascular dysfunction in experimental diabetes mellitus. Br J Pharmacol. 2009;156:909–919. doi: 10.1111/j.1476-5381.2008.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafighi Z, Shiva A, Arab S, Mohd Yousof R. Association of dietary vitamin C and e intake and antioxidant enzymes in type 2 diabetes mellitus patients. Glob J Health Sci. 2013;5:183–187. doi: 10.5539/gjhs.v5n3p183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, et al. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart. 2005;91:806–810. doi: 10.1136/hrt.2003.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Bátkai S, Mukhopadhyay B, Patel V, Haskó G, et al. Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy. J Cell Mol Med. 2009;13:2330–2341. doi: 10.1111/j.1582-4934.2008.00564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Bátkai S, Kechrid M, Mukhopadhyay P, Lee WS, Horváth B, et al. Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes. 2012;61:716–727. doi: 10.2337/db11-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassaf T, Poll LW, Brouzos P, Lauer T, Totzeck M, Kleinbongard P, et al. Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J. 2006;27:1699–1705. doi: 10.1093/eurheartj/ehl096. [DOI] [PubMed] [Google Scholar]
- Rassaf T, Ferdinandy P, Schulz R. Nitrite in organ protection. Br J Pharmacol. 2014;171:1–11. doi: 10.1111/bph.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SN, Jayaram R, Nahar K, Antoniades C, Verheule S, Channon KM, et al. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation. 2011;124:1107–1117. doi: 10.1161/CIRCULATIONAHA.111.029223. [DOI] [PubMed] [Google Scholar]
- Reiter RJ, Tan DX, Korkmaz A. The circadian melatonin rhythm and its modulation: possible impact on hypertension. J Hypertens. 2009;27(Suppl. 6):S17–S21. doi: 10.1097/01.hjh.0000358832.41181.bf. [DOI] [PubMed] [Google Scholar]
- Rosenkranz AC, Hood SG, Woods RL, Dusting GJ, Ritchie RH. B-type natriuretic peptide prevents acute hypertrophic responses in the diabetic rat heart: importance of cyclic GMP. Diabetes. 2003;52:2389–2395. doi: 10.2337/diabetes.52.9.2389. [DOI] [PubMed] [Google Scholar]
- Ruiz-Hurtado G, Delgado C. Nitric oxide pathway in hypertrophied heart: new therapeutic uses of nitric oxide donors. J Hypertens. 2010;28:S56–S61. doi: 10.1097/01.hjh.0000388496.66330.b8. [DOI] [PubMed] [Google Scholar]
- Ruiz-Hurtado G, Fernandez-Velasco M, Mourelle M, Delgado C. La419, a novel nitric oxide donor, prevents pathological cardiac remodeling in pressure overloaded rats via endothelial nitric oxide synthase pathway regulation. Hypertension. 2007;50:1049–1056. doi: 10.1161/HYPERTENSIONAHA.107.093666. [DOI] [PubMed] [Google Scholar]
- Saraiva RM, Minhas KM, Zheng M, Pitz E, Treuer A, Gonzalez D, et al. Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric Oxide. 2007;16:331–338. doi: 10.1016/j.niox.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwemmer M, Sommer O, Bassenge E. Blockade of angiotensin signaling improves myocardial function in hypercholesterolemia independent of changes in eicosanoid release. Cardiovasc Drugs Ther. 2000;14:317–327. doi: 10.1023/a:1007838809551. [DOI] [PubMed] [Google Scholar]
- Shah AM, MacCarthy PA. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol Ther. 2000;86:49–86. doi: 10.1016/s0163-7258(99)00072-8. [DOI] [PubMed] [Google Scholar]
- Simko F, Pechanova O. Remodelling of the heart and vessels in experimental hypertension: advances in protection. J Hypertens. 2010;28(Suppl. 1):S1–S6. doi: 10.1097/01.hjh.0000388487.43460.db. [DOI] [PubMed] [Google Scholar]
- Smith JM, Paulson DJ, Romano FD. Inhibition of nitric oxide synthase by L-NAME improves ventricular performance in streptozotocin-diabetic rats. J Mol Cell Cardiol. 1997;29:2393–2402. doi: 10.1006/jmcc.1997.0474. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123:2263–2273. doi: 10.1161/CIRCULATIONAHA.110.981738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockklauser-Färber K, Ballhausen T, Laufer A, Rösen P. Influence of diabetes on cardiac nitric oxide synthase expression and activity. Biochim Biophys Acta. 2000;1535:10–20. doi: 10.1016/s0925-4439(00)00078-8. [DOI] [PubMed] [Google Scholar]
- Stokes KY, Dugas TR, Tang Y, Garg H, Guidry E, Bryan NS. Dietary nitrite prevents hypercholesterolemic microvascular inflammation and reverses endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2009;296:H1281–H1288. doi: 10.1152/ajpheart.01291.2008. [DOI] [PubMed] [Google Scholar]
- Sutra T, Oiry C, Azay-Milhau J, Youl E, Magous R, Teissèdre PL, et al. Preventive effects of nutritional doses of polyphenolic molecules on cardiac fibrosis associated with metabolic syndrome: involvement of osteopontin and oxidative stress. J Agric Food Chem. 2008;56:11683–11687. doi: 10.1021/jf802357g. [DOI] [PubMed] [Google Scholar]
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- Tang L, Wang H, Ziolo MT. Targeting NOS as a therapeutic approach for heart failure. Pharmacol Ther. 2013;S0163-7258:00255-6. doi: 10.1016/j.pharmthera.2013.12.013. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Tirziu D, Simons M. Endothelium-driven myocardial growth or nitric oxide at the crossroads. Trends Cardiovasc Med. 2008;18:299–305. doi: 10.1016/j.tcm.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toblli J, Cao G, Rivas C, Munoz M, Giani J, Dominici F, et al. Cardiovascular protective effects of nebivolol in Zucker diabetic fatty rats. J Hypertens. 2010;28:1007–1019. doi: 10.1097/hjh.0b013e328337598c. [DOI] [PubMed] [Google Scholar]
- Tribulova N, Ravingerova T, Okruhlicova L, Gabauer I, Fickova M, Pancza D, et al. Modulation of cAMP level by tedisamil in guinea pig heart. Mol Cell Biochem. 2000;210:75–80. doi: 10.1023/a:1007133322295. [DOI] [PubMed] [Google Scholar]
- Tsai TH, Chai HT, Sun CK, Yen CH, Leu S, Chen YL, et al. Obesity suppresses circulating level and function of endothelial progenitor cells and heart function. J Transl Med. 2012;10:137. doi: 10.1186/1479-5876-10-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turko IV, Marcondes S, Murad F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am J Physiol Heart Circ Physiol. 2001;281:H2289–H2294. doi: 10.1152/ajpheart.2001.281.6.H2289. [DOI] [PubMed] [Google Scholar]
- Varga ZV, Kupai K, Szűcs G, Gáspár R, Pálóczi J, Faragó N, et al. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J Mol Cell Cardiol. 2013;62:111–121. doi: 10.1016/j.yjmcc.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Varma A, Das A, Hoke NN, Durrant DE, Salloum FN, Kukreja RC. Anti-inflammatory and cardioprotective effects of tadalafil in diabetic mice. PLoS ONE. 2012;7:e45243. doi: 10.1371/journal.pone.0045243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrankova S, Barta A, Parohova J, Pechanova O. Could NF-kappa B play a role in blood pressure regulation? Focused on L-NAME-induced hypertension. J Hypertens. 2009;27(Suppl. 4):S140. [Google Scholar]
- Wang XF, Zhang JY, Li L, Zhao XY, Tao HL, Zhang L. Metformin improves cardiac function in rats via activation of AMP-activated protein kinase. Clin Exp Pharmacol Physiol. 2011;38:94–101. doi: 10.1111/j.1440-1681.2010.05470.x. [DOI] [PubMed] [Google Scholar]
- Ward NC, Croft KD. Hypertension and oxidative stress. Clin Exp Pharmacol Physiol. 2006;33:872–876. doi: 10.1111/j.1440-1681.2006.04457.x. [DOI] [PubMed] [Google Scholar]
- Wayman NS, Hattori Y, McDonald MC, Mota-Filipe H, Cuzzocrea S, Pisano B, et al. Ligands of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J. 2002;16:1027–1040. doi: 10.1096/fj.01-0793com. [DOI] [PubMed] [Google Scholar]
- Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, et al. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension. 2009;54:338–344. doi: 10.1161/HYPERTENSIONAHA.108.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CS. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol Ther. 2010;126:119–145. doi: 10.1016/j.pharmthera.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids. 2009;37:1–17. doi: 10.1007/s00726-009-0269-0. [DOI] [PubMed] [Google Scholar]
- Wu G, Bazer FW, Davis TA, Kim SW, Li P, Marc Rhoads J, et al. Arginine metabolism and nutrition in growth, health and disease. Amino Acids. 2009;37:153–168. doi: 10.1007/s00726-008-0210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Su G, Brown SN, Chen L, Ren J, Zhao P. Peroxisome proliferator-activated receptors gamma and alpha agonists stimulate cardiac glucose uptake via activation of AMP-activated protein kinase. J Nutr Biochem. 2010;21:621–626. doi: 10.1016/j.jnutbio.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Ye Y, Lin Y, Manickavasagam S, Perez-Polo JR, Tieu BC, Birnbaum Y. Pioglitazone protects the myocardium against ischemia-reperfusion injury in eNOS and iNOS knockout mice. Am J Physiol Heart Circ Physiol. 2008a;295:H2436–H2446. doi: 10.1152/ajpheart.00690.2008. [DOI] [PubMed] [Google Scholar]
- Ye Y, Martinez JD, Perez-Polo RJ, Lin Y, Uretsky BF, Birnbaum Y. The role of eNOS, iNOS, and NF-kappaB in upregulation and activation of cyclooxygenase-2 and infarct size reduction by atorvastatin. Am J Physiol Heart Circ Physiol. 2008b;295:H343–H351. doi: 10.1152/ajpheart.01350.2007. [DOI] [PubMed] [Google Scholar]
- Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, et al. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes. 2002;51:2587–2595. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ohmori K, Chen Y, Sato C, Kiyomoto H, Shinomiya K, et al. Effects of pravastatin on progression of glucose intolerance and cardiovascular remodeling in a type II diabetes model. J Am Coll Cardiol. 2004;44:904–913. doi: 10.1016/j.jacc.2004.04.050. [DOI] [PubMed] [Google Scholar]
- Yue ZJ, Yu ZB. Cardioprotection by the inhibitory effect of nitric oxide. Sheng Li Xue Bao. 2011;63:191–197. [PubMed] [Google Scholar]
- Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients: the Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- Zakula Z, Koricanac G, Tepavcevic S, Stojiljkovic M, Milosavljevic T, Isenovic ER. Impairment of cardiac insulin signaling in fructose-fed ovariectomized female Wistar rats. Eur J Nutr. 2011;50:543–551. doi: 10.1007/s00394-010-0161-4. [DOI] [PubMed] [Google Scholar]
- Zanfolin M, Faro R, Araujo EG, Guaraldo AM, Antunes E, De Nucci G. Protective effects of BAY 41–2272 (sGC stimulator) on hypertension, heart, and cardiomyocyte hypertrophy induced by chronic L-NAME treatment in rats. J Cardiovasc Pharmacol. 2006;47:391–395. [PubMed] [Google Scholar]
- Zaobornyj T, Ghafourifar P. Strategic localization of heart mitochondrial NOS: a review of the evidence. Am J Physiol Heart Circ Physiol. 2012;303:H1283–H1293. doi: 10.1152/ajpheart.00674.2011. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Zou AP, Li PL. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am J Physiol Heart Circ Physiol. 2003;284:H605–H612. doi: 10.1152/ajpheart.00697.2002. [DOI] [PubMed] [Google Scholar]
- Zhang FJ, Ma LL, Wang WN, Qian LB, Yang MJ, Yu J, et al. Hypercholesterolemia abrogates sevoflurane-induced delayed preconditioning against myocardial infarct in rats by alteration of nitric oxide synthase signaling. Shock. 2012;37:485–491. doi: 10.1097/SHK.0b013e318249b7b6. [DOI] [PubMed] [Google Scholar]
- Zhang H, Morgan B, Potter BJ, Ma L, Dellsperger KC, Ungvari Z, et al. Resveratrol improves left ventricular diastolic relaxation in type 2 diabetes by inhibiting oxidative/nitrative stress: in vivo demonstration with magnetic resonance imaging. Am J Physiol Heart Circ Physiol. 2010;299:H985–H994. doi: 10.1152/ajpheart.00489.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Janssens SP, Wingler K, Schmidt HH, Moens AL. Modulating endothelial nitric oxide synthase: a new cardiovascular therapeutic strategy. Am J Physiol Heart Circ Physiol. 2011;301:H634–H646. doi: 10.1152/ajpheart.01315.2010. Erratum in: Am J Physiol Heart Circ Physiol (2012) 303(2): H241. [DOI] [PubMed] [Google Scholar]
- Zhen J, Lu H, Wang XQ, Vaziri ND, Zhou XJ. Upregulation of endothelial and inducible nitric oxide synthase expression by reactive oxygen species. Am J Hypertens. 2008;21:28–34. doi: 10.1038/ajh.2007.14. [DOI] [PubMed] [Google Scholar]
- Zhou MS, Schulman IH, Raij L. Nitric oxide, angiotensin II, and hypertension. Semin Nephrol. 2004;24:366–378. doi: 10.1016/j.semnephrol.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000;97:1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109:817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]