

Abstract
Background and Purpose
Hypertension is an important mediator of cardiac damage and remodelling. Hydrogen sulfide (H2S) is an endogenously produced gasotransmitter with cardioprotective properties. However, it is not yet in clinical use. We, therefore, investigated the protective effects of sodium thiosulfate (STS), a clinically applicable H2S donor substance, in angiotensin II (Ang II)-induced hypertensive cardiac disease in rats.
Experimental Approach
Male Sprague Dawley rats were infused with Ang II (435 ng kg min−1) or saline (control) for 3 weeks via s.c. placed osmotic minipumps. During these 3 weeks, rats received i.p. injections of either STS, NaHS or vehicle (0.9% NaCl).
Key Results
Compared with controls, Ang II infusion caused an increase in systolic and diastolic BP with associated cardiac damage as evidenced by cardiac hypertrophy, an increase in atrial natriuretic peptide (ANP) mRNA, cardiac fibrosis and increased oxidative stress. Treatment with NaHS and STS prevented the development of hypertension and the increase in ANP mRNA levels. Furthermore, the degree of cardiac hypertrophy, the extent of histological fibrosis in combination with the expression of profibrotic genes and the levels of oxidative stress were all significantly decreased.
Conclusions and Implications
Ang II-induced hypertensive cardiac disease can be attenuated by treatment with STS and NaHS. Although BP regulation is the most plausible mechanism of cardiac protection, the antifibrotic and antioxidant properties of released sulfide may also contribute to their effects. Our data show that H2S might be a valuable addition to the already existing antihypertensive and cardioprotective therapies.
Linked Articles
This article is part of a themed section on Pharmacology of the Gasotransmitters. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-6
Table of Links
| TARGETS | LIGANDS |
|---|---|
| H2S | Angiotensin II |
| KATP channel | Atrial natriuretic peptide |
| AT1 receptor | Galectin-3 |
| Fibronectin | |
| Nitric oxide | |
| NaHS |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a, b, c).
Introduction
Hypertensive heart disease is a major global health problem and a considerable cause of cardiovascular morbidity and mortality. Hypertension is a key contributory factor in cardiovascular disease, especially cerebrovascular disease and heart failure (Drazner, 2011). The heart's response to increased stress such as hypertension is captured by the term ‘cardiac remodelling’, which encompasses myocyte hypertrophy, formation of cardiac fibrosis and vascular rarefaction. Remodelling is associated with activation of various stress responses including the induction of neurohormonal and inflammatory pathways. Progressive remodelling may perpetuate into left ventricular dysfunction and heart failure (Mann, 2002; Jia et al., 2012). Angiotensin II (Ang II) is a well-established mediator in the pathogenesis of hypertensive heart disease. In addition to its hypertensive effect, Ang II induces cardiac remodelling by directly exerting prohypertrophic effects through enhancing inflammatory responses and activation of several profibrotic factors via a number of signalling pathways (Wynn, 2008; Shahbaz et al., 2010). Blockade of the renin-angiotensin-aldosterone system (RAAS) is able to regress cardiac remodelling and has evolved into the cornerstone of cardiovascular disease management. Despite the use of RAAS intervention and other antihypertensive treatments, many patients with hypertensive heart disease still develop heart failure (Koitabashi and Kass, 2011). Therefore, additional modes of intervention are warranted.
Hydrogen sulfide (H2S) is recognized as a biologically important gaseous signalling molecule with a myriad of physiological functions (Wang, 2002). H2S is endogenously produced from the degradation of L-cysteine by cystathionine β-synthase (CBS) and cystathionine γ–lyase (CSE) (Abe and Kimura, 1996; Singh et al., 2009) and from D-cysteine by 3-mercaptopyruvate sulfurtransferase (Shibuya et al., 2013). H2S is serially oxidized to persulfide, thiosulfate (TS, S2O3), sulfite and sulfate (Hildebrandt and Grieshaber, 2008; Kabil and Banerjee, 2010). Similar to NO, H2S functions as an endothelial cell-derived relaxing factor via direct activation of ATP-sensitive potassium (KATP) channels (Mustafa et al., 2011). Accordingly, deprivation of endogenously produced H2S contributes to the development of hypertension (Yang et al., 2008; Sen et al., 2010). Furthermore, H2S has beneficial effects on oxidative stress (Bos et al., 2013), inflammation and fibrosis (Snijder et al., 2013). In various cardiovascular disease states, a reduction in H2S plasma and tissue levels was observed, suggesting a possible role for H2S in its pathogenesis (Jiang et al., 2005; Zhao et al., 2008; Kovacic et al., 2012; Kondo et al., 2013). Exogenous treatment with H2S is cardioprotective in various experimental models of cardiac injury. The pathways implicated in the cardioprotective actions of H2S are multiple. While most of the studies focus on acute myocardial protection, there are some elegant studies investigating the protective effects of H2S in experimental models for chronic heart failure (Szabo et al., 2011). The effects of H2S on Ang II-induced hypertensive heart disease have not been investigated.
For the administration of H2S in experimental models, soluble sulfide salts, such as NaHS and Na2S, are often used. Other ways of delivery include H2S gas or slow-release H2S donors. A novel approach is the use of sodium thiosulfate (STS, Na2S2O3), an endogenous player in the enzymatic pathways of H2S production in mammalian cells. Increasing evidence supports the idea that a dynamic conversion exists between STS and H2S (Ubuka et al., 2001; Olson et al., 2013). In humans, the short-term therapeutic use of STS has been proven safe for the treatment of calciphylaxis (Singh et al., 2011), providing us with the possibility of using H2S related therapies in a clinical setting.
The vasodilating and cytoprotective features of H2S make it an attractive candidate for therapeutic reduction of the cardiovascular alterations associated with hypertension. We investigated the protective properties of sulfide-releasing compounds in Ang II-induced hypertensive heart disease in rats.
Methods
Animals
Male Sprague Dawley rats (n = 27, 240–280 g, Harlan, Zeist, the Netherlands) were housed under standard conditions with a 12 h light-dark cycle at the animal research facility with ad libitum access to food and water. Experimental procedures were in agreement with institutional and legislator regulations and approved by the local committee for animal experiments (DEC 6412B). With regard to the execution of our animal experiments, we followed the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010).
Ang II infusion and NaHS or STS treatment
Osmotic minipumps (model 2004, Alzet, Cupertino, CA, USA) were placed s.c. under general anaesthesia (2% isoflurane/O2) for continuous administration of Ang II (435 ng kg−1 min−1, n = 7 per group; Bachem, Weil am Rhein, Germany) or vehicle (0.9% NaCl, n = 6). To check the depth of the anaesthesia a pain stimulus was administered. Postoperatively, all rats received a s.c. injection of 50 μg kg−1 buprenorphin (Schering-Plough, Houten, the Netherlands) for analgesic purposes and were allowed to recover from the surgery at 37°C in a ventilated incubator. During placement of the pumps, Ang II-infused rats were randomized to either STS (1 g kg−1 day−1; Sigma, Zwijndrecht, the Netherlands), NaHS (5.6 mg kg−1 day−1; Sigma) or 0.9% NaCl treatment. Rats receiving 0.9% NaCl via osmotic minipumps received treatment with 0.9% NaCl. During the 3 weeks of infusion, rats received i.p. injections with one of the compounds twice a day. At baseline, blood was collected via orbital puncture. On a weekly basis, body weight was measured. After 3 weeks, BP was measured under general anaesthesia (2% isoflurane/O2) via an intra-aortic probe (Cardiocap/5, GE Healthcare, Little Chalfont, Buckinghamshire, UK). Subsequently, rats were terminated and blood was collected. Hearts were perfused with 0.9% NaCl, removed and then weighed. The hearts were dissected and mid-papillary slices were fixed in 4% paraformaldehyde and paraffin embedded for immunohistochemical analysis, upper parts were immediately snap frozen in liquid nitrogen and stored at −80°C for molecular analysis.
Cardiomyocyte size
To investigate cardiac hypertrophy, we visualized the membrane of the cardiomyocytes and measured its size. After deparaffinization, mid-papillary cardiac sections were incubated with wheat germ agglutinin (WGA)-FITC (1:100, L4895-2 mg, Sigma) for 30 min in room temperature. Nuclei were stained with 4,6-diamino-2-phenylindole (6-H-1200, Vector Laboratories, Brunschwig Chemie, Amsterdam, The Netherlands). Images were captured with a fluorescence microscope. For each slide, the size (short axis) of 75–100 WGA-stained cardiomyocytes was measured with NIH ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Cardiac fibrosis
To investigate cardiac fibrosis Masson's trichrome staining was used on paraffin-embedded mid-papillary cardiac sections. Subsequently, cardiac sections were scanned using an Aperio Scanscope GS (Aperio Technologies, Vista, CA, USA). The extent of collagen deposition was determined using the Aperio positive pixel analysis v9.1 algorithm. The ratio between the fibrotic surface area and the total cardiac surface area was calculated. Histopathological analysis was performed in a blinded fashion.
Qualitative real-time PCR (qRT-PCR)
Rat cardiac tissue was homogenized in lysis buffer and total RNA was extracted using the TRIZOL method (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized using Superscript II with random hexamer primers (Invitrogen). Gene expression (Applied Biosystems, Foster City, CA, USA) was determined by qRT-PCR based on the Taqman methodology. GAPDH was used as a housekeeping gene. The primers of atrial natriuretic peptide (ANP) and Cybb (Nox2) were obtained from Applied Biosystems as Assays-on-Demand (AOD) gene expression products. The AOD IDs used were: Cybb (Nox2) Rn00576710_m1 and Nppa (ANP) Rn00561661_m1. The primers used for fibronectin and galectin-3 were: FN1 (fibronectin) Forward – GTACCACTGGCCACACCTAC Reverse – TGTCAGCCTGCACGTCCAAC; Lgals3 (galectin-3) Forward – CCCGCTTCAATGAGAACAAC Reverse – ACCGCAACCTTGAAGTGGTC (both from Sigma). For Cybb (Nox2) and ANP, the qRT-PCR reaction mixture contained 20 ng cDNA template and 5 μL PCR mastermix. Nuclease-free water was added to a total volume of 10 μL. For fibronectin and galectin-3, qRT-PCR was performed in a volume of 20 μL containing 10 ng cDNA and 15 μL PCR mastermix (SYBR GREEN Applied Biosystems; 5 mL P/N 4309155). All assays were performed in triplicate. The thermal profile was 2 min at 50°C, 15 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The average Ct values for target genes were subtracted from the average housekeeping gene Ct values to yield the δCt. Results were expressed as 2−ΔCt.
Plasma nitrite and nitrate (NOx)
Measurement of combined NOx was used as a marker for NO production. We colorimetrically measured plasma NOx applying the Griess reaction after reduction of nitrate to nitrite.
Urinary malondialdehyde (MDA) measurements
MDA is a major breakdown product of lipid peroxides and generated after oxidative stress. Twenty microlitres of urine was incubated with 90 μL of 3% SDS and 10 μL of 0.5 M butylated hydroxytoluene followed by the addition of 400 μL 0.1 N HCl, 50 μL 10% phosphotungstic acid and 200 μL 0.7% 2-thiobarbituric acid. The reaction mixture was incubated for 30 min at 95°C. After adding 800 μL of 1-butanol, the samples were centrifuged at 960 g for 10 min. Two hundred microlitres of the 1-butanol phase was fluorescently measured using 530 nm excitation and 590 nm emission wavelengths.
Plasma and urinary calcium measurements
Plasma and urinary calcium levels were determined by standard assays (Roche Diagnostics GmbH, Mannheim, Germany) in our clinical chemical laboratory.
Statistical analysis
Data were analysed and graphed using GraphPad Prism 5.0 software (GraphPad, San Diego, CA, USA). All data are expressed as the mean ± SEM unless otherwise indicated. Normality was tested using the Kolmogorov–Smirnov test. Statistical analyses were performed using t-tests, Mann–Whitney U-tests, two-way anova, one-way anova or Kruskal–Wallis tests where appropriate. Bonferroni or Dunn's post-correction was applied where multiple comparisons where made. Statistical significance was accepted at P < 0.05.
Results
STS and NaHS treatment attenuates Ang II-induced hypertension
Infusion with Ang II significantly increased systolic BP (SBP; 211 ± 9 mmHg vs. 143 ± 2 mmHg, P < 0.001) and diastolic BP (DBP; 127 ± 10 mmHg vs. 84 ± 2 mmHg, P < 0.01) compared with controls. Simultaneous treatment with either STS or NaHS decreased SBP by 18% (173 ± 7 mmHg, P < 0.001) and 22% (164 ± 3 mmHg, P < 0.001), and DBP by 30% (89 ± 7 mmHg, P < 0.01) and 26% (93 ± 8 mmHg, P < 0.05) respectively (Figure 1, Table 1996).
Figure 1.
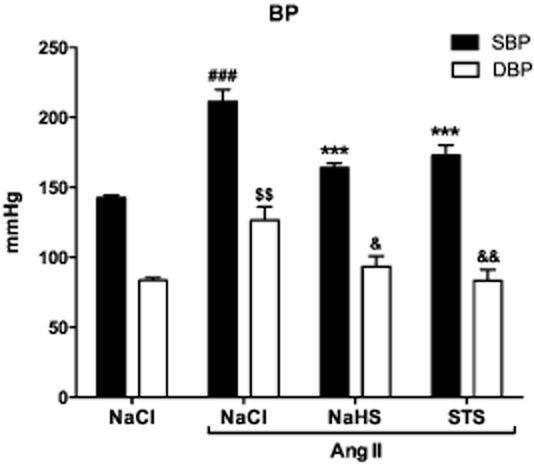
Treatment with STS and NaHS prevents the development of Ang II-induced hypertension. Three weeks of Ang II infusion caused hypertension in the vehicle treated animals (n = 7). SBP was increased by 48% and DBP by 51%. Treatment with STS (n = 7) and NaHS (n = 6) prevented the development of hypertension by Ang II. (SBP: ###P < 0.001 vs. control, ***P < 0.001 vs. Ang II + NaCl; DBP: $$P < 0.01 vs. control, &&P < 0.01 vs. Ang II + NaCl, &P < 0.05 vs. Ang II + NaCl).
Effects of STS and NaHS on cardiac hypertrophy
After 3 weeks, Ang II-infused rats treated with vehicle had a lower body weight compared with NaCl-infused controls (P < 0.001). Treatment with NaHS, but not with STS, resulted in a higher bodyweight compared with treatment with vehicle (NaHS: P < 0.01; Figure 2A). The heart-to-body weight ratio of vehicle-treated Ang II-infused rats was significantly increased compared with 0.9% NaCl-infused controls (P < 0.001) (Figure 2B), suggesting that these animals developed hypertrophy subsequent to Ang II infusion. To confirm this, we investigated the cardiomyocyte size by analysing WGA-stained cardiac sections. Indeed, we observed cardiomyocyte hypertrophy in the vehicle-treated Ang II-infused rats as evidenced by an increased cardiomyocyte size (P < 0.001) (Figure 2C and D). Treatment with STS and NaHS prevented the development of cardiac hypertrophy as evidenced by a preserved heart-to-body weight ratio (STS: P < 0.05, NaHS: P < 0.01) and cardiomyocyte size (P < 0.001) (Figure 2, Table 1).
Figure 2.
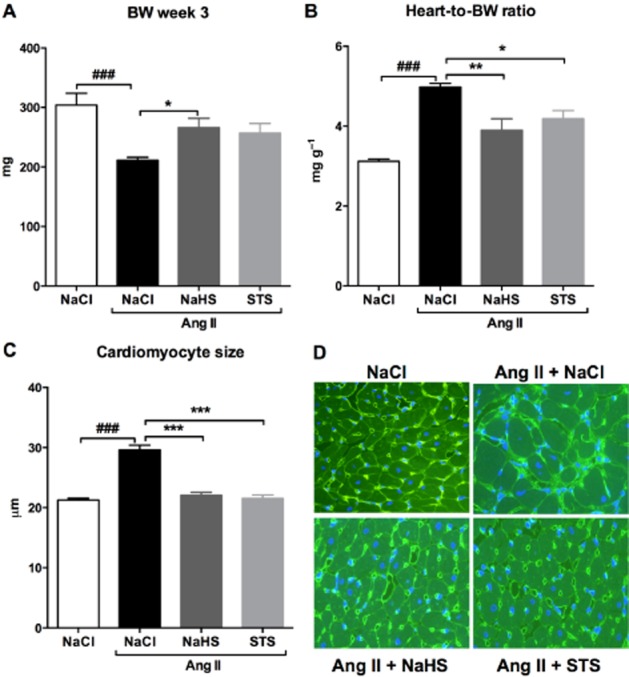
Treatment with STS and NaHS attenuated the cardiac hypertrophy induced by Ang II. (A) Ang II-infused vehicle-treated animals (n = 7) had a lower body weight than controls (n = 6). Simultaneous treatment with NaHS (n = 6), but not with STS (n = 7), resulted in a higher body weight compared with Ang II-infused vehicle-treated animals. (B) After 3 weeks of Ang II infusion the heart-to-body weight ratio was increased in vehicle-treated rats (n = 7) compared with 0.9% NaCl-infused controls (n = 6). Treatment with STS (n = 7) and NaHS (n = 6) attenuated the increase in heart weight. (C) To confirm this finding, we measured the average cardiomyocyte size in WGA-stained cardiac sections. STS (n = 7) as well as NaHS (n = 6) prevented the Ang II-induced increase in cardiomyocyte size. (D) Representative photomicrographs of WGA-stained cardiac sections. (###P < 0.001 vs. control, ***P < 0.001, **P < 0.01, *P < 0.05 vs. Ang II + NaCl).
Table 1.
Overview of all parameters
| NaCl (%) | Ang II + NaCl (%) | Ang II + NaHS (%) | Ang II + STS (%) | |
|---|---|---|---|---|
| SBP | 100 (1) | 148 (6)### | 115 (2)*** | 121 (5)*** |
| DBP | 100 (2) | 146 (11)## | 108 (9)* | 103 (10)** |
| Body weight week 3 | 100 (7) | 70 (2)### | 88 (5)* | 85 (5) |
| Heart-to-body weight ratio | 100 (2) | 160 (3)### | 125 (9)** | 134 (7)* |
| Cardiomyocyte size | 100 (1) | 139 (4)### | 104 (2)*** | 101 (3)*** |
| ANP mRNA | 100 (10) | 928 (255)## | 332 (103)* | 274 (63)* |
| NOx plasma | 100 (2) | 171 (13)### | 119 (25)** | 140 (6)* |
| Fibrosis | 100 (20) | 222 (43)# | 108 (11)* | 107 (9)** |
| Fibronectin mRNA | 100 (12) | 429 (33)### | 229 (57)** | 230 (59)* |
| Galectin-3 mRNA | 100 (16) | 320 (27)### | 178 (29)** | 228 (22)* |
| Nox2 mRNA | 100 (7) | 201 (26)## | 121 (17)** | 140 (11)* |
| MDA excretion | 100 (14) | 237 (18)### | 154 (25)* | 167 (10)* |
| Plasma calcium | 100 (4) | 104 (6) | 103 (5) | 109 (3) |
| Calcium excretion | 100 (44) | 567 (44)# | 342 (110) | 698 (150) |
The results are presented as % change relative to control (NaCl).
P < 0.001
P < 0.01
P < 0.05 vs. NaCl
P < 0.001
P < 0.01
P < 0.05 vs. Ang II + NaCl. SEM is shown in parentheses.
Treatment with STS and NaHS reduces ANP mRNA levels
ANP mRNA expression, a hallmark of the reactivation of the fetal gene programme, was significantly increased in hearts of Ang II-infused rats treated with vehicle compared with 0.9% NaCl-infused controls (P < 0.01). In hearts of STS- and NaHS-treated rats, the relative ANP expression was significantly reduced compared with vehicle-treated rats (P < 0.05) (Figure 3, Table 1).
Figure 3.
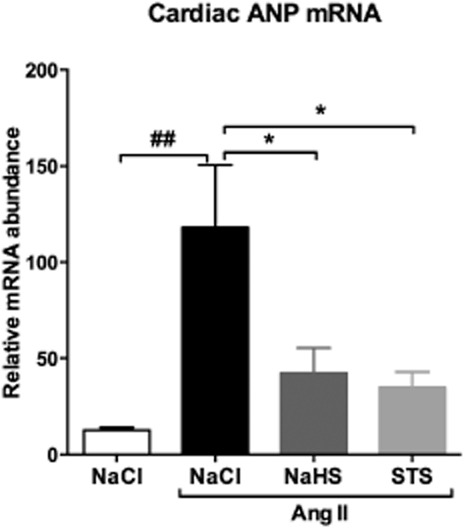
Reduced cardiac mRNA levels of ANP in STS- and NaHS-treated rats. Cardiac mRNA levels of ANP were ninefold increased after 3 weeks of Ang II infusion (n = 7) compared with 0.9% NaCl-infused controls (n = 6). Treatment with STS (n = 7) and NaHS (n = 6) reduced ANP mRNA expression by 70% and 64% respectively. (##P < 0.01 vs. control, *P < 0.05 vs. Ang II + NaCl). ANP mRNA levels were normalized to GAPDH.
Rise in plasma NOx after infusion with Ang II
After 3 weeks of infusion with Ang II, plasma NOx levels were increased by 71% compared with 0.9% NaCl-infused control animals (23.2 ± 1.7 μmol L−1 vs. 13.6 ± 0.3 μmol L−1, P < 0.001). In the STS- and NaHS-treated groups, plasma NOx levels were lower compared with NOx levels in Ang II-infused rats treated with vehicle (STS: 19.0 ± 0. 9 μmol L−1, NaHS: 16.3 ± 1.2 μmol L−1) (Figure 4, Table ).
Figure 4.
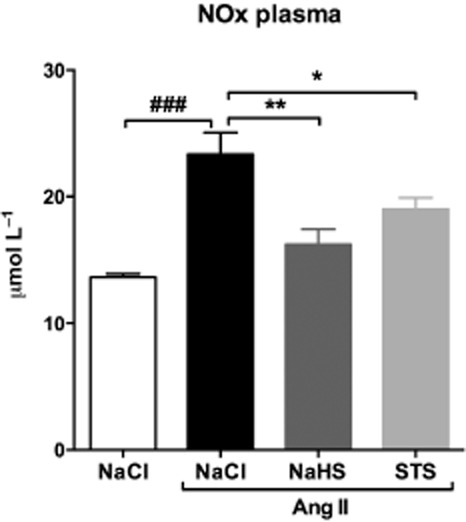
Ang II infusion induces a rise in plasma NOx levels. Plasma NOx levels were increased in Ang II-infused animals treated with vehicle (n = 7). In STS- (n = 7) and NaHS-treated (n = 6) animals, we observed no significant increase in plasma NOx levels. (###P < 0.001 vs. control, **P < 0.01, *P < 0.05 vs. Ang II + NaCl).
Protective effects of STS and NaHS against cardiac fibrosis
Fibrosis, as measured by collagen deposition in Masson stained sections 3 weeks after Ang II infusion, was markedly increased in animals treated with vehicle compared with 0.9% NaCl-infused controls (P < 0.05). Treatment with STS or NaHS reduced collagen deposition by 52% for both compounds (STS: P < 0.01, NaHS: P < 0.05) (Figure 5, Table 1). Cardiac mRNA levels of the profibrotic genes fibronectin and galectin-3 were assayed. Fibronectin was massively increased in Ang II-infused animals treated with vehicle (P < 0.001), while only a moderate increase was detected in animals of the STS- and NaHS-treated groups (Figure 6A, Table 1). Galectin-3 mRNA expression was increased 3.2-fold in hearts of vehicle-treated Ang II-infused rats compared with control (P < 0.001). In the hearts of STS- and NaHS-treated mice, the relative galectin-3 expression was reduced by 29% (P < 0.05) and 44% (P < 0.01) respectively (Figure 6B, Table 1).
Figure 5.
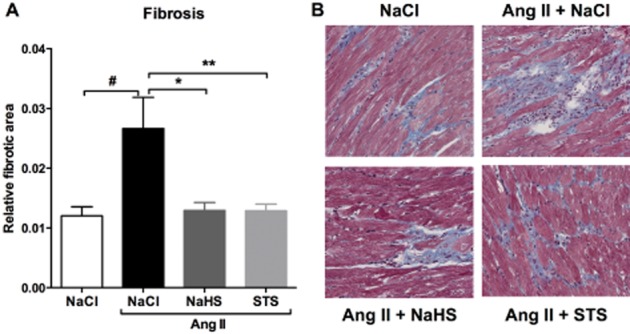
STS and NaHS treatment prevented the development of Ang II-induced cardiac fibrosis. (A) The relative fibrotic area was 2.2-fold increased in hearts of Ang II-infused animals treated with vehicle (n = 7) compared with 0.9% NaCl-infused control animals (n = 6). Treatment with STS (n = 7) and NaHS (n = 6) attenuated the deposition of collagens. (B) Representative photomicrographs of Masson stained cardiac sections. (#P < 0.05 vs. control, **P < 0.01, *P < 0.05 vs. Ang II + NaCl).
Figure 6.
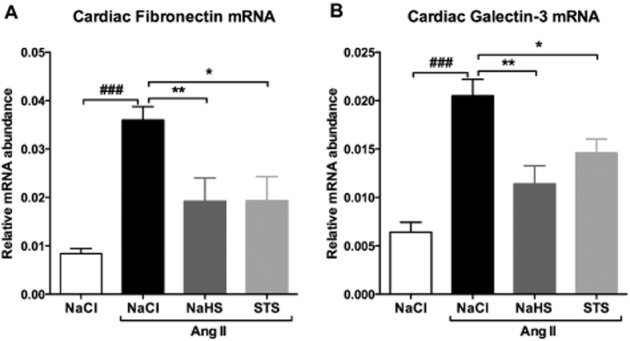
Treatment with STS and NaHS prevents the up-regulation of fibronectin and galectin-3. (A) Fibronectin and (B) galectin-3 mRNA levels were 4.3-fold and 3.2-fold increased in Ang II-infused animals treated with vehicle (n = 7) respectively. In STS- (n = 7) and NaHS-treated (n = 6) animals, no increase in fibronectin and galectin-3 mRNA levels were observed. (###P < 0.001 vs. control, **P < 0.01, *P < 0.05 vs. Ang II + NaCl). Fibronectin and galectin mRNA levels were normalized to GAPDH.
Treatment with STS and NaHS reduces oxidative stress
Cybb (Nox2) is a member of the NADPH oxidase family that is responsible for the formation of reactive oxygen species (ROS). Cybb (Nox2) mRNA expression is increased twofold after Ang II infusion (P < 0.01) (Figure 6A). Also, the urinary excretion of MDA, which is considered a biomarker of oxidative stress, is increased more than twofold in Ang II rats treated with vehicle (P < 0.001) (Figure 6B). Simultaneous treatment with either STS or NaHS resulted in a decrease in Cybb (Nox2) mRNA expression by 30% (P < 0.05) and 40% (P < 0.01), and urinary MDA levels by 30% (P < 0.05) and 35% (P < 0.05) respectively (Figure 7, Table 1).
Figure 7.
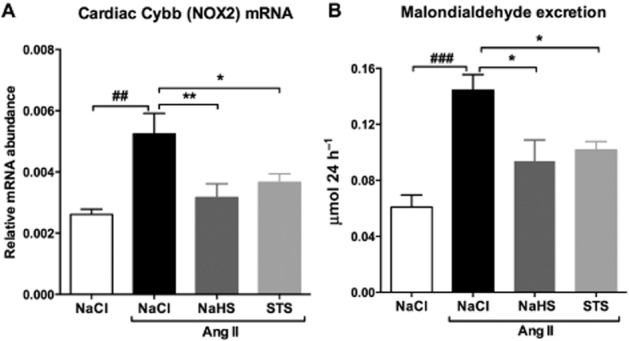
Treatment with NaHS and STS reduces oxidative stress. (A) Cardiac levels of Cybb (Nox2) mRNA and (B) urinary MDA levels were increased in vehicle-treated animals after 3 weeks of Ang II infusion (n = 7). Simultaneous treatment with either STS (n = 7) or NaHS (n = 6) prevented the development of oxidative stress, as evidenced by near-control Cybb (Nox2) mRNA and urinary MDA levels. (###P < 0.001, ##P < 0.01 vs. control, **P < 0.01, *P < 0.05 vs. Ang II + NaCl). Nox2 mRNA levels were normalized to GAPDH.
Unchanged plasma and urinary calcium levels
No significant differences between groups were observed in plasma calcium levels. Urinary excretion of calcium was increased in Ang II-infused rats treated with vehicle compared with 0.9% NaCl-infused controls (P < 0.05). In both STS- and NaHS-treated groups, urinary calcium levels were not different from vehicle-treated Ang II-infused animals (Figure 8, Table 1).
Figure 8.
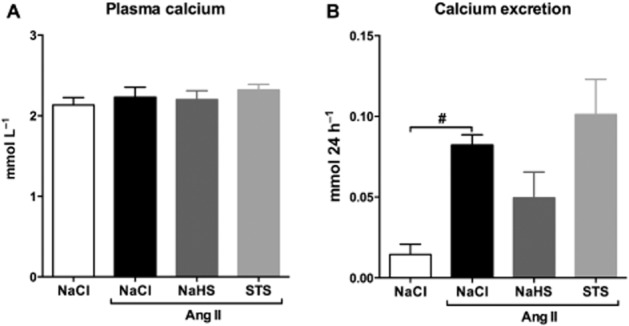
Unchanged urinary calcium levels upon STS treatment. (A) Plasma calcium levels were unaltered upon infusion with Ang II (n = 7) and simultaneous treatment with NaHS (n = 6) and STS (n = 7). (B) After 3 weeks of Ang II infusion (n = 7), the calcium excretion was increased compared with controls (n = 6). Urinary calcium levels from STS- (n = 7) and NaHS-treated (n = 6) animals were not different from Ang II-infused animals treated with vehicle. (#P < 0.05 vs. control).
Discussion
In the present study, we demonstrated that exogenous administration of STS and NaHS attenuates the development of hypertension and associated hypertensive heart disease. BP reduction is the probable mechanism of action, but secondary effects such as reducing oxidative stress and fibrosis might also play a role. Furthermore, we are the first to show in a hypertensive rat model that the clinically applicable compound STS has promising cardioprotective properties.
For the administration of H2S in experimental models, soluble sulfide salts are often used, while occasionally gaseous H2S is administered. Recently, slow-release H2S donors such as GYY4137 are becoming more popular. TS is an old player in the field, but recently rediscovered as a H2S producing substance. TS is an intermediate of sulfur metabolism and a metabolite of H2S that can also lead to the production of H2S (Ubuka et al., 2001; Olson et al., 2013). In this study, we used STS because this is a non-toxic substance that is associated with the production of H2S. A recent study showed that inhalation of H2S markedly increased TS levels during endotoxaemia in mice. In addition, administration of STS dose-dependently improved survival after LPS challenge in mice (Tokuda et al., 2012). The dosage of STS used in this study is based on previous experimental studies (Asplin et al., 2009; Tokuda et al., 2012; Shirozu et al., 2014) and STS usage in humans (Cicone et al., 2004; Farese et al., 2011). The dosage range of STS in experimental studies is 0.5–2 g kg−1 given daily, three times a week; in humans even 12.5 and 25 g of STS have been infused without adverse effects. In humans, the effect of i.v. STS infusion on plasma TS concentrations has been studied (Farese et al., 2011). In our rat study, we did not measure plasma TS concentrations, to our knowledge there are no studies available that have done so in rats. It has to be taken into account that in our rat model, STS was given i.p. and not i.v., the kinetics of this route is not known. The clinical use of H2S has not been attained, but STS has been used in clinical practice for decades. To date, short-term treatment with i.v. STS is safely used in patients for the treatment of calciphylaxis (Singh et al., 2011). However, one of the disadvantages for long-term administration might be the route of delivery, as orally administrated STS is quickly degraded in the stomach and a validated oral form of the compound has not been developed yet. Furthermore, the long-term effects of STS administration should be further explored.
A substantial body of literature demonstrates the cardioprotective effects of H2S. Many of these studies focused on the beneficial effects of H2S in acute myocardial infarction induced by coronary artery ligation (Szabo et al., 2011). There is only one study describing beneficial effects of STS in a hypotensive high-cardiac output model (Sen et al., 2008). In humans, the effects of STS on BP have not been demonstrated extensively; however, there are indications that STS exhibits vasodilating effects (Thomas and McGinnis, 2002; Hayden et al., 2005; Hayden and Goldsmith, 2010). We observed that STS has similar protective effects as NaHS thereby providing us with exciting possibilities for the translation into clinical use.
The reduction in BP observed in this study is in line with previous findings showing the vasorelaxing properties of H2S. Homozygous CSE-knockout and heterozygous CBS-deficient mice both develop hypertension (Yang et al., 2008; Sen et al., 2010), suggesting a role for endogenous H2S production in the pathogenesis of hypertension. Furthermore, exogenous administration of H2S causes vasodilatation in experimental models of hypertension (Zhong et al., 2003; Mustafa et al., 2011; Roy et al., 2012). A proposed mechanism is the activation of KATP channels through sulfhydration (Mustafa et al., 2011), but it is also suggested that the increased bioavailability of NO might play a role in the vasodilator effects of H2S (Zhao and Wang, 2002). However, we observed that the antihypertensive effect of H2S was not accompanied by an increase in plasma NOx as compared with Ang II-infused animals suggesting that the vasodilator effects of H2S are not related to an increased bioavailability of NO in our model. The increased plasma levels of NOx in the Ang II-infused animals treated with vehicle might be compensatory and due to the high BP. The ability of H2S to reduce renin and the activity of ACE also contributes to its BP reducing effects (Lu et al., 2010). However, the contribution of this mechanism in this particular model is probably low or undetectable because of continuous Ang II infusion and its negative feedback effect on the production of renin.
In our model, the prevention of hypertension by STS and NaHS is probably the basis of the attenuation of cardiac hypertrophy. In the STS- and NaHS-treated groups, we observed a concordant smaller cardiomyocyte size and a lower heart-to-body weight ratio compared with untreated animals. The lower expression of ANP mRNA in these groups further supports the unloading of the heart. In overload-induced models of heart failure, treatment with H2S attenuated the development of hypertrophy. Conversely, CSE-knockout mice with little endogenous H2S production showed an increase in cardiac hypertrophy compared with wild-type mice (Kondo et al., 2013).
Although we postulate that BP regulation is the primary mechanism of action in our model, it is known that Ang II causes damage independent of an elevated BP (Mervaala et al., 2000). This enables additional protective modes of action of H2S. We observed that treatment with H2S influences cardiac fibrotic pathways as evidenced by a reduction in fibronectin mRNA and Masson-positive surface area. It is not certain if H2S has any direct effects in fibrosis. Recent studies have demonstrated a potential link between the Ang II-associated fibrotic response and NADPH oxidases in cardiac fibroblasts (Lassegue and Griendling, 2010). Therefore, the antifibrotic effects of H2S might be related to the reduction in Cybb (Nox2) we observed in this study. However, we also observed a reduction in galectin-3 mRNA, a protein with established fibrotic properties (de Boer et al., 2009). Recently, it was shown that genetic and pharmacological inhibition of galectin-3 prevents cardiac remodelling by interfering with myocardial fibrogenesis (Yu et al., 2013). Ang II-induced cardiac fibrosis was significantly decreased in galectin-3 knockout mice (Yu et al., 2013).
Ang II induces oxidative stress by activating NADPH oxidases via the angiotensin-1 receptor (Agarwal et al., 2004). H2S is a known antioxidant and can directly scavenge ROS, increase intracellular glutathione levels and reduce the amount of ROS produced through modulation of mitochondrial ROS production (Kimura and Kimura, 2004; Kimura et al., 2010; Bos et al., 2013). In concordance with previous findings, we observed that treatment with NaHS reduced oxidative stress as evidenced by low Cybb (Nox2) mRNA and urinary MDA levels. These antioxidant effects may also have contributed to the protective effects of STS and NaHS in this study. The production of ROS because of the abundant presence of Ang II causes endothelial dysfunction and can therefore also contribute to the development of hypertension (Rathaus and Bernheim, 2002). Accordingly, treatment with antioxidants is able to reduce BP in experimental models of hypertension (Mullan et al., 2002; Racasan et al., 2004).
From our data, we are unable to deter whether the protective effects of STS can be completely attributed to the conversion of H2S or if there are direct effects of STS. One of the proposed direct effects of STS is calcium chelation based on the fact that there are studies showing that calcium chelation inhibits some effects of Ang II (Brinson et al., 1998; Du et al., 1999). However, in contrast to what has been published repeatedly, other studies show that STS is not a strong calcium chelator (Pasch et al., 2008; O'Neill and Hardcastle, 2012). Our data indicate that calcium chelation did not play a major role in the protective effects of STS in this model. However, the calcium excretion measurements (Figure 8B) are possibly underpowered, so we must be careful in drawing firm conclusions from these data.
In this model, the beneficial effects are probably related to a reduction in BP, but also the attenuation of oxidative stress and fibrosis may contribute to the protection against cardiac remodelling. We have extended these findings by demonstrating that STS has marked protective properties, which brings us closer to translation into clinical use. Whether the protective effects of STS are solely mediated by the conversion to H2S or by additional direct effects of STS is currently not known. H2S is a promising agent in the battle against hypertensive heart disease and might be a valuable addition to the already existing cardioprotective therapies.
Acknowledgments
The authors would like to express their gratitude towards Sippie Huitema, Marian Bulthuis, Pieter Klok, Petra Ottens, Jacco Zwaagstra and Martin Dokter for their excellent technical support. This work was supported by two grants from the Dutch Kidney Foundation (NSN C08–2254 and IP13–114). The study was elaborated with COST Action BM1005 (European Network on Gasotransmitters).
Glossary
- Ang II
angiotensin II
- ANP
atrial natriuretic peptide
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- Nox2
NADPH oxidase 2 (also known as Cybb, cytochrome b (558) subunit β)
- DBP
diastolic BP
- H2S
hydrogen sulfide
- KATP channels
ATP-sensitive potassium channels
- MDA
malondialdehyde
- NaHS
sodium hydrosulfide
- NOx
nitrite and nitrate
- RAAS
renin-angiotensin-aldosterone system
- ROS
reactive oxygen species
- (S)TS
(sodium) thiosulfate (Na2S2O3)
- SBP
systolic BP
- WGA
wheat germ agglutinin
Author contributions
P. M. S., A. R. F., R. A. d. B., A. P., J. L. H., H. G. D. L. and H. v. G. designed the research; P. M. S., A. R. F., H. G. D. L. and H. v. G. performed the research; R. A. d. B., A. P. and J. L. H. contributed the new reagents/analytic tools; P. M. S., A. R. F., R. A. d. B., A. P., J. L. H., H. G. D. L. and H. v. G. analysed the data; P. M. S., A. R. F. and H. v. G. wrote the paper.
Conflicts of interest
A. P. has support from an unrestricted research grant provided from Köhler Chemie. The other authors declare no conflicts of interest.
References
- Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal R, Campbell RC, Warnock DG. Oxidative stress in hypertension and chronic kidney disease: role of angiotensin II. Semin Nephrol. 2004;24:101–114. doi: 10.1016/j.semnephrol.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013b;170:1607–1646. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asplin JR, Donahue SE, Lindeman C, Michalenka A, Strutz KL, Bushinsky DA. Thiosulfate reduces calcium phosphate nephrolithiasis. J Am Soc Nephrol. 2009;20:1246–1253. doi: 10.1681/ASN.2008070754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer RA, Voors AA, Muntendam P, van Gilst WH, van Veldhuisen DJ. Galectin-3: a novel mediator of heart failure development and progression. Eur J Heart Fail. 2009;11:811–817. doi: 10.1093/eurjhf/hfp097. [DOI] [PubMed] [Google Scholar]
- Bos EM, Wang R, Snijder PM, Boersema M, Damman J, Fu M, et al. Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J Am Soc Nephrol. 2013;24:759–770. doi: 10.1681/ASN.2012030268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinson AE, Harding T, Diliberto PA, He Y, Li X, Hunter D, et al. Regulation of a calcium-dependent tyrosine kinase in vascular smooth muscle cells by angiotensin II and platelet-derived growth factor. Dependence on calcium and the actin cytoskeleton. J Biol Chem. 1998;273:1711–1718. doi: 10.1074/jbc.273.3.1711. [DOI] [PubMed] [Google Scholar]
- Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43:1104–1108. doi: 10.1053/j.ajkd.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–334. doi: 10.1161/CIRCULATIONAHA.108.845792. [DOI] [PubMed] [Google Scholar]
- Du J, Peng T, Scheidegger KJ, Delafontaine P. Angiotensin II activation of insulin-like growth factor 1 receptor transcription is mediated by a tyrosine kinase-dependent redox-sensitive mechanism. Arterioscler Thromb Vasc Biol. 1999;19:2119–2126. doi: 10.1161/01.atv.19.9.2119. [DOI] [PubMed] [Google Scholar]
- Farese S, Stauffer E, Kalicki R, Hildebrandt T, Frey BM, Frey FJ, et al. Sodium thiosulfate pharmacokinetics in hemodialysis patients and healthy volunteers. Clin J Am Soc Nephrol. 2011;6:1447–1455. doi: 10.2215/CJN.10241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258–262. doi: 10.1111/j.1525-139X.2010.00738.x. [DOI] [PubMed] [Google Scholar]
- Hayden MR, Tyagi SC, Kolb L, Sowers JR, Khanna R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: the emerging role of sodium thiosulfate. Cardiovasc Diabetol. 2005;4:4. doi: 10.1186/1475-2840-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt TM, Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- Jia L, Li Y, Xiao C, Du J. Angiotensin II induces inflammation leading to cardiac remodeling. Front Biosci (Landmark Ed) 2012;17:221–231. doi: 10.2741/3923. [DOI] [PubMed] [Google Scholar]
- Jiang HL, Wu HC, Li ZL, Geng B, Tang CS. Changes of the new gaseous transmitter H2S in patients with coronary heart disease. Di Yi Jun Yi Da Xue Xue Bao. 2005;25:951–954. [PubMed] [Google Scholar]
- Kabil O, Banerjee R. Redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- Koitabashi N, Kass DA. Reverse remodeling in heart failure–mechanisms and therapeutic opportunities. Nat Rev Cardiol. 2011;9:147–157. doi: 10.1038/nrcardio.2011.172. [DOI] [PubMed] [Google Scholar]
- Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, et al. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic D, Glavnik N, Marinsek M, Zagozen P, Rovan K, Goslar T, et al. Total plasma sulfide in congestive heart failure. J Card Fail. 2012;18:541–548. doi: 10.1016/j.cardfail.2012.04.011. [DOI] [PubMed] [Google Scholar]
- Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Liu YH, Goh HS, Wang JJ, Yong QC, Wang R, et al. Hydrogen sulfide inhibits plasma renin activity. J Am Soc Nephrol. 2010;21:993–1002. doi: 10.1681/ASN.2009090949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- Mervaala E, Muller DN, Schmidt F, Park JK, Gross V, Bader M, et al. Blood pressure-independent effects in rats with human renin and angiotensinogen genes. Hypertension. 2000;35:587–594. doi: 10.1161/01.hyp.35.2.587. [DOI] [PubMed] [Google Scholar]
- Mullan BA, Young IS, Fee H, McCance DR. Ascorbic acid reduces blood pressure and arterial stiffness in type 2 diabetes. Hypertension. 2002;40:804–809. doi: 10.1161/01.hyp.0000039961.13718.00. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–1268. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR, Deleon ER, Gao Y, Hurley K, Sadauskas V, Batz C, et al. Thiosulfate: a readily accessible source of hydrogen sulfide in oxygen sensing. Am J Physiol Regul Integr Comp Physiol. 2013;305:592–603. doi: 10.1152/ajpregu.00421.2012. 10.1152/ajpregu.00421. [DOI] [PubMed] [Google Scholar]
- O'Neill WC, Hardcastle KI. The chemistry of thiosulfate and vascular calcification. Nephrol Dial Transplant. 2012;27:521–526. doi: 10.1093/ndt/gfr375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasch A, Schaffner T, Huynh-Do U, Frey BM, Frey FJ, Farese S. Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney Int. 2008;74:1444–1453. doi: 10.1038/ki.2008.455. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racasan S, Braam B, van der Giezen DM, Goldschmeding R, Boer P, Koomans HA, et al. Perinatal L-arginine and antioxidant supplements reduce adult blood pressure in spontaneously hypertensive rats. Hypertension. 2004;44:83–88. doi: 10.1161/01.HYP.0000133251.40322.20. [DOI] [PubMed] [Google Scholar]
- Rathaus M, Bernheim J. Oxygen species in the microvascular environment: regulation of vascular tone and the development of hypertension. Nephrol Dial Transplant. 2002;17:216–221. doi: 10.1093/ndt/17.2.216. [DOI] [PubMed] [Google Scholar]
- Roy A, Khan AH, Islam MT, Prieto MC, Majid DS. Interdependency of cystathione gamma-lyase and cystathione beta-synthase in hydrogen sulfide-induced blood pressure regulation in rats. Am J Hypertens. 2012;25:74–81. doi: 10.1038/ajh.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen U, Vacek TP, Hughes WM, Kumar M, Moshal KS, Tyagi N, et al. Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology. 2008;82:201–213. doi: 10.1159/000156486. [DOI] [PubMed] [Google Scholar]
- Sen U, Munjal C, Qipshidze N, Abe O, Gargoum R, Tyagi SC. Hydrogen sulfide regulates homocysteine-mediated glomerulosclerosis. Am J Nephrol. 2010;31:442–455. doi: 10.1159/000296717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbaz AU, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, McGee JE, et al. Fibrosis in hypertensive heart disease: molecular pathways and cardioprotective strategies. J Hypertens. 2010;28(Suppl. 1):S25–S32. doi: 10.1097/01.hjh.0000388491.35836.d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, et al. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun. 2013;4:1366. doi: 10.1038/ncomms2371. [DOI] [PubMed] [Google Scholar]
- Shirozu K, Tokuda K, Marutani E, Lefer D, Wang R, Ichinose F. Cystathionine γ-lyase deficiency protects mice from galactosamine/lipopolysaccharide-induced acute liver failure. Antioxid Redox Signal. 2014;20:204–216. doi: 10.1089/ars.2013.5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RP, Derendorf H, Ross EA. Simulation-based sodium thiosulfate dosing strategies for the treatment of calciphylaxis. Clin J Am Soc Nephrol. 2011;6:1155–1159. doi: 10.2215/CJN.09671010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem. 2009;284:22457–22466. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder PM, de Boer RA, Bos EM, van den Born JC, Ruifrok WP, Vreeswijk-Baudoin I, et al. Gaseous hydrogen sulfide protects against myocardial ischemia-reperfusion injury in mice partially independent from hypometabolism. PLoS ONE. 2013;8:e63291. doi: 10.1371/journal.pone.0063291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Veres G, Radovits T, Gero D, Modis K, Miesel-Groschel C, et al. Cardioprotective effects of hydrogen sulfide. Nitric Oxide. 2011;25:201–210. doi: 10.1016/j.niox.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JE, McGinnis G. Safety of intraventricular sodium nitroprusside and thiosulfate for the treatment of cerebral vasospasm in the intensive care unit setting. Stroke. 2002;33:486–492. doi: 10.1161/hs0202.103410. [DOI] [PubMed] [Google Scholar]
- Tokuda K, Kida K, Marutani E, Crimi E, Bougaki M, Khatri A, et al. Inhaled hydrogen sulfide prevents endotoxin-induced systemic inflammation and improves survival by altering sulfide metabolism in mice. Antioxid Redox Signal. 2012;17:11–21. doi: 10.1089/ars.2011.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubuka T, Abe T, Kajikawa R, Morino K. Determination of hydrogen sulfide and acid-labile sulfur in animal tissues by gas chromatography and ion chromatography. J Chromatogr B Biomed Sci Appl. 2001;757:31–37. doi: 10.1016/s0378-4347(01)00046-9. [DOI] [PubMed] [Google Scholar]
- Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Ruifrok WP, Meissner M, Bos EM, van Goor H, Sanjabi B, et al. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ Heart Fail. 2013;6:107–117. doi: 10.1161/CIRCHEARTFAILURE.112.971168. [DOI] [PubMed] [Google Scholar]
- Zhao W, Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol. 2002;283:H474–H480. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]
- Zhao X, Zhang LK, Zhang CY, Zeng XJ, Yan H, Jin HF, et al. Regulatory effect of hydrogen sulfide on vascular collagen content in spontaneously hypertensive rats. Hypertens Res. 2008;31:1619–1630. doi: 10.1291/hypres.31.1619. [DOI] [PubMed] [Google Scholar]
- Zhong G, Chen F, Cheng Y, Tang C, Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens. 2003;21:1879–1885. doi: 10.1097/00004872-200310000-00015. [DOI] [PubMed] [Google Scholar]